
4
The Carbon Cycle—Controls on Atmospheric CO2 and Climate in the Geologic Past
MICHAEL A.ARTHUR
University of South Carolina
INTRODUCTION
The chemical-biogenic sediments deposited in marine settings act as a major buffer for geologically short-term and long-term excursions in atmospheric and ocean chemistry, particularly those of the gases CO2 and O2. The chemical and stable isotopic composition of the chemical-biogenic sediments also reflects to some extent the chemistry of the seawater from which the sediments were precipitated. Therefore, the marine sedimentary record can be studied to obtain a record of the chemical history of seawater. This record can be compared to changes in other phenomena such as sea level, positions of land masses, tectonic events, and particularly climate (e.g., Fischer and Arthur, 1977; Berger, 1977, 1979; Berggren and Hollister, 1977).
A common assumption in studies of geochemical cycles is that the ocean reservoir maintains a relatively constant composition (chemical uniformity as opposed to a chemical steady state with respect to equilibria) through time. Although this is a sometimes necessary and simplifying assumption, in the absence of data to the contrary, it is in reality not too satisfactory. There is undoubtedly a complex interplay among atmospheric composition, climate, continental weathering, and the riverine flux of dissolved chemical species to the oceans. In turn, these factors influence or are influenced by changes in rates and mechanisms of ocean circulation and by changes in biological and nonbiological extraction and storage of chemical constituents in marine sediments. The chemical loops just described are to various degrees interdependent. Major perturbations in one flux into, or out of, the system, because of climatic or tectonic forcing (including relative changes in sea level), will spread to the others through a series of feedback mechanisms. The marine sedimentary record—changes in lithology, chemistry, stable isotopic composition, and the biotic constituents of pelagic sediments—is a monitor of changes in ocean chemistry, and through consideration of the feedback mechanism one can deduce possible variations in global climate. The extent to which we can recognize these variations and their causes or effects is dependent on the completeness of the sedimentary and fossil record, on a stratigraphic framework and absolute-time scale adequate for correlations and estimation of the leads and lags in the system, and on the geochemical tools that we have available to us.
The purpose of this chapter is to outline briefly the role of
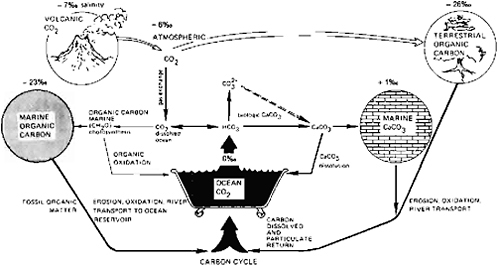
ocean chemistry, mainly through its participation in the carbon cycle (Figure 4.1) in buffering changes in pCO2 and consequent changes in climate. The discussion deals first with the extent of possible excursions in atmospheric pCO2 and their causes, the operation of the geologic carbon cycle, and the feedback mechanisms that appear to help damp pCO2 fluctuations (i.e., long- and short-term buffers); then briefly with the record of Cenozoic paleo-oceanography and climate and the possible influence of pCO2 changes; and finally with geologically sudden events, such as the isolation and evaporation of an ocean basin and their possible influence on global climate.
CARBON DIOXIDE AND CLIMATE CHANGE
It has been difficult to detect any climatic effect of the increased atmospheric CO2 over the last several decades due to burning of fossil fuels, largely because the predicted effects are within the limits of natural climatic noise (e.g., see Madden and Ramanathan, 1980). However, a CO2-greenhouse effect is expected, and various climatic models have been constructed to estimate the magnitude of climatic warming associated with excursions in atmospheric pCO2 (see Marland and Rotty, 1979; NRC Climate Research Board, 1979, for recent reviews). The commonly accepted range for the average global-temperature rise associated with a doubling of atmospheric pCO2 is about 1.5–3.0°C, although estimates of 0.7–9.6°C have been published. The major problem of determining the potential magnitude of the effect by climate modeling is that feedback mechanisms, such as changes in cloudiness and associated albedo changes, may be improperly modeled. Manabe and Wetherald (1975, 1980), for example, used a sophisticated three-dimensional general-circulation model, but in their early model the degree of cloudiness was fixed, topography was idealized, and there was no seasonal variation imposed. For doubling of atmospheric CO2 they predicted a 2.9°C increase in global-mean surface temperatures (greater at high latitudes) and an increase in evaporation-precipitation.
These models would not apply well to the Cretaceous or early Cenozoic Earth, when continental configurations were different. global-mean temperatures were much higher, and the oceans were much warmer overall. All in all, the effects of increased pCO2 in the geologic past are difficult to estimate, but for the sake of discussion we will adopt a change of several degrees for a doubling of pCO2. This temperature change may, of course, be grossly in error, as there is still difficulty in discerning climatic change because of variations in atmospheric CO2 versus other factors, such as changes in the latitudinal distribution of continents and their effects on the distribution of albedo, on patterns of surface- and deep-ocean currents, or on both. These problems are briefly dealt with below after an examination of possible natural variations in atmospheric CO2.
NATURAL SOURCES AND VARIABILITY OF CO2
We also must consider natural sources of CO2 to the atmosphere. Berner et al. (1981) have demonstrated glacial-interglacial pCO2 changes on the basis of changes in the CO2 gas pressure in ice. The pCO2 apparently was lower during the last glacial. In this regard, Berger (Chapter 3) has discussed the short-term (i.e., 10,000 yr) sudden expulsion of CO2 that could result from glacial-interglacial changes in the residence time of deep water. Sudden or more rapid overturn of “old” deep water that might occur during the transition from glacial to interglacial periods would inject large amounts of CO2 into the atmosphere [1014 to 1018 grams of carbon (g of C)]. Atmospheric CO2 also can change as the result of changes in overall temperature and salinity (as well as volume) of seawater. Cooling, a decrease in salinity, or both increase the solubility of CO2 in seawater and thereby reduce the pCO2 of the atmosphere (Table 4.1), at least on the short term. The glacial to interglacial warming of surface water would have had nearly twice the effect that the salinity decrease would have had, such that the pCO2 changes from this cause would be minor. However, a change from warm, saline surface- and deep-ocean water in the Eocene to colder, less saline water masses in the Oligocene (Berger, 1977) may have resulted in a more impor-
tant atmospheric CO2 decrease. Atmospheric CO2 must also have varied during glacial-interglacial cycles because of the changes in terrestrial biomass and soil carbon (Shackleton, 1977). The increased oxidation of soil carbon or humus could provide a large source of CO2 to the atmosphere. Broecker (1981) recently suggested that burial of massive amounts of organic carbon in shelf sediments during sea-level rise following glacial retreat could also be a mechanism for rapid lowering of pCO2 and removal of phosphate from the ocean. A pCO2 increase would follow because organic carbon burial rate would then decrease. The buried organic carbon could be oxidized during subsequent glacial lowering of sea level, in such a way that phosphate and CO2 are returned to the atmosphere. Models by Berger (Chapter 3), Shackleton (1977), and Broecker (1981) each are supported by variations in δ13C of pelagic microfossils. Currently, there is no available evidence to distinguish between the effects of the three models. These types of short-term CO2 pulses are not restricted to the Pleistocene, although they may be amplified at that time by the large cyclic variation in climate. We see evidence of similar possible exchanges of carbon dioxide and burial of organic matter reflected in δ13C values of carbonate across 105-yr cycles in the Cretaceous, for example (Figure 4.6). Could the intensity and rapidity of change between glacial and interglacial cycles be, in part, controlled or reinforced by these exchanges of CO2 (Berger, Chapter 3; Broecker, 1981)?
Oceanic fertility [the availability of phosphorous and nitrogen (Figure 4.2)] also may have changed over time periods of 1–10 million years (m.y.). Decreased fertility could drastically affect the rate of burial of marine organic matter and the ability of the ocean system to absorb “excess” atmospheric CO2 in this way. Atmospheric CO2 may have risen during low-fertility episodes (e.g., Tappan, 1968; Berger, 1977). Conversely, times of apparent high fertility [such as those during the deposition of large phosphate deposits (Arthur and Jenkyns, 1981)] would likely result in decreased atmospheric pCO2 because of increased burial of organic carbon in marine sediments.
