
6
Reporting Foodborne Threats: The Case of Bovine Spongiform Encephalopathy (BSE)
OVERVIEW
The rapid reporting of foodborne threats is essential to reducing the burden of foodborne illness, but it also carries direct and indirect costs to individuals, communities, industries, and national economies. The balance of costs and benefits associated with reporting foodborne threats is clearly illustrated by the world’s recent and ongoing experience with bovine spongiform encephalopathy (BSE, or mad cow disease). A series of workshop presentations by contributors to this chapter explored the biology of BSE and its implications for food safety, international perspectives on BSE surveillance and prevention, and public health lessons learned from this disease and its consequences.
A member of the family of diseases known as transmissible spongiform encephalopathies (TSEs; also known as prion diseases), BSE was first identified in 1986 in the United Kingdom and has since been detected in 26 countries (GAO, 2005). In the early 1980s, Stanley Prusiner, the author of this chapter’s first paper, proposed that the pathogens that cause two TSEs—Creutzfeldt-Jakob disease (CJD) and scrapie, a disease of sheep—consist entirely of an infectious form of protein that he termed the prion; in 1997, he was awarded the Nobel Prize in Physiology and Medicine for his work on prion biology. Researchers have since learned that in addition to scrapie and CJD, prions cause BSE and its human variant, vCJD, as well as chronic wasting disease in deer and elk.
Prusiner presents experimental evidence for the prion model of TSE and describes the etiology and diagnosis of vCJD and other human prion diseases. He emphasizes the differences between prion and viral illnesses—most notably, that prions can arise spontaneously—and observes that the mistaken equation of the
two can impede the development of effective preventions against invariably fatal prion diseases. “The only rational strategy is to test all cattle for prions and eliminate those harboring prions from the food supply,” Prusiner concludes.
The second paper, by Steven Collins, codirector of the Australian National CJD Registry, describes his country’s approach to TSE surveillance in both animals and humans. The vast majority of CJD cases in the Australian Registry are sporadic; no cases of vCJD or endogenous cases of either BSE or scrapie have been reported to date in the country. Collins recounts the range of measures Australia has adopted to protect commercial livestock from BSE and scrapie, which include bans on the importation of meat and bone meal and of live cattle from any country reporting BSE, as well as the prohibition of ruminant-to-ruminant feeding of meat and bone meal.
Maura Ricketts, who has directed prion disease surveillance and research for Health Canada and the World Health Organization (WHO), discusses BSE and vCJD from a public health perspective in the chapter’s third paper. After defining and applying relevant core principles of public health, Ricketts identifies and explores key issues that underlie the development and implementation of health policy to address BSE and vCJD. She concludes with an overview of possible public health actions that reflect the importance of controlling the risk of BSE exposure.
In the chapter’s final contribution, Wil Hueston of the Center for Animal Health and Food Safety at the University of Minnesota shares insights gained from 16 years of involvement with BSE and the interface between animal and human health. He distills this experience into seven lessons that lead to a series of key actions that could be taken to address key issues raised by BSE and, more generally, to improve the response to infectious disease.
PRIONS AND THE SAFETY OF THE FOOD SUPPLY
Stanley B. Prusiner, M.D.1
University of California San Francisco
In December 2003, mad cow disease made its U.S. debut when federal officials announced that a Holstein cow from Mabton, Washington, had been stricken with bovine spongiform encephalopathy (BSE). Although the U.S. government acted surprised by the finding, they should have expected such cattle based on the biology of the prion diseases. Perhaps the novel principles of disease that have emerged over the past two decades from investigations of prions (Prusiner, 2004b) are still too new and different for many people to grasp easily the implications of this discovery. Prions are unprecedented infectious pathogens that are composed
solely of protein; they are devoid of nucleic acid—DNA and RNA. The absence of a nucleic genome sets prions apart from all other infectious pathogens including viruses, viroids, bacteria, fungi, and parasites.
Because prions multiply and cause disease in the host, scientists thought for many years that prions must be slow-acting viruses. Moreover, the identification of varieties or strains of prions made the argument that prions must be viruses even more appealing. Because so many attempts to detect a prion-specific nucleic acid genome failed (Alper et al., 1967; Bellinger-Kawahara et al., 1987a,b; Safar et al., 2005b), some scientists argued that the nucleic acid must be quite small (Bruce and Dickinson, 1987; Chesebro, 2004; Kimberlin, 1982; Weissmann, 1991). Although the identification of micro RNAs has given new life to such arguments in recent years, the production of synthetic prions from recombinant prion protein (PrP) is likely to end the quest for an auxiliary molecule within the prion (Legname et al., 2004, 2005).
Based on a wealth of data including the production of synthetic prions in mammals and fungi, it is reasonable to define prions as infectious proteins. Prions multiply by forcing the precursor protein to acquire a second conformation. Different conformations of proteins in the prion state encipher distinct strains and are prone to aggregation. In mammals, prions accumulate to high levels in the nervous system where they cause dysfunction and fatal degeneration.
Both mammalian and fungal prions have been produced in cell-free systems (Brachmann et al., 2005; Maddelein et al., 2002; Sparrer et al., 2000; Tanaka et al., 2004). Synthetic PrP peptides and recombinant PrP fragments have been used to form mammalian prions, and N-terminal regions, called prion domains, that are rich in glutamine and asparagine have been used to form fungal prions.
In mammals, prions cause a group of invariably fatal, neurodegenerative diseases. No human or animal has ever recovered from a prion disease once neurologic dysfunction has manifested. Prion diseases may present as genetic, infectious, or sporadic disorders, all of which involve modification of normal, cellular PrP, designated PrPC. The tertiary structure of PrP is profoundly altered as prions are formed, and as such, prion diseases represent disorders of protein conformation. In the sporadic and genetic forms of prion disease, prions arise spontaneously. In contrast, infectious prion diseases result from exposure to an exogenous source of prions. Although the incidence of sporadic prion disease in humans is low (1–5 cases per 106 people), it is the most common form, accounting for approximately 90 percent of all cases. The genetic, or inherited, forms of prion disease account for approximately 10 percent of all human cases and the infectious forms for less than 1 percent. Whether or not the infectious forms of human prion disease are underestimated and low levels of animal prions in the food supply are responsible for 10–20 percent of the sporadic cases is unknown.
The prion diseases in humans include CJD, which generally presents as a progressive dementia, as well as kuru and Gerstmann-Sträussler-Scheinker disease (GSS), both of which frequently present as ataxic maladies. Like kuru and
GSS of humans, scrapie of sheep and BSE of cattle usually manifest as ataxic illnesses. Deer, elk, and moose with chronic wasting disease (CWD) appear emaciated and ataxic.
In these diseases, mammalian PrPC is recruited and converted into the disease-causing isoform (PrPSc). PrPC has a high -helical content and little -sheet structure, whereas PrPSc has less -helical structure and a high -sheet content. Comparisons of secondary structures of PrPC and PrPSc were performed on proteins purified from Syrian hamster (SHa) brains (Pan et al., 1993). Limited proteolysis of PrPSc produces a protease-resistant core, designated PrP 27–30, which retains prion infectivity; under these conditions, PrPC is completely hydrolyzed.
Prion Disease Paradigm
Despite some similarities between prion and viral illnesses, these disorders are very different. Viral diseases are infectious illnesses that begin with infection by exogenous virions. In contrast, the vast majority of prion diseases are initiated from within the host, in which prions arise spontaneously. Often the term prion infection is used synonymously with prion disease because once prions are formed spontaneously they can be transferred to another host and thus, are infectious. Unlike viral infections, no host defenses are mounted in response to prion infection: no humoral immunity, no cellular immunity, and no interferons are elicited to the replicating prion.
Molecular genetic studies have been crucial in deciphering the novel features of the prion disease paradigm. In the sporadic form of prion disease, the sequence of the PrP gene is wild-type (wt); whereas, in the inherited prion diseases, the sequence of the PrP gene harbors a nonconservative substitution or insertion. Generally, the PrP genes of humans and animals with infectious prion disease are wt.
Before the discovery of mutations in the PrP gene as the cause of familial prion disease, geographic clusters of prion disease were thought to be due to common source exposures to exogenous prions. For example, Libyan Jews with a very high incidence of CJD were thought to have contracted the disease by eating lightly cooked sheep brains (Alter and Kahana, 1976). Molecular genetic investigations showed that every Libyan Jew developing prion disease carried a PrP gene mutation resulting in an EK substitution at position 200 (Goldfarb et al., 1991; Hsiao et al., 1991). Risk analysis studies revealed that every Libyan Jew carrying the E200K mutation would eventually develop prion disease if he or she did not die of some other illness (Chapman et al., 1994; Spudich et al., 1995).
Synthetic Prions and Spontaneous Disease
Investigations of humans with PrP gene mutations were extended to transgenic (Tg) mice harboring the analogous mutation causing GSS in humans. Tg
mice expressing high levels of MoPrP(P101L) developed neurodegeneration spontaneously (Hsiao et al., 1990; Telling et al., 1996). Extracts prepared from the brains of these mice transmitted disease after approximately 250 days to other Tg mice (designated Tg196) expressing low levels of MoPrP(P101L) (Hsiao, 1994). Subsequently, a synthetic PrP peptide of 55 residues carrying the P101L mutation, designated MoPrP(89–143,P101L), was produced and inoculated into the Tg196 mice (Kaneko et al., 2000). The Tg196 mice developed central nervous system (CNS) dysfunction approximately one year after inoculation, and brain extracts from the ill mice were found to produce disease on serial passage (Tremblay et al., 2004). The MoPrP(89–143, P101L) peptide produced disease in the Tg196 mice only if it was folded into a -rich conformation (Kaneko et al., 2000).
An approach similar to the one used in the studies with MoPrP(89– 143,P101L) peptide was employed with wt PrP. In those studies, wt MoPrP(89– 230) was produced in E. coli, purified by chromatography, and polymerized into amyloid fibrils (Legname et al., 2004). The amyloid fibrils were injected into Tg mice expressing MoPrP(89–231) and produced neurodegeneration after approximately 500 days. Brain extracts from the ill Tg mice contained protease-resistant PrPSc and produced disease on subsequent passage into both wt and Tg mice (Legname et al., 2005). These studies demonstrated that only PrP is required to generate prion infectivity, and as such, spontaneous forms of prion disease can occur in any mammal as PrPC seems to be ubiquitous among this class of vertebrates.
Spontaneous prion disease contrasts with viral disorders, for which exogenous infection is required except in the case of latent retroviral genomes. For example, after infection with exogenous HIV, the virus may disappear but often its RNA genome has been reverse-transcribed into DNA, and the DNA copies may remain dormant for years.
