
2
Regulatory Assessment
This section summarizes the presentations and discussions regarding the benefit–risk assessment that occurs in the premarket regulatory phase. Participants’ discussion focused on the value of formal benefit–risk assessment and the best ways to conduct it, while also considering the public perception of the benefit–risk assessment process.
PROBLEMS AND POTENTIAL SOLUTIONS FOR REGULATORY ASSESSMENT OF BENEFIT AND RISK1
Panelists discussed how benefit and risk data are currently collected and evaluated prior to a drug gaining regulatory approval from the FDA. The challenges to completing the assessment were identified as follows:
-
Lack of a systematic, consistent, and transparent approach to benefit–risk analysis;
-
Uncertainty regarding how to balance risk and benefit;
-
Insufficient knowledge about the risks of drugs at the time of their launch;
-
Conflict of interest (for example, experts consult with industry and
-
government on the same products) and involvement of stakeholders (for example, scientists, industry) in evaluating benefit and risk;
-
Lack of involvement of prescribing physicians and the public in the FDA regulatory process; and
-
Confusing and inconsistent terminology in benefit–risk assessment.
Value of a Quantitative Approach to Benefit–Risk Assessment
Dr. Tollman explained that federal agencies, such as the Environmental Protection Agency (EPA) and the Federal Motor Carrier Safety Administration (FMCSA) use quantitative evaluations to more rigorously assess benefit and risk, as well as cost. While parallels to pharmaceuticals are not conclusive, they suggest that quantitative approaches to drug benefit–risk assessment may be viable. Quantification, however, has its own set of challenges:
-
Quantitatively capturing a complex drug benefit–risk profile;
-
Quantitatively characterizing drug benefit–risk for individuals because of variation among patients in terms of both physiology and preferences;
-
Updating benefit–risk assessments with new information through the drug life cycle;
-
Addressing the inherent uncertainty in benefit–risk measurement;
-
Addressing disagreement about the role that cost should play in benefit–risk calculations;
-
Addressing the cost of adopting a quantitative framework and its potential adverse effect on innovation; and
-
Effectively presenting and communicating quantitative information.
Dr. Tollman suggested that adopting a quantitative approach could be beneficial in that it could objectively combine clinical trial data and information on patient preferences. Dr. Tollman noted that the academic community has a number of simple, powerful frameworks and utility weighting methods that could feasibly be adapted to the drug approval process. He cautioned, however, against quantitative elements being too simplistically or narrowly interpreted.
Dr. Tollman further argued that a more structured, transparent, and quantitative approach would be advantageous for all constituents— patients, regulators, and industry. For patients, advantages include the fact that the approval decision incorporates patient preferences, ultimate drug choice is based on individual response and preferences, and more differentiated treatment options become available to patients. For regulators, advantages include decisions that are grounded in a preagreed
framework that is consistently maintained from year to year and across divisions of the FDA. For industry, advantages include more predictable results and ultimately less attrition, more opportunities to innovate and differentiate products, and an ability to better align internal portfolio planning with a drug candidate’s true likelihood of success.
Challenges in Developing a Common Methodology
Dr. Galson argued that developing a common methodology for assessing efficacy and safety throughout the regulatory life cycle of a drug poses an enormous challenge because our understanding can change substantially over its course. While knowledge about efficacy grows exponentially during clinical testing, it continues to increase after approval when drugs are put to new uses. Also, exponential growth in our knowledge of safety doesn’t occur until after the drug has reached the market, when sample size increases and more data become available (Figure 2-1).
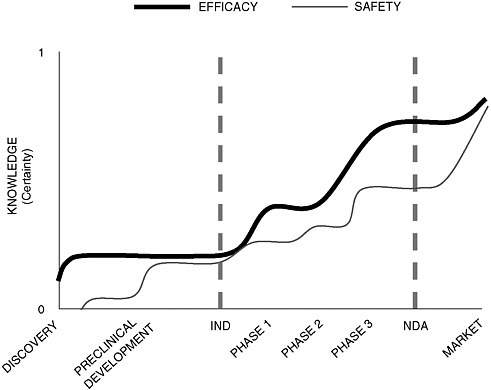
FIGURE 2-1 The time paths of knowledge related to drug efficacy and drug safety through the drug development process.
SOURCE: Adapted from Steven Galson’s presentation.
