
Biological and Biomimetic Polypeptide Materials
TIMOTHY J. DEMING
University of California, Los Angeles
Fibrous structures act as load-bearing components in vivo in many natural proteins besides enzymes, which are soluble globular molecules. Natural evolutionary processes have produced structural proteins that surpass the performance of man-made materials (e.g., mammalian elastin in the cardiovascular system that lasts half a century without loss of function and spider webs composed of silk threads that are tougher than any synthetic fiber) (Kaplan et al., 1994; Mobley, 1994; Viney et al., 1992). These biological polypeptides are all complex copolymers that derive their phenomenal properties from precisely controlled sequences and compositions of the constituent amino acid monomers, which, in turn, lead to precisely controlled self-assembled nanostructures. Recently, there has been a great deal of interest in the development of synthetic routes for preparing natural polymers, as well as de novo designed polypeptide sequences for applications in biotechnology (e.g., artificial tissues and implants), biomineralization (e.g., resilient, lightweight, ordered inorganic composites), and analysis (e.g., biosensors and medical diagnostics) (Capello and Ferrari, 1994).
To be successful, these materials must be “processible,” or better yet, capable of self-assembly into precisely defined structures. Peptide polymers have many advantages over conventional synthetic polymers because they are able to assemble hierarchically into stable, ordered conformations (Voet and Voet, 1990). This ability depends in part on the exact structures of protein chains (i.e., defined molecular weight, monodispersity, stereoregularity, and sequence and
composition control at the monomer level). The inherent conformational rigidity of polypeptide chains also contributes to their ability to self-assemble. Depending on the amino acid side-chain substituents, polypeptides are able to adopt a multitude of conformationally stable, regular secondary structures (e.g., helices, sheets, and turns), tertiary structures (e.g., the β-strand-helix-β-strand unit found in β-barrels), and quaternary assemblies (e.g., collagen microfibrils) (Voet and Voet, 1990). Conformational rigidity and precise chain structures work together to produce hierarchically ordered, three-dimensional materials from linear macromolecules.
Beyond laboratory replication of natural structural biopolymers, the synthesis of polypeptides that can self-assemble into nonnatural structures is an attractive challenge for polymer chemists. Synthetic-peptide-based polymers are not new materials; homopolymers of polypeptides have been available for many decades, but their use as structural materials has been limited (Fasman, 1967, 1989). New methods in biological and chemical synthesis have made possible the preparation of increasingly complex polymer sequences of controlled molecular weight that display properties far superior to ill-defined homopolymers.
SYNTHESIS OF BIOLOGICAL POLYPEPTIDES
The advent of recombinant DNA methodologies has provided a basis for the production of polypeptides with exact sequences that can be controlled by design of a suitable DNA template. The most common technique for biosynthesis of protein polymers has been to design an artificial peptide sequence that can be repeated to form a larger polymer and then to construct the DNA polymer that encodes this protein sequence (McGrath et al., 1990; Tirrell et al., 1991). The DNA sequences are then cloned and expressed, typically in yeast or bacterial hosts, to produce the designed polypeptides, which are then either secreted or isolated by lysing the microorganism cells. This methodology has been used for both small- and large-scale production of biomimetic structural proteins, such as silks and elastins, as well as de novo sequences designed to fold into ordered nanostructures (Kaplan et al., 1994; Mobley, 1994; Viney et al., 1992).
The steps required for synthesizing polypeptides using genetic engineering are shown in Figure 1. Once a sequence of amino acids has been chosen, it is encoded into a complementary DNA sequence, which must be chemically synthesized. Solid-phase synthesis has made significant advances in recent years, and it is now possible to synthesize polynucleotides that can encode chains of hundreds of amino acids (McBride and Caruthers, 1983). By cloning these DNA sequences into circular plasmid DNA, the sequences can be incorporated into host cells (e.g., bacteria). In these cells, the plasmids are amplified through replication and can then be sequenced to check for mutations or deletions before final cloning into an expression plasmid. The expression plasmid contains a promoter sequence that drives transcription. A strong promoter, such as the one in the pET
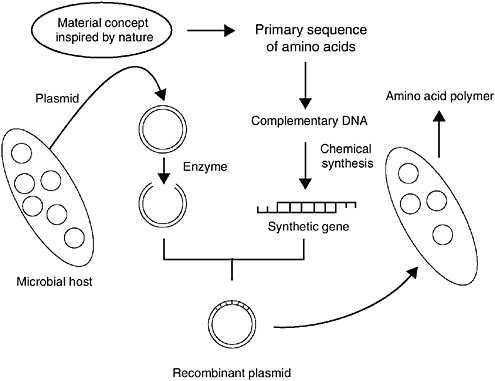
FIGURE 1 Genetic engineering of polypeptides. Source: van Hest and Tirrell, 2001. Reprinted with permission from The Royal Society of Chemistry.