Another possible major source of changes in atmospheric CO2 is volcanism. Vogt (1972, 1979) and Kennett and Thunell (1975) have suggested a major periodicity in volcanism during the Cretaceous-Cenozoic. Could this periodicity result in fluctuations of pCO2 and changes in global climate? At present, it is estimated, and this is a difficult estimate to make, that degassing of the Earth through volcanism emits 0.09×1015 g of C/yr as CO2 to the atmosphere (Holland, 1978). This amount makes up a predicted deficit caused by operation of the carbon cycle (see Table 4.2 and next section). A doubling of the rate of CO2 degassing would double atmospheric CO2 in about 10,000 yr if the CO2 is not compensated for by other feedback mechanisms. This flux is about 50 times less than the rate of CO2 addition from the burning of fossil fuels over the past few decades. Thus, over time periods of several million years, increased volcanic discharge could be important to climate. However, the major question here is whether the cooling effect of aerosols ejected into stratosphere during episodes of explosive volcanism would offset the warming effect of increased pCO2 (e.g., Pollack et al., 1976; Pollack, 1979).
TABLE 4.1 Estimated Flux Rate, Mass, and Isotopic Changes in the Ocean during CO2 Transfersa
|
Type of Flux |
Rate and/or Amount of Carbon Transferred (Duration)b |
Δ δ13C Ocean (Total Dissolved Carbon)b |
|
Volcanic CO2 addition (doubling of estimated steady-state degassing rate) |
0.8×1014 g of C/yr (over 8×104 yr) |
−1 ‰ (for −6×1018 g of C addition) |
|
Oxidation of soil carbon (or burning of fossil fuels) |
5.0×1014 g of C/yr (over 3×103 yr) |
−1 ‰ (for ~1.5×1018 g of C addition) |
|
Net transfer of ocean TDC to marine organic carbon burial |
3.0×1014 g of C/yr (over 5×103 yr) (<4.3% of ocean TDC; or ~60% of annual TDC input by rivers) |
+1 ‰ (for 1.5×1018 g of C depletion) |
|
Net transfer to and from carbonate reservoirs |
~0 |
~0 |
|
aValues determined in order to equal a 1‰ change in ocean TDC over steady-state flux rates (estimated from Tables 4.2 and 4.3). bChanges in total C content of atmosphere as CO2 induced by changes in temperature and salinity assuming constant surface seawater volume equal to present and 300×10−6 atm of CO2: ΔpCO2/ΔC (for cooling 1°C)=−24×1015 g of C (~4% of pCO2)/°C; ΔpCO2/Δ salinity (for salinity decrease of 1‰)=−30×1015 g of C (~5% of pCO2)/‰ salinity. |
||
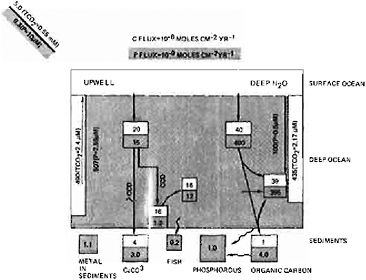
FIGURE 4.2 Linkage between the carbon and phosphorous cycles in the ocean. The availability of phosphorous as a nutrient limits the amount of organic carbon and carbonate that can be produced and buried in the oceans (after Froelich et al., 1981). The importance of different phosphorous (and organic carbon-carbonate) sinks may have varied significantly in the past.
TABLE 4.2 Approximate Present-Day Sizes and Isotopic Compositions of Carbon Reservoirsa
|
Reservoir |
Mass of Carbon in 1017 g |
Average δ13C (‰) |
|
Ocean-dissolved carbon (primarily HCO3) |
350.0 |
0 |
|
Annual nct marine carbonate (biogenic CaCO3) |
0.0014 |
+1.0 |
|
Carbonate sediment reservoir |
610,000 |
+1.0 |
|
Annual net marine organic carbon (biogenic |
0.00073 |
−23 |
|
0.018 |
−23 |
|
|
Organic carbon sediment reservoir |
130,000 |
−23 |
|
Atmospheric CO2 |
6.0 |
−7 |
|
Land-plant biomass (Corg) soil “humus” |
8.4 |
−26 |
|
12.0 [10.5–30] |
−25 |
|
|
Stream flux (annual) |
|
|
|
Dissolved inorganic carbon |
0.0047 |
−6.5 |
|
Dissolved organics |
0.0032 |
−26 |
|
Particulate organics |
0.0007 |
−26 |
|
Volcanic gases (mantle carbon) |
(?) 0.0008 |
−7 |
|
(see Table 4.3) |
|
|
|
aModified from Scholle and Arthur, 1980. |
||
LONG-TERM STATUS AND FEEDBACK MECHANISMS IN THE CYCLING OF CARBON
A number of researchers have suggested that the chemistry of the Earth’s atmosphere and oceans has remained constant within certain limits to account for the continuity of life and the relatively narrow excursions in the composition of chemical sediments through geologic time (e.g., Holland, 1972, 1974, 1978; Garrels and Perry, 1974). To maintain this long-term geochemical status there must be a variety of efficient feedback mechanisms that buffer expected variations in geochemical cycles due to changes in intensity of tectonism, in continental area and mean altitude, and in climate. These buffering mechanisms may be short term, long term, or both. The operation of the carbon cycle is one of the most important in this regard as it ultimately controls levels of oxygen and carbon dioxide in the atmosphere. The carbon cycle is responsive to changes in climate and oceanography and is coupled to nutrients cycles. If climate is influenced by varying levels of atmospheric CO2, for example, controls on atmospheric pCO2 levels are therefore important to understand. This understanding must include adequate knowledge of CO2 sources and source strengths, the allowable excursions of pCO2 levels within the limitations imposed on the operation of global carbon cycle by nutrient availability, net productivity and burial of organic carbon, carbonate sedimentation and dissolution, weathering rates and reactions (Figures 4.1 and 4.2), and the rates of CO2 excursions versus the kinetics of CO2-consuming processes (see Table 4.3).
We know, for example, that the increase in atmospheric CO2 levels from burning of fossil fuels and other activities of man has not been immediately taken up [perhaps 48 percent of the total CO2 released by burning of fossil fuels has been removed from the atmosphere by various mechanisms (Oeschger et al., 1975)]; the CO2 is produced at a rate of about 5×1015 to 6× 1015 g of C/yr (Rotty, 1977) and increases by about 4 percent per year. Thus, the feedback mechanisms that must act to restore balance are not entirely efficient on the order of a few tens of years. The major control mechanisms on time scales of 102 to 106 yr are probably weathering reactions, carbonate dissolution in the oceans, and burial of organic carbon in sediments (through changes in net primary productivity or enhanced preservation in sediments). The efficiency of all of these mechanisms is limited by one or more factors. We have only a partial understanding of the carbon-cycle system. Parts of the system can be isolated, but the whole is difficult to integrate. The next section gives an example of how we might approach the CO2 problem as a subsystem of the global carbon cycle. Short-term buffering mechanisms, such as uptake as dissolved CO2 in seawater and uptake in increased terrestrial biomass, will not be considered.
Weathering Reactions as a Feedback Mechanism
Weathering reactions, such as decomposition of silicates and carbonates by acid soil, groundwaters, and surface runoff rich in CO2, may be a major long-term sink for atmospheric CO2.