The dramatically different principles that govern prion biology from those underpinning the viral diseases are frequently misunderstood. This lack of understanding has led to some regrettable decisions of great economic, political, and possibly public health importance. For example, scrapie and BSE have different names, yet they are the same disease in two different species. Scrapie and BSE differ in only two respects: first, the PrP sequence in sheep differs from that of cattle at seven or eight positions of 270 amino acids (Goldmann et al., 1990, 1991), which results in different PrPSc molecules. Second, most scrapie strains of prions seem to be different from the BSE strains.
The Mad Cow Epidemic
The world awoke to the dangers of prion disease in cows after the BSE outbreak began ravaging the British beef industry in the mid-1980s. The truly novel concepts emerging from prion science forced researchers and society to think in
unusual ways and made coping with the epidemic difficult. Investigators eventually learned that prions were being transmitted to cattle through meat-and-bone meal (MBM), a dietary supplement prepared from the parts of sheep, cattle, pigs and chickens that are processed, or rendered, for industrial use. High heat eliminated conventional pathogens, but PrPSc survived and went on to infect cattle.
As infected cattle became food for other cattle, BSE began appearing throughout the UK cattle population, reaching a high of 37,280 confirmed fied cases in 1992 (Phillips, 2000). The British authorities instituted some feed bans beginning in 1989, but it was not until 1996 that a strict ban on cannibalistic feeding finally brought BSE under control in the United Kingdom; the country saw 612 cases in 2004. Overall, the United Kingdom has identified approximately 180,000 mad cows, and epidemiologic models suggest that another 1.9 million were infected but went undetected (Anderson et al., 1996).
For many people, the regulations came too late. Despite the British government’s early assurances to the contrary, mad cow disease proved transmissible to humans. In March 1996, Robert Will and his colleagues reported that 11 British teenagers and young adults had died of a variant of Creutzfeldt-Jakob disease (vCJD) (Will et al., 2004, 1996). In these young patients, the patterns of PrPSc deposition in the brain differed markedly from that found in typical CJD patients.
Many scientists, including myself, were initially dubious of the presumed link between BSE and vCJD. I eventually changed my mind, under the weight of many studies. The most compelling of these studies used Tg mice genetically engineered to resemble cattle, at least from a PrP point of view. These mice became ill approximately 250 days after receiving injections of prions either from cattle with BSE or people with vCJD, and the resulting disease looked the same whether the prions originated from diseased cows or vCJD patients (Scott et al., 1999).
Since the detection of mad cow disease in the United Kingdom, two dozen other nations have uncovered cases. Canada and the United States are the latest entrants to the list of countries affected. On May 20, 2003, Canadian officials reported BSE in an eight-year-old cow that had spent its life in Alberta and Saskatchewan. (The country’s only previous mad cow had arrived as a UK import 10 years earlier.) Although the animal had been slaughtered in January 2003, slow processing meant that officials did not test the cow remains until April. By then, the carcass had been turned into pet food and exported to the United States.
Seven months later, on December 23, 2003, the U.S. Department of Agriculture (USDA) announced the country’s first case of BSE in Washington state. The six-year-old dairy cow had entered the United States at the age of four. The discovery meant that U.S. officials could no longer labor under the misconception that the nation is free of BSE. Like Canada, U.S. agricultural interests want the BSE problem to disappear. Financial woes stem primarily from reduced beef exports: 58 other countries are keeping their borders shut, and a $3 billion export
market has largely evaporated. At the time of writing, six more cases of BSE in Canada and two additional cases in the United States have been reported.
Infectious Human Prion Diseases
Prions from different sources have infected humans. Human prions have been transmitted to others both by ritualistic cannibalism and iatrogenic means. Kuru in the highlands of New Guinea was transmitted by ritualistic cannibalism, as people in the region attempted to immortalize their dead relatives by eating their brains (Alpers, 1968; Gajdusek, 1977; Glasse, 1967). Iatrogenic transmissions include prion-tainted human growth hormone (HGH) and gonadotropin, dura mater grafts, and corneal transplants from people who died of CJD. In addition, CJD cases have been recorded after neurosurgical procedures in which ineffectively sterilized depth electrodes or instruments were used.
Variant Creutzfeldt–Jakob Disease (vCJD)
The first cases of vCJD in teenagers and young adults were identified in Great Britain in 1994 (Will et al., 1996). More than 170 teenagers and young adults have died of vCJD in Britain, France, Ireland, Italy, Japan, Portugal, and the United States. Although the average age of vCJD patients is 26 years of age, the youngest patient was 12 years old and the oldest was 74 years of age (Spencer et al., 2002). The median duration of the illness is 13 months, with the range from 6 to 69 months.
In addition to the young age of these patients (Bateman et al.,1995; Britton et al., 1995), vCJD is characterized by numerous PrP amyloid plaques surrounded by a halo of intense spongiform degeneration in the brain (Ironside, 1997). These unusual neuropathologic changes have not been seen in CJD cases in the United States, Australia, or Japan (CDC, 1996; Ironside, 1997). Both macaque monkeys and marmosets developed neurologic disease several years after inoculation with bovine prions (Baker et al., 1993), but only the macaques exhibited numerous PrP plaques similar to those found in vCJD (Lasmézas et al., 1996).
The majority of vCJD patients present with psychiatric symptoms, including dysphoria, withdrawal, anxiety, insomnia, and loss of interest (Spencer et al., 2002; Will et al., 2004). Generally, neurologic deficits do not appear until at least four months later; these neurologic changes consist of memory loss, paresthesias, sensory deficits, gait disturbances, and dysarthria. Most vCJD cases have been reported from Britain, and 10 have been found in France. The one U.S. case was a 23-year-old woman, who is thought to have been exposed to bovine prions while living in Britain during the first 12 years of her life. From both epidemiologic and experimental studies, the evidence is quite compelling that vCJD is the result of prions being transmitted from cattle with BSE to humans through consumption of prion-contaminated beef products.
Transmission of vCJD Prions by Blood Transfusion
vCJD has been identified in two patients who received blood transfusions from donors that later died of vCJD. In one case, the recipient was a 69-year-old male who was transfused 6.5 years before the onset of neurologic dysfunction (Llewelyn et al., 2004). Many details of the second case are not published, but the patient is known to have died of a nonneurologic disease (Peden et al., 2004). Although vCJD prions were found in the spleen and cervical lymph nodes of this patient, none were found in the brain.
A glimpse of future vCJD cases caused by prion-tainted transfused blood may come from a survey of tissues collected during appendectomies and tonsillectomies. Such a survey from the United Kingdom reports that of the 12,674 appendectomy specimens examined, three were positive for PrPSc by immunohistochemistry (IHC) (Hilton et al., 2004). This finding argues that as many as 3,800 people in the United Kingdom may be replicating vCJD prions in their lymphoid tissues. Considering that immunohistochemistry (IHC) is considerably less sensitive than the conformation-dependent immunoassay (CDI), the number of Britons harboring vCJD prions in their lymphoid tissues may approach 20,000 (Safar, 2005a).
Approaches to Prion Diseases
Because prion diseases have aspects that resemble illnesses caused by viruses as noted above, many people use analogies to viruses when thinking about prions. But these analogies can sow confusion. One example is the presumed origin of the mad cows in Canada and the United States. Although it is true that BSE first appeared in the United Kingdom and then spread elsewhere through exported prion-contaminated feed, approaches from a traditional bacterial or viral epidemic are only partly helpful. In such situations, quarantines or bans can curb the spread of disease. But prions can arise spontaneously, which is an extremely important characteristic that distinguishes prions from viruses. In fact, any mammal is capable of producing prions spontaneously.
Spontaneous prion disease is thought to have triggered the epidemic of kuru, which decimated a group called the Fore in New Guinea in the past century. According to one theory, sporadic (s) CJD occurred in an individual whose brain was then consumed by his or her fellow Fore in a funerary rite involving cannibalism. The continued practice created a kuru epidemic. Ceasing the practice of this funerary rite also resulted in the decreased incidence of kuru.
Similarly, a feed ban that prevents cattle from eating the remains of other animals is crucial in containing BSE. But such bans will not eliminate the presence of mad cows when pathogenic prions arise spontaneously. If every year, 1– 5 people per million spontaneously develop prion disease, why not the same incidence for cows? Indeed, I suspect that the North American BSE cases are likely to have arisen spontaneously and that afflicted animals have occasionally ap-
peared unrecognized in herds ever since humans started cattle ranching. We have been extraordinarily lucky that a spontaneous case did not trigger an American BSE epidemic. Or perhaps small epidemics did occur but were undetected.
Still, many prefer the idea that the mad cows in North America acquired prions from their feed. Such reasoning allows people to equate prions with viruses—that is, to think of prions only as infectious agents (even though most of time, they arise spontaneously)—and to offer a seemingly plausible plan to eradicate BSE by quarantining herds. But ignoring the revolutionary concepts that govern prion biology can only hamper efforts at developing an effective program to protect the American public from exposure to these deadly agents. We must think beyond quarantine and bans, and test for prions even in the absence of an epidemic.
Diagnosis of Prion Diseases
The clinical diagnosis of human prion disease is often difficult until the patient shows profound signs of neurologic dysfunction (Roos et al., 1973; Will et al., 2004). In humans with sCJD, the most common clinical presentation is a progressive dementia. Approximately 10 percent of sCJD patients present with a progressive ataxia.
It is widely accepted that the clinical diagnosis must be provisional until a tissue diagnosis either confirms or rules out the clinical assessment. Prior to the availability of antibodies to PrP, a tissue diagnosis was generally made by histologic evaluation of neuropil vacuolation. IHC using anti-glial fibrillary acidic protein antibodies in combination with hematoxylin and eosin (H&E) staining preceded the use of anti-PrP antibody staining.
Postmortem Tissue Detection of Prions
The role of IHC in the diagnosis of scrapie was challenged after a study of the brains from eight clinically affected goats inoculated with the SSBP1 prion isolate (Foster et al., 2001). Thalamic samples taken from seven of eight goats with scrapie were positive for PrPSc by Western blotting but negative by IHC. The eighth goat was negative by both Western blotting and IHC. Consistent with these findings in goats are the results of a study of humans who died of sCJD or familial (f) CJD. In this study, IHC of formalin-fixed, paraffin-embedded human brain samples was substantially less sensitive than the conformation-dependent immunoassay (CDI) (Safar, 2005a).