expression vectors developed by Studier et al. (1990), results in high levels of DNA transcription into messenger RNA, which can result in high levels of polypeptide production.
Once the material has been formed, the artificial protein must be isolated from cellular by-products and other proteins. In some cases, the polypeptides form insoluble aggregates in the cells that can be purified by simply extracting all the cellular debris into suitable solvents and recovering the aggregates by filtration. In cases where polypeptides are soluble after cell lysis, they can typically be purified using affinity chromatography by incorporating an affinity marker into the polypeptide sequence (Dong et al., 1994; Zhang et al., 1992).
Recombinant DNA methods have been used to prepare a variety of polypeptide materials. Tirrell’s group has prepared lamellar crystals of controlled thickness and surface chemistry (Creel et al., 1991; Krejchi et al., 1994; McGrath et al., 1992), smectic liquid crystals from monodispersed, rod-like α-helical polypeptides (Zhang et al., 1992), and hydrogels from helix-coil-helix triblock copolypeptides (Petka et al., 1998). In related work, van Hest has prepared polypeptide β-sheet fibrils with the recombinant polypeptide chemically coupled
to synthetic poly(ethylene glycol) chains to improve solubility (Smeenk et al., 2005). The genetically engineered polypeptide yields well-ordered fibrils of controlled thickness and width.
Chaikof and coworkers have used recombinant DNA methods to prepare protein-based thermoplastic elastomers (Nagapudi et al., 2005). They prepared triblock copolymer sequences that mimic the natural protein elastin; the sequences were modified in such a way that the outer segments were plastic, and the central segment was elastomeric, similar to purely synthetic thermoplastic elastomers, such as styrene-isoprene-styrene rubber. Overall, with genetic engineering we can prepare polypeptides with the complexity of natural proteins. Hence, it is possible to prepare materials with polymer properties that can be manipulated with exquisite control.
SYNTHESIS OF CHEMICAL POLYPEPTIDES
To circumvent tedious protein purification, large investments of time in gene synthesis, and difficult artificial amino acid incorporation, it would be advantageous if we could synthesize complex copolypeptides chemically from inexpensive amino acid monomers. However, there is a large gap between biologically produced materials and the polypeptides that can be produced synthetically. The most common techniques used for polypeptide synthesis are solid-phase synthesis and polymerization of α-aminoacid-N-carboxyanhydrides (NCAs) (Kricheldorf, 1987, 1990). Solid-phase synthesis, which originated in the pioneering work of Merrifield, involves the stepwise addition of N-protected amino acids to a peptide chain anchored to a polymeric support (Figure 2) (Bodanzsky and Bodanzsky, 1994; Wunsch, 1971). The products of this method are sequence-specific peptides that can be isolated as pure materials. Like solid-phase synthesis of oligonucleotides, the number of peptide residues that can be correctly added to the chains depends on the efficiency of each individual step. However, the capability of strict sequential control in short sequences has made it possible to prepare peptide-based materials.
Solid-phase synthesis has been used to prepare a variety of polypeptide materials. Ghadiri and coworkers used the natural peptide gramacidin-A as a model to develop cyclic peptides that self-assemble into hollow peptide nanotubes (Ghadiri et al., 1993; Khazanovich et al., 1994). The key component of their assembly is the pattern of alternating stereochemistry in the amino acid sequence, which leads to a barrel-like π-helical structure. Numerous other groups have prepared short peptide sequences that assemble into well-defined nanofibers, either through β-sheet or α-helical coiled-coil assembly (Aggeli et al., 1997; Lashuel et al., 2000; Niece et al., 2003; Schneider et al., 2002; Zimenkov et al., 2004).
