
4
Designing a Successful Metagenomics Project: Best Practices and Future Needs
As the number and diversity of metagenomics studies have grown, so too has an appreciation of the challenges that these studies present as compared with genome-based analysis of single organisms. Many of the challenges are likely to diminish with the development of new technologies and mathematical tools. Nonetheless, many of the criteria of success in metagenomics studies will remain unchanged by new knowledge or methods. This chapter is devoted to the key steps in developing a metagenomics project, the decision points along the way, and the issues that need to be considered at each step.
PARALLELS WITH TRADITIONAL MICROBIAL GENOME SEQUENCING
Metagenomics-based approaches share many features with traditional genome sequencing of cultured bacteria but also present a number of unprecedented challenges. This chapter describes a number of complementary approaches to the study of microbial communities (see Figure 4-1) which, depending on the goals of a particular project, can be applied individually or together to obtain a new understanding of the numbers and abundance of microbial community members, their metabolic capabilities, and how these parameters change in response to external stimuli. This chapter identifies the advantages and limitations of each approach and explores the research needed to overcome barriers to understanding microbial communities.
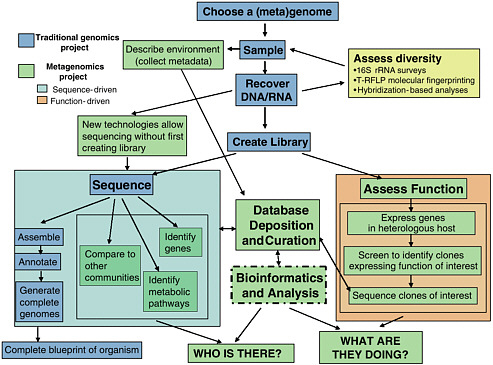
FIGURE 4-1 Metagenomics differs from traditional genomic sequencing in many ways. The dark blue boxes show the typical steps in the sequencing of a single organism’s genome. Metagenomics requires greater attention to sampling, and assessing the diversity of the sample by various means (yellow box) is necessary to ensure that the sample is representative. Extracting the appropriate nucleic acids from the sample is another step that can be challenging in a metagenomics project. Preparation of a library is often the next step, but new sequencing technology can bypass this step. The DNA from metagenomics samples can then either be sequenced (blue box) or assessed for the functions it encodes (orange box). The sequence can sometimes be assembled into complete genomes of community members, but can also be analysed in other ways (light blue box). Data storage and computational analyses are critical steps in metagenomics projects and must be integrated throughout the project. Overall, a metagenomics project can answer the questions “Who is there?” and “What are they doing?” in addition to assembling genomes.
The development, about 15 years ago,of methods for rapid and efficient sequencing and assembly of large segments of DNA was critical for the revolution in microbial genomics and has led to the completion of more than 460 bacterial and archaeal genome sequences by January 2007.1 For
most of the projects, the starting material has been DNA extracted from pure cultures of organisms grown in the laboratory or in association with animal or plant cells. Regardless of the organism selected for sequencing, the goal of the projects has been the same: to generate a complete or nearly complete genome sequence that can serve as the substrate for genome annotation and analysis (see Figure 4-2). For metagenomics projects, it will be important to accumulate additional complete genome sequences, especially for currently under-represented taxa. Such sequences should help in the identification of otherwise unidentifiable open reading frames in metagenomic fragments, and facilitate scaffolding of metagenomic data.
Metagenomics projects differ from traditional microbial-sequencing projects in many respects. The starting material is a mixture of DNA from a community of organisms that may include bacterial, archaeal, eukaryotic, and viral species at different levels of diversity and abundance. Most of the organisms will elude attempts at cultivation. In some projects, sample collection may be confounded by the presence of limited amounts of DNA or the presence of contaminating DNA or other compounds that interfere with DNA extraction. These factors make it much more challenging to think about the generation of complete or nearly complete genome sequences from metagenomics projects. Often, generating complete genomes will not
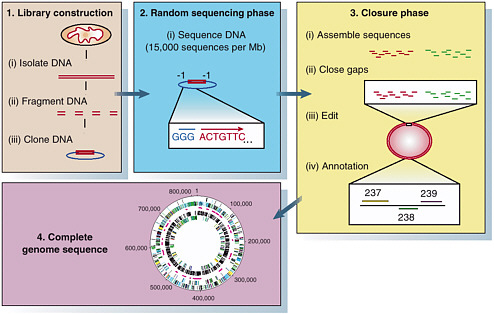
FIGURE 4-2 Steps in a traditional microbial genome project. SOURCE: Fraser et al. Reprinted by permission from Macmillan Publishers Ltd: Nature 406:799-803, copyright 2000.
be the focus—not so much because of the difficulty as because the real goal, understanding community composition and function, does not require it. In the study of complex communities, it is often necessary to address the question of how much sequence is enough to understand a community and to carry out comparative analyses of related communities. In many cases, this information can be obtained by applying various methods based on 16S rRNA sequence that can reveal a tremendous amount of information about microbial diversity and abundance. In other cases, whole-genome shotgun-sequencing data generated by the traditional Sanger sequencing methods, by newer very-high-throughput methods, or by a combination of the two approaches will provide additional information about the gene content of a community and its metabolic potential. Finally, function-driven metagenomic analysis, like the soil resistome project described in Chapter 3, which starts with functional expression of an activity in a surrogate host, followed by sequencing and phylogenetic analysis provides another measure of community potential. Regardless of the methods employed to answer questions about community structure and function, the composition of any microbial community is likely to be profoundly affected by the habitat from which the sample was obtained. Detailed knowledge about the habitat is essential for meaningful biological interpretation of the sequence data.