TABLE 4.3 Annual Gains and Losses of Atmospheric Carbon (in units of 1014 g/yr) Due to Weathering and Sedimentationa
An estimated 10 percent (0.43×1015 g of C/yr) of the present annual CO2 flux from the atmosphere is consumed in weathering reactions (Holland, 1978). If atmospheric CO2 levels increased greatly, rainfall possibly would become more acidic and the amount of CO2 in soil horizons would increase, thereby leading to higher rates of weathering. However, the increased weathering would lead to a rise in alkalinity and carbonate ion concentration of river water carried to the ocean, which, depending on the concentration of total CO2 and carbonate ion in seawater, will precipitate and deposit carbonates eventually. Carbonate deposition results in a net flux of CO2 back to the atmosphere. At present, the net deposition of carbonates results in an estimated gain of about 0.22× 1015 g of C/yr to the atmosphere and deposition of silicate minerals results in a gain of about 0.16×1015 g of C/yr, so that there appears to be a net loss of CO2 (about 0.05×1015 g of C/yr) from the atmosphere owing to the cycle of weathering reactions to silicate and carbonate reconstitution (see Table 4.3).
Weathering may be an important sink for CO2, but it must operate on longer time scales (i.e., a million years) to control atmospheric CO2 levels, and it is dependent on such things as continental area, rainfall, temperature, and soil carbon and moisture. However, there is opposition to this idea (E. Sundquist, U.S. Geological Survey, personal communication, 1980), and most weathering reactions may be dependent entirely on the concentration of soil CO2. If higher pCO2 results in global warming, possibly in increased rainfall at some latitudes, and in CO2 fertilization of higher plants, then this combination of effects might ultimately result in larger amounts of soil carbon and greater production of soil CO2, which, in turn, would promote higher rates of weathering. The controls on soil-CO2 concentrations and the concept of increasing acidity of rainfall with increasing atmospheric CO2 require more study,
Organic Carbon Production and Burial
The biosphere also exerts a tremendous influence on atmospheric CO2 levels. All the atmospheric CO2 is probably cycled through plants once in every 10 years or less. There is less carbon locked up in the biosphere, including soil humus (a total of more than 2000×1015 g of C), than in CO2 dissolved in oceans (Table 4.2) but more than in the atmosphere. The net burial of organic carbon in sediments accounts for a loss of CO2 from the atmosphere (and a gain in oxygen). Today, this burial accounts for the net withdrawal of about 0.12×1015 g of C/yr of atmospheric CO2 (Holland, 1978). This transfer of CO2 more than offsets the estimated gain in CO2 by oxidative weathering of old organic matter of about 0.09×1015 g of C/yr (Holland, 1978). Short-term increases in the size of the marine or terrestrial biota and soil carbon could accommodate short-term increases in atmospheric CO2 (e.g., Broecker et al., 1979), whereas longer-term control would have to be exerted by increasing the rate of organic carbon burial in sediments. The net carbon-burial increase could occur in peatlands, by an increase in forest-litter accumulation, in coal swamps and lakes, or in marine settings—especially in estuarine environments and on continental slopes. The terrestrial sinks, with the exception of coal (McLean, 1978a), would be relatively short-term reservoirs because of their general susceptibility of subaerial exposure and oxidation. The marinesediment reservoir is the probable long-term stable sink for organic matter. The major control in productivity of organic matter is nutrient availability.
Changes in nutrient supply may lead, in part, to changes in the burial rate of organic matter. The burial rate of organic matter is also a function of oxygen availability and circulation rates of deep-water masses. Burial of organic matter removes at least part of the nutrients used in organic synthesis, and, without sufficient available nutrients (mainly phosphorous and nitrogen), the organic part of the carbon cycle cannot respond to a CO2 increase. A large part of the nutrient flux in the oceans today is regenerated from organic-matter oxidation in the water column and within sediments (see Figure 4.2). Burial of terrestrial-organic matter (land plants) is a more efficient CO2-fixing mechanism in terms of nutrient usage; the average C:N:P ratio is about 510:4.2:1 as opposed to 106:16:1 in unoxidized average marine-organic matter, although much of the P and N may be regenerated before burial. Thus, times of widespread coal deposition (e.g., the Carboniferous) would have efficiently fixed much atmospheric CO2 into sediments. Burial of terrestrial organic matter in marine sediments is also important. Evidence suggests that, at present, relatively little land-derived organic matter reaches the deep sea beyond the shelf (e.g., Hunt, 1970; Sackett, 1964; Sackett and Thompson, 1963; Sackett et al., 1965; Rogers and Koons, 1969), except in the anoxic Black Sea, which is a large sink for terrigenious organic carbon (e.g., Simoneit, 1977). However, this lack of deposition of terrigenous organic carbon in marine environments may not be the norm because the rapid Holocene rise in sea level has influenced trapping of terrigenous organic matter in nearshore settings. There also may be difficulty in recognizing some terrestrially derived organic matter in marine
sediments after early diagnosis on the basis of δ13C values alone. Evidence from Deep Sea Drilling Project (DSDP) drill sites suggests that during the Early Cretaceous much more terrigenous organic carbon was preserved in deeper marine sediments than at present.
There is some evidence, as yet inconclusive, that greater ambient CO2 concentrations may fertilize higher plants and may either increase net primary productivity or decrease rates of transpiration (see Strain, 1978, for review). This evidence suggests that land plant productivity might increase with increasing pCO2, but, as Lemon (1977) pointed out, all data bearing on this problem have come from controlled experiments, and there is no available evidence that natural ecosystems could respond in this way; the process is nutrient-limited. However, this speculation is intriguing, and the mechanism may be of great importance.
In general, the operation of the organic portion of the carbon cycle is critical to controlling pCO2. Nutrients must be available to allow relatively unrestricted changes in biomass and organic carbon burial in response to increased pCO2. There is some evidence that nutrient levels in the sea, rates of cycling of nutrient phosphate and nitrogen through ocean waters, or both may have varied significantly in the past (e.g., Piper and Codispoti, 1975; Arthur and Jenkyns, 1981) and that the circulation patterns, supply to surface waters by upwelling, and productivity changed as well (e.g., van Andel et al., 1975; Berger, 1979). Thus, the evidence is not clear that sufficient nutrients are available always to allow the biota to buffer pCO2 increases. There is possibly a feedback link that provides an increased nutrient flux to the oceans by increased rates of weathering during pCO2 increase. However, this mechanism involves a time lag, possibly on the order of several million years, and just how weathering rates change with increased atmospheric CO2 is not yet clear. Times of increased elastic sediment flux to the oceans, possibly From increased tectonic activity and erosion of high-standing land masses, also may result in increased net organic carbon burial because high sedimentation rates enhance organic-matter preservation (Muller and Suess, 1979).
Effectiveness of Carbonate Dissolution in CO2Buffering
A final major mechanism of atmospheric CO2 buffering, and one that could be expected to operate over a time scale of 100,000 years or less (the residence time of carbon in the ocean is about 10,000 years) is dissolution of carbonate minerals in the deep sea. This mechanism has been discussed at length in the literature (e.g., Broecker and Takahashi, 1978; Broecker et al., 1979) and will not be dealt with in detail here. The main area of carbonate dissolution is below the lysocline, a level of increased undersaturation and increased rate of dissolution of calcite in deep-water masses. It is estimated that there are today at least 3600×1015 g of C as carbonate readily available for dissolution to about 10-cm depth in deep-sea sediments, with a probable addition of 1-2×1015 g of C/yr from annual production. However, the efficiency of increased oceanic dissolved CO2 levels in dissolution of carbonate depends on the rate-limiting process in dissolution (e.g., burrow-stirring, breakdown of organic coatings, and saturation of pore waters) and on the rate of overturn of oceanic deep waters (currently with a residence time of about 1200 years) that controls the rate of delivery of dissolved CO2 from the atmosphere to the depths for buffering by carbonate dissolution.