Recently, Woolfson’s group has shown that well-defined kinks can be placed in these fibrils by careful design of the peptide sequence (Ryadnov and Woolfson,
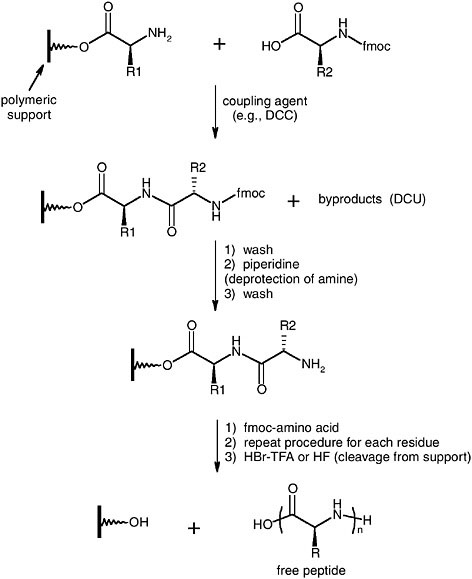
FIGURE 2 Solid-phase peptide synthesis. FMOC = fluorenylmethoxycarbonyl, DCC = dicyclohexylcarbodiimide, DCU = dicyclohexylurea.
2003). Zhang’s group has also shown that small, amphiphilic peptides can be designed to form fibrils that create hydrogels that can be used as cell scaffolds (Zhang et al., 2002), as well as membranes that can close off into spherical and tubular vesicles (Vauthey et al., 2002). Solid-phase peptide synthesis has also been used extensively in the preparation of hybrid copolymers, in which a small peptide sequence can have a big effect on overall materials properties (Klok, 2002).
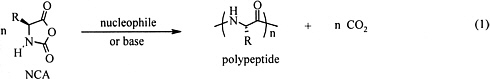
Equation 1
The most common technique currently used for large-scale preparation of polypeptides is NCAs (eq. 1). However, these materials are almost exclusively homopolymers, random copolymers, or graft copolymers that do not have the sequence specificity and monodispersity of natural proteins (Kricheldorf, 1987, 1990). Therefore, the physical properties of NCAs do not meet the requirements for most applications, even though they have long been available and the monomers are inexpensive and easy to prepare. Recently, a methodology has been developed using transition-metal complexes as active species to control the addition of NCA monomers to polymer chain ends. The use of transition metals to control reactivity has been proven to increase both reaction selectivity and efficiency in organic and polymer synthesis (Boor, 1979; Webster, 1991; Wulff, 1989).
Using this approach, substantial advances in controlled NCA polymerization have been realized in recent years. Highly effective zerovalent nickel and cobalt initiators (i.e., bpyNi(COD) [Deming, 1997] and (PMe3)4Co [Deming, 1999]) developed by Deming allow the polymerization of NCAs in a controlled manner without side reactions into high molecular weight polypeptides via an unprecedented activation of the NCAs into covalent propagating species (eq. 2). These cobalt and nickel complexes are able to produce polypeptides with narrow
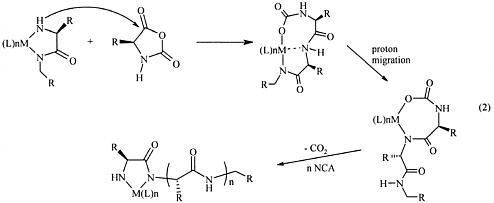
Equation 2
chain-length distributions (Mw/Mn < 1.20) and controlled molecular weights (500 < Mn < 500,000) (Deming, 2000). Because this polymerization system is very general, controlled polymerization of a wide range of NCA monomers can be produced as pure enantiomers (D or L configuration) or as racemic mixtures. By adding different NCA monomers, the preparation of block copolypeptides of defined sequence and composition is also feasible (Deming, 2000).
