
3
From Genomics to Metagenomics: First Steps
Genomics as a discipline is at most three decades old. The notion that it might be possible to sequence the genome of our own species began to be discussed in the early 1980s and was seriously considered at federally sponsored workshops in 1984 and 1985; pilot projects began in 1986 (Lambright 2002). The completion of the Human Genome Project (HGP) in 2000 has not only greatly accelerated biomedical science, it has also transformed it. Many questions first asked at the level of individual molecules and genes have better and more complete answers at the level of genomes and systems. And this is true not only for humans: it is now nearly unthinkable to launch a major comprehensive initiative in the biology of any species without sequencing its genome. We have genome sequences for many species of fungi; for nematodes, fruit flies and zebrafish (all highly useful models for human biology); for Arabidopsis (a model plant) and rice; for dog, cow, chimpanzee, and many more eukaryotes; and for almost five hundred bacteria and archaeans.
Furthermore, the many associated “omic” sciences (transcriptomics, proteomics, structural genomics, and metabolomics), all using similar high-throughput systems approaches, have revolutionized understanding of what genes do and how they work together (see Box 1-1, page 14). Genomic scientists have returned to hypothesis-testing, making predictions about the behavior of biological systems that can be tested through the acquisition of genome-level data by comparative sequencing and the application of the new “omic” methods.
The field of metagenomics would not be possible without the technological advances and bioinformatics tools that grew out of the HGP.
Descriptions of some of the earliest metagenomics projects illustrating different approaches to characterizing microbial communities are presented later in this chapter.
SEQUENCING IS JUST ONE KIND OF METAGENOMICS
As with genomics, many early metagenomics projects concentrated on gathering enough sequence information to characterize complete genomes. For metagenomics projects, the assembly of complete genomes from samples that are not pure cultures requires the physical recovery of organism-specific clones from environmental-DNA libraries or the computational recovery from environmental-DNA sequence databases of overlapping target-organism-specific sequences (“contigs”). For environments of low complexity, such as the acid mine drainage described below, it is possible to assemble several genomes simultaneously from an environmental sequence database by using various sophisticated “binning” methods (see Box 3-1). Other early metagenomics efforts, including the ones that first applied the term metagenome, used the term to describe a resource (all the genes in a particular community) to be mined for specific genes by assessing biochemical functions performed by large-insert clones in suitable hosts (Rondon et al. 2000). This kind of project is now called functional metagenomics, but that term is also sometimes taken to have a meaning analogous to functional genomics. In functional genomics, the goal is to determine not just the sequence of the genome but each gene’s function in the organism in which it is found. The metagenomic analogue would assess functions of the genes found in a community (or a sampling thereof) rather than in an individual species.
Many other “omics” techniques can be borrowed across disciplines. DNA microarrays, when bearing multiple rRNA (or other phylogenetic marker) gene sequences as probes, can be used to track variations in population structure and thus (indirectly) in community function over time and space. Microarrays based on selected genes (and gene variants) involved in processes of particular interest can be used to assess a community’s ability to perform a collective function (such as biodegradation of contaminants) and monitor changes in it over relevant periods (for example, during bioremediation). Community transcriptomics and metabolomics are still subdisciplines in their infancy because of the lability of mRNA and the complexity of communities, but metaproteomics (separation and identification through mass-spectrometric methods of many of the proteins in an environmental sample) is surprisingly well-advanced. And in communities where several genomes are known, it is beginning to be possible to develop community-interaction maps. Meta-omic monitoring of microbial communities as they function and change with time—for instance, genetic,
|
