
2
Scientific Session II: Epigenetics
Moderated by Rowena Matthews, G. Robert Greenburg Professor of Biological Chemistry, Research Professor in the Life Sciences Institute, Research Professor in the Biophysics Research Division, and Professor in the Department of Chemistry, University of Michigan
CRITICAL EVENTS: GENOMIC PROGRAMMING AND REPROGRAMMING
Presented by Rudolf Jaenisch, Professor of Biology, Massachusetts Institute of Technology
Genomics studies can identify the specific genes involved in the body’s response to nutrients and pinpoint the genetic variations responsible for differences in a given response among individuals, but such studies have little to say about how nutrient molecules interact with genes to modify their expression and why nutrients can affect genetic expression long after the interaction has occurred. To address these questions, a different approach is needed, one that delves into the molecular mechanisms underlying the genetic response to nutrients.
Rudolf Jaenisch, who is a founding member of the Whitehead Institute, offered three definitions of epigenetics that provide different but complementary ways of viewing the same event. First, epigenetics is “the transmission of information through meiosis or mitosis that is not based on the DNA sequence.” That is, during cell division (which accompanies meiosis or mitosis) information encoded in the DNA sequence is passed from one stage of cell division to the next; epigenetics concerns itself with the remainder of that information. Second, epigenetics is “a mechanism for the stable maintenance of gene expression states that involve physically marking the DNA or its associated proteins.” Because gene expression depends not just on the state of the DNA but also on the state of the
entire chromatin complex, including both the DNA and its protein scaffolding, epigenetics takes all of this into consideration. Third, epigenetics is “mitotically or meiotically heritable changes in gene expression that are not coded in DNA itself.” Gene expression states can be passed on from one cell generation to the next, which happens, for example, during embryonic development, when cells differentiate and then reproduce to form lines of specialized cells (e.g., nerve cells and muscle cells), each of which is defined by specific genes. These genes can be either activated or silent. Gene expression states are heritable, making it possible to express traits that are not dependent on the expression of a given gene per se.
In short, epigenetics describes the way in which cells store and pass on information that is not coded in the DNA sequence itself but rather in various modifications made to the DNA and, more generally, to the chromatin complex containing it.
Epigenetics is thus an important tool for nutrigenomics because it offers a way of understanding how nutrients interact at the molecular level with the genome to create long-lasting effects. “Epigenetic regulation is a mechanism that allows the genome to integrate intrinsic signals and environmental signals. It is a way the genome interacts with the environment. So, what you ate for lunch has found its way to change in some very subtle way the epigenetic state of your DNA.” That, in turn, is related to how diet can affect health and, in particular, the risk of certain diseases. When gene-environment interactions alter the epigenetic state of the genome, they may affect the incidence of diseases with long latencies or late-stage onset, such as cancer and neurodegenerative diseases.
Relatively little research that has applied the tools of epigenetics to nutrigenomics has been conducted to date; but epigenetics is a rapidly developing field, one that has been invigorated by the sequencing of the human genome. The growing arsenal of tools and techniques available from the study of epigenetics thus offers new and revolutionary ways of studying how nutrients interact with the genome.
The Agouti Mouse Paradigm
The agouti mouse can express a number of different phenotypes. It can be yellow and obese or brown and slim. It can have a mottled yellow or a brown coat. These differences, however, are not genetic in origin. These mice are genetically identical. The differences arise from variations in the expression of the agouti gene; and coat color expression can be controlled by varying the mother’s diet before, during, and after pregnancy. The agouti allele is normally expressed only in a mouse’s skin, creating a yellow fur wherever it is expressed, but in agouti mice the gene is expressed throughout the body. In the mouse’s brain, for example,
|
BOX 2-1 Methylation Methylation is one of the primary mechanisms of regulating gene expression. Methylation occurs when a methyl group is attached to a CpG site in the DNA strand. DNA methyltransferase attaches the methyl group to the cytosine, converting it to 5-methylcytosine. CpG sites are relatively rare in human and vertebrate genomes, and they are most frequently found in promoter regions. Methylation can be associated with increased or decreased gene expression, although, in general, when a promoter is hypomethylated the gene can be expressed, whereas when it is hypermethylated gene expression is repressed or turned off. |
the agouti protein blocks a feeding control center, leading the animal to overeat and become fat. The reason for the ubiquitous expression is that the gene has an IAP (Intracisternal A-particle) proximal enhancer (IPE) element inserted next to the gene. The IPE element acts as a promoter and switches on the gene everywhere, not just in the skin.