Relatively soluble carbonate minerals such as aragonite and high-Mg calcite deposited in shallow-water environments might provide another small and short-term buffer for increased pCO2 (e.g., Broecker et al., 1979), but the surface ocean is at present everywhere at least 1.7 times saturated with respect to aragonite. According to Broecker et al. (1979) the atmospheric CO2 would have to increase at least 5.3 times and 8.5 times that of today to cause undersaturation in ocean surface waters for aragonite and calcite, respectively. Therefore, dissolution of shallow-water carbonate sediments is not likely to be too significant as a sink for CO2 at present and was relatively insignificant in the past unless major pCO2 increases were allowed. Also, if high-Mg calcite, aragonite, or both were dissolved and reprecipitated as low-Mg calcite, then there is no net CO2 consumption.
Because dissolution of 1 mole of carbonate consumes 1 mole of CO2, only about 40,000 yr would be required to dissolve the estimated available deep-water carbonate if the estimated rate of CO2 degassing by volcanism were to double (e.g., an increase of about 0.09×1015 g of C/yr). However, an increase in dissolution rate equal to about 10 percent of the carbonate produced in surface waters each year would also balance the aforementioned CO2 flux increase. Thus, all other factors being equal, we might expect to see a decrease in the net deep-sea carbonate-accumulation rate with an increase in atmospheric CO2, as well as an apparent shallowing of the carbonate saturation horizons [the lysocline and calcium carbonate compensation depth (CCD), e.g., Berger, (1977)].
Carbon Isotopes in Pelagic Carbonates as Constraints on Carbon Cycling
Consideration of the possible past excursions of atmospheric CO2 has been made by examining constraints on changes in ocean chemistry imposed by the composition of marine chemical sediments—mainly evaporites and carbonates— through time and by examining changes in the carbon isotopic composition (δ13C values) of limestones through time. Holland (1972, 1974, 1978) has shown that during the Phanerozoic, surface seawater has always been saturated or supersaturated with respect to calcite and aragonite. The probability is that the concentration of Ca2+, SO42−, and HCO3− have never varied by more than a factor of 2 to 3 in either direction from their present values. Holland has shown that all of these factors constrain pCO2 variations only by about a factor of 103 to 104—that is, that pCO2 has probably always remained between 101.5 and 105.5 atm (currently 103.5 atm).
The δ13C values of marine limestones are presumed to reflect the carbon isotopic composition of total dissolved carbon in the ocean reservoir (e.g., Broecker, 1974). The average isotopic composition of the reservoir generally represents the balance between deposition of carbonates and organic carbon. The two
have very different carbon isotopic compositions (average organic carbon=−23 ‰ and average carbonate=+1 ‰; see Figure 4.1). Limestones are isotopically similar to total carbon in the oceanic reservoir. This model assumes an input of dissolved carbon to the oceans of constant mass and isotopic composition. This assumption is necessary because we have no data to substantiate large variations, but in reality the riverine flux may vary as a function of rates of weathering and the proportion of limestone versus organic carbon weathered (or oxidized).
Making the assumption of constant input, we can state the following. Excursions to more positive δ13C values indicate perhaps that relatively more organic matter is being buried, while burial of carbonate decreases or remains constant. More negative isotopic values imply a shift to larger values in the burial ratio of carbonate to organic carbon. Junge et al. (1975), Veizer and Hoefs (1976), Garrels and Perry (1974), and Garrels et al. (1976) have suggested that δ13C values in limestones through time have stayed relatively constant (about 0±2.5‰) implying that there has been little change in the partitioning of carbon between the organic and carbonate reservoirs during the Phanerozoic. This relative constancy suggests an efficient long-term control against major excursions in atmospheric and ocean chemistry. However, these considerations use average values from long time periods (i.e., 30 m.y. to 50 m.y.) and do not have the resolution necessary to detect short-term perturbations. For example, Scholle and Arthur (1980) have detected large and rapid excursions of as much as 4 ‰ in δ13C in both positive and negative directions of pelagic carbonates of Cretaceous and early Cenozoic age. These excursions may occur in less than a million years and imply rapid changes in the production and burial of organic matter and carbonate (see also Bender and Keigwin, 1979; Vincent et al., 1980). Shackleton (1977) has demonstrated cyclic changes in δ13C of less than 1 ‰ in benthic foraminifers over a few thousand years during the Late Pleistocene. He attributed these variations to transfer of carbon to and from the terrestrial biosphere to the ocean-atmosphere during glacial-interglacial climatic changes in the amount of about 1014 g of C/yr (a total of 1018 g of C in 10,000 years or so). Note that this amount is perhaps only a few percent of the rate of addition of CO2 to the atmosphere by man’s activities today. Tree-ring carbon-isotope data suggest that a 1 to 1.5 ‰ decrease has resulted in the δ13C value of atmospheric CO2 from burning of isotopically light organic fuels.
DETECTING ATMOSPHERIC CO2 EXCURSIONS AND CLIMATE CHANGE IN THE CENOZOIC
Is there any hope of detecting variations in atmospheric CO2 and establishing these as a cause of climate change in the geologic past? The problem amounts to one having several parts: (a) constructing realistic models that demonstrate the significance of climate change related to pCO2 variations; (b) designing more sophisticated models that incorporate all important factors in the carbon cycle in the land-ocean-atmosphere system (as discussed earlier), and plugging in a variety of possible variations in inputs and outputs in order to provide constraints on interpretation of past geologic data; and (c) collection and integration of geologic and climatic data for comparison with (b) above. Part (a) has already been discussed briefly in previous sections. Part (b) is a difficult enterprise, but various attempts to model the system are in progress (e.g., Bolin et al., 1979; E.Sundquist, U.S. Geological Survey, personal communication, 1980).
Data for part (c) have been slowly accumulating, mainly through paleontologic, lithologic, and geochemical analysis of DSDP cores. Berger (1977) previously inferred atmospheric CO2 changes during the Cenozoic by examining the known fluctuations in various parameters of the carbon system derived from this data base. However, climate change seems dependent on so many factors that we cannot definitely state that a given climatic event is dependent on a single cause. Many of the factors possibly causing climate changes during the Cenozoic, for example, are coincident (see the other chapters in this volume; Berger, 1977; Berggren and Hollister, 1977; Fischer and Arthur, 1977; Frakes, 1979), and their relative effects must be carefully modeled.
Because of the variety of mechanisms that can induce climate change, a given warming in the geologic past is difficult to attribute to a greenhouse effect resulting from increased atmospheric CO2. However, there are several signals that, taken together along with evidence of climate warming, might point to increased levels of CO2 as a cause (e.g., Berger, 1977). These signals are the following: (1) evidence of increased dissolution of carbonate in the deep sea and possible decreases in the rate of accumulation in deep-sea sediments; (2) changes in the δ13C values of total carbon in the oceanic reservoir as reflected in analyses of pelagic-carbonate bulk samples, or preferably both benthic and planktonic organisms, through the time interval in question; (3) a change in the rate of accumulation of organic matter in marine and/or nonmarine sediments; and (4) negative evidence of a major change in ocean circulation that might result in any of the preceding signals.
The following discussion deals briefly with the Cenozoic climatic and paleo-oceanographic record and some evidence of possible pCO2 excursions. This is a qualitative treatment only and is intended to illustrate the type of approach necessary and the difficulties in isolating atmospheric pCO2 variations as causes of climate change. References and discussion of climate changes based on oxygen isotope and paleontologic data can be found in a number of papers in this volume (e.g., Chapters 12, 13, 16, and 18). The paleo-oceanographic data base comes largely from Figures 4.3–4.5, which are compiled from numerous sources (see also Berger, 1979; Arthur, 1979).