BOX 3-1 Organizing Metagenomic Sequence Data Clustering: An approach to data analysis in which a large dataset is divided into distinct subsets based on some specific measure. In analyzing DNA or protein sequences, clustering is used to identify groups of sequences that share an evolutionary origin (families) but can also identify larger sets, such as genomes (see binning). Genome annotations can be viewed as form of clustering, where individual genes are assigned to well-characterized (or at least previously known) gene families. In metagenomics, direct clustering of DNA sequences is likely to remain a primary annotation method, as most of these sequences will not be easily assigned to any known gene family. In direct clustering, the nucleotide (or predicted protein) sequence itself is the basis of the grouping of sequences. Binning: A clustering method that uses composition and/or other characteristics of DNA contigs (overlapping individual reads) to divide them into groups (clusters) that belong to specific genomes or groups of genomes. Examples of characteristics that can be used for binning are GC content and codon use. In metagenomic projects in which genome assembly is a goal, this is used as a preliminary step. Gene annotation: A process of classifying predicted genes into known and well-characterized gene families. In metagenomics, where a substantial percentage of sequences cannot be easily classified, annotations often remain at the preliminary stage of clustering the sequences into groups (families) that are otherwise uncharacterized. Gene prediction: A process of analyzing genomic DNA sequences to predict which encode biological functions, such as coding for proteins, structural and regulatory RNA, and other regulatory elements. Gene prediction is important for determining the functional repertoire of a microbial community and for comparing the capabilities of different communities. |
population, and metabolic processes that affect methane generation in the permafrost as it experiences global warming—is in principle not different from monitoring such changes in a culture of saccharomycetes as it adapts to a new substrate, in a fruit fly embryo as it develops, or in a human tumor as it progresses. Structural genomics—the systematic expression and structural characterization of the products of all the uncharacterized genes in a genome—will also be a boon; so far, this approach has been applied in the organismal context, but all the highly expressed but unidentified genes in a community metagenome would be an ideal target.
New concepts and methods will be developed for metagenomics that will expand the general genomic repertoire. Metagenomics captures microdiversity, or variation among strains of the same species, thereby producing
a more nuanced view of microbes. For instance, a comparison of sequenced genomes of Prochlorococcus in the Sargasso Sea shotgun-clone database facilitates identification of “genomic islands” that are highly variable within these genomes; in contrast, genomics on a pure culture would typically generate the sequence of only one of the variants, and the subtlety of population variation would be lost (Coleman et al. 2006). The use of such enormous databases to identify regions and mechanisms of variation within genomes or individual genes is a novel contribution of the metagenomic approach. So is DNA-based stable-isotope probing, in which specific incorporation of substrate containing a stable-isotope (such as C13) by cells in a community that can use it allows specific separation (by density) and identification (by sequencing or with microarrays) of their genes (Dumont and Murrell 2005).
PIONEERING PROJECTS IN METAGENOMICS
We illustrate below, through discussion of a few pioneering achievements and projects now under way, what the metagenomic research paradigm embraces and how its practitioners have begun to combine data collection and hypothesis-testing in sophisticated ways. Five types of projects are discussed: a simple community analyzed in depth, a large-scale sequencing survey in an environmental setting, a functional genomic project, a project focused on a microbial community living in a host, and a project focused on viruses.
The Acid Mine Drainage Project
Microbes in collaboration with humans have wreaked havoc on some geologic sites. One example is the production of extremely acidic outflows from metal mines around the world. The acid is produced by oxidation of sulfide minerals that are exposed to air as a result of mining activity. The acidic solutions that form as a result of mining activities are referred to as acid mine drainage (AMD) (see Figure 3-1). The microbial communities that drive the acidification have formed the basis of some remarkable metagenomic analyses designed to explore the distribution and diversity of metabolic pathways involved in AMD (for example, nitrogen fixation, sulfur oxidation, and iron oxidation), to understand the mechanisms by which the microbes tolerate the extremely acidic environment, and to evaluate how the tolerance mechanisms affect the geochemistry of the environment (Allen and Banfield 2005; Tyson and Banfield 2005; Ram et al. 2005; Tyson et al. 2004).
The AMD project has been paradigm-setting in part because the community exhibits just the right level of complexity—only five major players

(three bacterial and two archaeal species) reproducibly form a dense biofilm at the sites under study—and in part because it has been studied in great depth. Shotgun sequencing of community DNA enabled the nearly complete assembly of two genomes and partial recovery of three others. The challenge of simultaneously assembling multiple genomes was met by several binning procedures that allow provisional assignment of contigs (sequences that have been generated by computer assembly of overlapping individual DNA fragment reads) to different genomes on the basis of such overall characteristics as base composition and frequency of recovery (see Box 3-1).