Dr. Galson used the FDA’s experience with Lotronex, a serotonin receptor antagonist indicated for treatment of irritable bowel syndrome, to illustrate how the use pattern of an approved drug may change after clinical trials and how our assumptions about risk change over time. The premarketing safety database, which included approximately 3,000 patients in two dose-ranging trials, revealed only limited dose-dependent adverse events. After launch, however, when the population of patients who were exposed to the drug increased rapidly, there was a severe increase in labeled gastrointestinal (GI) events. The drug was withdrawn, the clinical trial data were reassessed, and a relaunch was initiated after the implementation of a risk management plan. Dr. Galson argued that the misprescribing of Lotronex suggests that even the strongest predictive benefit–risk methodology can be defeated by knowledge gaps in the development program. Even if a common methodology for assessing benefit–risk ratios is developed, he noted, communication strategies for rollout and proper drug use must also be improved.
Dr. Galson further noted the lack of unanimity about the ideal benefit–risk balance for therapeutic products—there is still no consensus about what a good benefit–risk ratio is. While the FDA’s responsibility is to evaluate the benefits and risks of a drug for the population at large (or for the population for which the drug is being developed), providers are responsible for balancing benefits and risks for individual patients, and patients are responsible for making final benefit–risk decisions based on their own information and values (Figure 2-2).
Dr. Galson emphasized that drug safety is a societal issue, and the best benefit–risk assessment methodology will take us only so far. He presented two case examples to illustrate the societal context of the problem. First, the American Psychiatric Association harshly criticized FDA efforts to improve the labeling of SSRI (selective serotonin reuptake inhibitor) antidepressants, arguing that the increased level of warning and the better description of benefit and risk may scare away patients who could benefit from the drug. Dr. Galson remarked that it is difficult to see how improving benefit–risk assessment methodology will resolve this type of challenge.
As a second example, although the FDA has implemented several risk management plans to make sure that pregnant women do not take Accutane, the FDA still receives e-mails arguing that even one congenital malformation is not worth the benefits of this cosmetic drug. He suggests that no amount of benefit–risk assessment methodology can resolve this conflict. He concluded by stating that while the challenge in balancing pharmaceutical product benefit and risk is multidisciplinary, successful adoption of methodological improvements would nonetheless benefit all stakeholders.
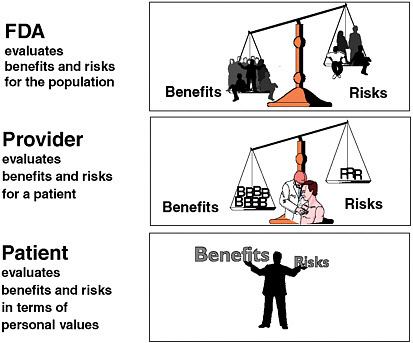
FIGURE 2-2 Alternative perspectives on the benefit–risk relationship.
SOURCE: Adapted from Steven Galson’s presentation.
QUANTITATIVE APPROACHES
A QALY-Based Approach to Benefit–Risk Modeling
Committees often take a piecemeal approach when weighing benefit and risk evidence and making approval decisions. Dr. Garrison suggested that it would be useful to have a more systematic framework for integrating and evaluating the tremendous amount of complex information that must be sifted in the process of evaluating safety. He commented on how pharmaceutical benefits and risks are measured in different units and how there is no clear approach for their quantification. He suggested that a more transparent, structured model based on quality-adjusted life years (QALYs)2 could be a basis for developing that framework.
There is general consensus, he argued, that QALYs are a useful tool. Health technology assessment (HTA) and health outcomes researchers express health effects in terms of QALYs, and a recent Institute of Medicine (IOM) panel recommended that regulatory cost-effectiveness analyses (CEAs) use QALYs to represent net health effects (IOM 2006). However, it must be decided whether QALY-based utility analysis and other outcomes research tools (e.g., integrative modeling of long-term health outcomes) provide a useful methodology for a more formal, explicit, transparent, and quantitative process for assessing pharmaceutical drug benefit–risk than the current approach.
Dr. Garrison explained that for most new drugs, estimating QALYs requires using models to synthesize information and extrapolate beyond what is traditionally collected in Phase III trials. While this method is being applied across a wide spectrum of diseases, usually for purposes of reimbursement, it has limits. He asserted that the FDA and physicians either do not fully understand the method, or do not believe that it is scientifically valid; it may be inappropriate for physicians to use the method at their level of decision making; it is usually applied more to benefits than to adverse events (risks); and QALYs are risk-neutral and do not explicitly take into account risk aversions (although there are ways to adjust for this).