The climatic events near and following the Cretaceous-Tertiary boundary may be evidence of an atmospheric CO2 excursion at that time. The possibility was suggested by McLean (1978b) in an elaborate explanation for the biotic extinctions occurring at the boundary. He suggested that pCO2 increased because of a failure of marine photosynthetic organisms. This pCO2 increase resulted in a warming across the boundary during the earliest Paleocene. This warming of a few degrees
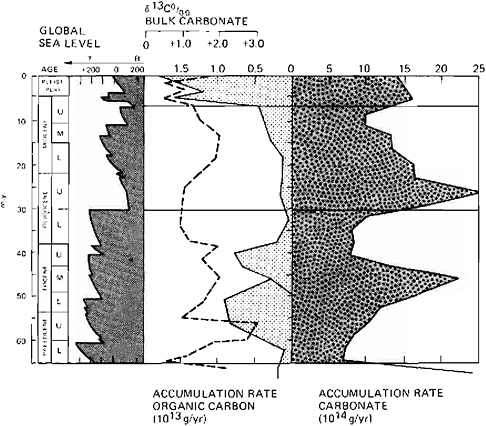
FIGURE 4.3 Accumulation rates of organic carbon (M. A. Arthur, unpublished data) and carbonate (after Worsley and Davies, 1981) in the deep sea during the Cenozoic as compared with average δ13C values of bulk carbonate and global sea level. The fluctuations reflect changing fertility and possibly changes in atmospheric CO2.
Celsius has been detected in oxygen isotopic and paleontologic data. Hsü (1980) has also suggested a large increase in pCO2 at this time associated with an impact of an extraterrestrial object.
The early Paleocene warming lasted 3–5 m.y. The δ13C values of carbonate were low (average+1 ‰) at this time, as were δ13C gradients from surface to deep water inferred from the average difference between planktonic and benthic foraminifers (Figures 4.3 and 4.4). The accumulation rates of carbonate in the deep sea were also at a low point in the early Paleocene, as were those of organic carbon. The CCD was relatively high, especially at the boundary. The early Paleocene (65–62 Ma) is therefore a possible candidate for a possible pCO2 excursion that leads to climatic warming. A sudden injection of up to 90×1018 g of CO2 into the ocean-atmosphere is suggested by the rapid negative δ13C shift of 1.5 ‰ at the Cretaceous-Tertiary boundary, assuming that the CO2 had a δ13C value equivalent to that of volcanic emanations. The effects of this event lasted perhaps 3–5 m.y., suggesting that feedback mechanisms to adjust to the pCO2 excursion were relatively efficient on a geologic scale. A slight cooling occurred during the mid to early Late Paleocene. It is not clear, however, what caused the amelioration of the possible pCO2 increase at the Cretaceous-Tertiary boundary. Because deep-sea organic carbon accumulation rates were fairly low and the extent of shallow shelf seas was apparently small, much of the proposed pCO2 increase might have been accomodated by dissolution of pelagic carbonate (e.g., Worsley, 1974).
A second major warming, not explained by other factors such as continental positions and opening of ocean “gateways,” occurred in Late Paleocene-Early Eocene time.
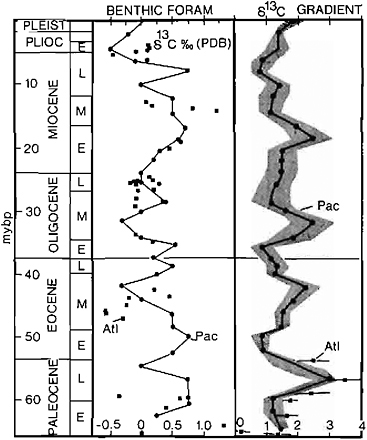
FIGURE 4.4 δ13C values of calcite of benthic foraminifers and 13C gradients between surface and deep water through the Cenozoic (compiled from Kroopnick et al., 1977; Letolle et al., 1979; Boersma et al., 1979).
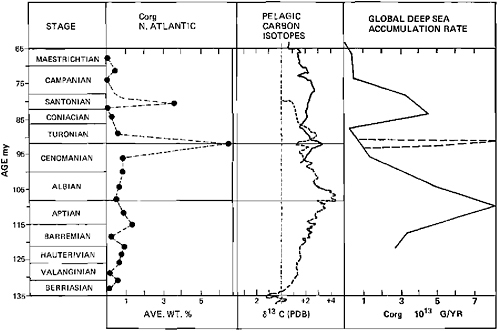
FIGURE 4.5 Accumulation rates of organic matter in the deep sea (M.A.Arthur, unpublished data from DSDP Sites) and δ13C values of pelagic carbonates (from Scholle and Arthur, 1980). Note major change in both parameters near Aptian-Albian, possibly related to the evaporite episode in the South Atlantic.
There is first an increase then a sudden decrease in bulk pelagic carbonate δ13C values amounting to over 2 ‰. The δ13C gradient was highest during the Late Paleocene and also decreased greatly into the Early Eocene. The CCD remained high, and dissolution gradients were also relatively high (van Andel, 1975; van Andel et al., 1975). The accumulation rate of carbonate in the deep sea was only slightly higher than that in the early Paleocene. Organic carbon accumulation rates, however, increased greatly in the Early Eocene. There was certainly a Late Paleocene-Early Oligocene increase in oceanic fertility as evidenced by large phosphorite deposits and high accumulation rates of organic carbon both on the shelves and in the deep sea (Arthur and Jenkyns, 1981). This combination of evidence again seems to be a possible candidate for warming because of an atmospheric CO2 increase. It is intriguing that Vogt (1979) has suggested a substantial peak in volcanicity that occurred in the Late Paleocene {centered at about 56 Ma). Could this volcanicity have been the cause of the climatic warming and changes in paleo-oceanographic parameters? The Early Eocene was also one of the warmest periods, as interpreted from terrestrial floras (see, e.g., Chapter 16),
The aforementioned pCO2 excursion and climatic optimum possibly induced increased weathering rates on land, and this feedback loop brought increased dissolved Ca+2, bicarbonate, silicates, and phosphate to the oceans. The Middle Eocene is marked by cooling, possibly because of decreased pCO2, by a gradual increase in pelagic carbonate δ13C values; by widespread biogenic silica-rich sediments, by low organiccarbon accumulation rates; and by the highest accumulation rates of carbonate in the deep sea (even with a relatively high CCD) of any time in the Early Cenozoic. Again, about 5 m.y. seems to be the response time of the carbon cycle to dampen the effects of a possible pCO2 rise.
Climatic cooling in the Oligocene generally has been explained by the increased isolation of the Antarctic and by the development of the circum-polar current (see Chapter 13). The cooling may also be due to low atmospheric CO2, although there is no good evidence for this. A drop in the deep-sea accumulation rate of organic carbon and carbonate accompanied a deepening of the CCD, a decrease in carbonate-dissolution rates, and low δ13C values, which suggest low fertility and a high ratio of preservation of carbonate to organic matter. These relations suggest, but do not prove, a period of relatively low-atmospheric CO2 in latest Eocene through probably Late Oligocene. In fact, deep-sea carbonate accumulation rates reached a maximum in the Late Oligocene. However, part of this increase may have been due to greater supply of carbonate to the oceans during major sea-level regression (e.g., Worsley and Davies, 1981).
However, an early to middle Miocene warming has not been satisfactorily explained by other mechanisms. This warming episode peaked at about 17-15 Ma and coincides with a second major peak in volcanism shown by Vogt (1979). This peak also coincided with a sharp rise in the CCD in nearly all ocean basins, an apparent increase in dissolution rates (e.g., van Andel et al., 1975), and a decrease in deep-sea carbonate accumulation rates, as well as an elevated δ13C gradient and more positive δ13C values. Deep-sea accumulation rates of organic carbon remained low, as in the Oligocene, but were high around the continental margins and began to increase in the deep sea in the Late Miocene. Pelagic carbonate δ13C values also dropped sharply in the Late Middle Miocene, and δ13C gradients were lower. These factors again suggest that atmospheric CO2 increase could have been responsible for climatic warming. By Late Miocene time, again on the order of about 5-7 m.y. later, the system recovered and cooling began. Both deep-sea organic carbon and carbonate accumulation rates picked up at about 7 Ma.
These relationships are highly speculative but suggest something about the role of p CO2 in climate change in the geologic past. Contrary to this, however, the Plio-Pleistocene peak in volcanism noted by Vogt (1979) and Kennett and Thunell (1975) appears to have had the opposite effect—that is, inducing cooling—or no effect at all.