Bioinformatic analyses have shown how individual community members might collectively interact biochemically, and the sequences themselves have provided evidence of more long-term genetic interaction at the level of recombination and lateral gene transfer. Nitrogen fixation could be assigned (because of infrequent recovery of relevant genes) to a minor “keystone” species, and metagenomic information guided the later cultivation of this species in pure form; one benefit of metagenomics will be that it will allow the cultivation of more currently “uncultivatable” organisms. Metaproteomic analyses of the same biofilms have now been performed. Many (about half) of the proteins predicted from the genomes of the dominant organism could be found, and their relative abundances say much about how the consortium functions bioenergetically. Proteins involved in coping with protein refolding and oxidative stress are highly expressed, and this reflects how difficult it is to live in acid mine drainage. Many abundant proteins appear to be novel (hitherto unknown) and peculiar to this harsh environment; these proteins will be key targets for a structural genomics approach.
The AMD project moved quickly and relatively easily, partly because of the very low complexity of the microbial assemblage studied. However, most microbial assemblages in nature are not nearly so simple, and this AMD biofilm assemblage represents a rare exception rather than the rule. Therefore, many of the challenges and opportunities of microbial-community genomics cannot be fully appreciated from this single, exceptionally simple example.
Further exploration of diverse microbial communities now demonstrates that shotgun sequencing alone cannot easily be used to complete whole microbial genomes, even in communities that are only moderately complex. Newer methods and approaches now being developed (for example, single-cell genome amplification) are likely to be necessary to dissect and compare the moderate- to high-complexity microbial communities common in natural settings. And completing whole genomes will often not be the goal of metagenomics projects: as the next examples will show, much can be learned about communities without identification of their individual members.
The Sargasso Sea Metagenomic Survey and Community Profiling
The world’s oceans harbor vast microbial populations that in part regulate the flux of energy, matter, and greenhouse gases in the sea. The biological properties of these globally distributed microbial communities are still only poorly described. In one approach to this problem, one of the largest metagenomic sequencing endeavors conducted to date employed a shotgun sequencing survey of microbial assemblages from the Sargasso Sea—an ocean environment thought to be relatively low in diversity (Venter et al. 2004). The project began by collecting microbial cells and viruses in different size fractions and extracting DNA from them. The single survey reported 1,214,207 identified putative protein-encoding genes, which represented almost 10 times more protein sequences than were present in all curated protein databases at the time (see Figure 3-2) (Coleman et al. 2006). The Sargasso Sea dataset is remarkable not only with regard to its new information, but also because of the sheer volume of data in it. The Sargasso Sea study was one among several recent studies that heralds a sea change in environmental microbiology efforts, and underscores the significant challenges and opportunities now associated with archiving, integrating, and analyzing massive metagenomic sequence datasets. It is becoming clear that metagenomic DNA sequences soon will outnumber all other types of DNA-sequence data combined.
The gene complement of the assembled Sargasso Sea microbial plankton included 1412 individual ribosomal RNA genes—a useful metric for taxonomic calibration. Counts of proteins useful as taxonomic markers were used to estimate species richness. The proteins indicated that there were about 1800 species (as usually defined) in the sample—which was in fair agreement with the total unique rRNA counts. The types of microbes encountered were generally consistent with those known to be prevalent in the ocean (with some exceptions, discussed below), on the basis of cultivation-independent surveys conducted in the sea over the last decade.