METAGENOMICS STEP BY STEP
Habitat Selection
The choice of the microbial community to study will be driven by the underlying scientific question being addressed. However, the more information one has about the study habitat—physical, chemical, and ecological—the more insight can be derived from the metagenomic data. Specific hypotheses can be posed and genes sought in genomic data from a well-characterized site. The acid mine drainage study is a case in point. The geochemical conditions that create and maintain that habitat were delineated before the researchers embarked on their metagenomics journey. As a result, the information gleaned in studying the genomes could be placed in a phylogenetic, biochemical, and physiological context. For example, knowledge of the nitrogen budget of the site impelled the researchers to seek nitrogen-fixation genes in the metagenome. When they did not find candidate genes in the dominant members, they examined the minor components of the community and discovered that one of the least abundant members of the community, Leptospirillum ferrodiazotrophum, carried the nif operon. They then cultured that bacterium by providing N2 as the only nitrogen source to ensure that only nitrogen-fixing bacteria would grow. The discovery of the keystone species (a community member whose signifi-
cance to the community is larger than its relative abundance) was made possible by an ecological inference that depended entirely on knowledge of the site.
Exploring habitats that have been well studied by other methods will accelerate progress in metagenomics.
-
Well-characterized habitats will leverage the value of metagenomic data.
-
Interdisciplinary collaborations with scientists studying the non-microbial aspects of the habitat will inform the analysis.
-
Different habitats require different depths of sequencing depending on their complexity and the degree of completeness needed to address the questions being posed. Pilot studies to determine the required depth of sequencing may be necessary.
Sampling Strategy
Sampling is fraught with challenges. Each decision about the type, size, scale, number, and timing of sampling shapes the conclusions and inferences that can be drawn. The labor intensity of producing and analyzing metagenomic libraries aggravates sampling issues that are inherent in all ecological studies. If conclusions about the habitat are to be drawn, the samples must be representative of the habitat. To obtain representative samples, it is critical to know the scale and amplitude of variation in the habitat environment. Soil communities, for example, change on a micrometer scale, following the physical and chemical heterogeneity of the mineral and biological materials that make up the soil. A 1-cm3 aggregate may contain aerobic and anaerobic regions; clay, silt, and sand particles; plant matter in various stages of decomposition; and a variety of invertebrates, each of which probably has its own associated microbiota. For such a habitat, what is the appropriate sample size? Is it possible to account for the minute microhabitats when 50 g of soil is needed to build a metagenomic library?
Habitat change over time is one of the most interesting aspects of communities. Their responses to changing conditions are central to understanding community structure, function, and robustness. Understanding the role of host-associated microbial communities in host development and health requires not only sampling from the same host over time, but also understanding host-to-host variation. For biodefense and forensics purposes, it may be important to determine whether threat organisms originated in nature or in a laboratory. But the variability of communities creates a sampling conundrum. How many samples are needed to represent the many conditions of a community? How are different types of change accounted
for—natural cycles versus catastrophic events, which might be a tooth-brushing in the case of oral microbiota and a flood in the case of soil? Even more challenging are the long-term changes, such as global climate change, that both affect and are affected by microbial communities. How much work is needed to differentiate baseline variability from real change?
The answers to most of these questions depend on the complexity of the community, the heterogeneity of the habitat over time and space, and the fineness of the distinctions that need to be made. As biological and computational methods become more efficient, it will be possible to draw more robust conclusions from more complex communities in more variable habitats. No matter the power of the methods now or in the future, it is essential to consider sampling issues and limitations at the beginning and throughout any metagenomics study of a complex community, and the sampling scheme must inform the interpretation of results.
-
Sampling strategy should be carefully considered and the variability in the experimental method assessed before sampling (see Table 4-1). If understanding factors that influence change in a community is a research goal, adequate controls should be in place to distinguish baseline variation from real change.
-
Pilot projects may be needed to assess diversity, variability, and the appropriateness of different technological approaches (such as targeting of different subgroups or type of sequencing technology) to enable optimization of the project plan.
Macromolecule Recovery
The quality and completeness of data obtained from metagenomic analysis of any community will be only as good as the procedures used for the extraction of DNA from a sample. A currently unanswered question is whether methods like those developed for the direct isolation of DNA from different types of soil are equally effective in recovering DNA from all members of other complex communities. For example, cells from different species differ in their susceptibility to lysis under various conditions, and some members of the same species differ in their susceptibility to lysis in different physiological states. Furthermore, the conditions necessary to lyse the more recalcitrant cells in a community may be sufficiently harsh to cause degradation of DNA from other community members. Another issue that has been tackled recently is the development of methods to distinguish between DNA from viable and dead cells in a given sample—a distinction that may be important in drawing conclusions about the overall metabolic capabilities of a microbial community. Results from a number of studies related to this question suggest that no universal approach is equally effi-
TABLE 4-1 Sampling Considerations in Metagenomic Analyses
|
Sampling Considerations |
Questions |
|
Scale |
What is the size of the habitat? What is the size of the sample of the habitat? How representative of the habitat is the sample? |
|
Biological variation |
How is biological variation in the site accommodated in the sampling scheme? On what scale is the variation (subsample to subsample, sample to sample, site to site)? How much replication is needed to represent the full variety of properties of the site? How flexible is the community? If very flexible, then what does it mean to take a snapshot in time? |
|
Experimental variability |
Where is the experimental variability in process sampling? In extracting DNA? In cloning? In storage of samples? How does the experimental design maximize replication to account for experimental variability? |
|
Reproducibility |
If patterns are detected, are they reproducible? |
|
Coordinates of place and time |
Is detailed information about the site and time of sampling recorded? |
|
Repository |
Can the samples be stored for future analysis? Can they be placed in a central repository? |
|
Singletons |
What is the significance of a singleton (a unique sequence or other data point)? If it is never found again, how should its relevance be assessed? |
cient for DNA extraction in all environments. These challenges are not insurmountable, and improvements in DNA-extraction methods should be vigorously pursued, but the limitations in DNA extraction methodologies as applied to specific projects should be acknowledged and addressed.
