
2
Current Adverse Event Reporting Systems
Participants discussed systems and structures currently in place for health-care providers, consumers, and others to report adverse drug events. Postmarket information on adverse drug events originates from patients and clinicians who are using and prescribing drugs. Reports of adverse drug events (ADEs) make their way to the FDA through various means—through pharmaceutical companies, which collect and report adverse events on their drugs; directly from providers and patients through the FDA’s MedWatch system; from patient databases of payers such as the Centers for Medicare and Medicaid Services and managed care companies; and from hospitals. There are numerous challenges associated with the collection of ADE reports that may lead to significant underreporting of adverse events.
MONITORING ADVERSE DRUG EVENTS
In an effort to monitor the effects of drugs after their release into the market, the Food and Drug Administration (FDA) instituted the Adverse Event Reporting System (AERS) in 1993. AERS is a computerized information database designed to support the FDA’s postmarketing safety surveillance program for all approved drug and therapeutic biologic products. AERS is supported by the FDA’s Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). Staff members evaluate reports made by drug manufacturers, physicians, and consumers to detect safety signals and monitor drug safety. The report-
ing of adverse events provides a signal or hypothesis that may be further evaluated by epidemiological methods and forms the basis for further epidemiological studies when appropriate (FDA, 2005b). Manufacturers must report to the FDA the following serious and previously unknown adverse events within 15 days of their occurrence and conduct a follow-up investigation (FDA, 2005c):
-
Events associated with drug use in a professional practice
-
Events resulting from accidental or intentional overdose
-
Events occurring from drug abuse
-
Events occurring from drug withdrawal
-
Any failure of expected pharmacological action
The analysis of serious adverse events identifies issues that should result in changes to drug labels or that require physician notification of adverse events. This information, as Anne Trontell, deputy director of the FDA’s Office of Drug Safety, noted, allows the FDA, in collaboration with the manufacturer, to ensure that there is effective product labeling to alert health-care practitioners and patients to possible safety risks and areas of risk prevention.
Daniel Troy, a partner at Sidley Austin LLP and former chief counsel at the FDA, added that after a drug is approved by the FDA, manufacturers must submit quarterly reports for the first three years and annual reports after this three-year period. The FDA can extend the quarterly reporting period upon written notice. The content of reports includes a narrative summary, analysis of information in the 15-day alert reports, and a listing of actions taken since the last report, such as labeling changes or any studies that were initiated. The FDA can withdraw the approval of a drug if the correct reporting has not occurred.
The FDA does not conclude from a submitted report that a drug is the direct cause of an adverse event, but rather that the event is associated with use of the drug. The FDA does not impose on physicians any legal requirements for reporting adverse events because it lacks authority to regulate the practice of medicine, a responsibility of individual state governments. Currently, 20 states have mandatory reporting systems, but according to Mr. Troy his experience has been that there are known cases of adverse reactions that are not reported. In states without mandatory reporting systems, report submission is completely voluntary and therefore dependent on the participation of health-care professionals. Possible reasons for underreporting include the time it takes to complete a report, fear that reporting events will have a negative effect on the practice of medicine, and liability concerns. Mr. Troy recommended that the AERS be streamlined to encourage reporting, for example, a single-page form
that is tested with physicians. There should also be no liability faced by health-care providers who report adverse events.
MEDWATCH
The FDA’s program for collecting data on ADEs is known as MedWatch. It has an outreach component, designed to facilitate public reporting of adverse events, and a reporting component, which provides the actual means for reporting, including an Internet portal for reporting drug event information. It also has standardized forms that can be downloaded and used to report ADEs (FDA, 2005d). In 2005, intake and acknowledgment of these reports were overseen by approximately five FDA staff who were assisted by contractors. Drug-related information that is collected through MedWatch ends up in the AERS if the adverse event is serious—i.e., if it results in a life-threatening event or hospitalization (or if it is for a new drug). The MedWatch system collects reports for other products as well—for example, medical devices and foods—and forwards those reports to the appropriate database.
The FDA reports that MedWatch captures only a small percentage of the total burden of adverse events (Trontell, 2004). Richard Platt of Harvard Medical School and Harvard Pilgrim Health Care stated that completing and filing forms for MedWatch (and the Vaccine Adverse Event Reporting System [VAERS]) requires clinicians to recognize that a medical problem may be an adverse reaction to a drug, remember how and where to obtain the forms, and then invest substantial time in providing the required information. These steps lead to substantial underreporting and incomplete reporting. In 2004, 48 percent of reports regarding adverse events were provided to the FDA through MedWatch by physicians and pharmacists (according to Trontell, predominantly pharmacists, with a lesser percentage from physicians) (see Figure 2-1). Consumers contributed only 17 percent.
Another concern about MedWatch is the variability of report quality. Although MedWatch is available to health-care professionals, these professionals are not necessarily taught how to use the system (Malenka et al., 2005). According to Mr. Troy, the FDA “generally assumes that only 1 in 10 adverse events is reported.” However, utilization of other reporting methods, such as registries, can result in higher reporting rates. One issue with voluntary reporting is that clinicians report only when they think something is both significant and drug related. The large majority of adverse events are either not recognized or not reported, and there are unknown biases in the reporting that does occur, according to Dr. Platt. There have been suggestions that anonymity of reporting and perhaps
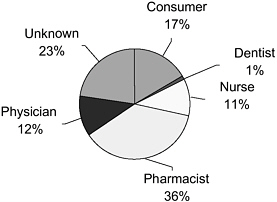
FIGURE 2-1 Sources of reports to MedWatch.