EVAPORITE DEPOSITION EVENTS AND GLOBAL CLIMATE CHANGE
Changes in ocean chemistry can modulate or change climate in other ways as well. Rapid and large-scale deposition of evaporites in isolated small ocean basins may have a substantial effect on ocean chemistry, and, in addition, may directly or indirectly influence global climate through its effects on the sulfur and carbon cycles. Garrels and Perry (1974) have pointed out that precipitation of major evaporite bodies may require large transfers in the sulfur and carbon reservoirs. Precipitation of calcium sulfate at higher than steady-state rates in an evaporite basin requires transfer of Ca2+ from the carbonate to the evaporite reservoir and results in a net gain of CO2 to the atmosphere. The CO2 gain, in steady state, should be compensated for by net gain in burial of organic carbon, by increased dissolution of carbonates. or in both. However, in the event that evaporite deposition is extremely rapid, these balancing processes may not be able to work effectively in the short term to remove the CO2 excess. The CO2 spike to the atmosphere may then lead to climate warming, depending on the extent of the CO2 anomaly.
Early Cretaceous Evaporites and a Global Warm Episode
An example of this type of “internal” control on global climate may have occurred during the early Cretaceous when the northern South Atlantic (Angola-Brazil Basin) became the site of massive evaporite deposition as it was effectively isolated from the rest of the world ocean at intermediate latitudes under high evaporation rates. Arthur and Kelts (1979) have suggested that the Angola-Brazil Basin was isolated for 2 m.y., and within that time period a basin 500 km wide by 2000 km long was filled with between 2 and 3 km of evaporites. Assuming at least 30 percent CaSO4 and 70 percent NaCl within the evaporites, they suggest that nearly 1.4×1021 g of CaSO4 and 3.0×1021 g of NaCl were deposited in the geologically brief 2 m.y. period. Assuming modern rates of river input of Na+, Cl−, Ca2+, and SO42−, and an initial oceanic reservoir of those elements equal to that of today, this chemical extraction means that oceanic salinity could have been decreased by 4 to 5 ‰ (see also Hay, 1979) and that the oceanic sulfate reservoir of sulfate and calcium would have been drawn down significantly. A trend to much lighter δ34S values during Aptian time (e.g., Claypool et al., 1980) may be evidence for this drawdown. The cycling of carbon would have been affected as well. An abrupt CCD rise in Aptian time (Thierstein, 1979) and a trend toward more positive δ13C in marine carbonates (Figure 4.6) may have occurred because of an increase in the rate of dissolution of carbonate resulting from a decrease in the oceanic Ca2+ concentrations and an increase in CO2, and because of an increase in the rate of burial of organic carbon. The increased rate of burial of organic carbon may have resulted from enhanced preservation under anoxic or nearanoxic conditions. Stable stratification related to salinity contrasts between surface and deep waters may have been one mechanism for the development of poorly oxygenated deep waters (Ryan and Cita, 1977; Roth, 1978; Thiersten and Berger, 1978; Arthur and Natland, 1979); some of the most saline deep water may have been derived from periodic spillage from the evaporatic northern South Atlantic and from epicontinental and shelf seas in low latitudes. This mechanism might essentially provide the feedback to rid the system of the supposed CO2 excess by enhancing burial of marine organic carbon. However, if the Aptian-Albian oceans were relatively nutrient depleted as proposed by Roth (1978) and Arthur and Kelts (1979), plankton productivity would be low, and this would not be an effective way to draw down atmospheric CO2. The burden of fixing this CO2 might have fallen on the terrestrial plants; in support of this theory it has been argued that a significant proportion of organic carbon buried in Aptian-Albian deep-sea sediments is of terrestrial derivation (Tissot et al., 1980). This is probably a more slowly operating feedback mechanism, thus pCO2 concentrations may have risen fairly rapidly and been only slowly lowered following the Aptian evaporite episode. The Albian climatic optimum (Savin, 1977) may have resulted from this CO2 excess. A similar scenario could be envisioned for the Permian evaporite episode.
The “Messinian Event” and Global Cooling
The preceding ideas are preliminary and are based on intuitive rather than rigorously systematic interpretations of the
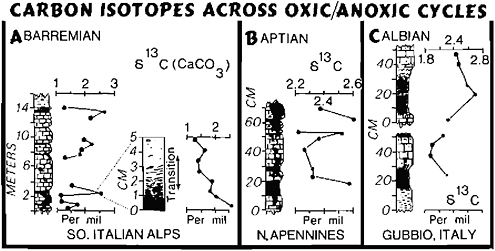
FIGURE 4.6 δ13C profiles across laminated organic-rich shale/marl and limestone cycles of Cretaceous age (A, after Weissert et al., 1979; B and C, M.A.Arthur, unpublished data).
linkages between major geochemical cycles. However, the discussion provides some insight as to how the carbon cycle and global climate may have responded to sudden perturbations of internal forcing mechanisms such as tectonic isolation of major ocean basins. But climatic warming may not be a necessary consequence of rapid and massive evaporite precipitation. The Messinian (latest Miocene) event may provide an illustration of the opposite effect, that is, climatic deterioration, because of the salinity crisis brought on by the isolation of the Mediterranean Tethys and the deposition of as much as 1.5× 106 km3 of evaporites within about 1 m.y. This amount is about three quarters of that deposited in the Aptian northern South Atlantic. Ryan (1973) has suggested that the overall change in ocean salinity may have led to the increased production of sea ice in high southern latitudes that precipitated a sudden latest Miocene cooling rather than a warming by a CO2 excursion. In fact, the high rate of burial of organic matter in deep-sea sediments just prior to and associated with the Messinian event may have led to a net atmospheric-CO2 drawdown. This drawdown may also have contributed to the latest Miocene cooling. However, there is still some doubt as to whether the evaporite deposition preceded the cooling or followed a pronounced regression related to increased glaciation on Antarctica (see Wright and Cita, 1979). Clearly, similar events may have different climatic consequences, depending perhaps on the global climate configuration at the time. The latitudinal distribution of continents during the Early Cretaceous contrasts greatly with that of the Late Miocene, a time when a greater proportion of the land mass was concentrated in higher latitudes. The position of Antarctica over the South Polar region is probably critical in the difference in global climate between the Early Cretaceous and the Late Miocene. Also, the Miocene oceans seem to have been much more fertile and possibly circulated more rapidly than the Early Cretaceous oceans. This apparent increase in nutrient availability may have allowed more rapid depletion of any pCO2 increase by burial of marine organic matter in the Miocene oceans.
CONCLUSIONS
At present, separation of cause from effect is difficult when examining possible changes in ocean chemistry and their relations to climate during the last 120 m.y., as is attributing any given climate change in the past to changes in atmospheric pCO2. The chemistry of the ocean plays a dominantly passive role in modulating climate largely through its thermal inertia and its part in the carbon cycle. Conversely, ocean chemistry and circulation can certainly change in response to climatic events. Abrupt changes in the depth distribution and accumulation rate of organic carbon and carbonate in the ocean basins, and in the carbon isotopic composition of pelagic-calcareous microfossils indicate major changes in ocean-water mass structure, rates of oceanic overturn, atmospheric CO2 flux, and fertility. These changes within the ocean system in turn appear related to those in global climate.
Excess atmospheric CO2 from volcanic or other sources may be titrated by dissolution of carbonate in deeper water. Increased rates of burial of organic carbon of either terrestrial or marine origin in marine sediment also facilitate removal of excess CO2. The ability of the ocean-chemical system to maintain steady-state atmospheric CO2 levels in this way, however, is largely dependent on overall nutrient availability and on rates of replenishment and levels of oxygenation of deep water, among other things. These factors bear a complicated and varying relation to climate as well.
Changes in ocean chemistry also may trigger climatic variation. For example, rapid and massive precipitation of evaporites in isolated basins involves major chemical transfers affecting the carbon cycle and perhaps leading to increased levels of atmospheric CO2. However, the relative influence of such events and the direction of resultant climatic changes appear to depend on the prevailing climatic regime at the time.