The effect of contaminants on the recovery of DNA or RNA of interest from many different environments presents another technical challenge. Because of the very large differences in genome size between eukaryotic and bacterial cells, even minor contamination of a sample with host (plant, animal, or human) DNA reduces the effective concentration of bacterial DNA available for sequencing and hence increases the cost of generating sufficient useful (nonhost) sequence. It also reduces the chances of recovering low-abundance members of the bacterial community. There appear to be no published reports comparing methods for removing host DNA for bacterial metagenomic analysis. One study used nucleases derived from the host, an insect gut, to digest the host DNA before lysing the bacteria; this was very effective but may not be generally applicable (Guan et al. 2006). One method of building metagenomic libraries from soil involves physically separating the bacteria from the rest of the soil matrix before lysis to mini-
mize contamination with the numerous inhibitors of the enzymes that are used for cloning that are found in soil (Akkermans et al. 1995; Berry et al. 2003). This method and alternatives used in other fields suggest that various filtration, centrifugation, or lysis methods could be adapted to the challenge of separating bacterial cells from eukaryotic cells before library construction. The use of subtractive hybridization (hybridization of the community DNA to immobilized or labeled copies of eukaryotic DNA, from which the unbound bacterial DNA can then be separated), or separation based on GC content (Holben and Harris 1995), will also, in theory, allow enrichment of bacterial DNA at the expense of eukaryotic DNA. Research is needed in more robust nucleic acid extraction procedures that have known effects on recovery of DNA from community members that are more difficult to lyse, that are in different physiological or physical states, or that are rare. Extraction procedures need to be standardized for all habitat types as much as possible to aid comparisons among habitats.
Even after extraction of DNA from the sample, all methods that rely on library construction, including metagenomic approaches, have potential for bias or skewing. Some cloned genes are toxic, and so may be underrepresented in typical clone libraries used for sequencing. Some types of DNA (for example, certain viral DNAs) are chemically modified in such a way that they are difficult to clone. Many of these challenges, however, are reasonably well known from prior molecular biological studies, and can be assessed and addressed by careful analyses that monitor recovery and quantitation issues using parallel independent approaches. In addition, newly evolving sequencing approaches that do not rely on cloning (discussed elsewhere) will alleviate some of the problems associated with cloning bias.
GETTING THE MOST OUT OF METAGENOMICS STUDIES
Obtaining the most information from metagenomics studies will continue to be a challenge primarily because the potentially disparate and incomplete datasets are so large. The approaches to analysis can be divided into three general categories, each of which has advantages and limitations.
16S rRNA-Based Surveys
The first category includes a set of methods based on analysis of 16S rRNA genes, which provide relatively rapid and cost-effective methods for assessing bacterial diversity and abundance. These types of assays are often used as a first step in larger metagenomics projects to evaluate bacterial diversity in potential samples of interest (soil samples from dif-
ferent locations in a defined area, fecal samples from various individuals, etc.) in order to choose the most appropriate samples for more in-depth analysis. These methods can also be used to monitor changes in community composition over time and space without the need to generate other types of sequence data.
One of the simplest ways to assess community structure is based on a method for molecular fingerprinting of microbial communities called terminal-restriction fragment length polymorphism (T-RFLP) analysis. The technique employs polymerase chain reaction (PCR) in which two differentially fluorescently labeled primers are used to amplify a selected region of the 16S rRNA gene from total community DNA. The mixture of dually labeled amplicons is digested with a restriction enzyme (MspI or HaeIII) releasing the labeled 5′ and 3′ ends—or terminal restriction fragments (T-RFs)—of each individual amplicon. These differentially labeled primer pairs combined with the two restriction enzymes result in six fluorescently labeled T-RFs. T-RFLP profiles can be determined using an automated capillary DNA sequencer and GeneScan® software (Applied Biosystems). T-RFLP profiles reflect differences in the numerical abundance of bacterial populations in the samples (Liu et al. 1997). Changes or differences in microbial community structure can be detected based on the gain or loss of specific fragments from the profiles (Engebretson and Moyer 2003; Forney et al. 2004; Osborn et al. 2000) and statistical clustering analysis of T-RFLP data can identify communities that have similar numerically abundant populations.
Significant insights into species richness, structure, composition, and membership of microbial communities have been gained through analysis of 16S ribosomal RNA (rRNA) gene sequences. PCR amplification with primers that hybridize to highly conserved regions in bacterial or archaeal 16S rRNA genes (or eukaryotic microbial 18S rRNA genes) followed by cloning and sequencing yields an initial description of a microbial community. Powerful computational tools have been developed to assess species richness (FastGroup and DOTUR) in a sample and the similarity between two communities in membership (SONS) or structure (AMOVA, LIBSHUFF, UNIFRAC, and TreeClimber). Analyses with these tools have revealed many challenges still to be resolved. Most communities have many members (that is, they are species-rich) whose abundance is uneven. This presents a sampling issue: how many samples need to be taken to find members of the sparser groups? However, recent estimates based on 16S rRNA sequencing and statistical modeling of soil communities indicate that with decreasing sequencing costs, it is possible to conduct a fairly complete census of soil communities even though these are the most species-rich and uneven in structure of communities studied so far.
The challenges associated with unknown community structure may soon become more manageable.
In addition to sampling challenges, the 16S-based approach to the study of microbial communities has other limitations. First, 16S rRNA sequences provide a phylogenetic framework into which community members can be placed, but that framework does not tell us much about the functional capabilities of the individual members or the entire community under study. In addition, PCR-based studies are inherently biased in that not all rRNA genes amplify equally well with the same “universal” primers. Indeed, in several published metagenomics studies there have been discrepancies between estimates of community diversity derived from PCR-based 16S rRNA gene surveys and those derived from whole-genome shotgun data, although in some studies the estimates are remarkably similar (Liles et al. 2003; Tyson et al. 2004). A third limitation of 16S rRNA gene surveys is that these genes occur in multiple, nonidentical copies in many bacterial and archaeal taxa, which may lead to overrepresentation of some species in 16S rRNA gene libraries; this limitation might be overcome through the use of additional, single-copy phylogenetic markers, such as recA or rpoB, for initial community surveys. Research is needed for the development of additional genetic markers of community diversity to enhance the phylogenetic and functional resolution of microbial communities.