The CDI detected PrPSc in all regions of the brain that were examined in 24 sCJD and 3 fCJD(E200K) cases. Comparative analyses demonstrated that the CDI was vastly superior to both histology and IHC. When 18 regions of 8 sCJD and 2 fCJD(E200K) brains were compared, it was discovered that both histology and IHC were unreliable diagnostic tools except for samples from a few brain
regions. In contrast, the CDI was a superb diagnostic procedure as it detected PrPSc in all 18 regions in 8 of 8 sCJD and 2 of 2 fCJD(E200K) cases (Safar, 2005a).
Concerned that limited digestion with proteinase K (PK) was hydrolyzing some or even most of the PrPSc, the CDI was developed so as not to require PK digestion to detect PrPSc. The CDI revealed that as much as 95 percent of PrPSc is protease sensitive (sPrPSc) and thus was being destroyed during limited proteolysis used to hydrolyze PrPC. sPrPSc comprises 80–95 percent of the PrPSc found in the frontal lobe and in the white matter of CJD patients (Safar, 2005a).
The CDI detected HuPrPSc with a sensitivity comparable to the bioassay for prion infectivity in Tg mice expressing chimeric human-mouse PrP. The high sensitivity achieved by the CDI is due to several factors including the use of phosphotungstic acid (PTA) that specifically precipitates sPrPSc and rPrPSc (Lee et al., 2005; Safar et al., 1998, 2005a). PTA has also been employed to increase the sensitivity of Western blots, enabling the detection of rPrPSc in human muscle and other peripheral tissues (Glatzel et al., 2003; Wadsworth et al., 2001). A comparison between the CDI and Western blotting on brain samples from sCJD and vCJD patients showed that the CDI is 50- to 100-fold more sensitive (Minor et al., 2004).
The CDI has also been used to study GSS caused by the P102L mutation. In mice expressing the GSS mutant PrP transgene, the CDI detected high levels of sPrPSc(P101L) as well as low levels of rPrPSc(P101L) long before neurodegeneration and clinical symptoms occurred (Tremblay, 2004). sPrPSc(P101L) as well as low concentrations of rPrPSc(P101L) previously escaped detection (Hsiao et al., 1994).
BSE Testing
The transmission of kuru prions to more than 2,500 Fore people in the highlands of New Guinea and the transmission of BSE prions to more than 170 teenagers and young adults who died of vCJD argues that oral prion infection can occur. The recognition that patients with vCJD were infected with BSE prions from cattle (Bruce et al., 1997; Collinge et al., 1996; Scott et al., 1997, 2005) prompted the European Union to institute testing of all cattle over 30 months of age at the time of slaughter. Currently, both Western blotting and ELISA tests for rPrPSc are being used on brainstems from cattle (Grassi et al., 2001; Kübler et al., 2003). The CDI test, which measures both protease-sensitive and protease-resistant PrPSc, has been adapted for bovine brainstems and is available for testing cattle.
The recent identification of BSE-positive cattle in Canada and the United States has prompted increased surveillance in these countries, but the number of cattle tested remains less than 2 percent of the annual slaughter (Prusiner, 2004a). Despite the small number of cattle being tested, new cases of BSE are being found. These new cases are attributed to tainted feed by agriculture authorities,
who continue to think of prion diseases as being similar to infectious illnesses caused by viruses or bacteria. These officials want to believe that BSE will disappear once the consumption of ruminant-derived feed ceases. They refuse to entertain the idea that most cases of prion disease are likely to be sporadic once contaminated feed is eliminated from the food supply. In Japan, 4 million cattle have been tested over the last four years, and close to 20 cases of BSE have been identified. One Japanese cow was 21 months old and another 23 months old (Yamakawa et al., 2003), younger than the animals tested in the European Union. It seems likely that most or all of these young animals developed sporadic BSE.
Determining how early in the incubation period BSE prions can be detected by bioassay is now possible due to the construction of Tg mice expressing bovine PrP, designated Tg(BoPrP)Prnp0/0 mice (Buschmann et al., 2000; Scott et al., 1997, 1999). Prior to the production of Tg(BoPrP)Prnp0/0 mice, cattle were used for bioassays of bovine prions. In a limited study using cattle bioassays, bovine prions were undetectable in the obex of the bovine brainstem until 26 months after oral inoculation (Wells, 2002). In these studies, prion infectivity was detected much earlier in the lymphoid tissue of the distal ileum.
Prions in Muscle
Animal meat products consumed by humans are predominantly muscle tissue. For many years, muscle tissue was thought to be devoid of prions. In studies of the hind limb muscles of mice, prions were found at a level of 5 percent of that in brain (Bosque et al., 2002); other muscle groups also had prions but at lower levels. PrPSc was found in virtually all muscles after prions were fed to hamsters (Thomzig et al., 2003). Investigations of prions in the tongue have shown high levels of both PrPSc and prion infectivity (Bartz et al., 2003). PrPSc was identified in the muscles of 25 percent of the sCJD patients analyzed (Glatzel et al., 2003). In livestock, PrPSc was found in myocytes of the fore and hind limbs of sheep with both natural and experimental scrapie (Andreoletti et al., 2004), and prion infectivity was reported recently in extracts prepared from the muscle of BSE-infected cattle. In the latter studies, prions were detected by transmission to Tg mice expressing bovine PrP (Buschmann and Groschup, 2005).
The Only Rational Strategy
The only rational strategy is to test all cattle for prions and eliminate those harboring prions from the food supply. No reasonable human would knowingly expose himself or herself to prions as prion diseases are invariably fatal.
In Europe, a policy was instituted four years ago of prion testing for all cattle destined for human consumption that are over 30 months of age. The 30-month cutoff point was chosen for surveillance by the Office International des Epizooties (OIE; also known as the World Organization for Animal Health), but it was never intended for food safety. Some European countries have arbitrarily adopted a 24-
month cutoff. It is irrational to believe that all cattle younger than 24 months of age are free of prions and that those older than 24 months are potentially infected. Initially, the Japanese government proposed adopting the European Union’s testing protocol, but consumer advocates forced the government to change its policy and test every slaughtered animal.
Rapid prion tests used in Europe vary in their sensitivity and reliability. Until now, the tests have not been sufficiently sensitive. Whether or not one or more of the newer tests can provide the desired sensitivity is unclear. Further, confining testing to brain tissue may be imprudent because other tissues such as muscle and lymphoid cells can harbor substantial levels of prions.
Given that seemingly healthy animals can carry prions, I believe that testing all slaughtered animals is the only rational policy. But this policy needs to be accompanied by a systemic approach to reward food suppliers for identifying livestock harboring prions. To maximize the protection from ingesting prions, we must aim to eliminate prions from the food supply by using the most sensitive and reliable test. The current system of using the least sensitive of the government-approved tests to minimize the number of prion-positive livestock is an unacceptable, dangerous common practice that must be terminated immediately. Providing the most safe food supply is a critical responsibility of every government—it is not an optional, incidental activity.
Acknowledgments
This work was supported by grants from the National Institutes of Health (AG02132, AG10770, and AG021601) as well as by a gift from the G. Harold and Leila Y. Mathers Charitable Foundation. S. B. P. has financial interests in InPro Biotechnology, Inc.
Correspondence should be addressed to: Institute for Neurodegenerative Diseases, 513 Parnassus Ave, HSE-774, University of California, San Francisco, CA 94143-0518; Tel: (415) 476-4482; Fax: (415) 476-8386. E-mail: stanley@ind.ucsf.edu.
SURVEILLANCE AND PREVENTION OF vCJD AND BSE: THE AUSTRALIAN PERSPECTIVE
Steven Collins, M.D.2
University of Melbourne, Parkville, Australia
As an introduction to bovine spongiform encephalopathy (BSE) and its consequent zoonosis, variant Creutzfeldt-Jakob disease (vCJD), I will briefly review the first human TSE epidemic, kuru, which contains a number of important in-
sights and lessons. Kuru occurred endemically among the Fore linguistic group of the eastern highlands of Papua New Guinea (PNG). The first cases of kuru came to the attention of Western medicine in the middle to late 1950s as Australian patrols were gradually reaching the more remote areas of PNG (Gajdusek and Zigas, 1957). Predominantly manifesting as an inexorably progressive cerebellar ataxia with later onset dementia (Gajdusek, 1962), the etiology and spread of the disease were eventually linked to cannibalistic rites of mourning for deceased relatives (Gajdusek, 1977). Not long after noting neuropathologic and other similarities between kuru and scrapie (Hadlow, 1959), kuru was proven to be a transmissible spongiform encephalopathy (Gajdusek et al., 1966). The predilection of kuru for women and children was related to their more usual consumption of the highly infectious central nervous system tissues. It is believed that ritualistic endocannibalism was successfully eradicated by the end of the 1950s.
Detailed epidemiologic studies of kuru suggested a mean incubation period of around 12 years, but ongoing contemporary field surveillance supports the likelihood that the most recent cases have incubation periods spanning up to 50 years (Collinge, 1999; Collinge et al., 2006). Consequently, based on these observations and those from various animal models of prion disease (Dickinson et al., 1975; Hill et al., 2000), there may not be a finite incubation period. Once exposed, the risk of developing a TSE may persist lifelong. Further, kuru indicated that despite the very low transmissibility of TSEs compared with many conventional microbes, high levels of disease can arise, including through an oral route of inoculation, if unique circumstances prevail to support “unnatural” intraspecies recycling of highly infectious tissues. At the height of the epidemic, annual mortality from the disease approached 50 percent in some Fore villages, with an annual incidence of approximately 10 percent in a number of Fore tribes (Gajdusek and Zigas, 1957).
Bovine Spongiform Encephalopathy and Variant Creutzfeldt-Jakob Disease
By the mid-1980s, United Kingdom veterinary authorities had confidently recognized BSE as a new form of cattle disease. In retrospect, the early years of the BSE epidemic evinced undue reassurance from the scrapie precedent. The lack of documented evidence of scrapie transmission to humans from farmed sheep and the generally low levels of endemic scrapie over the few hundred years that scrapie had been recognized in sheep flocks suggested BSE would perhaps behave similarly. In discomfiting contrast, the incidence of BSE rapidly escalated and by 1992 at the height of the UK epidemic, some 3,500 cases per month were confirmed; overall, an estimated 2 million contaminated cattle are believed to have entered the human food chain (Donnelly et al., 2002). Given the dramatic increase in BSE, in accordance with one of the recommendations of the Southwood enquiry, national surveillance for human prion diseases was prudently re-
commenced in the United Kingdom in 1990. Particular interest was to be given to ascertaining the occurrence of any new forms of disease that might have arisen zoonotically from BSE. In 1996, a new or variant form of CJD (vCJD) was reported by the National CJD Surveillance unit in Edinburgh, Scotland, with 10 younger adults and adolescents manifesting a phenotype hitherto not described (Will et al., 1996). A range of subsequent research has confirmed the likely causal link between vCJD and BSE (Hill et al., 1997).
