
3
Active Surveillance Systems
Active surveillance systems screen claims data and notify healthcare providers who then determine if follow-up or adverse event reporting is required. These systems can scan for known adverse events or facilitate adverse event reporting. Analysis of claims data is required to examine suspected new adverse events or modulation in the frequency of common events. The frequency of events and the timing of the event associated with a particular drug will impact the surveillance system’s detection ability (Brewer and Colditz, 1999). Currently no standard alert system is universally accessed and used.
Workshop participants discussed types of surveillance systems, current technology used by these systems, and challenges in obtaining quality data. According to Peter Kilbridge of Duke University, there is a great need for standard alert systems; however, the number of people working to build and connect such databases is relatively small. Each current approach usually focuses on the hospital or facility level. Historically, adverse drug event (ADE) surveillance has focused more on the inpatient than on the outpatient setting. A 2004 survey of hospitals in Missouri and Utah found that only 34.1 percent of these facilities reported implementation of computerized physician order entry systems for medications (Longo et al., 2005). The current emphasis in developing alert systems is on assessing drug interactions rather than adverse drug reactions.
TYPES OF SURVEILLANCE
Dr. Anne Trontell of the Food and Drug Administration (FDA) gave three examples of surveillance: drug-based, setting-based, and outcome-based. Drug-based surveillance occurs when clinicians prescribe a new drug product and actively report on patient safety. This approach examines populations of interest after the launch of a product. It is similar to the United Kingdom’s prescription event monitoring system that follows the first 10,000 users of a new product. Japan’s health-care system also engages in active surveillance in the first six months of a product’s marketing. The UK and Japanese systems could serve as potential models for the United States in postmarketing surveillance.
Dr. Trontell discussed setting-based surveillance as another way to capture ADEs. Drug-associated adverse events may present or otherwise be concentrated in certain health-care facilities. Setting-based surveillance systems in hospitals, emergency room departments, or pharesis centers may help detect relevant drug-related events. Dr. Trontell also noted that the FDA, in collaboration with the Centers for Disease Control and Prevention (CDC), the Consumer Product Safety Commission (CPSC), and the National Electronic Injury Surveillance System (NEISS), looks at its ability to detect drug-related injuries that present to emergency departments.
The National Electronic Injury Surveillance System: Cooperative Adverse Drug Events Surveillance (NEISS-CADES) is a nationally representative subsample of 64 of 98 NEISS hospitals selected as a sample of U.S. hospitals (CDC, 2005). At each of the hospitals, coders review all emergency department charts for ADEs. The case definition excludes drug withdrawal, drug abuse, self-harm attempts, lack of therapeutic effect, and effects of medications administered in the emergency department. This system captures prescription and over-the-counter medications, vaccines, vitamins, and nutritional supplements.
The final area Dr. Trontell discussed was outcome-based surveillance of selected health outcomes that are often associated with drug toxicity. For example, the FDA is working with the Drug-Induced Liver Injury Network (a network of liver transplant centers) to solicit information about antecedent drug exposures in individuals who are listed for or require a liver transplant. Such a system may identify individual agents or combinations thereof that are associated with hepatotoxicity.
SURVEILLANCE TECHNOLOGY
Dr. Kilbridge reported that in the Duke University adverse event detection system, even with strong encouragement to report adverse events, approximately one out of every six events is logged into the
voluntary reporting system. In comparison, estimates are that the ratio in community hospitals is 1 in every 80 events. These data from Duke illustrate how voluntary reporting falls short of accurately reflecting the number of adverse events experienced by patients since 5 out of 6 events are missed.
Dr. Kilbridge pointed out that surveillance systems are constrained by the types of data available. Data are derived from many different sources and are highly variable in quality. He also indicated that the systems are resource intensive, consuming both financial and human capital. The low specificity of the alert system creates too many alerts for human staff to respond to each one. Dr. Kilbridge stated that the Duke system operates more than 60 triggers and sends the university hospital as many as 60 to 70 alerts per day, creating a substantial amount of work for health-care providers. Despite all this work, the system’s current logic leads to a true alert only about one out of six times that such events occur. Dr. Kilbridge expressed the hope that rules can be developed with a high enough predictive value so that they can be effective as a real-time intervention. “We need to balance the opportunity for real-time intervention with the practicalities of the providers’ work flow,” said Kilbridge.
Dr. Kilbridge stated that while the Duke University Pharmacy Group made approximately 128 reports to MedWatch in the past year, it received 1,500 automated detected ADE reports and 4,000 voluntary reports from Duke University Hospital alone. Many of these adverse events were not necessarily reportable to the FDA. The majority of Duke’s MedWatch reports originating in the pharmacy come from voluntary reporting by pharmacists who observe events they believe to be unusual. “We screen for things that we already know about as side effects of drugs,” stated Dr. Kilbridge. Robert Califf commented that in the future, most active surveillance systems will automatically report both to the relevant pharmaceutical company and to MedWatch. However, he raised the question of whether the FDA would be able to handle the increased volume of reports.