The native microbial Sargasso Sea sequence assemblies demonstrated that assembling large, accurate DNA contigs and scaffolds from shotgun-sequence datasets of complex mixed microbial populations is still a difficult problem. In the Sargasso Sea dataset for example, relatively few large contigs, and no whole genomes, could be assembled from the native microbial population. In retrospect, that is perhaps not too surprising in that it is widely recognized that native microbial populations harbor vast amounts of sequence variation. Inherent intrapopulation genetic complexity, combined with variable species richness and evenness, still poses extreme challenges for standard shotgun sequencing and assembly approaches developed for single microbial strains. One lesson learned from the Sargasso Sea analysis was that complementary approaches, including large-insert
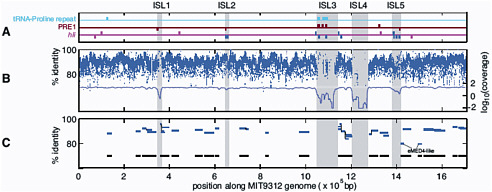
FIGURE 3-2 Genomic islands in the Prochlorococcus MIT9312 genome (ISL 1-5) compared to metagenomic data from the Sargasso Sea and the Pacific Ocean. A. PRE1, 48 bp repetitive element; hli, high light inducible genes. B. Blue dashes represent nucleotide identity of individual Prochlorococcus DNA fragments in the Sargasso Sea metagenomic dataset; the blue line represents average coverage; C. Blue lines represent individual Prochlorococcus large genome fragments (36 kbp) in a Pacific Ocean metagenomic dataset. Black lines represent total coverage of all the fragments across the Prochlorococcus MIT9312 genome. SOURCE: Coleman et al. (2006). Genomic islands and the ecology and evolution of Prochlorococcus. Science 311: 1768-70. Reprinted with permission from AAAS.
DNA sequencing and single-cell genomic approaches, will be useful and required for thorough characterization of all but the simplest of microbial communities.
Another serious issue with the initial Sargasso metagenomics effort was the microbial contamination that appears to have compromised a large portion of the largest sample, severely limiting its utility for ecological interpretations, and biasing it with non-indigenous microbial genes (DeLong and Karl 2005; Mahenthiralingam et al. 2006). This unfortunate result clearly indicates that metagenomics studies require much more integrated efforts, as opposed to simplistic cloning and sequencing of random environmental samples. Careful sampling and verification procedures, coordination with field experts, and independent sample validation are prerequisites for large-scale metagenomic sequencing efforts. Deep knowledge of the environment sampled, experience with the types and distributions of the indigenous microbes, and independent sample validation methods, will facilitate scientifically rigorous metagenomic studies. Skills from a wide variety of disciplines including environmental science, microbiology, molecular biology,
bioinformatics, mathematics, biochemistry, physiology, and ecology, are all necessary to properly gather and interpret metagenomics datasets.
Despite indigenous genomic complexities, potential sampling problems, and analytical challenges, the Sargasso Sea dataset has already proved a useful resource. Among previously discovered genes and proteins, including such photoproteins as proteorhodopsins, the Sargasso Sea dataset revealed new variations on a theme. The dataset’s value is evidenced in the large number of papers that have mined its taxonomic information, analyzing new gene sequences or even synthetically producing and characterizing never-before-studied genes and gene products. Detailed studies of whole-genome genomic variability, structural organization, and evolution in taxa that were well represented, including Prochlorococcus, have been greatly advanced by these new data. The theoretical and practical discoveries and applications already arising from this single dataset provide ample evidence of the value of large-scale, whole-microbial-community genomic analyses.
In lieu of full genome assemblies, comparative analyses of the Sargasso Sea and other datasets have demonstrated the utility of individual gene comparisons within and between samples. A new approach in microbial ecology, comparative community genomics, is emerging from such studies. A recent study, for example, compared, on a gene-by-gene basis, similarities and differences between community gene-sequence datasets from the Sargasso Sea, a sea-floor whale carcass, and the acid mine drainage community (Tringe et al. 2005). By taking a “gene-centric” approach, as opposed to an assembly-driven “genome-centric” approach, it was possible to compare the patterns of occurrence of specific gene categories and assemble “community profiles” of functional-gene content. The overrepresented specific categories of “environmental gene tags” (EGTs) in different samples (for instance, a disproportionate representation of photosynthetic genes and rhodopsins in the Sargasso Sea sample) verified the utility of the approach for inferring metabolic features associated with specific microbial communities. Judicious sampling can greatly facilitate such comparisons by allowing comparison of communities along well-validated environmental gradients. Another recent study in the Pacific Ocean compared microbial communities along a depth gradient, from surface waters to 4 km deep. Genomic characteristics that covaried with the environment were evident and suggested depth-specific functional and evolutionary themes in microbial communities and genomes (DeLong et al. 2006). This recent work highlights the future promise and utility of comparative community genomic studies.
