
6
Drug Labels
The drug label serves as an important source of information for physicians when making prescribing decisions (Ray and Stein, 2006). There are, however, concerns about the usefulness of information contained in drug labels. Leander Fontaine of Pharmiceutics LLC noted that the drug label uses precisely defined regulatory terms such as adverse events or adverse reactions that might not be interpreted correctly by physicians and patients. Ed Staffa of the National Association of Chain Drug Stores stated that pharmacists worry that they will miss major drug interactions by having to sift through all the minor or theoretical ones. As an example of the volume and impracticality of this information, in the year 2000 edition of the Physician’s Desk Reference, the entry for cisapride occupied more than 10 pages and contained more than 470 facts about the drug (Woosley, 2000).
Furthermore, changes to labels and black boxes often do not occur in a timely fashion. One study found that only 50 percent of new adverse drug reactions are documented in the Physicians Desk Reference within a seven-year period (Lasser et al., 2002). “Labeling, by itself, although it is considered a principal tool for communicating drug information, generally does poorly. The FDA guidance for revising labeling goes some way toward improving the process, but not far enough,” said Sidney Kahn of Pharmacovigilance & Risk Management, Inc. According to Dr. Kahn, labeling information should be available, up-to-date, and easy to navigate and should contain trusted information about suspected adverse reactions.
The greatest problem in drug labeling is how to improve the quality of the data presented to the prescriber, particularly information concerning drug-drug interactions. Currently, the information available is not helpful in managing patients in real time, and drug interactions are found only on newer product labels, not the older ones, said Dr. Kahn.
In the present regulatory environment, all possible adverse reactions are included on labeling because exclusion of information could potentially represent great legal liability. “There is a common misconception that FDA regulations require that every single potential adverse reaction report be listed on the label, but that is not the case.” said Rachel Behrman of the FDA. Processing excess information is an inconvenience experienced not only by prescribers. The burden of so much content impacts pharmacists as well. “Part of the reason for the information overload in pharmacies sometimes is the concern of the pharmacy owner or operator hearing that if they don’t let the pharmacists see all of the information, all of the potential interactions, all of the theoretical interactions, that they will somehow be held liable for missing something down the road,” said Dr. Staffa.
NEW LABEL REQUIREMENTS
The FDA estimates that 300,000 preventable adverse events occur each year in the United States because of confusing medical information (FDA, 2006). The development of content for the label is a result of collaboration among the FDA, industry, and U.S. Pharmacopeia (USP) standards. Although companies write label drafts, only the FDA has authority over the final content. After approval, a label change can be requested by the FDA, but the company is not required by law to comply (Ray and Stein, 2006), although the FDA does have absolute authority to withdraw the drug.
To address concerns about labeling, the FDA has recently changed the format of drug labels in an attempt to make the information more useful. The new labels have a brief highlights section (see Figure 6-1) that summarizes information contained in the boxed warning, indications and usage, and dosage and administration. It also refers the health-care professional to the appropriate section of the full prescribing information (FDA, 2006).
Manufacturers must add any new information learned in the preceding year to the highlights section in an effort to keep physicians updated on new indications and interactions. A toll-free number and Internet address will be provided on the label to make reporting incidents more convenient. A table of contents and the date of initial drug approval will also appear on the product labeling. As of December 2006, all marketed

prescription drugs had to have electronic copies of their labels on the Medline website and be accessible free of charge. This information is to be kept up-to-date within 24 hours of approval or the FDA’s receipt of information, added Dr. Behrman. Up-to-date information on the FDA labeling initiative can be found at http://www.fda.gov/cder/regulatory/physLABEL/default.htm.
COMMUNICATION OF RISK TO CONSUMERS AND PHYSICIANS
Drug labels do not communicate the likelihood that a particular adverse event will occur when taking the drug. Dr. Fontaine suggested there should be approaches to inform consumers and physicians about the level of causal certainty in labeling, as well as severity or relevance categories of adverse reactions based on their expected probability, when this information can be obtained. He proposed that the adverse reactions section of U.S. labeling provide only lists of (suspected) adverse reactions (i.e., a causal relationship between product use and type of event is considered at least a reasonable possibility). Tables of mere adverse events from clinical trials, to the extent they are considered valuable information for prescribers, should be provided in a scientific background document to the labeling, comparable to the scientific section of Canadian drug labels.