A very recently reported case-control study of vCJD has highlighted diet as a principal risk factor, especially the consumption of greater amounts of products likely to contain bovine mechanically recovered meat (MRM) and bovine head meat (sausages, burgers, and pies) (Ward et al., 2006). These bovine meats are more prone to contamination by brain, spinal cord, and dorsal root ganglia, and the study results are therefore consistent with the hypothesis that people developing vCJD were exposed to greater amounts of those bovine meat products more likely to be contaminated by the highly infectious central nervous system tissue (summarized in Figure 6-1). One caveat concerning this data is the possibility of recall bias, suggested by the observation that the respondents for patients initially suspected to be manifesting vCJD but eventually proven to have a different illness also reported the same dietary associations.
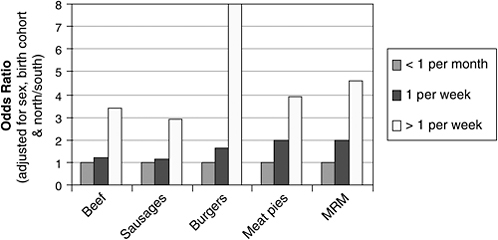
FIGURE 6-1 Reported frequency of food consumption, odds ratio of vCJD cases versus general population controls. Results of this case-controlled study suggest that people with vCJD consumed foods likely to contain bovine mechanically recovered meat (MRM) and bovine head meat (sausages, burgers, and pies) more frequently than as compared with the general population. Such meats are more prone to contamination with central nervous system tissue, and therefore—if the bovine source was infected with BSE—higher concentrations of prions. It should be noted, however, that because consumption rates were self-reported, they may reflect recall bias (see text).
SOURCE: Adapted from Ward et al. (2006).
As of July 2006, a total of 161 cases of vCJD have been diagnosed in the United Kingdom. Remarkable uniformity in the clinical illness has been maintained: it affects young people (the median age at onset is 26 years) with a median survival of 14 months. France has the second highest number of vCJD cases (18 in total), with case numbers and the temporal profile behaving as predicted from modeling based on transmissions through beef imported from the United Kingdom (Valleron et al., 2001). The primary United Kingdom vCJD epidemic is thought to be in decline, and most experts expect that the total number of cases will not exceed 200 (Valleron et al., 2001). However, the recent reports of three cases of vCJD related to transfusion of blood products and the higher than expected potential prevalence of subclinical or preclinical vCJD suggested by the retrospective study of archival appendix and tonsil specimens raises the sobering possibility of a secondary wave of iatrogenic vCJD (Hilton et al., 2004; Llewelyn et al., 2004). Based on animal transmission studies involving “species barriers” and subsequent strain adaptation, such human-to-human transmissions may prove far more efficient than the primary cross-species infections from cattle to humans through oral consumption of contaminated beef. Further, not only must we be alert to a possible secondary wave of vCJD, but given the unprecedented calamitous transmission of BSE to humans, we must also keep vigilant to the possibility of other cross-species transgressions from TSEs now apparently confined to animals, such as chronic wasting disease.
Australia’s National CJD Registry
The Australian National CJD Registry (ANCJDR) is a federally funded surveillance unit that commenced operation in 1993, with the initial mission to ascertain further cases of human pituitary hormone-related CJD. At the inauguration of ANCJDR activities, five persons with pituitary hormone-related CJD were known, and no additional cases have been detected. The scope of ANCJDR human TSE monitoring has expanded with the passage of time, and as of 1996, following the discovery of vCJD, surveillance was broadened to include that disease (Collins et al., 2002). The ANCJDR has also participated in the European surveillance consortium (EUROCJD) since 1997. As of 2004, the Communicable Diseases Network Australia agreed to make human TSEs notifiable diseases in all Australian states and territories. Primary case review of suspect notified cases involves the ANCJDR, with evaluation outcomes a collaborative exercise between the appropriate state or territory and the registry.
Case definitions used by ANCJDR for classification purposes are those endorsed by EUROCJD. A range of standard surveillance methods are employed by the ANCJDR, including the use of semiannual surveys sent to all neurologists, neuropathologists and pathologists within Australia. Respondents are prompted to report any suspect, probable, or confirmed cases of CJD they have seen or been aware of in the preceding six months. Follow-up is undertaken for positive re-
sponses, and we also receive unprompted personal communications regarding suspect and confirmed CJD from these same medical practitioner groups. The ANCJDR annually reviews state and territory morbidity separation codings for citings of CJD or CJD-like illnesses, and conducts national death certificate searches as well. Since 1997, the most important mechanism of case notification has been through the national cerebrospinal fluid (CSF) 14-3-3 protein diagnostic testing service that the registry offers (Collins et al., 2000). For case confirmation, CSF 14-3-3 protein detection serves as a reasonably reliable and specific marker of sporadic CJD and is now an accepted component of case definitions for surveillance classification purposes (Zerr et al., 2000).
Neuropathologic examination of the brain remains necessary for confirmation and classification as a definite case of CJD (Collins et al., 2004), and given the problems frequently encountered with brain biopsies, postmortem examination is preferred and pursued in all patients with suspect CJD. Very occasionally routine autopsy will detect a completely unsuspected case of TSE. Unfortunately, however, postmortem rates in major teaching hospitals have declined dramatically throughout Australia over the past 10–15 years and are now running at around 12 percent. Further, fewer medical centers are willing to conduct autopsies on patients with suspected CJD. Over recent years the ANCJDR has maintained a postmortem rate of around 60 percent, but the aforementioned combination of factors is militating against our ability to obtain neuropathologic confirmation, which remains the gold standard for diagnosis.
Similar to all previous and ongoing human TSE surveillance, the vast majority of cases in the ANCJDR are sporadic; there are several familial cases and a small number of iatrogenic cases, but no cases of vCJD so far (see Table 6-1) (Collinge, 1999). Measures have been undertaken to try to ensure that vCJD has not been overlooked in Australia.
TABLE 6-1 Cases of Transmissible Spongiform Encephalopathies (TSE) Reported to the Australian National CJD Registry (ANCJDR), January 1, 1970 Through June 30, 2005
|
Classification |
Sporadic CJD |
Familial CJF/GSS |
Iatrogenic CJD |
Variant CJD |
Unclassified |
Total |
|
Definite |
265 |
29 |
5a |
|
00 |
299 |
|
Probable |
178 |
9 |
4 |
0 |
0 |
191 |
|
Possible |
7 |
0 |
1 |
0 |
0 |
8 |
|
Incomplete |
0 |
98b |
98 |
|
|
98 |
|
Total |
450 |
38 |
10 |
0 |
98 |
596 |
|
aIncludes one definite iatrogenic case who received pituitary hormone treatment in Australia but disease onset and death occurred while a resident overseas. This case is not included in statistical analysis since morbidity and mortality did not occur within Australia. bIncludes 59 living cases. SOURCE: Adapted from Klug et al. (2005). |
||||||
Reaffirmation of Absence of vCJD in Australia
The likelihood of vCJD in a given country appears to reflect either the levels of endemic BSE, the amounts of imported contaminated United Kingdom beef products through 1980–1996, or—as in the experience of Canada, the United States, and perhaps Japan—its citizens’ travel and migration patterns. Because the travel and migration profile of Australians appears similar to that of Canadians and Americans, the ANCJDR undertook a reexamination of the clinical files and neuropathologic reports of all cases referred to the registry who died between January 1, 1992, and June 30, 2003, and which had been classified as either sporadic CJD or non-CJD. None of the 365 reexamined cases fulfilled case definitions for possible, probable, or definite vCJD, and the molecular (PrP glycotyping) profile typical of vCJD was not seen in any of the 37 cases for which frozen tissues was available for Western blot analysis (Lewis et al., 2005).
Animal TSEs in Australia
To date, there have been no endogenous cases of either BSE or scrapie in Australia. Scrapie was inadvertently introduced in 1952 from the United Kingdom, but was quickly recognized and the affected flock slaughtered. There have been two cases of TSEs in felines in Australia: an imported cheetah and an Asiatic golden cat, both of which were believed to have contracted their disease through contaminated feed prior to their importation to Australia.
Protective Measures
For the protection of domestic commercial livestock, Australia has adopted a range of measures to lessen the risk of BSE and scrapie, which can be found at www.aahc.com.au. Since 1966, there has been a ban on the importation of meat and bone meal and any stock feed containing ruminant materials from anywhere in the world except New Zealand; this was initially imposed to avoid the risk of anthrax but was maintained thereafter. From 1988, live cattle from the United Kingdom and any other country reporting BSE cannot be imported into Australia. Owners of animals imported from countries in which BSE is subsequently discovered can choose to place the imported animals under lifelong quarantine or have them slaughtered.
A voluntary ban on ruminant-to-ruminant feeding of meat and bone meal was established in 1996 but became compulsory through Commonwealth legislation in October 1997. This was then expanded in June 1999 and again in March 2001, whereby vertebrate materials (except for milk, gelatin, and tallow and oils from fish and poultry) are prohibited from use in ruminant feeds (www.aahc.com.au). To ensure compliance with the feed bans, national audits are conducted. These include onsite visits to producers, animal feed manufacturers, and render-
ing plants, as well as PCR screening of meat and bone meal intended for feed. To date, three such audits have been completed.
Animal TSE Surveillance
Australia’s National TSE Freedom Assurance program oversees and coordinates the National TSE Surveillance Program (NTSESP), education strategies, and enforcement of the ruminant feed ban (www.aahc.com.au). Prior to establishment of the national audits, more than 3,300 brains of cattle were examined histopathologically for BSE; no disease was found. In 1998, NTSESP was initiated to satisfy OIE requirements concerning the surveillance and monitoring of BSE and scrapie. Under this program, every animal (cow or sheep) showing signs of nervous system disease must be examined by designated veterinarians, who also take brain specimens for histopathologic examination. If disease is suspected, the samples are subjected to Western blot analysis. Approximately 450 sheep and 400 cattle per year with neurologic illness are examined through NTSESP. The program is designed to achieve a 99 percent confidence level, which means that it can detect one case of BSE per one million cattle. Following a change in the OIE terrestrial animal health code, an additional 400 sick animals (including downer animals, those that die on the farm, and those slaughtered to contain a disease outbreak) are now tested for BSE by Western blot of brain specimens.
Education
Education of stakeholders and the public is the final important component of Australia’s effort to control BSE. Various media are used to introduce a broad understanding of legislation on this issue and its rationale with dissemination through agencies ranging from the Environmental Protection Agency, the State Farmer’s Association, the Australian Veterinary Association, and Animal Health Australia, as well as through various state and territory education programs.