Agouti gene expression can be silenced, however, by methylation (see Box 2-1) of the IPE element at a cytosine-phosphate-guanine (CpG) site. This methylation shuts down the promotion effects of the IPE element. Methylation can inhibit expression of the agouti gene altogether, resulting in a mouse with brown fur and normal weight.
A key point is that the methylation state can be passed from one replicative generation to the next or from parent to offspring, as in the case of the agouti mouse, causing the differences in phenotypic expression. In a 2003 experiment performed by Waterland and Jirtle at Duke University,1 female mice were fed one of two different diets before, during, and after their pregnancies. Those mice on the normal (control) diet produced pups that had a yellow coat, developed obesity, and were susceptible to a number of cancers. Those given a diet high in vitamin B12, folic acid, and other supplements that promote methylation produced pups with a brown coat that maintained a normal weight and had no increased cancer risk.
This experiment demonstrated that the diet modulated methylation of the IPE element in the mouse genome, which repressed expression of the agouti gene for the lifetime of the offspring. “I think this is a very interesting experiment,” Jaenisch said. “It tells us that the environmental effect at a certain short stage early in life affects the gene expression pattern throughout life and has an enormous effect on phenotype.” It is, in short, a prototype for the sort of epigenetic actions important to nutrig-
enomics: diet, acting through the mechanism of methylation, affects gene expression in a stable, lasting way with clear consequences for the health of the individual.
Epigenetics and Cancer
One of the most exciting areas in which epigenetics has been applied is the study of cancer. The development of cancer depends upon a complex series of events. Some of these events are genetic, involving genetic mutations, whereas others are epigenetic, involving changes in DNA methylation or in the chromatin state, such as histone modifications. Epigenetic events are particularly interesting because they can, in principle, be reversed by therapeutic intervention. Genetic events, by contrast, cannot be reversed and thus offer limited potential for intervention.
One of the best experimental models of cancer is the APC mouse, which has a mutation in the APC (adenomatous polyposis coli) tumor suppressor gene. Mice with this mutation develop colon polyps that become tumors in a progression that parallels the development of colon cancer in humans with familial adenomatous polyposis. In the 1980s, researchers studying the APC mouse discovered that the DNA in colon polyps was hypomethylated, which led to the hypothesis that such hypomethylation might be a necessary step in the development of colon cancer.2 To investigate the role that methylation plays in the development of cancer in the APC mouse, Jaenisch and colleagues manipulated the level of methylation in various ways and observed its effects. If hypomethylation was indeed a necessary step in the development of colon cancer, decreasing the level of methylation should increase the number of polyps. However, the opposite effect was found.
Typically, an APC mouse develops about 130 polyps by 6 months of age. When Jaenisch and colleagues knocked out one allele of the methyltransferase gene, DNMT1, in these mice, it reduced the level of methylation activity by half, and the mice in which the DNMT1 gene was knocked out developed only a third as many polyps as expected. When the APC mice were treated with the anticancer drug 5-aza-D-cytosine, it reduced the levels of DNMT1 in the mice and reduced the number of polyps by nearly 90 percent. When the knockout of one DNMT1 allele was combined with the 5-aza-D-cytosine treatment, the number of polyps dropped by two orders of magnitude. “So this is the opposite of what was thought,” Jaenisch said. “In this case DNMT1 acts like an oncogene: the more you have, the more tumors you have.”
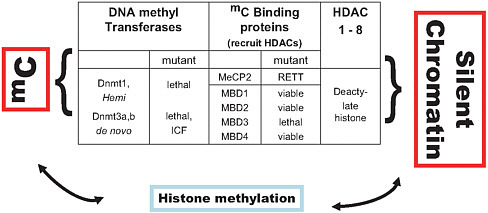
FIGURE 2-1 Epigenetic regulation: DNA methylation, chromatin conformation, and gene expression. MC–methylated cytosine; HDAC–histone deactelyase; MeCP2–methyl-CpG-binding protein; RETT–Rett syndrome phenotype; Hemi –hemi-methylated DNA; ICF–Immunedeficiency, Centromeric region instability, and Facial abnormalities syndrome.