Dr. Garrison noted the importance of including subgroup analyses and calculating benefit–risk ratios separately for different populations. He also noted two key challenges of measuring risk in terms of QALYs. First, clinical trial signals are often weak, making it difficult to measure known adverse events in quality terms without making certain assumptions. Second, it is difficult to measure the potentially serious side effects that occur at rates of less than 1 in 10,000 or 1 in 100,000.
Dr. Garrison recommended that the feasibility and usefulness of an explicit, transparent process of benefit–risk measurement relying on QALY-based outcome models be more fully evaluated, with the recognition that it may not be reasonable to apply the full methodology to every product and that patient preferences vary.
Patient Differences
Dr. Garber outlined the difficulties of creating a single measure that sums up benefits and risks. Not only does every drug have multiple health effects, but people perceive these effects differently. Patient differences pose a tremendous challenge for benefit–risk assessment. Dr. Garber identified two types of patient heterogeneity: physiological (e.g., genetic variation) and preference variation (different people attaching different values to risks and benefits). He remarked that no federal agency has a formal process for weighing preference variation, and there is no consen-
sus in the literature for how it should be done. He explained the impossibility of relying on a purely objective framework “that is independent of patient preferences or some kind of element of human judgment” and discussed problems that can arise when decisions are based on population averages rather than patient preferences. The challenges will increase as we deal more frequently with treatments whose effects are not only life extension, but also improvement in function or quality of life.
Dr. Garber suggested that some variables in a cost-effectiveness analysis might also be important to include in a more general analysis. He discussed the results of a study published in 2003, before Vioxx was withdrawn, of cyclooxygenase-2 (COX-2) inhibitors versus naproxen, and explained how that study demonstrates the usefulness of QALYs as a metric (Speigel et al. 2003). The study makes it very clear that what can be a “bad” drug for some people (people not at risk of GI bleeding) can be a very “good” drug for others (people at risk of GI bleeding). He remarked that the types of information that would be needed for a preference-based benefit–risk analysis are not very demanding, compared to what has historically been required for FDA approval.
Participants noted that the calculation of QALYs requires modeling and extrapolation beyond the information typically collected in Phase III trials. Since modeling requires making assumptions—for example, how surrogate outcomes translate into clinical benefit—the uncertainty surrounding those assumptions must be addressed. Dr. Garber argued that while addressing these uncertainties complicates such models, the alternatives are almost invariably worse. Recommendations based on one-year outcomes in a clinical trial often reflect a range of informal assumptions about what is going to happen beyond the end of the trial. A formal model, on the other hand, requires that assumptions be explicit and transparent.
Additional questions on uncertainty were discussed. For example, given that there are different levels of uncertainty in benefits and risks across disease areas, what should drug companies have to achieve at the end of Phase III, and might it be more than just a certain benefit-minus-harm difference? Could it instead be a commitment to do Phase IV studies or to spend a certain amount to reduce uncertainty about risks and benefits? Is it worth spending $10 million or $20 million to narrow the confidence interval (that is, uncertainty) about risks and benefits only slightly, and when might this greater certainty suggest that an approval decision be changed?
While discussing variability among subgroups and individual patients, a participant noted that the balance of risks and benefits may be specific to particular populations, and that unless you can identify such populations, the effort to assess risks and benefits breaks down. Accurately identifying such populations involves significant physician
involvement, over which the FDA has little control. While the FDA can define benefits and risks for different populations, it cannot prevent the inappropriate prescribing by physicians once a drug is on the market. Dr. Garber remarked that while it is inappropriate for a regulatory agency to be a source for improving physician practice, sometimes there comes a point where the likelihood of misuse is so great that it does change the FDA’s thinking about approval.
Other Ways to Think About Risk
Dr. Slavin introduced the notion of tolerability of risk (TOR), which he noted is widely used in evaluating occupational and environmental standards in both the United States and Europe. It is particularly useful in situations where the incidence of the risks is unknown and the understanding of the hazards are ambiguous. Rather than generating a single number for comparison against a standard (because there is insufficient knowledge for true certainty), a TOR framework describes a pre-agreed, bounded area of risk, or a box, against which a probabilistically derived footprint of the true risk can be compared. Over time, as more data are collected and knowledge increases, the area of that footprint shrinks, and any decisions that need to be made about a drug become easier to make.