The Soil-Resistome Project
The discovery of antibiotics transformed medicine in the middle of the 20th century by providing effective treatments for infections that were
previously untreatable and often fatal. After decades of use, the power of many antibiotics has diminished because populations of many human pathogens have evolved resistance to them and left us with no retaliatory weapons for some of the most virulent infectious agents. Although many of the genes that enable pathogens to resist antibiotics have been identified, little is known about where they originate in nature. Most of the antibiotics in clinical use were discovered in soil bacteria, so it seems likely that resistance genes also arose there. That the vast majority—perhaps as many of 99.9%—of the microbes in soil are not readily culturable, invites the use of metagenomics to assess the suite of antibiotic-resistance genes, or the “resistome,” in soil (D’Costa et al. 2006). The soil-resistome project takes a functional metagenomics approach: fragments of DNA are cloned from soil, and the clones are screened for expression of antibiotic resistance. This differs from the metagenomic studies discussed so far in that the genes are recognized by their activity—antibiotic resistance—rather than by their sequence, and this provides the opportunity to detect genes that might be unrelated to any known resistance genes.
The soil-resistome project has led to isolation of new groups of antibiotic-resistance genes. The strategy has been to clone metagenomic DNA from soil in temperate sites with natural vegetation, mixed grassland in Wisconsin, and a boreal forest in central Alaska. One of the advantages of studying antibiotic resistance is that it provides a selectable phenotype; only the clones of interest will grow in the presence of the antibiotic, so it is possible to screen libraries that contain millions of clones. This is impossible with most screens because they usually require addressing each clone individually and recording some feature of interest. In the soil-resistome project, the libraries are cultured in the presence of each antibiotic of interest, clones that grow are retested, and clones that are confirmed are saved for further study.
The resistome study revealed aminoglycoside resistance genes that encode a group of enzymes called acetyltransferases that are more closely related to each other than any previously described members of the family (Riesenfeld et al. 2004) and genes that encode resistance to β-lactam antibiotics (penicillin-like compounds) that were phylogenetically distinct from previously described enzymes. A gene that encodes an acetyltransferase was also discovered in the Alaskan forest soil, and its closest homologues in the sequence database were the genes discovered in the Wisconsin soil. This result raises intriguing questions: Are the resistance genes seen in clinical isolates the most abundant in the environment? Will some of the new genes find their way to clinical settings? If so, by what route? Will tools used in the past to inhibit the activity of resistance genes work in the future if these “wild” resistance genes reach the clinic?
The greatest challenge in metagenomic analysis based on functional
screens is gene expression. Success generally depends on expression, in a laboratory strain of E. coli, of exotic genes from exotic organisms. The differences in gene-expression mechanisms among species are likely to prevent detection of many genes by this method. Using multiple host species and tweaking the gene expression machinery of E. coli are mechanisms for achieving expression of a wider array of genes that deserve further study to enhance the utility of the function-based approach to metagenomics.
The Human-Microbiome Project
Microbes thrive on us: we provide wonderfully rich and varied homes for our 100 trillion microbial (bacterial and archaeal) partners. Considering that we contain perhaps 10 times more microbial than human cells and at least 100 times more microbial than human genes, it is inescapable that we are superorganisms composed of both microbial and human parts. Bacterial communities play an important role in health and disease in a variety of anatomical locations, such as the female reproductive tract, the skin, the oral cavity, and the respiratory tract. Even after completion of the first reference human genome, our view of the “human” genetic landscape is quite incomplete. We know little about how our microbial component has evolved or about the forces that are shaping it as our biosphere, our lifestyles, and our technologies change. What aspects of our microbiome are uniquely “human,” or mammalian? Are we undergoing a form of “micro-evolution” because of changes in our microbial ecology that is affecting our biology and our predispositions to diseases?