In parallel with efforts in 16S rRNA sequencing, several groups have been pursuing the development of 16S rRNA-based microarrays (or microarrays based on other phylogenetic marker genes) for high-throughput compositional analysis of microbial communities (Wu et al. 2006). Such phylogenetic oligonucleotide arrays typically carry hundreds to thousands of spots bearing synthesized oligonuceotides as probes matching rRNA gene sequences that are found in databases or are expected to be present in samples. The arrays can be designed to include probes that target bacterial species at different taxonomic levels, from species to phyla. This approach makes it feasible to assess bacterial diversity in large numbers of samples; this facilitates continuous monitoring of microbial communities, and the content of the microarrays can be expanded as new species or phylotypes are revealed in metagenomic studies. One disadvantage of microarraybased approaches to metagenomic analysis is that the information that can be obtained is limited by known bacterial phylogeny represented on the array—that is, the arrays are blind to species that have yet to be discovered. Microarray-based approaches also suffer from a technical challenge that plagues other studies of diverse microbial communities: it may be difficult to distinguish a hybridization signal from a low-abundance community member from background. These caveats aside, microarraybased assays have the potential to provide valuable complementary information in metagenomics projects. For example, the current version (2.0)
of the Affymetrix-based PhyloChip targets over 30,000 unique database sequences, totaling almost 9,000 distinct taxonomic groups, with each group represented by a set of 11 or more perfectly matching probes and a corresponding mismatch control probe. The PhyloChip has been successfully used to characterize complex environments such as soil, aquifers, and urban air (Brodie et al. 2007; Desantis et al. 2007). As expected, the PhyloChip detects broader diversity than typical clone library sequence analysis (Brodie et al. 2007; Desantis et al. 2005). Depending on the microbial diversity of the sample, the PhyloChip detects on average twice as many taxa as 16S rRNA gene sequencing (Desantis et al. 2005). The quantitative power of microarray-based assays is somewhat limited to sample comparison at this stage (community dynamics), but the combination of 16S rRNA gene sequencing and arrays is a unique and powerful tool for the characterization of any microbial community because it allows both the discovery of novel phyla and extensive cataloguing of each taxonomic unit present in a given environment.
16S rRNA Phylogenetic and Functional Anchors: A Hybrid Approach
Metagenomic clones can be given a context, or “anchored,” by looking for a gene that characterizes the clone or the organism that it came from. The genes most commonly used as anchors are such phylogenetically informative ones as those encoding 16S rRNA or RecA protein. Sequencing all clones that are derived from one phylogenetic group may help to stitch together a picture of the group even in the absence of cultured members. Functional anchors have also been used to collect clones that share a characteristic, in this case an expressed function. The clones expressing the function of interest can be sequenced to search for phylogenetically informative genes to begin to piece together a slice of the community that is related to a particular function.
-
Research is needed to develop phylogenetic and functional anchors for use in different microbial communities to advance the process of linking community membership and function.
-
New physical methods are needed to enhance the yield of inserts bearing such anchors.
-
Novel strategies to obtain gene expression of genes from a wider range of organisms will facilitate this work.
Generation of Large-Scale DNA Sequence
A second important approach to studying microbial communities is based on generating large amounts of DNA sequence using well-described
shotgun-sequencing strategies. This is an excellent method for obtaining information on the gene content and functional capabilities of mixed microbial communities. The sequence data, which can potentially provide information on “what are communities doing?” are most informative when coupled with other analyses that help to determine “who is there?” A random shotgun strategy for studying communities can reveal information on community diversity (e.g., bacteriophage and other viruses, eukaryotic species, novel bacteria and Archaea) that is not captured with 16S rRNA gene surveys. Bacteriophage, in particular, are thought to play a critical role in shaping microbial membership and evolution and their abundance and diversity cannot be assessed using the previously described approaches. The fundamental limitation of this approach is the vast number of genes that do not have homologs of known function in the databases.
Assembling Whole Genomes
If the goal of a metagenomics study is to determine the complete genome of some or all of a community’s members, many challenges must be overcome. Given that environmental samples contain DNA from many species that are present in different abundance and differ from each other in genome size, the final depth of sequence coverage for each organism at a given level of sequencing will vary. Piecing together all the separately sequenced fragments of a genome is a substantial bioinformatics challenge. In a simple community like the acid mine drainage system described in Chapter 3, there are enough overlapping fragments of the dominant members to assemble their entire genomes. In a more complex community, even the sequence fragments from the dominant members will be sufficiently diluted to preclude assembly, and species of low abundance may be represented by only a few sequences. These differences in sequence coverage can provide information on relative species abundance. It is important to take differential species representation into account in selecting assembly strategies for metagenomic data to avoid classifying sequences from the most abundant species as repeats and throwing them out of assembly algorithms.
In highly diverse microbial communities, even when very large amounts of DNA sequence data are generated (several billion to a trillion base pairs of DNA), it will be difficult to generate assembled genomes, and the less abundant members of any community might be represented only by singleton sequences. New sequencing technologies, now being introduced by a number of companies (see Table 4-2), provide alternative strategies for generating substantially more DNA sequence at a lower cost than current Sanger-based capillary sequencing methods. The new technologies will go a long way toward achieving sequence depth that extends beyond the
TABLE 4-2 New Sequencing Technologies
|
Applied Biosystems 3730 xl |
454 GS FLX Pyrosequencer |
Solexa 1G Genome Analyzer |
Applied Biosystems 1G SOLiD Analyzer |
|
1-2 Mbp per day/machine |
100 Mbp per day/machine |
800 Mbp per run/ machine (25 bp) |
1200 Mbp per run/ machine (Frag Library) |
|
|
|
960 Mbp per run/ machine (30 bp) (assumes 32M features) |
2400 Mbp per run/ machine (Mate Pair Library) |
|
Long sequence reads (600-900 bp) |
Medium sequence reads (200-300 bp) |
Short sequence reads (25-40bp) |
Short sequence reads (25-30 bp, 25x2 for mate pair libraries) |
|
Mate pair informationa |
No mate pair information |
No mate pair information (promised for future versions) |
Mate pair information |
|
Libraries subject to cloning bias |
No library cloning bias |
No library cloning bias |
Libraries may show cloning bias |
|
Can resolve homopolymers |
Cannot easily resolve homopolymers |
Can resolve homopolymers |
Can resolve homopolymers |
|
aMate pairs are two sequencing reads derived from the same clone, or molecule, one from each end. If the length of the clone, or molecule, is known, mate pair information constrains where these sequencing reads can be placed in an assembly. |
|||
most abundant members of microbial communities in shotgun sequencing projects, but some applications may still need additional methods (such as normalization, subtraction, or physical separation methods) that will ensure better representation of the lower-abundance community members. Furthermore, the new technologies are still vexed with issues such as shorter read lengths than those that have become routine with Sanger sequencing. The limitations have obvious consequences for assembly, particularly for metagenomics applications in which assembly is already complex and difficult, but considerable effort is also being devoted to figuring out solutions to these technical challenges.