SOURCE: FDA (2005e).
overcoming the barrier of liability concerns would motivate people to file more adverse event reports, said Dr. Trontell.
Most adverse events are reported through spontaneous reporting that places a burden on health-care providers. The FDA budget is not adequate for the investment in information technology that could help with the collection of and access to quality data (Gottlieb, 2005). Information is becoming increasingly available in medical claims databases and clinical databases that could be used in reporting adverse drug events. According to Dr. Platt, “These databases provide relatively complete and unbiased information for many drug and vaccine exposures and for important outcomes, along with substantial comorbidity information. Databases are particularly useful when combined with review of the full-text medical records of a very small number of people to confirm the database information and to gather additional data.”
REPORTING ADVERSE EVENTS TO INSTITUTIONAL REVIEW BOARDS
Current systems for reporting adverse events to institutional review boards (IRBs) are problematic because these reports rarely contain adequately detailed information and the data are reported sporadically and are not easily aggregated for analysis. In addition, the FDA has limited authority to regulate IRBs.
IRBs must review clinical investigations annually, at a minimum, and must maintain records of continuing review activities. According to Mr.
Troy, the FDA requires investigators to report an unanticipated problem to the IRB within 10 days. There are many reports received in a year that consist of raw and unanalyzed information. IRBs would be better served if information about agents used in a study was collated into summary reports. What is needed is a “manageable signal-to-noise ratio,” said Mr. Troy, who further stated that without specific regulatory guidance in this environment, overreporting is the best option from the legal liability perspective. Bernard Schwetz, of the Office for Human Research Protections (OHRP) in the Department of Health and Human Services (HHS) and a Forum member, echoed Mr. Troy’s comments about reports to IRBs not being synthesized. OHRP has created a draft guidance document titled Guidance on Reporting and Reviewing Adverse Events and Unanticipated Problems Involving Risks to Subjects or Others. It is intended to assist IRBs, investigators, research institutions, HSS agencies that conduct or sponsor human subjects research, and other interested parties. OHRP believes that there will be more collaborative efforts by federal agencies that are involved in human subjects research to create a new approach to handling adverse events (HHS Office for Human Research Protections, 2005).
REPORTING ADVERSE EVENTS TO THE CENTERS FOR MEDICARE AND MEDICAID SERVICES
The Centers for Medicare and Medicaid Services (CMS) compiles medical data on patients it covers in order to manage reimbursement. The databases created are also used to collect safety data about medications and devices, although claims data must be validated with medical records. CMS has examined 65,000 charts. David Hunt of the CMS Quality Improvement Group reported that this chart review was initiated to detect errors in payment. However, it became apparent that these reviews could be used to monitor clinical events. A CMS program, the Medicare Patient Event Safety Monitoring System, will examine ADEs in the near future. The first adverse event areas to be assessed are those associated with two classes of drugs, anticoagulants and hypoglycemics; one specific drug, digoxin; and a specific class of reactions, antibiotic-associated diarrhea. These areas are also being examined by Medicare quality improvement organizations. Data are gathered from chart reviews based on a random sample of Medicare enrollees. Dr. Hunt stated that CMS plans to examine adverse drug events captured by Part D data as they relate to inpatients, because that existing mechanism already works reasonably well. Plans to examine outpatient data are under way, and CMS is working to promote electronic recording practices. Medicare part D data should provide important insight. However, it will take a few years before clean data from
Part D are available. CMS and the FDA will share this information, which will inform both organizations’ quality improvement projects
CMS issued a guidance document in April 2005 supporting a system of postmarketing data collection for drugs for which national coverage decisions must be made under Medicare Part B (CMS, 2005). Reimbursement is a powerful driver for safety studies. Although Medicare will be collecting data on Part D drugs, it will have no authority to make coverage decisions based on that information (Gottlieb, 2005).
NEW APPROACHES FOR IMPROVING REPORTING SYSTEMS
Alastair Wood of Vanderbilt Medical School discussed new approaches to improve the current adverse event reporting systems. He described MedWatch and other reporting systems as being set up to detect rare events and said that what is needed is the capture of “high-frequency, high-impact” cases that are not detected with the current systems. “One approach,” said Dr. Wood, “is to have incentives for long-term safety studies.” This, however, would cause a long delay before drug approval and would not be cost-efficient. It would also entail discussion about which drugs would be subject to long-term study. Another approach is to conduct long-term safety studies after approval. This again requires consideration of which drugs would be chosen for study or whether all drugs should be studied.
One way to ensure the completion of safety studies is to offer extended exclusivity to companies that have acquired data to demonstrate that their drug is safe in the long term, offered Dr. Wood. This makes the drug more valuable to consumers and to the company. Most importantly, Dr. Wood said, “We need to move to a reward-based system that rewards demonstrated safety.” In his proposal, drugs that lacked long-term safety data would be clearly identified as such. In this way, physicians could make the appropriate choice of medication with their patients. A fundamental issue would be the design of these safety studies, which under Dr. Wood’s proposal would require FDA approval. In other words, extended exclusivity would be offered only for well-designed studies structured to answer important clinical questions.
Forum member Robert Califf proposed a clinical trial “light” system, in which new users of drugs would be notified about known drug risks and benefits. The system would indicate that the drug being prescribed had recently been approved but that information concerning both risks and benefits was being developed. Patients would be provided information to report any adverse events and asked to participate in follow-up studies concerning the medication.