Quantitative Assessment of Pre-Approval Risk: Zometa as a Case Example
Dr. Lesko argued that quantitative tools are attractive because they are complementary to conventional tools, which have certain limitations. For example, when Phase III data are analyzed with conventional tools, the change in response from baseline to end of study is compared between treatment and placebo groups. Measurement of response to treatment between baseline and end of study are not always considered in conventional studies, even though they are often the most interesting since they reflect disease progression or treatment over time. Quantitative analyses allow us to look at those measurements. Also, most Phase III conventional data analyses treat doses as categorical, not continuous, variables.
Dr. Lesko presented a case study on Zometa, a drug indicated for the treatment of hypercalcemia of malignancy and also for multiple myeloma and bone metathesis from solid tumors. The initial review was conducted by the FDA’s Office of Clinical Pharmacology with input from the Pharmacometrics Team (quantitative clinical pharmacology). For Zometa, the safety endpoint was renal toxicity. Dr. Lesko demonstrated how the use of quantitative tools revealed more accurate information about risk. Examining the difference between placebo and Zometa
categorically—normal versus abnormal renal function—was misleading in terms of the risk for any given patient of developing renal deterioration. Postmarketing reports began to reveal that the drug was associated with renal deterioration when used in a wider population than was defined in the clinical trial population. Quantitative tools were then employed to examine renal deterioration by using creatinine clearance (an indication of renal deterioration, which in turn is a predictor of renal toxicity risk) as a continuous variable.
Through data obtained from multiple quantitative analyses, Dr. Lesko and his team learned that a patient’s baseline renal functioning affected their risk of renal deterioration. They gained a better understanding of the drug’s toxicity and were able to inform the warning section and dosing recommendation on the product label. They decided to select doses of Zometa according to baseline creatinine clearance. It was demonstrated through quantitative analysis that linking kidney function exposure and dose adjustment would provide a reduction in the risk in individual patients and subgroups of patient.
While the learning process with Zometa was not used to design additional clinical trials (via simulations), this did occur with some of the other case examples that the Pharmacometrics Team has studied. In conclusion, Dr. Lesko argued that although there are some risks that we can do nothing about (e.g., age), others can be addressed and reduced through the use of quantitative analyses of treatment data.
Dr. Lesko first discussed the “learn–confirm paradigm” (Sheiner 1997) for delivering news to patients about benefits and risks. This construct separates drug development into two sets of concepts: (1) Learn—benefit– risk is not well defined and is assessed by looking across all of the clinical data for such things as dose-response and variables that influence exposure and its relationship to toxicity (he reported that the set of quantitative tools used in the case of Zometa were associated with the learning aspect of drug development); and (2) Confirm—efficacy is well defined, and rigorous, randomized control trials using pre-specified statistical analyses have been designed to answer efficacy (yes or no) questions in a general population.
An Argument Against Benefit–Risk Analysis in Drug Approval
Dr. Strom argued that quantitative benefit–risk ratios should not be used to make drug approval decisions, citing the following reasons:
-
Understanding of the benefit and risk relationship will change throughout the life of a drug and may vary between individual patients or populations.
-
Many important qualitative variables would not be adequately addressed—for example, marketing practices, prescribing patterns, patient compliance when taking the drug, and experimental design.
-
Despite decades of effort, assessment of a drug’s benefits presents enormous challenges—for example, quantifying pain and comparing unrelated outcomes, such as pain and heart attack or gastrointestinal bleed.
-
Measuring risks often requires the use of surrogate measures because we can’t wait for the ultimate outcome.
-
Benefits and risks are measured in different units and vary by context, for example: How many patients with pain relief (the benefit) are needed to balance one heart attack (the risk)? How much short-term risk is acceptable for long-term benefit (e.g., statins and blood pressure medications that yield long-term benefit)? How much individual risk is acceptable for societal benefit (e.g., vaccines)? How much societal risk is acceptable for individual benefit (e.g., antibiotics)?
Dr. Strom argued that plugging benefit and risk measures into a single equation is not feasible. While other speakers suggested standardizing units using a measure of utility, he argued that utility is a subjective judgment that varies among individuals. He questioned the practice of imposing average subjective judgment on individuals. He reasoned that, given such wide variation, the decision should be made by the person needing the benefit and taking the risk.