Polypeptide block copolymers prepared via transition-metal-mediated NCA polymerization are well defined, with the sequence and composition of block segments controlled by order and quantity of monomer, respectively, added to initiating species. These block copolypeptides can be prepared with the same level of control as in anionic and controlled radical polymerizations of vinyl monomers, which greatly expands the potential of polypeptide materials. The unique chemistry of these initiators and NCA monomers also allows NCA monomers to be polymerized in any order, which has been a problem in most vinyl copolymerizations. In addition, the robust chain ends allow preparation of copolypeptides with many block domains (e.g., > 4).
The self-assembly of these block copolypeptides has also been investigated (e.g., to direct the biomimetic synthesis of ordered silica structures [Cha et al., 2000], to form polymeric vesicular membranes [Bellomo et al., 2004 (Figure 3); Holowka et al., 2005], and to prepare self-assembled polypeptide hydrogels [Nowak et al., 2002]). Furthermore, poly(L-lysine)-b-poly(L-cysteine) block copolypeptides have been used to generate hollow, organic/inorganic hybrid microspheres composed of a thin inner layer of gold nanoparticles surrounded by a thick layer of silica nanoparticles (Wong et al., 2002). Using the same procedure, hollow spheres were also prepared; these consisted of a thick inner layer of core-shell CdSe/CdS nanoparticles and a thicker outer layer of silica nanoparticles (Cha et al., 2002). The latter spheres are of interest, because they allowed for microcavity lasing without the use of additional mirrors, substrate spheres, or gratings.
CONCLUSIONS
Many approaches are being investigated for synthesizing new polypeptide materials. Biological approaches have been demonstrated to be extremely powerful for preparing materials with the precision of natural proteins. Chemical techniques are being developed that might be used to prepare polypeptides of any amino acid monomer. The two methodologies complement each other very well. The biological approach is most useful for preparing polypeptides in which monomer sequence and composition must be controlled at the monomer level. The chemical approach is best suited for the preparation of high molecular weight polypeptides in which sequence and composition must only be controlled on length scales of many monomer units (i.e., homopolymer blocks). Future advances in both the biological and chemical arenas are obviously targeted toward
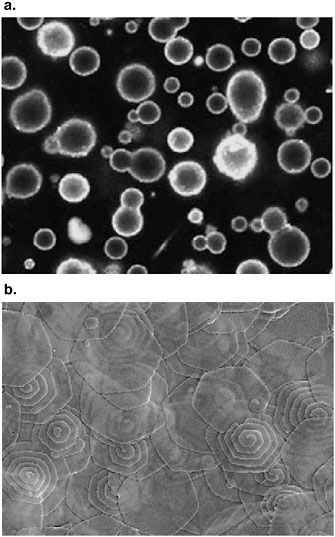
FIGURE 3 (a) Self-assembled vesicles from synthetic diblock copolypeptides. Source: Bellomo et al., 2004. Reprinted with permission from Macmillan Publishers Ltd. (b) Silica-coated plates of α-helical polypeptide single crystals. Source: Bellomo and Deming, 2006. Copyright 2006 American Chemical Society. Reprinted with permission.
conquering the shortcomings of each method—incorporation of artificial amino acids and simplification of preparation/purification for biological synthesis and control of monomer sequence and composition in chemical synthesis. Progress in these areas could expand both methodologies to the point where they might meet and overlap, thus providing scientists with the means of synthesizing any conceivable polypeptide structure.
REFERENCES
Aggeli, A., M. Bell, N. Boden, J.N. Keen, P.F. Knowles, T.C.B. McLeish, M. Pitkeathly, and S.E. Radford. 1997. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature 386: 259–262.
Bellomo, E.G., and T.J. Deming. 2006. Monoliths of aligned silica-polypeptide hexagonal platelets. Journal of the American Chemical Society 128: 2276–2279.
Bellomo, E., M.D. Wyrsta, L. Pakstis, D.J. Pochan, and T.J. Deming. 2004. Stimuli-responsive polypeptide vesicles by conformation-specific assembly. Nature Materials 3: 244–248.
Bodanzsky, M., and A. Bodanzsky. 1994. Practice of Peptide Synthesis, 2nd ed. New York: Springer-Verlag.
Boor, J. 1979. Zeigler-Natta Catalysts and Polymerizations. New York: Academic Press.
Capello, J., and F. Ferrari. 1994. Microbial Production of Structural Protein Polymers. Pp. 35–92 in Plastics from Microbes. Cincinnati, Ohio: Hanser/Gardner.