This is just one piece of a very large and complex puzzle. Methylation has various effects on the development of tumors, depending on the tissue and the situation. In the thymus and soft tissue, for example, decreasing the level of methylation enhances tumor formation by increasing genome instability, increasing the probability that mutations that repress the expression of tumor suppressor genes will occur. In short, there is no simple way to characterize the role of methylation in tumorigenesis. Rather, it is important to understand the steps involved in tumorigenesis in specific tissue types. Figure 2-1 shows the interrelationships between methylation, chromatin conformation, and gene expression.
In another set of experiments, the Jaenisch laboratory examined the relationship between genomic imprinting and tumorigenesis. In imprinting, offspring inherit two active copies of a gene from each parent; for a small subset of genes, however, only one copy is active. Imprinting is an epigenetic event, commonly mediated by methylation of either the gene or its regulatory sequence. The loss of imprinting is associated with the development of various cancers, including leukemia, lymphomas, and liver cancer, so the Jaenisch laboratory examined whether the loss of imprinting was a cause or a consequence of the cancer. The strategy was to remove all imprints from a mouse model and watch the development of cancer in the imprint-free mice. This required manipulation of the DNMT1 gene, which codes for the methyltransferase responsible for maintenance of the existing methylation pattern. (Other methyltransfer-
ase genes, such as DNMT3a and DNMT3b, are responsible for de novo methylation, such as that which occurs during early embryonic development.) Without DNMT1, the imprints would disappear.
It is impossible to create mice that are free of DNMT1, however, as DNMT protein deficiency is lethal in somatic cells, and embryos without DNMT1 die after the gastrulation stage of development. Embryonic stem cells can survive without DNMT, and so embryonic stem cells were generated with two alleles of DNMT1, both of which were conditional—that is, they could be turned on or off experimentally. One of the alleles was made conditional with CRE-Lox technology and the other was made conditional with FLP-FRT. Therefore, if the stem cells were exposed to CRE, it would deactivate the first DNMT1 copy, which was otherwise active, and if they were exposed to FLP, it would activate the second DNMT1 copy, which was otherwise inactive. The approach, then, was to expose the stem cells first to CRE to deactivate DNMT1 and cause the entire genome to be demethylated. They would then be exposed to FLP, which would turn on the other DNMT1 copy and remethylate everything except the imprinted genes. Control cells were exposed first to FLP and then to CRE, so that at least one copy of DNMT1 was always active and the cells never went through demethylation.
When microarrays were used to compare the imprint-free cells with control cells, the pattern of gene expression was as expected. Some genes in the imprint-free cells had double the expected level of expression; these were the genes in which imprinting involved methylation that silenced one of the copies. Others genes were not expressed at all in the imprint-free cells; thus, the imprinted genes were inactive unless they were methylated. The imprint-free stem cells grew well in culture and were immortal, so they could be injected into developing mouse embryos to produce chimeras. In these chimeras some of the tissue would descend from the imprint-free stem cells, whereas the rest would develop from the cells of the original embryo.
The animals were aged, and once they reached 12 months of age, every animal developed multiple tumors. Tumor types included hepatic and intestinal cancer, one seminoma, and leukemia and lymphomas. The conclusion was that the loss of imprinting played a causal role in the formation of multiple tumor types.
The last question addressed was the possibility of reversing those cancer states that are epigenetic in origin. A series of experiments were conducted in which somatic cell nuclear transfer techniques were used to remove the nuclei from tumor cells and inject them into enucleated eggs. The eggs were then allowed to develop to the blastocyst stage, which were then explanted in culture to derive cloned embryonic stem cells. The cells were injected into normal blastocysts to generate chimeric animals.
In these animals the embryonic stem cells cloned from the original tumor cells contributed to all tissues, suggesting that the cancer genome had been “reprogrammed” to a pluripotent epigenetic state. Thus, by monitoring the development of cancer in the chimeras, it was possible to determine whether transplantation of the cancer nucleus into the normal eggs would influence tumor developmental potency and rate of development.