Dr. Fontaine proposed that labeling inform consumers and physicians about the level of causal certainty of suspected adverse reactions. The likelihood that an adverse reaction will occur when taking a drug should, he suggested, be illustrated both by listing reporting rates of adverse events observed in clinical studies (as it is done currently) and by grouping reactions in frequency categories based on an estimate of attributable risk (applying the methodology used in the European Union); the latter would provide value-added, relevant information for the authors of patient information texts. To make risk communication effective, the medical and scientific community, users, media, industry, and regulators must have a common understanding and acceptance of the decision-making principles behind risk labeling; must agree on the meaning of terms such as adverse reaction, adverse event, and risk; and must use such terminology transparently and with discipline.
Dr. Fontaine also noted that to ensure that labeling is the primary trusted source of information about the risk of a product, any addition of new adverse reactions and interactions should be accompanied by a public summary of the evidence that supports the new information and by a discussion of the level of uncertainty about a causal association (see Figure 6-2). “The information consumers need should be from a single source
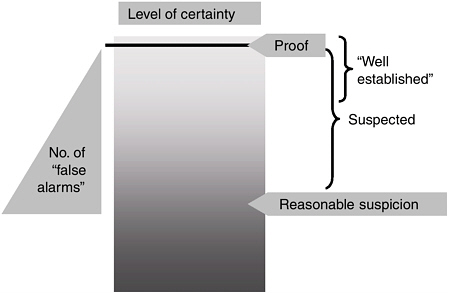
FIGURE 6-2 Level of certainty in association between a drug and an adverse event.
SOURCE: Leander Fontaine, workshop presentation.
and uniform in content for each drug or drug class. It should include all the information necessary for optimum drug use,” said Dr. Lipman.
DRUG LABELING AND DRUG-DRUG INTERACTIONS
Labels are not being utilized effectively to communicate a drug’s potential side effects or interactions with other substances. “We need to think about putting information in formats that are more clinically directed and useful, although a way of testing such formats … has yet to be established,” said Jeffrey Drazen. Dr. Kahn added that it is extremely necessary to devise a categorization or standard terminology to uniformly evaluate interactions for their clinical importance. He noted that the FDA and industry are restricted by the liability concerns surrounding labeling. As discussed earlier, parties have incentives to cite every possible safety hazard associated with drug use, resulting in too much information on the label for it to be useful. All reported safety information must be included on the label because there is no framework for excluding it. “Correct and accurate is good and is not good enough. Information has to be actionable,” stated Dr. Kahn in reference to information included on drug labels.
He added, “The end result is that information that is currently available does not help prescribers in the management of patients in real time.”
Since DDIs involve at least two drugs, interaction information may be placed on the label for the newer product in an interactive pair, but not for the older drug. The side effects listed on the label give no indication that the likelihood of experiencing these symptoms was higher when taking the drug or the placebo. Robert Califf emphasized the need for a third party (neither the FDA nor the pharmaceutical industry) to decide what information is relevant to consumers and useful to prescribers and therefore should be included in labeling. “I wouldn’t want the label content to be either a market-driven issue or an opinion poll,” said Dr. Califf.
Mr. Teagarden proposed the United States Pharmacopeia (USP) as a good medium for developing an official list of drug interactions derived from its official monograph for content. USP has already defined medication error through its National Coordinating Council for Medication Error Reporting and Prevention (NCC MERP). USP founded NCC MERP in 1995, and its membership includes 22 patient safety groups (NCC MERP, 2005). NCC MERP claims its definition of medication errors as a successful development. According to NCC MERP, “A medication error is any preventable event that may cause or lead to inappropriate medication use or patient harm while the medication is in the control of the health-care professional, patient, or consumer. Such events may be related to professional practice, health-care products, procedures, and systems, including prescribing; order communication; product labeling, packaging, and nomenclature; compounding; dispensing; distribution; administration; education; monitoring; and use” (NCC MERP, 2005).