Cha, J.N., G.D. Stucky, D.E. Morse, and T.J. Deming. 2000. Biomimetic synthesis of ordered silica structures mediated by block copolypeptides. Nature 403: 289–292.
Cha, J.N., M.H. Bartl, M.S. Wong, A. Popitsch, T.J. Deming, and G.D. Stucky. 2002. Microcavity lasing from block peptide hierarchically assembled quantum dot spherical resonators. Nano Letters 3: 907–911.
Creel, H.S., M.J. Fournier, T.L. Mason, and D.A. Tirrell. 1991. Genetically directed syntheses of new polymeris materials. Efficient expression of a monodisperse copolypeptide containing fourteen tandemmly repeated—(AlaGly)4ProGluGly—elements. Macromolecules 24: 1213– 1214.
Deming, T.J. 1997. Facile synthesis of block copolypeptides of defined architecture. Nature 390: 386–389.
Deming, T.J. 1999. Cobalt and iron initiators for the controlled polymerization of α-amino acid-N-carboxyanhydrides. Macromolecules 32: 4500–4502.
Deming, T.J. 2000. Living polymerization of a-amino acid-N-carboxyanhydrides. Journal of Polymer Science, Part A: Polymer Chemistry 38: 3011–3018.
Dong, W., M.J. Fournier, T.L. Mason, and D.A. Tirrell. 1994. Bifunctional hybrid artificial proteins. Polymer Preprints 35(2): 419–420.
Fasman, G.D. 1967. Poly α-Amino Acids. New York: Dekker.
Fasman, G.D. 1989. Prediction of Protein Structure and the Principles of Protein Conformation. New York: Plenum Press.
Ghadiri, M.R., J.R. Granja, R.A. Milligan, D.E. McRee, and N. Khazanovich. 1993. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature 366: 324–327.
Holowka, E.P., D.J. Pochan, and T.J. Deming. 2005. Charged polypeptide vesicles with controllable diameter. Journal of the American Chemical Society 127: 12423–12428.
Kaplan, D., W.W. Adams, B. Farmer, and C. Viney. 1994. Silk Polymers. American Chemical Society Symposium Series 544. Washington, D.C.: American Chemical Society.
Khazanovich, N., J.R. Granja, D.E. McRee, R.A. Milligan, and M.R. Ghadiri. 1994. Nanoscale tubular ensembles with specified internal diameters. Design of a self-assembled nanotube with a 13-Å Pore. Journal of the American Chemical Society 116: 6011–6012.
Klok, H.-A. 2002. Protein-inspired materials: synthetic concepts and potential applications. Angewandte Chemie International Edition 41: 1509–1513.
Krejchi, M.T., E.D.T. Atkins, A.J. Waddon, M.J. Fournier, T.L. Mason, and D.A. Tirrell. 1994. Chemical sequence control of beta-sheet assembly in macromolecular crystals of periodic polypeptides. Science 265: 1427–1432.
Kricheldorf, H.R. 1987. α-Aminoacid-N-Carboxyanhydrides and Related Materials. New York: Springer-Verlag.
Kricheldorf, H.R. 1990. Polypeptides. Pp. 1–132 in Models of Biopolymers by Ring-Opening Polymerization, edited by S. Penczek. Boca Raton, Fla.: CRC Press.
Lashuel, H.A., S.R. LaBrenz, L. Woo, L.C. Serpell, and J.W. Kelly. 2000. Protofilaments, filaments, ribbons, and fibrils from peptidomimetic self-assembly: implications for amyloid fibril formation and materials science. Journal of the American Chemical Society 122: 5262–5277.
McBride, L.J., and M.H. Caruthers. 1983. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Letters 24: 245–248.
McGrath, K.P., D.A. Tirrell, M. Kawai, M.J. Fournier, and T.L. Mason. 1990. Chemical and biosynthetic approaches to the production or novel polypeptide materials. Biotechnology Progress 6: 188–192.
McGrath, K.P., M.J. Fournier, T.L. Mason, and D.A. Tirrell. 1992. Genetically directed syntheses of new polymeric materials. Expression of artificial genes encoding proteins with repeating—(AlaGly)3ProGluGly—elements. Journal of the American Chemical Society 114: 727–733.