When these experiments are done with different types of cancer, some prove to be reversible and others do not, depending on the tumor type. Thus, there appears to be two extremes in tumorigenesis: “one where the phenotype is largely determined by epigenetic changes which are reversible and another where it is all genetic. Most tumors are probably somewhere in between, and one of the issues is to determine what is epigenetic and what is genetic.”
Finally, Jaenisch spoke of some of the issues facing those who would apply epigenetics tools to nutrigenomics. One of the key questions to be answered, he said, is how diet affects long-latency diseases. “I think diet strongly affects cancer incidence, but does it have an effect on neurodegenerative diseases, such as Alzheimer’s disease or Parkinson’s?” To be able to answer these sorts of questions, he said, one of the key tools will be the ability to determine the methylation state of individual CpG sites across the entire genome. “We don’t have these methods; that is a major issue. We can struggle to do it for a few genes, but we need it for the whole genome.” Similarly, he said, tools are needed to determine other sorts of chromatin modifications across the entire genome as well, because methylation of CpG sites is only one type of modification that is important in epigenetics. There are a number of others, and they may all play a role in how nutrients interact with the genome.
FOLATE METABOLISM AND THE FETAL ORIGINS OF ADULT DISEASE
Presented by Patrick Stover, Professor and Director of the Division of Nutritional Sciences, Cornell University
In 1986, David Barker of the University of Southampton proposed what would come to be known as the fetal origins hypothesis (Figure 2-2). Noticing that coronary heart disease was the most common cause of death among a group of men who had none of the usual risk factors, such as obesity or smoking, he noticed a pattern of low birth weight and suggested that the increased risk for heart disease might have its origins in nutritional deprivation in the womb decades earlier. Since then not only has Barker’s hypothesis been borne out, but epidemiological evidence
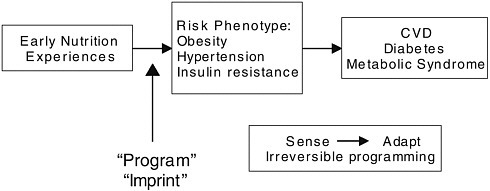
FIGURE 2-2 The Barker hypothesis of the fetal origins of adult disease. Fetal environmental exposures, especially nutrition, act in early life to program risk for adult health outcomes.
has linked low birth weight to a host of other diseases, from breast and prostate cancer to diabetes and depression.
As Patrick Stover explained, the underlying concept is that the nutritional environment in utero must somehow program a risk phenotype that later manifests itself in an increased risk for various diseases. “Implicit in this hypothesis is the idea that the fetal genome can sense its environment [and] make the necessary adaptations to survive in that environment and that those adaptations would be irreversible.” Unfortunately, the programming events that increase the chances of the fetus surviving its time in the womb prove deleterious later in life.
The question that arises is how the nutritional environment in utero programs the genome epigenetically and, in particular, how a nutritionally deficient environment results in programming that increases disease risk later in life. “We know that programming is associated with chromatin modification,” Stover noted, “whether it be DNA methylation or demethylation, acetylation, ADP ribosylation, or biotinylation of histone proteins. We also know that nutrients can influence some of these processes. There is increasing evidence, for instance, that both nutrients and metabolism can modify the probability of certain chemical modifications occurring, such as methylation.” These nutrient-driven changes can be transient, but according to the Barker hypothesis, if they occur within a critical window in development, the result may be a meta-stable (irreversible) modification that persists throughout life.
Research from the Stover laboratory focuses on one particular aspect of this issue: folate metabolism and its role in epigenetic modification of the genome. Folate, which refers to a family of chemically related com-
pounds and which is one of the basic B vitamins, has a variety of metabolic roles, particularly in DNA replication and genome maintenance, but the role that is most relevant to genome programming is as the source of the methyl groups that are used to modify chromatin and, hence, gene expression.
The end points of folate-mediated one-carbon metabolism include purines and thymidylate, which are necessary for DNA synthesis, and methionine, which can be adenylated and function as a cofactor in methylation reactions, including the methylation of DNA and histone proteins in chromatin. Once these reactions occur, the spent cofactor, S-adenosylhomocysteine, can be reused in additional methylation cycles. “This methylation cycle is key because it is a segue, if you will, between metabolism and chromatin modifications.”