BSE AS A CASE STUDY OF PUBLIC HEALTH AND THE PUBLIC GOOD
Maura N. Ricketts, M.D., M.H.Sc., F.R.C.P.C.3
Health Canada
The case of BSE represents a special challenge for public health professionals because the initiative and the interventions necessary for the control of BSE
lie within the animal health sector. As a direct result of this, it is to be expected that debate, disagreements, and occasionally dispute will populate the arena when animal health experts and public health experts talk about BSE. Regardless, the last 20 years of experience with BSE have refined the analysis of risk, and there is increasingly large overlap in policy recommendations with a notable shift toward recognition of the importance of risk perception, ethics and societal values in shaping public policy, the implications for broader healthcare practice, and other aspects of public health. This paper will examine BSE from the perspective of public health in the expectation that the paradigm of public health might be seen to be one that is particularly sensible when the protection of human health is ultimately the goal of public policy.
What Is Public Health and What Does It Do?
The definition of public health has proven that it is, like other branches of medicine, both an art and a science. The examination of a definition of public health provides the foundation for understanding public health-based recommendations:
Public health is one of the efforts organized by society to protect, promote, and restore the people’s health. It is the combination of sciences, skills, and beliefs that are directed to the maintenance and improvement of the health of all the people through collective or social actions…. Public health activities change with changing technology and social values, but the goals remain the same: to reduce the amount of disease, premature death, and disease-produced discomfort and disability in the population. Public health is thus a social institution, a discipline and a practice (Last, 1983).
The science and art of promoting health, preventing disease, prolonging life, and improving the quality of life through the organized efforts of society (Last, 2001).
A key component of the definition is the reference to “the organized efforts of society.” In Table 6-2 are the functions of public health that have been nationally adopted in Canada. Even a superficial review of the functions will confirm the extent of societal commitment that is required.
The functions of public health map themselves into core program areas (see Table 6-3). These are the core programs that must be sustained by any functional public health organization.
When examined in matrix format (see Table 6-4), the public health decisions taken regarding vCJD become clearer.
What About vCJD?
How can public health principles be applied to the case of BSE? The first and foremost function of public health is health protection. Table 6-5 lays out a well-
TABLE 6-2 The Functions of Public Health
|
Population Health Assessment |
|
|
Health Surveillance
|
|
Health Promotion
|
|
Disease and Injury Prevention
|
|
Health Protection and Emergency Preparedness |
|
SOURCE: National Advisory Committee on SARS and Public Health (2003). |
accepted series of steps that are followed when an outbreak of an unknown disease is found among human populations.
It is to be noted that an “outbreak” could be a single case of a disease whenever that disease is sufficiently unusual, as is the case with vCJD or BSE. The reasoning for this is similar to the use of canaries in mines—as soon as the first canary dies, it is time to act before the first person dies. In the case of vCJD, the canary is the first case of BSE—as was so clearly illustrated in the United Kingdom (see Figure 6-2).
TABLE 6-3 Functions and Core Programs
|
Public Health Functions |
Core Program Areas |
|
|
|
SOURCE: National Advisory Committee on SARS and Public Health (2003). |
|
TABLE 6-4 Activity Map for vCJD
|
|
Public Health Functions |
||||
|
Core Program Areas |
Population Health Assessments |
Public Health Surveillance |
Disease and Injury Prevention |
Health Promotion |
Health Protection |
|
Communicable disease prevention and control |
X |
X |
X |
|
X |
|
Prevention of chronic diseases and injuries |
|
|
|
|
|
|
Health development through life cycle |
|
|
|
X |
|
|
Environmental health |
X |
X |
X |
X |
X |
|
Emergency preparedness |
|
|
|
|
|
|
SOURCE: Ricketts (2005). |
|||||
TABLE 6-5 Health Protection and the Management of an Outbreak
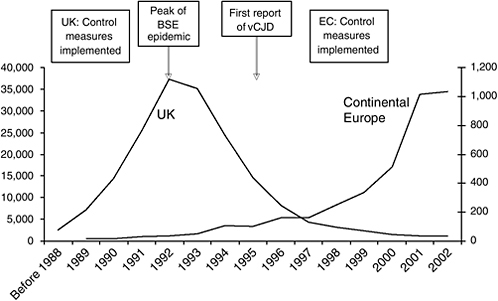
FIGURE 6-2 BSE and vCJD outbreak development in the UK; Epidemic curve of BSE in Europe (UK n = 180,845; continental Europe n = 3,286).
SOURCE: OIE (2006).
Population Health Assessment and Decision Making in Uncertainty
Since the first case of BSE and the subsequent first case report of vCJD some 10 years later, science and public policy have come a long way. Assumptions made early in the BSE epidemic, and even some made later, proved to be inaccurate or misplaced. It might have been said in the early years that those involved in developing policy to control BSE were, figuratively speaking, flying the plane while building it.
In public health, as in other arenas of public policy, it is necessary to make decisions even in the face of uncertainty. When uncertainty prevails, many other aspects of societal influence enter into the decision making, such as economic stability or principles such as risk avoidance or the precautionary principle. In the case of BSE, there were enormous economic and trade implications that directly impacted public policy.
At an entirely practical level, there are questions that will underlie the decisions finally taken by society. Where there are no answers, then it is required that assumptions are made. In an open society, the assumption should be stated. Based upon my experience in both public health and BSE, I propose the following core issues:
-
How do people get vCJD?
-
Is there any immunity? Treatment?
-
Are there people at higher risk?
-
Is there a test that can tell people they already are infected? Or will they get the disease?
-
Are there ways to stop people from getting BSE? Or from getting sick?
-
Do people die or recover?
How Do People Get vCJD?
The principal source of route and exposure for humans is through the consumption of food of bovine origin, just as for other animals. However, it can be seen that the risk must be very low because there are very few cases despite very large amounts of exposure. As of March 2006, 160 cases of vCJD (National CJD Surveillance Unit UK, 2006) had been reported in the United Kingdom—this in a country where it has been estimated that over 1 million infected cattle entered the human food chain (Ghani et al., 2000). It is now known that most of the BSE infectivity is found in particular parts of the infected cattle, and that the location of the infectivity shifts somewhat with age. Collectively, the contaminated tissues are referred to as specified bovine offals; those tissues that must be removed in order to protect human populations are the specified risk materials (SRM). Additionally, some kinds of mechanically recovered meat may become contaminated with neural tissue. The relative resistance of humans to BSE suggests that the removal of SRM and the modification of methods to mechanically recover meat would enormously reduce risks to human populations.
Human-to-human transmission is a possible secondary route of transmission of vCJD, but in this case, the risk must be very low because very few cases of human-to-human transmission have been recognized despite intense interest in this problem. All cases reported thus far are from the United Kingdom, and all involve transfusion of blood (Ward et al., 2003). In medical practice, it is anathema to cause disease during the course of treatment; despite the nearly immeasurable risk, extensive measures have been undertaken to protect against iatrogenic vCJD.
Is There Any Immunity? Treatment?
From the perspective of public health, the answer to both questions is no. Despite the possible advances in clinical medicine and proposed clinical trials, it is not possible to amend the public health risk assessment by making allowances for immune populations or for the potential impact of treatment.
Are There People at Higher Risk?
Clearly, there are. People residing in countries with a high incidence of BSE and where BSE control interventions have not completely removed the potential exposure of humans to high-risk tissues are at the highest risk. Additionally, it is recognized that people carrying the homozygous allele at codon 129 (Methionine/methionine) are overrepresented among vCJD cases. The number of people in the UK who have this gene combination and have not developed vCJD far exceeds the number of those who have. Finally, for the purposes of this paper, it must be noted that there is a risk based upon age; the age distribution is skewed toward younger adults. The reason for this remains obscure—could it be due to specific exposures? Is it because of particular physiologic characteristics of the age group? Could it be that everyone is at equal risk, but that the incubation period varies? These questions are asked, but not answered. Resultantly, if one was searching for a screening test that could identify high-risk populations, perhaps to offer them special interventions, these characteristics are not useful because very, very many more people with these patterns are not ill than are.
Is There a Test?
There is, to date, no test to detect the agent in food or by testing easily available biologic tissues of living animals, including humans. Testing in humans is completely limited to tests that are conducted after the appearance of symptoms.
In cattle, after slaughter, specific areas of cattle brains can be tested for BSE—these tests are being used more and more extensively globally. However, testing is comparatively expensive and requires special facilities and cannot be conducted on site at the time of slaughter. Detailed analysis of the impact of testing and other interventions can be found on the Canadian Food Inspection Web site, as on other Web sites (European Union, Switzerland, United Kingdom, France, and the United States for example).
Are There Ways to Prevent BSE Infection in Humans? Are There Ways to Prevent People Who Are Infected from Becoming Sick?
To both questions, the answer is no. None of the most time-tested interventions of public health (vaccination, prophylaxis) are useful. By implication, it is essential that humans must avoid exposure to the source of infection (e.g., contaminated cattle tissue).
Do People Die? Recover?
To date, no person with diagnosed vCJD has survived. It is not yet known if infection leads inevitably to disease, in part because there is no existing preclini-
cal diagnostic test. Once the symptom complex is diagnosed, the disease leads, inevitably, to death. Preventing vCJD can only be accomplished, at this time, by preventing exposure to BSE.
Unlike many other population health issues, the principal tools for protecting the public are actually not in the control of either clinical or public health physicians. Animal and food security is the first line of defence. With the lessons of the past, it is possible to make good public policy decisions regarding vCJD and BSE. There is not a recipe for controlling or eradicating vCJD or BSE. Table 6-6 outlines the key activities to control BSE; thorough discussion is beyond the intended scope of this paper.
From the perspective of public health, a series of problems can be anticipated even in those areas where public health has established authority. Principal among them is the need for a highly sensitive and accurate surveillance system that can identify all cases of human TSE and accurately distinguish vCJD. Surveillance may be supported through legislative authorities where necessary and with the financial commitment of government where needed. Surveillance capacity will have to be centralized since the collection, analysis, and dissemination of information for action requires expertise in the disease complex as well as in handling pathogenic tissues. The very rarity of the disease and the implications of each
TABLE 6-6 Controlling BSE
case mean that a high, uniform standard must be set, without risk of duplication of case reports. In order that cases are identified, there is a need for specialized clinical knowledge. The awareness, interest, and participation of neurologists and neuropathologists through collaborative networking is essential. Additionally, there is a need for access to specific laboratory-based diagnostic capacity, including EKG, CSF testing, MRI, and neuropathology, all within a context of high autopsy rates.