In conclusion, Dr. Strom remarked that subjective judgments are being made throughout the entire benefit–risk assessment life cycle and it is naïve to think that we can quantify these subjective judgments. Pharmaceutical companies make subjective judgments about whether to develop a drug; advisory boards and regulators make subjective judgments about whether to approve a drug; physicians make subjective judgments about whether to prescribe a drug; and patients make subjective judgments about whether to take a drug. The current system is flawed by its subjectivity, but it is probably the best there is. He cautioned that we risk forcing wrong answers by being overly quantitative and precise.
Dr. Strom’s recommendation that benefit–risk assessment not be quantified elicited much discussion. Dr. Leiden elaborated on two reasons for quantification. First, he reported that some lower-level reviewers in the FDA told him they feel pressure to not make mistakes. A more standardized, quantitative system would give FDA reviewers ammunition to explain their decisions to legislators and to change their decisions if the data suggest that they should. Second, there is tremendous anxiety in pharmaceutical companies trying to understand what it is that the FDA wants. Part of this anxiety stems from different expectations among divi-
sions within the agency and sometimes even among reviewers within divisions. Having more standardized, quantifiable methods would encourage industry to pursue innovative drug development programs that they might not otherwise pursue. Dr. Leiden remarked that the system is “moving in the wrong direction … of squashing innovation further.” Others agreed that the fear-based push for larger clinical samples sizes is leading to “an overly conservative system.”
Dr. Tollman argued that using a formula to assess benefit and risk may create false precision. On the other hand, to the extent that there is quantitative information and a valid statistical analysis of the benefits and risks of a drug that he is considering, he as a patient would like to see it. Finding a way to present the quantitative information in a way that would be helpful for patients would be very valuable. He said, “It would help me inform my choice, and from my point of view, that’s the overall objective of the process.”
Implementing Benefit–Risk Assessment
Dr. Throckmorton argued that integrating quantitative benefit–risk assessment into the early drug development process would enable all stakeholders to make better decisions. Companies could make earlier and more informed go or no-go decisions, the FDA and other regulators could make more informed early approval decisions, and patients could make better decisions about treatment options. He made three assertions regarding the development of a better approach to benefit–risk assessment:
First, benefit–risk assessment must be patient-centered, providing the best possible information to patients so that they can make the best possible choices.
Second, benefit–risk assessment must be integrated into the drug development process without reducing either safety or efficacy standards. Dr. Throckmorton proposed utilization of “model-based drug development” as a platform for achieving this. He noted that it would allow easy updating of benefit–risk assessments as new data become available. He also emphasized the importance of early dialogue between pharmaceutical companies and the FDA. Early discussion and agreement may provide regulatory clarity and reduce sponsor uncertainty, thereby resulting in more efficient product development. This requires that all assumptions and uncertainties are openly stated and discussed.
Third, a successful benefit–risk analysis must rely on standards. While there is no one-size-fits-all methodology, we need to identify and adopt best practices and build greater familiarity and expertise within the regulatory agencies and also in industry and academia. The Voluntary Genomics Data Submissions mechanism, whereby sponsors provide
genomic information for nonregulatory, data-sharing discussions, could serve as a model for facilitating this kind of information exchange.
Dr. Throckmorton identified key first steps in the implementation of this type of a system, analysis and application. Analysis should include some examples, ideally from prospective use during drug development. An analysis of nuanced benefit–risk decisions that have been made in the past would also be informative (e.g., drugs that may not have been lifesaving but had symptomatic benefits). Analysis could assist in developing appropriate methodologies for evaluating risk and benefit data. Then, these methodologies need to be applied and integrated into early drug development. Ideally, drug developers would propose what they believe to be the best assessment of risk and benefit, and regulators would have the expertise to discuss this assessment in a meaningful, forward-thinking way.