Because the human microbiota has not yet been extensively explored, much of what is known of the contributions of organisms’ microbial partners has come from comparisons of germ-free animals (reared with no microbes) with their counterparts that have been colonized with defined components of the mouse or human microbiota (Turnbaugh et al. 2006, 2006; Samuel and Gordon 2006). Comparisons of germ-free and colonized animals have shown, for example, that the gut microbiota regulates energy balance, directs myriad biotransformations (including detoxification of carcinogens), modulates the maturation and activity of the innate and adaptive immune systems, and affects the cardiovascular system. On the basis of these and other observations, the gut microbiota has been invoked as a factor that determines susceptibility to diseases ranging from obesity and diabetes to gastrointestinal and other malignancies, atopic disorders (such as asthma), infectious diarrhea, and various immunopathologic states, including inflammatory bowel diseases.
Initial results of 16S rRNA gene-based enumerations of the microbial communities of a small number of humans have revealed remarkable diversity in a number of habitats, including the gut (Eckburg et al. 2005; Ley et
al. 2006). That raises the question of whether there is a core microbiome associated with all humans and, if a shell of diversity surrounds such a core, what it contributes to the differences between individual physiologic properties.
The first truly metagenomic survey of a component of the human microbiota appeared in 2006 (Gill et al. 2006). It involved sequencing the microbial communities harvested from the colons of two healthy adults. Analysis of 78 million base pairs of unique DNA sequence disclosed that, compared with the human genome and previously sequenced microbial genomes, the gut metagenome is enriched in genes involved in the breakdown and fermentation of otherwise indigestible plant-derived polysaccharides that form an important part of modern diets, the detoxification of xenobiotics consumed intentionally or inadvertently, and the synthesis of essential amino acids and vitamins. These findings emphasize that the human metabolome is actually a composite of human and microbial attributes; they also point to a future in which it may be possible to optimize the nutritional status of the overfed or underfed on the basis of knowledge of their gut microbial ecology, or to predict the bioavailability of orally administered drugs, or to forecast the susceptibilities of individuals or populations to particular types of cancer. Greater knowledge of the microbial communities of the oral cavity, skin, and female reproductive tract will similarly improve our ability to prevent, diagnose, and treat diseases at those sites.
We are also host to countless viruses. A recent survey reported that human feces contain about a billion RNA viruses per gram, representing 42 viral “species” (Zhang et al. 2006). Most were plant viruses (probably originating in food), and the RNA of the most abundant (pepper mild mottle virus) was still infectious to its host (the pepper plant); this suggests a role for humans as agricultural-disease vectors.
What remains to be learned about the human microbiome is enormous. The early projects hint at how rich and productive further study will be. The Human Genome Project had to be thoughtfully staged and coordinated, and any project aimed at understanding the human microbiome will need similar careful planning.
Viral Metagenomics
Despite the challenges, early applications of metagenomics to marine virus populations have already provided considerable insight. Studies of naturally occurring phage in aquatic systems first physically separated viruses from co-occurring microbial cells and then used amplification techniques to generate “shotgun” viral DNA libraries, which were then randomly sequenced. The first look at seawater confirmed its huge diversity of viral assemblages: 65% of all the sequences examined from the first
seawater viral libraries were novel and bore no significant similarity to any known genes in the databases. The approach appeared reasonably comprehensive for double-stranded DNA phages; it recovered most major families, including those with bacterial or algal hosts. Similar applications in marine sediments yielded parallel results, but with some interesting differences. Among sediment viral assemblages, even greater novelty was detected: more than 75% of the viral sequences recovered resembled nothing in the databases. The double-stranded viral DNA sequences identified in sediments suggested an important role for temperate phages, for example, viruses that can integrate into their host’s genome. A large comparative analysis of seawater viral assemblages collected from diverse locales recently indicated that marine viral species have a global distribution (Angly et al. 2006).
Applications for analyzing RNA-based virus assemblages from seawater have also been developed, and have revealed new groups of RNA-viruses that infect marine planktonic protists and animals. In total, the early viral metagenomic analyses provide a solid starting place for exploring and interpreting the genomic diversity in naturally occurring viruses. They also provide a foundation for the next steps in microbial-community genomics, the integrated analyses of viruses and their cellular hosts collected from the same metagenomics samples and analyzed simultaneously.