Because the assembled sequence data from metagenomics studies will often be incomplete and it may often be difficult to draw unambiguous conclusions about who is there and what each species is doing metabolically, any additional data that would help with assembly validation and making phylogenetic and functional inferences will be of great utility.
The availability of reference genomes is an example of a kind of data that make assembly easier. In response to the need for such data, the
National Human Genome Research Institute has undertaken a phase 1 human gut microbiome initiative (NIH 2007). The initiative will deliver deep draft genome sequences of 100 cultured bacterial reference species representing each of the divisions known to make up the distal gut microbiota. The strategy adopted for this initiative is to generate 20X sequence coverage of purified DNA with a 454 GS20 pyrosequencer (i.e., 20,000 bp of sequence will be generated for every 1000 bp in the organism’s genome) and to combine the resulting data with paired end reads from plasmid wholegenome shotgun subclones from each targeted species (5X coverage, produced with a conventional ABI 3730xl capillary machine). This approach will generate hybrid assemblies of each genome (“scaffolds”) with nearly complete gene coverage. The completed genome sequences will provide a key reference for metagenomic projects related to the human gut, which will be valuable because relatively few members of the human gut microbiota have been sequenced. In later human metagenomics projects, sequence data generated from microbial-community DNA can be readily aligned with these 100 microbial genome scaffolds to help to validate metagenomics assemblies, answer questions related to phylogeny and metabolism (what species are contributing what genes to the community genome) and assist in the evaluation of gene flow between community members (by providing evidence of lateral gene transfer among community members).
Several kinds of research are needed:
-
Although genome assembly is not feasible in complex communities or necessary for answering many questions, it is useful in some cases. Hence, new approaches are needed for such assemblies and for interpretation of consensus genomes or partial genomes of communities.
-
Research is needed at both the experimental and computational levels to simplify complexity in complex communities so that patterns are discernable or particular subgroups can be adequately resolved. This includes improvements in bioinformatics tools, sequence binning, normalization, and methods of physical separation, such as flow cytometry and single cell or colony sequencing.
-
Metagenomics must be done in concert with improved culture-based science, including improved culturing techniques; generation of complete genome sequences for reference microbes (e.g., the type strains); and the physiological and ecological characterization of these reference organisms.
Gene-Centric Analyses
Because of the current technical limitations and cost associated with generating large amounts of DNA sequence from complex environments
and because it will often not be possible to assemble complete or nearly complete genome sequences, it may be necessary in many metagenomics projects to adopt a gene-centric rather than a genome-centric view of microbial diversity and abundance. One example of a gene-centric approach is the use of environmental gene tags (EGTs), short sequences of DNA that contain fragments of functional genes. Each EGT in a metagenomics dataset may be derived from a different member of a given community, but those genes that are essential for community survival will in theory be represented more frequently, or at least more consistently, than ones that are nonessential or are highly specialized. The collective set of EGTs from a given sample represents a “fingerprint” that can be compared across multiple sites or habitats or over time in the same environment. EGTs that are overrepresented or underrepresented can provide insights into unique metabolic capabilities associated with a particular environment even if it is not possible to assign a given EGT to a particular species. Application of this approach to the metagenomics data from the Sargasso Sea revealed that the community is enriched in genes that encode rhodopsin-like proteins as compared with ocean environments that receive less sunlight (Venter et al. 2004). Newly developed sequencing technologies (see Table 4-2) that do not rely on cloning may be especially useful for identifying EGTs. The new sequencing technologies allow deep sequence coverage and are not subject to potential cloning biases. For identifying tags and quantifying relative gene stoichiometries, they may be particularly useful. To advance the use of gene-centric analysis:
-
Non-genome-based methods are needed for the analysis of metagenomic data to identify capabilities that are present in a microbial community and deduce the ecological selection and evolutionary outcomes of the community.
-
Improvements in bioinformatics tools, improvements in the ability to deduce function from sequence, and completion of more reference microbial genome sequences are needed.
Hybridization- and Array-Based Analyses
Specific functional gene arrays have also been designed with probes corresponding to genes of interest in an environment (see Figure 4-3) (Wu et al. 2006). They can indicate the diversity of genes performing specific functions at specific sites and assess levels of expression of those genes when community mRNAs (or cDNAs made from them) are the target. Research is needed to:

FIGURE 4-3 A functional gene array containing 27,000 probes covering 10,000 functional genes used to monitor microbial community dynamics in an aquifer undergoing uranium bioremediation. Image provided by Jizhong Zhou, University of Oklahoma.
-
Improve array approaches for metagenomics applications, including sensitivity, interpreting specificity, speed, cost, and data analysis.
-
Improve methods for sensitive and accurate representation and measurement of community mRNA populations.
-
Enhance the database of annotated sequences of genes that have important environmental functions, and provide software for easy use in the analysis of metagenomic data and for probe and primer development.