Mobley, D. P. 1994. Plastics from Microbes. Cincinnati, Ohio: Hanser/Gardner.
Nagapudi, K., W.T. Brinkman, J. Leisen, B.S. Thomas, E.R. Wright, C. Haller, X. Wu, R.P. Apkarian, V.P. Conticello, and E.L. Chaikof. 2005. Protein-based thermoplastic elastomers. Macromolecules 38: 345–354.
Niece, K.L., J.D. Hartgerink, J.J.J.M. Donners, and S.I. Stupp. 2003. Self-assembly combining two bioactive peptide-amphiphile molecules into nanofibers by electrostatic attraction. Journal of the American Chemical Society 125: 7146–7147.
Nowak, A.P., V. Breedveld, L. Pakstis, B. Ozbas, D.J. Pine, D. Pochan, and T.J. Deming. 2002. Peptides are a gel’s best friend. Nature 417: 424–428.
Petka, W.A., J.L. Harden, K.P. McGrath, D. Wirtz, and D.A. Tirrell. 1998. Reverse hydrogels from self-assembling artificial proteins. Science 281: 389–392.
Ryadnov, M.G., and D.N. Woolfson. 2003. Engineering the morphology of a self-assembling protetin fibre. Nature Materials 2: 329–332.
Schneider, J.P., D.J. Pochan, B. Ozbas, K. Rajagopal, L.M. Pakstis, and J. Gill. 2002. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. Journal of the American Chemical Society 124: 15030–15037.
Smeenk, J.M., M.B.J. Otten, J. Thies, D.A. Tirrell, H.G. Stunnenberg, and J.C.M. van Hest. 2005. Controlled assembly of macromolecular b-sheet fibrils. Angewandte Chemie International Edition 44: 1968–1971.
Studier, W.F., A.H. Rosenberg, J.J. Dunn, and J.W. Dubendorf. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods in Enzymology 185: 60–89.
Tirrell, D.A., M.J. Fournier, and T.L. Mason. 1991. Genetic engineering of polymeric materials. Materials Research Society Bulletin 16: 23–28.
van Hest, J.C.M., and D. Tirrell. 2001. Protein-based materials, toward a new level of structural control. Chemical Communications 1897–1904.
Vauthey, S., S. Santoso, H.Y. Gong, N. Watson, and S.G. Zhang. 2002. Molecular self-assembly of surfactant-like peptides to form nanotubes and nanovesicles. Proceedings of the National Academy of Sciences 99: 5355–5360.
Viney, C., S.T. Case, and J.H. Waite. 1992. Biomolecular Materials. Materials Research Society Proceedings 292. Pittsburgh, Penn.: Materials Research Society.
Voet, D., and J.G. Voet. 1990. Biochemistry. New York: John Wiley and Sons.
Webster, O. 1991. Living polymers. Science 251: 887–893.
Wong, M.S., J.N. Cha, K.-S. Choi, T.J. Deming, and G.D. Stucky. 2002. Assembly of nanoparticles into hollow spheres using block copolypeptides. Nano Letters 2: 583–587.
Wulff, G. 1989. Main-chain chirality and optical activity in polymers consisting of C-C chains. Angewandte Chemie International Edition 28: 21–37.
Wunsch, E. 1971. Synthesis of naturally occurring polypeptides, problems of current research. Angewandte Chemie International Edition 10: 786–795.
Zhang, G., M.J. Fournier, T.L. Mason, and D.A. Tirrell. 1992. Biological synthesis of monodisperse derivatives of poly(alpha-L-glutamic acid): model rodlike polymers. Macromolecules 25: 3601– 3603.
Zhang, S., D.N. Marini, W. Hwang, and S. Santoso. 2002. Design of nanostructured biological materials through self-assembly of peptides and proteins. Current Opinion in Chemical Biology 6: 865–871.
Zimenkov, Y., V.P. Conticello, L. Guo, and P. Thiyagarajan. 2004. Rational design of a nanoscale helical scaffold derived from self-assembly of a dimeric coiled coil motif. Tetrahedron 60: 7237–7246.