Computerized data can be used to identify a signal that indicates the potential presence of an adverse event, and then human practitioners can intervene (Bates et al., 2003). Several claims-based systems and approaches that provide this information were discussed (i3 Drug Safety, Department of Veterans Affairs [VA], and the Health Maintenance Organization Research Network [HMORN]). According to the VA’s Francesca Cunningham, these approaches have the benefit of understanding population characteristics as well as adverse events. Different recording systems track different patient information. All of this information must then be integrated to form complete health data for patients.
Arnold Chan of the Harvard School of Public Health emphasized the
need for an active surveillance system not only for drugs, but also for vaccines and devices. One advantage of active surveillance systems is that the data have both a numerator and a denominator, which allow confidence intervals to be placed around potential event rates, said Dr. Chan. There are several active surveillance projects under way. i3 Drug Safety developed a data system identifying all new drugs on the U.S. market since 2003. This information can be accessed when safety concerns arise and has been made available to the FDA by contract, said Dr. Chan. A pilot collaboration between the Critical Path Institute (C-Path) and community pharmacies established a pharmacy-based electronic registry of patients taking new drugs. This pilot project will gather and verify baseline information, then follow up with patients. However, there are many challenges to gathering useful information. As many as 29 percent of patients do not take their prescriptions, 40 percent take drugs not listed in their medical record, and a large percentage of ADEs are never recorded.
Health-care providers may track the incidence and nature of events among their patients for purposes of quality improvement. Computers search the clinical databases for telltale combinations of data suggestive of ADEs. However, these searches are constrained by the source, quality, and type of searchable data. Construction and maintenance of these systems is an expensive endeavor in terms of both time and resources. Hershel Jick of Boston University Medical Center and Forum member Garret FitzGerald of the University of Pennsylvania pointed out that the field of people qualified to use these data systems effectively is relatively small. Human capital is essential to the success of maintaining and utilizing any database, whether newly created or already in place. Trained and experienced researchers are vital components of the reporting system. Quick access to large pools of high-quality data resources that are required for quality research is also essential.
Micky Tripathi of the Massachusetts eHealth Collaborative emphasized the important role that technology plays in improving surveillance systems (see Figure 3-1). He stated that given current technology, the next step is to engage practitioners and patients in a meaningful way in order to involve patients in their own health care and information tracking. In his presentation, Dr. Tripathi discussed some of the limitations of paper chart reviews, citing in particular the difficulty and expense involved in gathering all relevant records and navigating through them to find pertinent case information. This creates a time lag between the occurrence of a drug event and the practitioner’s ability to review medical data. One key benefit of computerized patient medical data is that they can be used to detect the frequency of adverse events and help identify methods to avoid them (Bates et al., 2003). Citing an example from an Indiana hospital, Dr. Tripathi stated that computerized surveillance systems detect
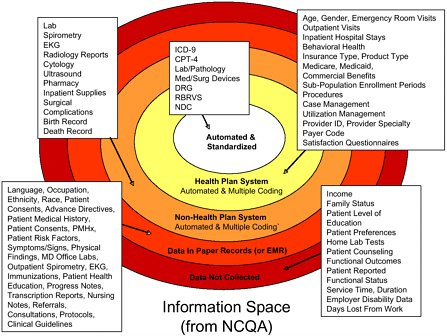
FIGURE 3-1 Levels of surveillance.
NOTE: CPT-4 = Current Procedural Terminology, 4th Edition; DRG = Diagnosis-Related Groups; EKG = electrocardiogram; ICD-9 = International Classification of Diseases, 9th Revision; NCQA = National Committeee for Quality Assurance; NDC = National Drug Code; RBRVS = Resource-Based Relative Value Scale.
SOURCE: Micky Tripathi, workshop presentation.
adverse drug event signals via the triggers found in patient medical records (see Table 3-1). As of 2004, 57.4 percent of hospitals surveyed used clinical codes from medical records to monitor patient safety, and 50.8 percent used quality improvement programs and discharge data to monitor patient injuries and adverse events (Longo et al., 2005).
Use of electronic medical records (EMRs) has developed in the healthcare field in an effort to improve data quality and the efficiency of data acquisition. The development of EMR systems is often piecemeal and focused primarily on billing and revenue. Even when systems are in place they are not always fully utilized. The VA’s EMR system is available free of charge but has not been widely adapted by other institutions. This is most likely due to challenges in integrating legacy systems and adaptation to varying institutional requirements. Implementation of EMR systems takes many years and requires substantial investments.