Function-Based Analyses of Microbial Communities
If the ultimate goal of metagenomics is to determine “who is doing what,” then sequencing alone is not the answer, because so many genes have unknown functions. Sequencing provides information that is limited by what is in the databases and by the available algorithms for linking sequence to function. Function is inferred when there is statistically significant sequence similarity between genes discovered by metagenomics and those in the databases. Computational tools that can predict secondary structure (how the protein will fold) and recognize a broader array of protein motifs based on the amino acid sequence alone are under development. Sequenced-based studies, however, will always be limited by the completeness of existing data and the accuracy of genome annotation. Furthermore, structural genomics projects that aim to improve the linking of sequence to function face a bottleneck: analysis can be done only on proteins that can be produced in large quantities, purified, and even crystallized. If a newly identified gene has only weak similarity to a gene whose product has been studied biochemically, if a similarity in sequence does not reflect a functional relationship, or if a particular gene can carry out multiple functions in the cell, sequence comparisons may lead to incorrect conclusions about function. Even if annotation and functional assignments were much improved, finding proteins with a defined function may be accomplished more efficiently by taking a functional approach to the metagenomic library.
One way to do this is to screen the metagenomic libraries directly for expressed functions. Function-driven metagenomics has unearthed many proteins that would not have been recognized by their sequences, including those coded for by genes involved in antibiotic biosynthesis, antibiotic resistance, biodegradation of environmental contaminants, and signal-transduction pathways. Finding these genes has increased what is known about the behavior of microbes, has enriched the databases, and has presented opportunities for biotechnology development. The potential for discovery is staggering: there are an estimated 1013 (10 trillion) genes in 1 g of soil; because these are derived from at least 103 species, there are at least 1 million different genes in 1 g of soil (Schloss and Handelsman 2006). Many of the genes will closely resemble genes in other organisms, including cultured and sequenced ones. But some will be novel—unrecognizable by sequence alone—and some will have dramatically new functions. This is one of the potential treasure troves of metagenomics.
Just as staggering as these potential riches are the barriers to discovery of genes by functional screening. The approach is grossly limited by the ability of the organism that is hosting the metagenomic library to express genes from anonymous organisms represented in the library. It is reasonable to imagine that most genes from members of the Enterobacteriaceae
will be expressed in E. coli and that some, but fewer, genes from diverse organisms, including other γ-Proteobacteria, Firmicutes, Actinobacteria, and Archaea, will also be expressed in E. coli. But the variation in geneexpression machinery among microbial groups makes it likely that most genes from the most exotic divisions (those distant from E. coli) will not be expressed. Therefore, it is essential to develop techniques that enable E. coli to express a greater array of genes (such as providing alternative sigma factors or tRNAs) and to screen libraries in bacteria from other divisions.
-
Functional-expression studies would be dramatically advanced by development of vectors and readily culturable host organisms from each phylum of Bacteria and Archaea.
-
Research to discern the rules that govern heterologous gene expression will advance this field.
-
Methods to expand the repertoire of genes expressed by surrogate hosts, such as E. coli, will contribute to function based metagenomics.
ADVANCING THE FIELD
The technical advances described in Table 4-3 need to be coupled with advances in bioinformatics (see Chapter 5) and basic microbiology. Genomic analysis to date has been valuable only because of five decades of comprehensive study of E. coli genetics, physiology, and biochemistry; intense study of many other organisms; and 150 years of microbial ecology research. It is imperative that microbiology remain strong and well funded to realize the potential of metagenomics (ASM 2007). This chapter concludes with a discussion of ways, both technological and scientific, in which progress will be most useful for advancing the field of metagenomics.
Sequencing Technology
One of the major challenges for metagenomics studies of complex environments is to capture the extent of bacterial diversity in a population with random-genome shotgun sequencing. As discussed above, without very extensive sequencing coverage of an environmental sample, the less abundant members of low-diversity bacterial communities will probably not be represented in any given dataset. With more complex communities, enormous amounts of DNA-sequence data will be required for assembly of even the most abundant members. Although advances leading to higher throughput and decreased costs for Sanger-based sequencing have occurred in the last 10 years, metagenomics projects will require new, higher-throughput, lower-cost sequencing technologies. For example, the pyrosequencing technology developed for the 454 Life Sciences Genome
TABLE 4-3 Technical Advances Needed in Functional Metagenomics
|
Current Limitation |
Enabling Technical Advances |
|
Not all clones can be expressed in current laboratory hosts; many functions are difficult to screen |
Novel gene-expression systems that represent a broader array of organisms |
|
|
Better high-throughput screens |
|
Inadequate number of reference genomes for many habitats. Habitat specific reference genomes would contribute to: |
Longer reads for 454 or Solexa sequencing |
|
Reference genome-sequence data for habitats of interest |
|
|
|
|
Inability to culture organisms from which gene of interest arose |
Further refine methods for culturing organisms |
|
Difficulty in associating functions with metadata, such as physical conditions |
Further development of methods for analysis of microbial community transcriptomes, proteomes, and metabolomes (which vary with physical conditions more directly than does sequence) |
|
Inadequate information about minor members of communities, which is needed, for example, to identify keystone species |
Development of improved methods for isolating single cells by microfluidics or cell sorting and for amplifying DNA and RNA from single cells; development of methods for subtraction and/or normalization of community DNA samples to facilitate the study of rare community members |
Sequencer FLX eliminates the need for library construction (in other words, the community DNA can be sequenced directly, without first being cloned into a laboratory host) and can generate more than 100 million base pairs of DNA sequence in a single run. That is equivalent to about 100 runs on the AB 3730xl instrument for approximately 20% of the cost. The Solexa 1G Genome Analyzer also does not require library construction and yields about 1 billion base pairs of sequence per run. Neither of these technologies yet offer the ability to sequence “mate pairs” (see Table 4-2 footnote), which greatly aid the assembly of sequences, but this will be a feature of the Applied Biosystems SOLiD Analyzer, which is projected to have a through-
put of about 3 billion base pairs per run when it comes to market in late 2007. Other technologies with similar throughputs are expected from, for example, Helicos, Intelligent-Bio-Systems, and Complete Genomics. In the longer run, a third generation of sequencing technologies, which use single DNA molecule substrates, are expected to reduce the cost even further and increase the throughput of DNA sequencing (Fan, Chee, and Gunderson 2006; Metzker 2005; Shendure et al. 2004).