The flow of accurate analysis to senior policy makers in government is required to ensure that a realistic and thorough BSE risk assessment is conducted and uses evidence from both human and animal health perspectives. The risk assessment model should examine modes of population exposure to the BSE agent and must do so with consideration of the scientific evidence and the multiple requirements of all stakeholders. There will be many interest groups and many different messages; by fostering trust and the honest prompt disclosure of information, nongovernmental organizations (NGOs) can support the overall public efforts and help prevent the dissemination of inaccurate or inflammatory messages. In all countries, one must consider that a trade-based economy is resistant to disclosure of risk and that competitive markets are affected by changes in import and export restrictions.
Finally, one must reflect upon the role of public opinion. vCJD is a highly dreaded disease accompanied by high levels of public outrage. People are outraged because they view the suffering and deaths as unnecessary; in fact, they may suggest that justice is not served because the death of a loved one is simply due to industry’s interest in profiting over human health concerns. In Canada, public opinion supported the smaller and less industrialized practice of cattle ranching seen in some Canadian provinces.
Experience in public health has demonstrated that there are a number of strategies that work to prevent the spread of communicable disease. vCJD is a particular challenge because there are no opportunities to prevent vCJD short of preventing BSE infection. In no particular order, the following illustrates the range of activities that would be pursued:
-
Healthy public policy should be implemented regarding exposure to BSE-contaminated tissues in all disciplines from occupational safety, infection control practices, biologics development and use, transplantation and transfusion safety, and food safety;
-
Inform people about health issues so that they can make their own decisions in an informed manner;
-
Prevent and respond to outbreaks and emergencies, with particular emphasis on the need to conduct surveillance for vCJD;
-
Help people develop personal skills (resilience), particularly those directly impacted by BSE through sickness or business;
-
Collaborate for intersectoral and community action, recognizing that the entire community is affected;
-
Enforce laws and regulations, notably in feed and food safety;
-
Reorient and assure the quality of health services, particularly recognizing that there will be confusion between other forms of human TSEs and vCJD. Additionally, given the experience in the United Kingdom, recognition that those who develop the disease are young;
-
Create supportive environments for all stakeholders; and
-
Evaluate the impact of interventions.
Many lessons were learned as a result of the outbreak of BSE in the United Kingdom and Europe. It is important to control the risk of BSE exposure; countries that have focused on the rate of BSE have been unable to prevent its importation and spread. As in a commonly used homily, a chain is only as strong as its weakest link. The key link for public health practitioners is that BSE and vCJD are the same agent. Simply put, if there were no further infections of cattle with BSE, there would be no further cases of vCJD.
INCENTIVES AND DISINCENTIVES FOR DISEASE SURVEILLANCE AND REPORTING: THE BSE CASE STUDY
Will Hueston, D.V.M., Ph.D.4
University of Minnesota
My contribution to this workshop comes in the form of seven lessons that I have learned from 16 years of involvement with bovine spongiform encephalopathy (BSE), followed by a brief list of recommendations derived from these lessons. The lessons and recommendations are drawn from my experience working as a private practice veterinarian, a resident veterinarian for an agribusiness enterprise, a university faculty member, a government animal health official, and an adviser and consultant to national and state government, national and international organizations, and food system companies from production to retail and food service.
Lesson 1: Detecting a New Animal Disease Is Extremely Difficult
Most individual animal diseases are treated on the farm following clinical diagnosis by the animal owner, farm manager, or in difficult cases, a private-practice veterinarian. If that clinical diagnosis is incorrect, and/or the animal does not recover, the animal is usually sold (culled), eaten, or buried. Most animal
diagnostic services accessible in such situations are provided on a fee-for-service basis. Only foreign animal disease investigations and diagnostics are provided free of charge by government veterinarians, as they are seen as a public good. Even then, however, the diagnostic workup is generally limited to ruling out a specific foreign animal disease (e.g., foot-and-mouth disease or BSE); there is no follow-up to determine the exact cause of illness if foreign animal diseases are ruled out. Consequently, it is very difficult to detect the signals of the emergence of a new animal disease.
Limited national monitoring and surveillance does occur, such as the National Animal Health Monitoring system and more recently the National Animal Health Laboratory Network. Creating inclusive national animal disease databases has been hampered by the lack of widely accepted standardized nomenclature for animal diseases and presenting signs. Unfortunately, most animal diagnostic laboratory and veterinary hospital record-keeping systems are designed to facilitate financial accounting and billing, not epidemiologic analysis.
By comparison with Europe, Canada, New Zealand, and Australia, the U.S. federal animal health laboratory system is quite limited. The federal government supports two national laboratories, one operated by the Department of Homeland Security on Plum Island, New York, and the other is operated by the USDA Animal and Plant Health Inspection Services Veterinary Services in Ames, Iowa. Research and diagnostics at these facilities focus on diseases for which there is a specific programmatic target, such as foreign animal diseases at Plum Island, and domestic program diseases such as brucellosis, tuberculosis, and BSE at Ames. These two laboratories provide reference services for state and private laboratories (confirmation of specific program diseases). The U.S. government does not have a national laboratory focused specifically on the detection or description of emerging animal diseases.
Animal disease diagnostics in the United States are performed by state, university, and private laboratories that vary greatly in terms of their quality and capacity. About 12 of the 50 state animal diagnostic laboratories are linked in a pilot version of a national animal health laboratory network. Limited funding has been provided to these laboratories to allow them to cooperate with the federal laboratories for foreign animal disease diagnostics but not for the identification and characterization of emerging diseases.
Lesson 2: Recognizing BSE in a Low-Incidence Country Is Difficult Even Under the Best Circumstances
BSE has no unique presenting clinical signs. Therefore, the disease can only be detected through specialized diagnostic testing of brain samples; it cannot be diagnosed by clinical evaluation of the live animal or by gross necropsy such as that carried out on a dead animal on the farm.
Most countries conduct passive surveillance for BSE and other animal diseases, providing diagnostic services for those animals voluntarily presented to the laboratory rather than actively searching for cattle demonstrating clinical signs compatible with BSE or dying of unknown causes. This focus on passive surveillance leads to confusion between the absence of evidence for a disease and the evidence of its absence. Countries with no “BSE suspects” presented for diagnostic workup claim that no BSE exists, even though the clinical signs associated with BSE (changes in mentation, sensation, and locomotion) are found in a number of commonly occurring cattle diseases that can only be differentiated from BSE by extensive diagnostic workup. In addition, adherence to the “disease present or absent” paradigm further reduces the effectiveness of passive surveillance by establishing a bias against detecting the disease so that a country can continue to represent themselves as “BSE-free.”
There are huge disincentives for expanding national surveillance for BSE. BSE surveillance is expensive, with the total costs for collecting and testing each sample usually in excess of US$20. Furthermore, it is not in the national interest to discover BSE unless there is a plan in place for addressing it. Reporting BSE can have devastating economic and political consequences; whereas, historically a country’s failure to detect the disease, or its lack of an adequate surveillance system, has been rewarded by continued trade.
Lesson 3: Most Farmers Are Honest, but Disincentives for Reporting BSE Greatly Outweigh the Incentives
Animal production has historically been measured by the number of animals produced, not the quantity and quality of food generated. Therefore, many producers see themselves as raising animals rather than as part of the food system. We are continuing to work to change that paradigm in order to foster a shared responsibility for the food system, from producer to consumer.
For years most countries in the world have pursued a cheap food policy, where the price of food has assumed paramount importance. Consequently, food producers throughout the food system (from the farm to the consumer’s table) strive to keep costs as low as possible. Although animal diseases impose costs on farmers, they recognize that absence of disease (100 percent prevention and control) is not always the optimal economic strategy. Producers weigh the costs of disease diagnostics, prevention, and control against the potential benefits they may ensure. They seek diagnostic support if they believe that understanding and preventing economically important diseases can reduce the cost of production more than the marginal cost of the diagnostics and prevention strategies, or if their products can be accorded a higher health status and, hence, a higher value, as a result of negative results on diagnostic tests where the risk of positive tests is low.
One is hard pressed to find individual producer incentives for reporting suspected cases of BSE in the United States. There is no treatment for BSE, and government-mandated controls increase production costs. Feed is the single greatest contributor to cost of production, and government feed regulations have removed a low-cost protein supplement. Removal and destruction of specified risk materials (those tissues where BSE agent accumulates in affected cows) has increased the costs of processing and reduced the value of each animal slaughtered. Additionally, the government certifies the nation’s BSE status, but not that of individual herds. Thus the individual producer gains no benefit from conscientious submission of suspect cattle where all the test results are negative.
At the same time, there are numerous disincentives for reporting BSE. Producers on whose farm a BSE cow is identified are ostracized by the rest of the industry, and their products are shunned by consumers and wholesale buyers. Their business (and personal life) is disrupted by the government, industry and media response. Finally, disposal of affected or suspect animals is difficult and often expensive, and government response to BSE diagnosis in a herd has all too often involved destruction of many more animals than epidemiologically necessary to control the disease.
Considering all the disincentives, a phrase uttered by the Prime Minister of Alberta was taken out of context as a new mantra for some cattle producers: “Shoot, shovel, and shut up” rather than report BSE suspects.
Lesson 4: Testing Can Become an End Unto Itself
Before implementing a widespread testing regime for disease surveillance or health monitoring, the purpose of the testing must be clarified. The purpose will change over the course of an epidemic, so it must always be clear why it is done, in order that appropriate sample size can be determined and the test results interpreted appropriately.
Testing alone cannot afford safety (defined by dictionaries as the “absence of risk”), and it is meaningless without the concurrent implementation of animal and public health measures. Testing the wrong populations can create a false sense of security that, while politically expedient, does not constitute a public health measure. For instance, controlled BSE challenge studies and accumulated BSE surveillance results demonstrate that young animals will test negative to all of our currently available tests even if exposed to BSE; this is because the disease takes years to create discernible damage to the central nervous system and for the disease agent, the prion protein, to accumulate to detectable levels. Consequently, testing only young cattle assures that all tests are negative but says nothing about the BSE status of a country. Similarly, testing all cattle in a country with BSE decreases the apparent prevalence of the disease because of all the young cattle testing negative regardless of the extent of BSE in the adult population.