More definite reconstructions of the relationship between changes in atmospheric CO2, ocean chemistry, and climate will depend on collecting and analyzing large amounts of data on accumulation rates of organic matter and carbonate in marine sediments through time, on the bulk chemistry of sediment, and on the carbon isotopic composition of organic matter and carbonate. Time series of such data, in conjunction with those of isotopic paleotemperatures and faunal studies, will aid in examining the interrelationships of climate and ocean chemistry and will allow better estimation of leads and lags in the system. These data must be integrated into sophisticated computerized models relating changes in climate to those in oceanic and atmospheric chemistry. But in order to evaluate the role of atmospheric CO2 changes in changing global climate, we must also be able to evaluate critically the climatic (and ocean chemical) effects of changing sea levels, continental distribution, tectonic and volcanic activity, and varying ocean gateways, At present, the number of degrees of freedom allow only a speculative and qualitative approach to the relationship between climate and atmospheric CO2 in the geologic past.
ACKNOWLEDGMENTS
I am grateful for the thorough and enlightening reviews of an earlier manuscript by George E.Claypool and Eric Sundquist and for discussions of aspects of the CO2 climate problem with them and with Eric Barron, Kemy Kelts, and Pete Scholle.
REFERENCES
Arthur, M.A. (1979). Paleo-oceanographic events—Recognition, resolution, and reconsideration, Rev. Geophys. Space Phys. 17, 1474–1494.
Arthur, M.A., and H.C.Jenkyns (1981). Phosphorites and paleoceanography, in Ocean Geochemical Cycles, W.H.Berger, ed., Oceanol. Acta, Supplement,
Arthur, M.A., and K.R.Kelts (1979). Evaporites, black shales and perturbations of ocean chemistry and fertility, Geol. Soc. Am. Abstr. Programs 11, p. 381.
Arthur, M.A., and J.H.Natland (1979). Carbonaceous sediments in the North and South Atlantic: The role of salinity in stable stratification of early Cretaceous basins, in Results of Deep Drilling in the Atlantic Ocean, Proceedings of the Second Maurice Ewing Symposium 3, M.Talwani, W.W.Hay, and W.B.F.Ryan, eds., American Geophysical Union, Washington, D.C., pp. 375–401,
Bender, M.L., and L.D.Keigwin Jr. (1979). Speculations about the upper Miocene change in abyssal Pacific dissolved bicarbonate δ13C, Earth Planet. Sci. Lett. 45, 383–393.
Berger, W.H. (1977). Carbon dioxide excurions and the deep sea record: Aspects of the problem, in The Fate of Fossil Fuel CO2in the Ocean, N.R.Andersen and A.Malahoff, eds., Plenum, New York, pp. 505–542.
Berger, W.H. (1979). The impact of deep sea drilling on paleoceanography, in Deep Drilling Results in the Atlantic Ocean, Continental Margins and Paleoenvironment, Maurice Ewing Series 3, M.Talwani, W.W.Hay, and W.B.F.Ryan, eds., American Geophysical Union, Washington, D.C., pp. 297–314.
Berggren, W.A., and C.D.Hollister (1977). Plate tectonics and paleocirculation: Commotion in the ocean, Tectonophysics 38, 11–48.
Berner, W., H.Oeschger, and B.Stauffer (1981). Information on the CO2 cycle from ice core studies, Radiocarbon 22, 227.
Boersma, A., N.J.Shackleton, M.Hall, and Q.Given (1979), Carbon and oxygen isotope records at DSDP Site 384 (North Atlantic) and some Paleocene paleotemperatures and carbon isotope variations in the Atlantic Ocean, in Initial Reports of the Deep Sea Drilling Project 43, U.S. Government Printing Office, Washington, D.C., pp. 695–718.
Bolin, B., E.T.Degens, S.Kempe, and P.Ketner, eds. (1979). The Global Carbon Cycle, SCOPE Rep. 13, Wiley, New York, 491 pp.
Broecker, W.S. (1974). Chemical Oceanography, Harcourt Brace Jovanovich, New York, 214 pp.
Broecker, W.S. (1981), Glacial to interglacial changes in ocean chemistry, in CIMAS Symposium, E.Kraus, ed., U. of Miami, Miami, Florida.
Broecker, W.S., and T.Takahashi (1978). The relationship between lysocline depth and in situ carbonate ion concentration, Deep Sea Res. 25, 65–95.
Broecker, W.S., T.Takahashi, H.J.Simpson, and T.H.Peng (1979). Fate of fossil fuel carbon dioxide and the global carbon budget, Science 206, 409–418.
Claypool, G.E., W.T.Holser, I.R.Kaplan, H.Sakai, and I.Zak (1980). The age curves of sulfur and oxygen isotopes in marine sulfate and their mutual interpretation, Chem. Geol.
Fischer, A.G., and M.A.Arthur (1977). Secular variations in the pelagic realm, in Deep Water Carbonate Environments, H.E.Cook and P.Enos, eds., Soc. Econ. Paleontol. Mineral. Spec. Publ. 25, pp. 19–50.
Frakes, L. (1979). Climates Throughout Geologic Time, Elsevier, Amsterdam, 310 pp.
Froelich, P.N., M.L.Bender, N.A.Luedtke, G.R.Heath, and P.Devries (1982). The marine phosphorous cycle, Am. J. Sci. 282, 474–511.
Garrels, R.M., and E.H.Perry (1974). Cycling of carbon, sulfur, and oxygen through geologic time, in The Sea, Vol. 5, E.D.Goldberg, ed., Wiley-Interscience, New York, pp. 303–336.
Garrels, R.M., A.Lerman, and F.MacKenzie (1976). Controls of atmospheric O2 and CO2: Past, present, and future, Am. Sci. 64, 306–315.
Hay, W.W. (1979). Impact of Deep Sea Drilling Project on paleooceanography (abs.) Am. Assoc. Petrol. Geol. Bull. 63, p. 464.
Holland, H.D. (1972). The geologic history of sea water: An attempt to solve the problem, Geochim. Cosmochim. Acta 36, 637–651.
Holland, H.D. (1974). Marine evaporites and the composition of sea water during the Phanerozoic, in Studies in Paleo-Oceanography, W.W.Hay, ed., Soc. Econ. Paleontol. Mineral. Spec. Publ. 20, pp. 187–192.
Holland, H.D. (1978). The Chemistry of the Atmosphere and Oceans, Wiley, New York, 351 pp.
Hsü, K.J. (1980). Terrestrial catastrophe caused by cometary impact at the end of Cretaceous, Nature 285, 201–203.
Hunt, J.M. (1970). The significance of carbon isotope variations in marine sediments, in Advances in Organic Geochemistry 1966, G.B.Hobson and G.C.Speers, eds., Pergamon, Oxford, pp. 27–35.
Junge, C.E., M.Schidlowski, R.Eichmann, and H.Pietrek (1975). Model calculations for the terrestrial carbon cycle: Carbon isotope geochemstry and evolution of photosynthetic oxygen, J. Geophys. Res. 80, 4542–4552.
Kennett, J.P., and R.C.Thunell (1975). Global increase in explosive volcanism, Science 197, 497–503.
Kroopnick, P.M., S.V.Margolis, and C.S.Wong (1977). δ13C variations in marine carbonate sediments as indicators of the CO2 balance between the atmosphere and oceans, in The Fate of Fossil Fuel CO2in the Ocean, N.R.Anderson and A.Malahoff, eds., Plenum, New York, pp. 295–322.
Lemon, E. (1977). The land’s response to more carbon dioxide, in The Fate of Fossil Fuel CO2in the Oceans, N.R.Anderson and A.Malahoff, eds., Plenum, New York, pp. 97–130.