A feature of the current generation of new sequencing technologies is short or relatively short read lengths (in comparison with Sanger capillary sequencing). Short read-lengths make it more difficult to assemble genomes. However, read-lengths will continue to increase with further development; moreover, the promise of sequencing paired-ends (“mate-pairs”) will reduce the disadvantage of short read-lengths dramatically. It is impossible today to predict the advances, and cost, of sequencing technologies; they are changing too fast. It is sufficient to say that the needs of clinical medicine are driving technology development at a rapid pace, and metagenomics sstudies will benefit enormously from the consequent increase in throughput and reduction in cost.
A recent report described the first metagenomic analysis of two samples taken from the Soudan Iron Mine in Minnesota with 454 sequencing technology (Edwards et al. 2006). The analysis revealed interesting differences in metabolic potential between the two sampled environments; but, just as important, it suggested that the 454 sequencing data are remarkably similar to those generated from the same sample with Sanger sequencing, at least in terms of 16S rRNA sequences. Additional studies to validate the utility of the short reads clearly are warranted, but the initial data support the role of new sequencing technologies in future metagenomics studies because they will facilitate deeper sampling of environmental samples than is currently possible. At the same time, it is important that alternative strategies for enrichment of the less abundant members of communities, such as suppressive subtraction hybridization or flow sorting of cells, continue to be developed and implemented.
Gene-Expression Systems
Function-based metagenomics is predicated on expression of genes from anonymous organisms in a surrogate host. The likelihood of gene expression is low in any one host; thus, function-based approaches will be greatly facilitated by the development of vectors that can be maintained in a number of host species.
Aside from E. coli, several bacterial hosts are being developed to serve as vehicles for gene cloning in metagenomics. For example, the actinomycetes—which include genera such as Corynebacterium, Myco-
bacterium, Nocardia, and Streptomyces—have genomes that are highly GC rich. This group is well known for the production of such natural products as antibiotics, herbicides, and other secondary metabolites. If metagenomics is to be used as a means to discover new antibiotics, it is imperative that efforts to develop gene-expression systems for this group be increased. Streptomycetes are of much promise because they are easy to grow in the laboratory and are useful for the expression of genes from other actinomycetes. The gene-expression machinery of Streptomyces is adapted to a genome of high GC content, and their use as hosts for cloning of community DNA may facilitate expression of genes from other high GC content genomes (Wang et al. 2000). Another well-developed bacterial system for heterologous gene expression is Bacillus subtilis, which is a better system than E. coli for the expression of extracellular proteins (Li et al. 2004). B. subtilis is easy to manipulate and grows quickly, although it is known to exhibit plasmid instability, low-level gene expression, and degradation, by its native proteases, of heterologously expressed proteins (Li et al. 2004; Stephenson and Harwood 1998). Attempts have been made to solve the protein-degradation problem by creating protease-deficient B. subtilis hosts. The availability of heterologous gene-expression systems for archaea is limited, although there are well-developed gene-manipulation systems for the euryarchaeotes Halobacterium salinarum (Peck et al. 2000), Methanosarcina acetivorans (Metcalf et al. 1997), and Methanococcus maripaludis (Gardner and Whitman 1999) and to some extent for some species of Sulfolobus (Worthington et al. 2003), which are representatives of the crenarchaeotes. Development of shuttle vectors for cloning or for heterologous gene expression of the components of large DNA inserts in multiple microbial hosts will accelerate the quest to reap the fruits of metagenomics.
Single-Cell Analyses
A problem that has haunted microbial ecologists since the beginning of the field is the inability to account for minor members of a community. The abundance of species varies so widely that it is unlikely that the least abundant members will be captured in a given metagenomic analysis. Their DNA may well be in the libraries, but the probability of identifying, out of the millions of sequence fragments, the relatively few that came from the same low abundance community member, is low. Another aspect of the natural microbial world revealed from several metagenomics studies is the presence of microheterogeneity among organisms that are now typically grouped as members of the same species. The concept of a microbial pan-genome suggests that species can be defined as a core set of genes that are shared by all members, together with variable genes that differ from
one isolate to the next. Thus, a single genome sequence is not sufficient to represent the range of diversity found within a single species. From a functional standpoint, such variability may be critically important in the overall metabolic potential of one community as compared to another. At first it may seem paradoxical to study single cells in order to understand communities, but in fact the function of any given community reflects the contributions of each of its members.
One novel technological development illustrated in Figure 4-4 is the ability to amplify DNA from single cells (or single members of a community) in order to study how they contribute to community function. The method is based on the multiple displacement amplification reaction that uses ![]() DNA polymerase and random primers for DNA amplification (Hellani et al. 2004). This technique alone, and with modification, has been applied successfully to the amplification of bacterial sequences from low abundance samples from natural environments with minimal amplifica-
DNA polymerase and random primers for DNA amplification (Hellani et al. 2004). This technique alone, and with modification, has been applied successfully to the amplification of bacterial sequences from low abundance samples from natural environments with minimal amplifica-
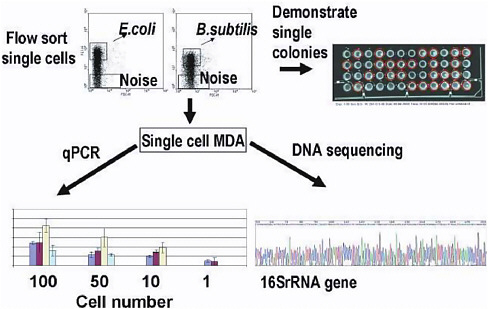
FIGURE 4-4 Molecular Displacement Amplification (MDA) of DNA from single cells. Flow sorted bacterial cells were plated to prove that single colonies could be reliably obtained. Then, having verified that flow sorting was reliable, the DNA from small numbers of cells (1-100) was amplified by MDA and tested in qPCR and DNA sequencing reactions (left panel). SOURCE: Roger S. Lasken, et al., Multiple Displacement Amplification from Single Bacterial Cells. In Whole Genome Amplification; Eds: Simon Hughes and Roger S. Lasken, Scion Publishing Ltd, Oxfordshire, UK.
tion bias. The ability to isolate single cells with microfluidics coupled with technology to amplify genomic DNA from single cells will revolutionize the study of unculturable species and the microheterogeneity within species. A number of approaches are being explored to facilitate the capture of information from individual bacteria or minority populations, including fluorescence-activated cell sorting, which can give useful information about size and functional chromophores (Zhang et al. 2006); growing mixed microbial communities in porous microbead columns so that each organism grows as a separate clone (Zengler et al. 2002; Green and Keller 2006); and single cell sequencing by dilution followed by amplification with strand displacement polymerase followed by debranching (Zhang et al. 2006).