Lesson 5: Focus on Risk, Not the Presence or Absence of Disease
The key for human and animal health protection is effective risk management, not the disease status of the country. Most countries have focused disproportionately on the reported presence or absence of disease rather than on the effectiveness of the risk management. Internally, the lack of positive diagnostic tests has propelled officials to proclaim disease freedom, thereby creating a false sense of security and reducing the imperative of prevention and control. Outside their borders, countries have tended toward implementation of total trade bans when a trading partner identifies BSE, a policy that ignores the fact that a variety of risk management measures exist that allow for the safe trade of animals and animal products from countries regardless of their disease status. Infectious diseases do not respect national borders, and yet we frequently hear the statement “we have sealed our borders,” all too often followed by the false reassurance that the disease of concern will “never” occur here. Not only are these statements factually inaccurate, but also they represent the ultimate risk communication error—providing absolute guarantees. Above all else, we usually fail to consider most zoonoses in the context of ecosystem risk management, and we develop national public policy rather than a regional or global approach.
Lesson 6: Take Opportunity Costs into Account
Every dollar spent on BSE testing, prevention, or control is unavailable to address a different risk or challenge. The cost of BSE testing may be disproportionate to the resulting public health benefit, as compared with addressing other pressing issues in protecting the global food system. Similarly, taking a zero-risk approach forecasts ever increasing costs as the precautionary principle tends toward taking preemptive actions on every identified hazard, no matter how small the risk. Developing animal health and public health priorities must be conducting from a holistic perspective in which all hazards are considered and the opportunity costs of various initiatives weighed. Similarly, surveillance priorities should be established through broad-based considerations of risk management, not simply from a desire to impress the public or trading partners by more tests. I was struck by the concern voiced by one of my international colleagues caught in a massive (and expensive) government response to a few BSE cases. “What will we tell our grandchildren,” he asked, “when we have spent all this money for a rare cattle disease with a relatively small human health impact, while at the same time we fail to take basic, proven public health measures for other diseases and conditions that impact the lives of millions because ‘we have no money.’”
Lesson 7: High Health Status Is a Curse
Once high health status is attained, the impetus for maintaining an animal health and public health infrastructure fades. We celebrate the successful eradica-
tion of a whole string of animal diseases and successful risk management of zoonoses that plagued our grandparents such as bovine tuberculosis and undulant fever. Having achieved this unique high health status for the entire nation, then we turn around and ask why we are maintaining an infrastructure for something that no longer exists. Anchoring our animal and public health infrastructure on a limited number of “program” diseases undercuts the overall system as successes are celebrated. As a result of the resulting budget cuts and retrenchment, we have few resources to commit to investigating emerging disease threats and little surge capacity when a significant infectious disease outbreak occurs. Replacing experienced professionals with unseasoned new recruits saves money in the short run but costs the nation (and world) in the long term. That is where we find ourselves today as we struggle to manage emerging issues with limited resources. Cutting both physical and human resources provides a rapid means for decreasing budgets, but rebuilding an effective animal and public health infrastructure is a monumental undertaking in terms of resources and time.
Strategies for Managing This Dilemma
-
Reframe surveillance discussions to focus on the purpose and the scientific basis for surveillance rather than the number of tests conducted;
-
Consider the entire farm-to-table food system when designing surveillance systems to support effective risk management for the end consumer and the nation;
-
Recognize that no one surveillance system fits all situations; the sampling design, sample size, and testing protocol must be adapted to the needs of each country in order to support optimal risk management;
-
Create more incentives for reporting disease to pull in more samples, rather than simply demanding testing through regulatory initiatives and penalties;
-
Develop a national animal identification system to support rapid response to disease outbreaks and long-term support for emerging disease detection;
-
Strengthen the national animal health laboratory system and increase its capacity, perhaps by providing federal resources to states tied to performance requirements and reporting so that all states can meet a minimum level of proficiency and quality;
-
Foster increased collaboration between biologic, medical, and social sciences in order to better understand the sociology and psychology of disease reporting and compliance. Biologic and medical sciences alone are not enough;
-
Focus on risk management rather than disease eradication or “zero risk”;
-
Adopt and implement science-based regulations for BSE and other emerging diseases building on international standards;
-
Build public-private partnerships to address emerging diseases on a global, rather than a national, scale;
-
Recognize that all animal health issues are public health issues because of
-
their direct effects on human heath and their indirect effects on human well-being through their psychologic, economic, and ecologic consequences; and
-
Break down the silos that separate various professions and different stages of the food system (agriculture, processing, distribution, retail, wholesale, and the consumer) in order to aggressively promote transdisciplinary approaches to animal and public health.
REFERENCES
Alper T, Cramp WA, Haig DA, Clarke MC. 1967. Does the agent of scrapie replicate without nucleic acid? Nature 214(90):764–766.
Alpers MP. 1968. Kuru: Implications of its transmissibility for the interpretation of its changing epidemiological pattern. In: Bailey OT, Smith DE, Eds. The Central Nervous System: Some Experimental Models of Neurological Diseases. Baltimore, MD: Williams and Wilkins Company. Pp. 234–251.
Alter M, Kahana E. 1976. Creutzfeldt-Jakob disease among Lybian Jews in Israel. Science 192 (4238):428.
Anderson RM, Donnelly CA, Ferguson NM, Woolhouse ME, Watt CJ, Udy HJ, MaWhinney S, Dunstan SP, Southwood TR, Wilesmith JW, Ryan JB, Hoinville LJ, Hillerton JE, Austin AR, Wells GA. 1996. Transmission dynamics and epidemiology of BSE in British cattle. Nature 382(6594):779–788.
Andreoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, Lugan S, Corbiere F, Ferre P, Foucras G, Laude H, Eychenne F, Grassi J, Schlecher F. 2004. PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nature Medicine 10(6):591–593.
Baker HF, Ridley RM, Wells GAH. 1993. Experimental transmission of BSE and scrapie to the common marmoset. Veterinary Record 132(16):403–406.
Bartz JC, Kincaid AE, Bessen RA. 2003. Rapid prion neuroinvasion following tongue infection. Journal of Virology 77(1):583–591.
Bateman D, Hilton D, Love S, Zeidler M, Beck J, Collinge J. 1995. Sporadic Creutzfeldt-Jakob disease in a 18-year-old in the UK [Letter]. Lancet 346(8983):1155–1156.
Bellinger-Kawahara C, Cleaver JE, Diener TO, Prusiner SB. 1987a. Purified scrapie prions resist inactivation by UV irradiation. Journal of Virology 61(1):159–166.
Bellinger-Kawahara C, Diener TO, McKinley MP, Groth DF, Smith DR, Prusiner SB. 1987b. Purified scrapie prions resist inactivation by procedures that hydrolyze, modify, or shear nucleic acids. Virology 160(1):271–274.
Bosque PJ, Ryou C, Telling G, Peretz D, Legname G, DeArmond SJ, Prusiner SB. 2002. Prions in skeletal muscle. Proceedings of the National Academy of Sciences 99(6):3812–3817.
Brachmann A, Baxa U, Wickner RB. 2005. Prion generation in vitro: Amyloid of Ure2p is infectious. EMBO Journal 24(17):3082–3092.
Britton TC, Al-Sarraj S, Shaw C, Campbell T, Collinge J. 1995. Sporadic Creutzfeldt-Jakob disease in a 16-year-old in the UK [Letter]. Lancet 346(8983):1155.
Bruce ME, Dickinson AG. 1987. Biological evidence that the scrapie agent has an independent genome. Journal of General Virology 68(Pt. 1):79–89.
Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389(6650):498–501.
Buschmann A, Groschup MH. 2005. Highly bovine spongiform encephalopathy-sensitive transgenic mice confirm the essential restriction of infectivity to the nervous system in clinically diseased cattle. Journal of Infectious Diseases 192(5):934–942.
Buschmann A, Pfaff E, Reifenberg K, Müller HM, Groschup MH. 2000. Detection of cattle-derived BSE prions using transgenic mice overexpressing bovine PrP(C). Archives of Virology 16(suppl): 75–86.
CDC (Centers for Disease Control and Prevention). 1996. Surveillance for Creutzfeldt-Jakob disease—United States. Morbidity and Mortality Weekly Report 45(31):665–668.
Chapman J, Ben-Israel J, Goldhammer Y, Korczyn AD. 1994. The risk of developing Creutzfeldt-Jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology 44(9):1683–1686.
Chesebro B. 2004. Biomedicine. A fresh look at BSE. Science 305(5692):1918–1921.
Collinge J. 1999. Variant Creutzfeldt-Jakob disease. Lancet 354(9175):317–323.
Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383(6602):685–690.
Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. 2006. Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet 367(9528): 2068–2074.
Collins S, Boyd A, Fletcher A, Gonzales M, NcLean CA, Byron K, Masters CL. 2000. Creutzfeldt-Jakob disease: Diagnostic utility of 14-3-3 protein immunodetection in cerebrospinal fluid. Journal of Clinical Neuroscience 7(3):203–208.
Collins S, Boyd A, Lee JS, Lewis V, Fletcher A, McLean CA, Law M, Kaldor J, Smith MJ, Masters CL. 2002. Creutzfeldt-Jakob disease in Australia 1970–1999. Neurology 59(9):1365–1371.
Collins SJ, Lawson VA, Masters CL. 2004. Transmissible spongiform encephalopathies. Lancet 363(9402):51–61.
Dickinson AG, Fraser H, Outram GW. 1975. Scrapie incubation time can exceed natural lifespan. Nature 256(5520):732–733.
Donnelly CA, Ferguson NM, Ghani AC, Anderson RM. 2002. Implications of BSE infection screening data for the scale of the British BSE epidemic and current European infection levels. Proceedings. Biological Sciences/The Royal Society 269(1506):2179–2190.
Foster J, Goldmann W, Parnham D, Chong A, Hunter N. 2001. Partial dissociation of PrPSc deposition and vacuolation in the brains of scrapie and BSE experimentally affected goats. Journal of General Virology 82(Pt. 1):267–273.
Gajdusek DC. 1962. Kuru: An appraisal of five years of investigation. Eugenics Quarterly 9:69–74.
Gajdusek DC. 1977. Unconventional viruses and the origin and disappearance of kuru. Science 197(4307):943–960.
Gajdusek DC, Zigas V. 1957. Degenerative disease of the central nervous system in New Guinea. The endemic occurrence of “kuru” in the native population. New England Journal of Medicine 257(20):974–978.
Gajdusek DC, Gibbs CJ Jr, Alpers M. 1966. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 209(25):794–796.
GAO (Government Accountability Office). 2005 (February). Mad Cow Disease: FDA’s Management of the Feed Ban Has Improved, but Oversight Weaknesses Continue to Limit Program Effectiveness. GAO-05-101. Washington, D.C.
Ghani AC, Ferguson NM, Donnelly CA, Anderson RM. 2000. Predicted vCJD mortality in Great Britain. Nature 406(6796):583–584.
Glasse RM. 1967. Cannibalism in the kuru region of New Guinea. Transactions of the New York Academy of Sciences 29(6):748–754.