Letolle, R., C.Vergnaud-Grazzini, and C.Pierre (1979). Oxygen and carbon isotopes from bulk carbonates and foraminiferal shells at DSDP Sites 400, 401, 402, 403 and 406, in Initial Report of the Deep Sea Drilling Project 48, U.S. Government Printing Office, Washington, D.C., pp. 741–755.
Madden, R.A., and V.Ramanathan (1980). Detecting climate change due to increasing carbon dioxide, Science 209, 763–768.
Manabe, S., and R.T.Wetherald (1975). The effects of doubling the CO2 concentration on the climate of a general circulation model, J. Atmos. Sci. 32, 3–15.
Manabe, S., and R.T.Wetherald (1980). On the distribution of climate change resulting from an increase in CO2 content of the atmosphere, J. Atmos. Sci. 37, 99–118.
Marland, G., and R.M.Rotty (1979). Carbon dixoide and climate, Rev. Geophys. Space Phys. 17, 1813–1824.
McLean, D.M. (1978a). Land floras: The major late Phanerozoic atmospheric carbon dioxide/oxygen control, Science 200, 1060–1062.
McLean, D.M. (1978b). A terminal Mesozoic “greenhouse,” lessons from the past, Science 201, 401–406.
Muller, P.J. and E.Suess (1979). Productivity, sedimentation rate, and sedimentary organic matter in the oceans—organic preservation, Deep Sea Res. 26A, 1347–1362.
NRC Climate Research Board (1979). Carbon Dioxide and Climate: a Scientific Assessment, National Academy of Sciences, Washington, D.C., 22 pp.
Oeschger, H., V.Siegenthaler, V.Schotterer, and A.Gugelmann (1975). A box diffusion model to study the carbon dioxide exchange in nature, Tellus 27, 168–192.
Piper, D.Z., and L.A.Codispoti (1975). Marine phosphate deposits and the nitrogen cycle, Science 188, 15–18.
Pollack, J.B. (1979). Climate change on the terrestrial planets, Icarus 37, 479–553.
Pollack, J.B., O.B.Toon, C.Sagan, A.Summers, B.Baldwin, and W.Van Camp (1976). Volcanic explosions and climatic change: A theoretical assessment, J. Geophys. Res. 81, 1071–1083.
Rogers, M.A., and C.B.Koons (1969). Organic carbon δ13C values from Quaternary marine sequences in the Gulf of Mexico: A reflection of paleotemperature changes, Trans. Gulf Coast Assoc. Geol. Soc. 19, 529–534.
Roth, P.H. (1978). Cretaceous nannoplankton biostratigraphy and oceanography of the northwestern Atlantic Ocean, in Initial Reports of the Deep Sea Drilling Project 44, U.S. Government Printing Office, Washington, D.C., pp. 731–759.
Rotty, R.M. (1977). Global carbon dioxide production from fossil fuels and cement, A.D. 1950-A.D. 2000, in Fate of Fossil Fuel CO2in the Oceans, N.R.Anderson and A.Malahoff, eds., Plenum, New York, pp. 167–181.
Ryan, W.B.F. (1973). Geodynamic implications of the Messinian crisis of salinity, in Messinian Events in the Mediterranean, C.W. Drooger, ed., K.Ned. Akad. Wet., Amsterdam, pp. 26–38.
Ryan, W.B.F., and M.B.Cita (1977). Ignorance concerning episodes of oceanwide stagnation, Mar. Geol. 23, 197–215.
Sackett, W.A. (1964). The depositional history and isotopic organic carbon composition of marine sediments, Mar. Geol. 2, 173–185.
Sackett, W.M., and R.R.Thompson (1963). Isotopic organic carbon composition of recent continental derived elastic sediments of Eastern Gulf Coast, Gulf of Mexico, Bull. Am. Assoc. Petrol. Geol. 47, 525.
Sackett, W.M., W.R.Eckelmann, M.L.Bender, and A.W.H.Be (1965). Temperature dependence of carbon isotope composition in marine plankton and sediments, Science 148, 235–237.
Savin, S.M. (1977). The history of the Earth’s surface temperature during the last 100 million years, Ann. Rev. Earth Planet. Sci. 5, 319–355.
Scholle, P.A., and M.A.Arthur (1980). Carbon isotopic fluctuations in pelagic limestones: Potential stratigraphic and petroleum exploration tool, Bull. Am. Assoc. Petrol. Geol. 64, 67–89.
Shackleton, N.J. (1977). Carbon-13 in Uvigerina: Tropical rainforest history and the equatorial Pacific carbonate dissolution cycles, in The Fate of Fossil Fuel CO2in the Ocean, N.R.Anderson and A. Malahoff, eds., Plenum, New York, pp. 401–427.
Simoneit, B.R.T. (1977). The Black Sea, a sink for terrigenous lipids, Deep Sea Res. 24, 813–830.
Strain, B.R. (1978). Report of the Workshop on Anticipated Plant Responses to Global Carbon Dioxide Enrichment, Dept. of Botany, Duke U., Durham, N.C.
Tappan, H. (1968). Primary production, isotopes, extinctions a the atmosphere, Paleogeogr. Paleoclimatol. Paleoecol. 4, 187–210.
Thierstein, H.R. (1979). Paleoceanographic implications of organic carbon and carbonate distribution in Mesozoic deep-sea sediments, in Deep Drilling Results in the Atlantic Ocean: Continental Margins and Paleoenvironment, Maurice Ewing Series 3, M.Talwani, W.W.Hay, and W.B.F.Ryan, eds., American Geophysical Union, Washington, D.C., pp. 249–279.
Thierstein, H.R., and W.H.Berger (1978). Injection events in Earth history, Nature 276, 461–464,
Tissot, B., G.Demaison, P.Masson, J.R.Delteil, and A.Combaz (1980). Paleoenvironment and petroleum-potential of middle Cretaceous black shales in Atlantic basins, Bull. Am. Assoc. Petrol. Geol. 64, 2051–2063.
van Andel, T.H. (1975). Mesozoic/Cenozoic calcite compensation depth and the global distribution of calcareous sediments, Earth Planet. Sci. Lett. 26, 187–195.
van Andel, T.H., G.R.Heath, and T.C.Moore, Jr. (1975). Cenozoic history and paleoceanography of the central equatorial Pacific Ocean, Geol. Soc. Am. Mem. 143, 1–134,
Veizer, J., and J.Hoefs (1976). The nature of 18O/16O and 13C/12C secular trends in sedimentary carbonate rocks, Geochim. Cosmochim. Acta 40, 1387–1395.
Vincent, E., J.S.Killingley, and W.H.Berger (1980). The magnetic Epoch-6 carbon shift: A change in the oceans 13C/12C ratio 6.2 million years ago, Mar. Micropaleontol. 5 185–203.
Vogt, P.R. (1972). Evidence for global synchronism in mantle plume convection and possible significance for geology, Nature 240, 338–342.
Vogt, P.R. (1979). Global magmatic episodes: new evidence and implications for the steady-state mid-oceanic ridge, Geology 7, 93–98.
Weissert, H., J.McKenzie, and Hochuli (1979). Cyclic anoxic events in the Early Cretaceous Tethys Ocean, Geology 7, 147–151.
Worsley, T.R. (1974). The Cretaceous-Tertiary boundary event in the ocean, in Studies in Paleo-Oceanography, W.W.Hay, ed., Soc. Econ. Paleontol. Min. Spec. Publ. 20, pp. 94–125.
Worsley, T., and T.Davies (1981). Paleoenvironmental implications of oceanic carbonate sedimentation rates, in Symposium and Results of Deep-Sea Drilling, R.Douglas, E.L.Winterer, and J. Warme, eds., Soc. Econ. Paleontol. Min. Spec. Spec. Publ. 30.
Wright, R., and M.B.Cita (1979), Geo and biodynamic effects of the Messimean salinity crisis in the Mediterranean, Palaeogeogr. Palaeoclimatol. Palaeoecol. 29, 215–222.