A number of different nutritional deficiencies can affect the metabolic pathway, for example, a deficiency in folate or vitamin B12, since the enzymatic activity of methionine synthase is dependent upon both of these vitamins. When the pathway is disrupted, all of the intermediate steps and cross-pathways will also be disrupted because they all compete for a limited pool of folate. Repercussions can occur, such as a decrease in the amount of thymidylate available to the nucleus, which then causes DNA polymerase to mistakenly incorporate uracil into replicating strands. Individuals who are marginally deficient in folate can have up to 10 times as much uracil as normal in their DNA. A decrease in the amount of 5-methyltetrahydrofolate will also decrease methionine synthesis, in turn reducing the rate of DNA methylation.
A common mutation that can negatively affect the folate pathway is present in the gene for methylenetetrahydrofolate reductase (MTHFR); carriers of this mutation have an increased risk for neural tube defects, miscarriage, and other fetal disorders. This common polymorphism in the gene for MTHFR causes the encoded proteins to have reduced activity and stability and, in particular, lessens the cell’s ability to accumulate methyltetrahydrofolate. This in turn results in a decreased capacity for methylation reactions, but because the methylation pathway is impaired, the competing thymidylate pathway actually becomes more efficient. This change in biochemistry confers both risks and benefits. The decreased methylation rate increases the risk for neural tube defects in the developing fetus, but in adults the increased level of thymidylate synthesis leads to a lower risk of colon cancer.
The Stover laboratory has investigated the enzyme serine hydroxymethyltransferase (SHMT), which is active only in certain tissues. SHMT converts serine and tetrahydrofolate to glycine and methylenetetrahydrofolate. In those tissues in which it appears, it has the effect of inhibiting the methylation cycle and increasing flux through the thymidylate pathway;
therefore, modifying the activity of the gene will affect both methylation status and DNA stability. Experiments with knockout mice have shown that if one of two copies of the gene is knocked out, which reduces the amount of SHMT in half, then the amount of uracil incorporated into DNA increases by as much as 10-fold. At the same time, the increased methylation affects the level of expression of some 200 other genes in the cell. Interestingly, the gene for SHMT functions as a “nutrient sensor,” in that it is highly responsive to zinc, retinoic acid, and ferritin. This exemplifies how the levels of several different nutrients can affect gene expression and DNA stability through a common pathway.
Clinical evidence also indicates that diet can affect epigenetic programming. A study in Italy evaluated seven patients with uremia, which resulted in elevated levels of homocysteine, the precursor to methionine in the folate-mediated one-carbon metabolism. The elevated homocysteine levels resulted in a reduction in methylation in the patients. When the patients were tested for the level of expression of a particular imprinted gene, H19, one copy of which is normally methylated and is thus inactivated, three of seven patients (who had the highest levels of homocysteine) had a second copy of the H19 gene that was also being expressed. Thus, the elevated homocysteine levels that resulted from uremia were reversing the imprinting of the gene. An effective treatment was to give the patients a folate supplement. This reduced their homocysteine levels and silenced the expression of the second copy of H19. Its imprinting had been restored. “So,” Stover concluded, “metabolism can influence reversibly imprinted gene expression once the imprint is already established.” This may be a property that is exclusive to stem cells.
Lastly, Stover offered examples of how folate can induce epigenetic changes to compensate for genetic shortcomings. In one group of mice that had been given a Hox gene knockout that caused a skeletal defect, putting the mothers on a high-folate diet prevented the skeletal defects. “In fact,” he said, “this has been shown for a number of mouse models: that you can reverse a variety of defects by giving high levels of folate or other methyl supplements.” In humans, giving folate supplements to mothers sharply reduces the risk of neural tube defects and can also reduce rates of spontaneous abortion.
The evidence, then, is that proper nutritional interventions can be a powerful tool for epigenetic reprogramming. However, a great deal of research must be done before the field begins to reach its potential. “This is an emerging area. There is lots of descriptive work. Mechanistic work is now emerging, and it is going to be absolutely critical in determining how nutrition can modify these epigenetic processes for long-term benefit,” Stover indicated. Research is needed to obtain an understanding of the effects of nutrient exposures on genome programming; identifying the
affected pathways, critical timing, and nutrient levels that alter programming; and determining the long-term risks and benefits associated with such nutrition interventions.