Glatzel M, Abela E, Maissen M, Aguzzi A. 2003. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. New England Journal of Medicine 349(19):1812–1820.
Goldfarb LG, Brown P, Mitrova E, Cervenakova L, Goldin L, Korczyn AD, Chapman J, Galvez S, Cartier L, Rubenstein R, et al. 1991. Creutzfeldt-Jacob disease associated with the PRNP codon 200Lys mutation: An analysis of 45 families. European Journal of Epidemiology 7(5):477–486.
Goldmann W, Hunter N, Manson J, Hope J. 1990. The PrP gene of the sheep, a natural host of scrapie. Proceedings of the VIIIth International Congress of Virology. Berlin, August 26–31. P. 284.
Goldmann W, Hunter N, Martin T, Dawson M, Hope J. 1991. Different forms of the bovine PrP gene have five or six copies of a short, G-C-rich element within the protein-coding exon. Journal of General Virology 72(Pt. 1):201–204.
Grassi J, Comoy E, Simon S, Creminon C, Frobert Y, Trapmann S, Schimmel H, Hawkins SA, Moynagh J, Deslys JP, Wells GA. 2001. Rapid test for the preclinical postmortem diagnosis of BSE in central nervous system tissue. Veterinary Record 149(19):577–582.
Hadlow WJ. 1959. Scrapie and kuru [Letter]. Lancet 2:289–290.
Hill A, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. 1997. The same prion strain causes vCJD and BSE. Nature 389(6650):448–450.
Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. 2000. Species-barrier-independent prion replication in apparently resistant species. Proceedings of the National Academy of Sciences 97(18):10248–10253.
Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW. 2004. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. Journal of Pathology 203(3):733–739.
Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. 1990. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 250(4987):1587–1590.
Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D, Scarlato G, Abramsky O, Prusiner SB, Gabizon R. 1991. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. New England Journal of Medicine 324(16):1091–1097.
Hsiao KK, Groth D, Scott M, Yang SL, Serban H, Rapp D, Foster D, Torchia M, DeArmond SJ, Prusiner SB. 1994. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proceedings of the National Academy of Sciences 91(19): 9126–9130.
Ironside JW. 1997. The new variant form of Creutzfeldt-Jakob disease: A novel prion protein amyloid disorder. Amyloid: International Journal of Clinical and Experimental Investigation 4:66–69.
Kaneko K, Ball HL, Wille H, Zhang H, Groth D, Torchia M, Tremblay P, Safar J, Prusiner SB, DeArmond SJ, Baldwin MA, Cohem FE. 2000. A synthetic peptide initiates Gerstmann-Sträussler-Scheinker (GSS) disease in transgenic mice. Journal of Molecular Biology 295(4): 997–1007.
Kimberlin RH. 1982. Scrapie agent: Prions or virinos? Nature 297(5862):107–108.
Klug GM, Boyd A, Lewis V, Kvasnicka M, Lee JS, Masters CL, Collins SJ. 2005. Creutzfeldt-Jakob disease: Australian surveillance update to 31 December 2004. Communicable Diseases Intelligence 29(3):269-271.
Kübler E, Oesch B, Raeber AJ. 2003. Diagnosis of prion diseases. British Medical Bulletin 66: 267–279.
Lasmézas CI, Deslys J-P, Demaimay R, Adjou KT, Lamoury F, Dormont D, Robain O, Ironside J, Hauw JJ. 1996. BSE transmission to macaques. Nature 381(6585):743–744.
Last JM, ed. 1983. A Dictionary of Epidemiology. Oxford: Oxford University Press.
Last JM, ed. 2001. A Dictionary of Epidemiology. Oxford: Oxford University Press.
Lee IS, Long JR, Prusiner SB, Safar JG. 2005. Selective precipitation of prions by polyoxometalate complexes. Journal of the American Chemical Society 127(40):13802–13803.
Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. 2004. Synthetic mammalian prions. Science 305(5684):673–676.
Legname G, Nguyen HO, Baskakov IV, Cohen FE, DeArmond SJ, Prusiner SB. 2005. Strain-specified characteristics of mouse synthetic prions. Proceedings of the National Academy of Sciences 102(6):2168–2173.
Lewis V, Hill AF, Klug GM, Boyd A, Masters CL, Collins SJ. 2005. Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology 65(1):113–118.
Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. 2004. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363(9407): 417–421.
Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. 2002. Amyloid aggregates of the HET-s prion protein are infectious. Proceedings of the National Academy of Sciences 99(11):7402–7407.
Minor P, Newham J, Jones N, Bergeron C, Gregori L, Asher D, van Engelenburg F, Stroebel T, Vey M, Barnard G, Head M; WHO Working Group on International Reference Materials for the Diagnosis and Study of Transmissible Spongiform Encephalopathies. 2004. Standards for the assay of Creutzfeldt-Jakob disease specimens. Journal of General Virology 85(Pt. 6):1777–1784.
National Advisory Committee on SARS and Public Health. 2003. Learning from SARS—Renewal of Public Health in Canada. Ottawa: Health Canada. [Online]. Available: http://www.phac-aspc.gc.ca/publicat/sars-sras/naylor/ [accessed July 25, 2006].
National CJD Surveillance Unit UK. 2006. CJD Statistics. [Online]. Available: http://www.cjd.ed.ac.uk/figures.htm [accessed March 17, 2006].
OIE (Office International des Epizooties). 2006. Bovine Spongiform Encephalopathy (BSE). [Online]. Available: http://www.oie.int/eng/info/en_esb.htm [accessed July 26, 2006].
Pan K, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. 1993. Conversion of a-helices into b-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences 90(23):10962–10966.
Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. 2004. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364(9433):527–529.
Phillips NA, Bridgeman J, Ferguson-Smith M. 2000. The BSE Inquiry. Vol 2. London: Stationery Office.
Prusiner SB. 2004a. Detecting mad cow disease. Scientific American 291(1):86–93.
Prusiner SB. 2004b. Prion Biology and Diseases. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Ricketts M. 2005 (October 26). Session VI: What Are the Incentives and Disincentives Associated with Disease/Contamination Reporting? Impacts on Human Health and International Trade—BSE as a “Case Study.” Presentation at the Forum on Microbial Threats Workshop Meeting, Foodborne Threats to Health: The Policies and Practices of Surveillance, Prevention, Outbreak Investigations, and International Coordination, Washington, D.C., Institute of Medicine, Forum on Microbial Threats.
Roos R, Gajdusek DC, Gibbs CJ Jr. 1973. The clinical characteristics of transmissible Creutzfeldt-Jakob disease. Brain 96(1):1–20.
Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. 1998. Eight prion strains have PrPSc molecules with different conformations. Nature Medicine 4(10):1157–1165.
Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller B, DeArmond SJ, Prusiner SB. 2005a. Diagnosis of human prion disease. Proceedings of the National Academy of Sciences 102(9):3501–3506.
Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D. 2005b. Search for a prion-specific nucleic acid. Journal of Virology 79(16):10796–10806.
Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M, Koehler R, Tremblay P, Walther D, Cohen FE, DeArmond SJ, Prusiner SB. 1997. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proceedings of the National Academy of Sciences 94(26):14279–14284.
Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. 1999. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proceedings of the National Academy of Sciences 96(26):15137–15142.
Scott MR, Peretz D, Nguyen HO, DeArmond SJ, Prusiner SB. 2005. Transmission barriers for bovine, ovine, and human prions in transgenic mice. Journal of Virology 79(9):5259–5271.
Sparrer HE, Santoso A, Szoka FC Jr, Weissman JS. 2000. Evidence for the prion hypothesis: Induction of the yeast [PSI+] factor by in vitro-converted Sup35 protein. Science 289(5479):595–599.
Spencer MD, Knight RS, Will RG. 2002. First hundred cases of variant Creutzfeldt-Jakob disease: Retrospective case note review of early psychiatric and neurological features. British Medical Journal 324(7352):1479–1482.
Spudich S, Mastrianni JA, Wrensch M, Gabizon R, Meiner Z, Kahana I, Rosenmann H, Kahana E, Prusiner SB. 1995. Complete penetrance of Creutzfeldt-Jakob disease in Libyan Jews carrying the E200K mutation in the prion protein gene. Molcular Medicine 1(6):607–613.
Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature 428(6980):323–328.
Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. 1996. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes & Development 10(14):1736–1750.
Thomzig A, Kratzel C, Lenz G, Kruger D, Beekes M. 2003. Widespread PrPSc accumulation in muscles of hamsters orally infected with scrapie. EMBO Reports 4(5):530–533.
Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB, Safar JG. 2004. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. Journal of Virology 78(4):2088–2099.
Valleron AJ, Boelle PY, Will R, Cesbron JY. 2001. Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. Science 294(5547):1726–1728.
Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358(9277):171–180.
Ward HJT, Everington D, Sanchez-Juan P, Smith A, Cooper S, Heath C, Knight RSG, Will RG. 2003 (October 8-10). Secondary Transmission of Variant Creutzfeldt-Jakob Disease? Presentation at the International Prion Conference, Munich.
Ward HJ, Everington D, Cousens SN, Smith-Bathgate B, Leitch M, Cooper S, Heath C, Knight RS, Smith PG, Will RG. 2006. Risk factors for variant Creutzfeldt-Jakob disease: A case-control study. Annals of Neurology 59(1):111–120.
Weissmann C. 1991. A “unified theory” of prion propagation. Nature 352(6337):679–683.
Wells GAH. 2002 (January 10–11). Report on TSE Infectivity Distribution in Ruminant Tissues (State of Knowledge, December 2001). Brussels: European Commission.
Will RG, Ironside JW, Zeidler M, Cousens S, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347(9006):921–925.
Will RG, Alpers MP, Dormont D, Schonberger LB. 2004. Infectious and sporadic prion diseases. In: Prusiner SB, Ed. Prion Biology and Diseases. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. Pp. 629–671.
Yamakawa Y, Hagiwara K, Nohtomi K, Nakamura Y, Nishijima M, Higuchi Y, Sato Y, Sata T; Expert Committee for BSE Diagnosis, Ministry of Health, Labor and Welfare of Japan. 2003. Atypical proteinase K-resistant prion protein (PrPres) observed in an apparently healthy 23-month-old Holstein steer. Japanese Journal of Infectious Diseases 56(5-6):221–222.
Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, Bernheimer H, Cardone F, Delasnerie-Laupretre N, Caudrado Corrales N, Ladogana A, Bodemer M, Fletcher A, Awan T, Ruiz Bremon A, Budka H, Laplanche JL, Will RG, Poser S. 2000. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 55(6):811–815.





