This definition has been adopted by the FDA, Centers for Medicare and Medicaid Services (CMS), and USP. The development of a common nomenclature has the potential to enhance the ability of different institutions to share information.
TOOLS FOR IMPROVEMENT
A working group, using the NCC MERP information as a model, could improve the information provided to the prescriber of a drug. The group could bring together experts from academia, practice, pharmacy, and industry, as well as regulators. It could “improve the tools that are already available to communicate the interactions between drugs and their likely consequences,” said Dr. Kahn.
The FDA has several related regulatory initiatives to improve drug safety. The electronic labeling rule requires industry to submit e-labels to the FDA beginning in June 2006. This rule requires bar-coding on all over-the-counter medications. MedWatchPlus will unify adverse event
reporting systems and expand communications. The FDA would “like to move into the paperless world,” according to Dr. Behrman. Paperless labeling will eliminate the requirement for paper package inserts, which cost companies about $1 million per year per product.
Electronic connectivity will be integral to improving safety, and patient involvement is absolutely necessary. E-prescribing orders drugs in a clear manner and also provides specific information about the patient for whom the medication was prescribed. Access to specific patient information allows a health-care practitioner to appropriately assess potential adverse events based on specific patient knowledge. A problem with databases is that they can only give general information about potential adverse events. Electronic prescribing is enabled in 85 percent of chain drug stores, said Dr. Staffa, but very few of these have the connections necessary to take full advantage of patient-specific information.
In addition to e-prescribing, pharmacists can obtain necessary information about individual cases by simply talking to patients. Pharmacists cannot rely on database information alone. In speaking with a patient, the pharmacist may learn that the doctor is already aware of a particular potential interaction but wants to utilize the combination despite the warnings against it. The pharmacist is also in a unique position to find out what other substances the patient uses that may interact with medications. Information about alcohol, food, and over-the-counter medication is not included in the databases. Dr. Staffa added, “We need to see the pharmacist in a greater service role rather than a product-fulfillment role.”
Board certification is a mechanism through which to engage physicians in education concerning the drug safety surveillance system. Cary Sennett, of the American Board of Internal Medicine (ABIM), discussed the ways in which the ABIM uses performance evaluation as part of recertification. The ABIM certifies approximately 180,000 doctors, almost one-third of all practicing physicians in the United States. Certification is not a one-time event; it is a lifelong process that begins with initial certification and requires physicians to maintain their performance through ongoing reevaluations. The certificate holder must pass a secure proctored exam and “demonstrate a commitment to maintain the currency of medical knowledge through ongoing self-assessment of practice performance,” added Dr. Sennett. The certificate holder can use practice improvement models (PIMs) to meet this requirement; although current PIMs do not address drug safety explicitly, they include a survey of practice and the infrastructure directly relevant to safe medication management.
“Physicians will continue to participate and will be looking for sources of information to help them meet the maintenance of certification requirement,” stated Dr. Sennett. PIMs permit the systematic collection of data relevant to practice performance, such as the way a physician conducts
analysis and plans responses to predictable events. To address the rising demand for better drug safety surveillance, the ABIM is developing a patient safety module that will address drug safety in physician certification and recertification. This project will be completed in late 2006 or early 2007. “Maintenance of certification is a hook through which other quality improvement exercises can be linked or to which they can be attached,” said Dr. Sennett.
Assuming that the current level of funding continues, the FDA planned to launch a complete inventory of prescription drugs in the winter of 2006 and an inventory of all marketed drugs by 2008 on the Internet site Facts@FDA/DailyMed. E-prescribing code sets are also planned to be complete and up-to-date by 2008. Further incorporation of drug safety in certification is an important approach to improving education in this area. These new approaches should give the public and health-care providers significantly improved information. Dr. Drazen concluded that in order to improve the system, clear, accurate, and immediately accessible prescribing information should be made available and should be accompanied by expanded data standards, improved passive surveillance, and robust postmarket active surveillance efforts.