THE ROLE OF MATERNAL AND INFANT NUTRITION IN GENETIC PROGRAMMING AND EPIGENETICS
Presented by Cutberto Garza, Provost and Dean of Faculties, Boston College
Public health will respond as nutrigenomics research progressively unravels the bases of associations between early diets and medium and longer-term health outcomes. Cutberto Garza described one particular area in which nutrigenomics may have major public health implications. Several retrospective and prospective studies from multiple countries support the possibility that low birth weight and rapid “catch-up” weight gain (defined by the upward crossing of centiles on growth grids) increase the risk of type II diabetes and, possibly, cardiovascular disease, especially among individuals who experience significant in utero growth retardation. Studies based on the Dutch famine are similarly intriguing. Men exposed to famine conditions in utero during the first trimester experienced a nearly twofold increased risk of obesity. Other studies have reported a U-shaped relationship between birth weight and the risk of diet-related chronic diseases. Low birth weight is also reported to enhance the risk of increased blood pressure. Positive associations between postnatal weight gains and adult systolic blood pressure have also been reported. Some investigations detected this relationship only among those who experienced rapid weight gain in early childhood but not during infancy. It is particularly relevant that many well-controlled studies with animals also report links between early perinatal nutritional experiences and an increased risk of chronic disease.
Human and animal studies such as these raise three questions that are especially relevant to public health: Are the long-term adverse effects of early prenatal nutrition experiences on chronic disease risk reversible? Do increased risks persist across generations? What are the implications of such possibilities to public health?
Animal experiments have examined both the reversibility of phenotypes apparently enabled by early prenatal nutrition experiences and the underlying mechanisms that may explain the putative reversibility of targeted phenotypic characteristics. Investigations with animals of methylation-based epigenetic mechanisms, perinatal experiences, and the functional consequences of adult onset may be particularly instructive. For example, pharmacologic and dietary agents have been used to
investigate the epigenetic methylation patterns associated with specific functional consequences and their reversibility. For the present, the significance of the findings from the available animal studies to providing an understanding of the links between early diets and the later onset of chronic disease remains difficult to assess.
The second question relates to the intergenerational potential of such associations; that is, do adverse effects of harmful nutritional conditions in one generation persist in succeeding generations exposed to improved nutrition? Garza identified two categories of transgenerational mechanisms. The first is limited to maternal transmission. This mechanism requires that dietary exposure somehow alter maternal reproductive structures or functions in a way that affects the mother’s progeny. The second enables both paternal and maternal transmission. The latter mechanism requires that exposure somehow alter the epigenetic development of the germ line.
The best human data relevant to the first category come from studies of the impact of maternal size on the progeny’s birth weight. Maternal stature, not uncommonly, is a good indicator of past nutritional status but is independent of current nutritional status; yet, an infant’s birth weight remains significantly dependent on maternal stature. Thus, associations between the progeny’s birth weight and various long-term outcomes often provide evidence for transgenerational effects of maternal in utero or early childhood experiences. These associations, however, provide limited insight into the underlying mechanisms.
Among the more interesting animal models used to examine the second category are those that study epigenetic inheritance in the agouti mouse and those that investigate epigenetic transgenerational effects of environmental toxins. Both may prove relevant to understanding the mechanisms underlying the functional long-term consequences of early diets.
Thus, studies with animals have explored both categories of trangenerational effects and their underlying mechanisms. Nonetheless, knowledge as to how early nutritional environments affect health in later life is limited. Importantly, there is a growing consensus that early nutrition remains relevant throughout life. Enhancing knowledge of early nutrition’s impact on longer-term and intergenerational health could play a large role in informing public health policies and thus are relevant to today’s research agenda. This relevance is likely to grow as the global population ages and life spans increase.
What are the practical implications of such information to public health, and what influence does such information have on contemporary public health practices? Over the longer term it is likely that such information will be relevant to a wide range of dietary manipulations designed
to enhance short-, medium-, and long-term outcomes, for example, food fortification policies, nutrient supplement use, and population-based chronic disease management. More immediately compelling, however, is the growing relevance of maternal care and growth-monitoring programs. Data from studies with humans and animals that support links between prenatal and early childhood growth patterns and multiple long-term disease risks underscore the need for promoting physiologic growth in utero and throughout childhood and the need for robust tools to enable those assessments.