The ability to amplify faithfully from single molecules has several applications and advantages relative to shotgun sequencing. Targeted amplification can be done in a way that retains quantitative information or can be normalized to emphasize the rarer species. As a result, information on haplotype or multiple chromosomes per cell can be retained, information about cells (and viruses) bound in pairs or larger aggregates can be captured (yielding data on symbiotic, parasitic, predatory, and other relations), and rare cells can be enriched with a simple single-amplified-cell prescreen (e.g., rRNA) followed by whole genome sequencing. Another rationale for developing methods for capturing and studying single cells in microbial communities reflects the fact that cells in communities are not randomly distributed, but in many cases form highly ordered assemblages of cells whose spatial orientation is essential for proper community function (biofilms on the tooth surface, for example).
Methods for Culturing Uncultured Species
Because the assembly of complete genome sequences is one of the major current limitations in metagenomics research, microbiologists are displaying renewed interest in the art of microbial cultivation. The most notable example is the cultivation of SAR11, a representative strain contained within a ubiquitous and dominant clade of marine heterotrophs, all of which had proved recalcitrant to cultivation for many years. Success in growing this organism was achieved with a combination of high-throughput (microtiter-plate) cultivation techniques using dilute media and rapid and sensitive screening using fluorescent probes specific for the SAR11 cluster (Rappe et al. 2002). Members of other ubiquitous microbial groups poorly represented in culture collections have since been isolated by tweaking of cultivation conditions—even by such simple adjustments as the use of solid vs liquid formulations, reducing nutrient and mineral concentrations, or increasing incubation time (Janssen et al. 2002; Rappe et al. 2002; Sait et al. 2002).
Basic Microbiology
The ultimate goal of metagenomics is to understand the structure and function of microbial communities. It depends on fundamental information about how microbial cells work in isolation and in populations and communities. The vast yield of information from metagenomic analyses conducted thus far has been built on more than a century of intensive study of microbes in pure culture. Recognition of genes based on sequence and making sense of genomic data, functional expression, and phylogenetic analysis depend on more detailed genetic, physiological, and ecological understanding of microbes in the laboratory. The many genes whose functions are not known, even in such well-understood microbes as E. coli, indicates the dearth of knowledge. Imperfect understanding of how gene expression machinery differs among species limits the power of function-based metagenomic analysis. And the lack of principles of microbial behavior in communities presents a wall that affects the depth of understanding that can be gleaned from metagenomics studies. Therefore, it is essential for the health and advancement of metagenomics that the study of microbes remains diverse and strong. Basic understanding of genetics, metabolism, gene regulation, cell structure, and responses to the environment needs to advance to aid in the design of metagenomic research strategies and the interpretation of metagenomic data.
Understanding Microbial Habitats and Collecting Metadata
Although seemingly a small component of Figure 4-1, the “Describe environment” box is perhaps the cornerstone of metagenomics studies. Information about the environment is the foundation of all analyses of the genetic and functional data obtained from organisms. Metadata collection, storage, retrieval, and analysis were not required in prior genome-sequencing projects. There were plenty of data in those studies—billions of nucleotides, in fact—but beyond inferring identity, function, and comparison between organisms, little was required of them. In contrast, the goal of metagenomics is to tease out the correlations between species abundance and environment, to link common gene functions to disparate environments, to determine whether the same organisms do the same things in different environments. All these comparisons require sophisticated and fast bioinformatics methods. How to “do it right” is still a fair question and will require creativity and detailed examination by many of the brightest bioinformaticians (discussed further in the next chapter).
Collaboration between microbiologists, geologists, chemists, oceanographers, meteorologists, clinicians, and a host of other scientists will bring rich rewards in the information that can be gleaned from metagenomics
datasets. Learning how to coordinate the different kinds of data collected and analyzed by scientists in so many disciplines is a major challenge, both conceptually and bioinformatically.
DOWNSTREAM DEVELOPMENT OF METAGENOMICS
Currently, metagenomics is heavily biased toward sequencing and its associated computational analyses, and pioneering functional analyses. While the current distribution of effort is appropriate for this initial exploratory phase, it will not be sufficient for the next phase of metagenomics, when more value will be desired from the sequence and its metadata. It is important to plan early for the mid- and longer-range development of the field so that both the researchers and agencies plan for and invest in new approaches and capabilities. Many of these downstream uses are difficult to predict in advance but the infrastructure for their encouragement and support can be established. It is important that the field not be slowed by overemphasis on massive sequencing without at least equivalent if not greater advances in other metagenomics approaches.
A 10-year trajectory of possible resource distribution would initially show a shift from emphasis on sequencing toward more computational development and analysis. Later, greater emphasis could be placed on such approaches as proteomics and transcriptomics, more in-depth analyses of metabolic and synthetic pathways (chemical bioinformatics, for example, could focus on detecting the genetic machinery for producing small biomolecules like signaling chemicals), and comprehensive knowledge building. The committee does not intend to deemphasize the importance of adequate sequencing resources, but to point out that the field will need to update its vision, tools, and goals continually, so that resources are appropriately divided between generating sequence and all of the other analytical and experimental approaches that comprise metagenomics.

