TABLE 3-1 Triggers in Patient Medical Records Used to Signal Adverse Events
According to Raymond Woosley of C-Path, an ideal surveillance database “should involve a hypothesis-based surveillance system, with accurate estimates of incidence. It should not only detect problems, but define the characteristics of adverse events, the associated risk factors, so we can identify methods of prevention.” Patient demographics, baseline medical history, and drug history are all key points of information to be included in such a system. Pharmacy networks are an underutilized information resource. Dr. Woosley stated that “[pharmacies] are electronically better networked than anyone else in our health-care system.” Dr. Woosley described a new research program at C-Path, an electronic registry into which pharmacies enter a patient’s drug history, a drug list, and complete contraindication and drug interaction screening and then arrange for personal follow-up. The program recommends that patients also register with a drug information center, which has trained pharmacists who are able to record adverse event occurrences and recommend treatment for ADEs. These pharmacists would also be trained to record adverse clinical outcomes from medications and manage them effectively.
The use of common data languages when communicating drug information would facilitate this higher level of knowledge about the safety of drugs. Robert Powell of the FDA reported that the Clinical Data Interchange Standards Consortium (CDISC) was instituted to increase more effective communication. The CDISC is a nonprofit organization dedicated to developing industry-wide standards for gathering and storing electronic information (CDISC, 2006). However, Dr. Powell noted that developing data standards for communicating information does not
seem to be a priority for either government or industry. In the year 2000, authors of a CDISC white paper estimated that the annual cost to industry for transferring data was $122.5 billion (Kush, 2001). Dr. Powell suggested that accelerating the CDISC standard for data submission would facilitate better communication of safety information.
THE VETERANS AFFAIRS HEALTH-CARE DATABASE SYSTEM
The Computerized Patient Record System (CPRS) is a drug alert program embedded in the VA health-care system. It is a powerful resource for surveillance and adverse event evaluation. According to Francesca Cunningham of the VA, the VA health-care system has more than 7 million enrollees and tracks prescriptions for more than 4.9 million patients in approximately 150 medical centers, 800 outpatient clinics, and 135 nursing homes. Its ongoing information systems monitor the VA’s population of high-risk patients. Elderly patients, patients with high medication use, patients exposed to new drugs shortly after approval, and patients with chronic health conditions are all at high risk for ADEs. Safety evaluations are routinely performed in which patients are categorized by demographic, medical history, medication, and treatment variables. They are then followed for 12 to 24 months. The system alerts providers to drug interactions, drug class duplications, and allergy warnings (Spina et al., 2005). The various databases can be integrated to give a more complete picture of the patient’s medical history than any one of the databases alone. This pooled information can be used to identify and track adverse events.
Within the VA’s database systems, the prescription database is merged with medical records, inpatient and outpatient files, and the mortality database. For specific projects, the VA has recently merged VA data with CMS databases for dual users, a move Dr. Cunningham reported helps the VA detect events that occur outside its own system. She provided a recent example of safety surveillance through the use of integrated VA databases. The VA found that the use of fluoroquinolones was associated with an elevated frequency of dysglycemia when compared to the use of azithromycin, particularly in diabetic populations. These findings led the VA to update the “VA Fluoroquinolone Criteria for Use,” an educational document for physicians explaining the need to monitor for dysglycemias more closely in certain patient populations. An important next step for the VA’s safety surveillance efforts will be to obtain provider feedback. “We want practitioner feedback. One of the things we are emphasizing is the evaluation of the impact that is made once this information is out,” said Dr. Cunningham.
DATA QUALITY
Nancy Santanello of Merck Research Laboratories emphasized the importance of obtaining information about risk factors and confounding influences on ADEs before and during clinical trials. Postmarketing trials and observational studies need sufficient power and length of follow-up on real-life populations with realistic end points. Concerns about observational studies include lack of randomization, lack of collection of important information for confounding variables, and the impacts of unknown cofounders. Data may be incomplete, missing, poorly measured, or invalid. Data also can be biased. Nonrandomized groups may be unequal, and unmeasured characteristics may be different. Even when risk factors for a particular disease are well known, adjustments for comorbid factors in an observational study may not be sufficient to remove the bias caused by differences between the comparison groups resulting from unmeasured risk factors. Exposures and outcomes also can be misclassified. These are not independent, and the resultant outcomes may be strongly biased in either direction even if the misclassification is nondifferential. Dr. Chan added that there is a need for studies to address comorbidity and co-medication use in real-life populations in order to improve postmarketing safety surveillance. Dr. Santello commented that “although a paradigm for the assessment of causality using observational studies exists, it must be applied cautiously and deliberately before definitive conclusions can be drawn.” She concluded that well-designed observational studies can supplement clinical trials and provide important additional information concerning the safety and effectiveness of therapeutic interventions.
Dr. Powell called for a more quantitative approach to the development of protocols and a higher level of learning throughout the entire development process. He noted that characterizing adverse events in terms of the time of onset, the relationship to when the dose is given, and how long it takes the adverse event to end is important. Unfortunately, this information is not found in many protocols. As a logical next step to resolve these issues, Dr. Powell recommended discussing safety and efficacy in the early stages of protocol development.