Flexibility for Products That Address Large Unmet Need
Dr. Leiden emphasized that it would be tremendously beneficial to biotech and pharmaceutical companies to have a clear understanding early on regarding the FDA’s efficacy and safety requirements, particularly for diseases with large unmet need where the lack of a clear regulatory path discourages companies from proceeding. Novel drugs for diseases with large unmet need have less well-defined regulatory paths, longer development times, less well-defined and often smaller markets, less well-defined safety issues, and high liability risks. These challenges make pharmaceutical executives more reluctant to initiate clinical development programs for novel drugs. Further, they create a much lower threshold for halting programs when the first hint of a safety or efficacy problem surfaces. Dr. Leiden proposed flexible regulatory, intellectual property (IP), and liability approaches for these products. This would require several steps: agreeing on a predefined list of high-priority diseases with large unmet need; significantly modifying the requirements for clinical trials for such agents (e.g., strong Phase I signals allowing for fast transition to Phase II–III with relatively small numbers of patients); offering provisional approval; offering a designated period of market exclusivity, so that the company is guaranteed minimum market time no matter how long development takes; and reduced liability exposure.
Dr. Leiden proposed that in exchange, pharmaceutical companies would agree to pursue a limited launch of products that obtained such a provisional approval. This would involve a limited launch to a carefully defined physician and patient population; explicit labeling with clear explanations of the known benefits and risks at the time of approval; limited marketing and promotion, with no direct-to-consumer or journal advertis-
ing; and prospectively defined Phase IV requirements. Dr. Leiden suggested that only after successfully completing Phase IV trials should companies be allowed to expand their marketing efforts and broaden their launch.
Dr. Leiden argued that both patients and industry would benefit from a flexible system as described above. Patients would benefit from: 1) the availability of more novel medicines for diseases with unmet need, 2) more drugs for less-prevalent diseases, and 3) a better understanding of benefits and risks made possible by the extensive efficacy and safety data collection in Phase IV. Industry would benefit from the incentive to pursue innovative products for smaller markets (for example, by not spending $100 million or $200 million on products that would be too risky and expensive to push through the regulatory process); guaranteed market exclusivity; and further R&D made possible by the revenue from provisionally approved products.
Dr. Leiden noted that his proposed system may sound heretical, given so much focus on the risks incurred because of the limits of what we know from testing a drug on only a few thousand patients. Here, he is advocating testing drugs in even fewer patients. He pointed to HIV/AIDS as a successful example of this kind of system having worked in the past. In the early and mid-1990s, patient advocacy groups pushed pharmaceutical companies and the FDA for early access to new treatments. FDA responded by allowing and establishing expedited review, with the first HIV/AIDS drugs being approved less than four years after the initial discovery. Not only did HIV/AIDS evolve from a fatal to a chronic illness in less than 10 years, at least in regions of the world where access to drugs is unlimited, but there have been no product withdrawals due to unexpected safety issues. Dr. Leiden stated that if the current system is not improved, “we will essentially strangle innovation, at least from large pharmaceutical companies.”
There was some question about how difficult it would be to adopt “model-based drug development” as a platform for integrating additional data into the development process, as Dr. Throckmorton proposed, and whether these data would add much value. Dr. Throckmorton responded that his point was that if we limit ourselves to only some aspect of the data and do not use all of the available information—if sponsors do not communicate about the animal models, biomarkers, internal benefit–risk decision making, or other tools that they are using—then we risk losing a chance to understand how decisions are being made. He argued that open communication about choices being made provides clarity and understanding and has value in and of itself.
Dr. Leiden agreed with Dr. Throckmorton’s concern and noted that the end-of-Phase II(A) meetings that the FDA has begun offering have been tremendously beneficial to industry and have opened up a whole
new avenue of discussion at a time when critical decisions are being made about very expensive Phase II(B) and III programs. That kind of interaction is extremely helpful not just to the FDA but also to industry, providing clarity and transparency. He also commented on a notion that Dr. Throckmorton addressed in his presentation: that we can explore some of these new benefit–risk methodologies in a penalty-free way (without adversely impacting approval of the drug), as was done with pharmacogenomic information before the FDA really understood how that information was going to influence regulatory approval. It would allow us to put a database together and gain a better understanding of how to use benefit–risk information.
The discussion turned to uncertainty. Dr. Throckmorton’s suggestion that benefit–risk data be used in clinical trial simulation early during development raised questions about how the uncertainty surrounding unknown risks would be handled. Dr. Throckmorton agreed that while we know very little about safety until later in development, class effects could be extrapolated and used to find safety signals.
Dr. Leiden argued that the proposal he put forward begins to address the issue of uncertainty. For example, there is a very explicit hypothesis about the unique benefits of COX-2s with respect to decreasing GI bleeding, but the cardiovascular risk signals during Phase I, II, or III were not recognized. Had Vioxx been developed according to the paradigm that he suggested, the drug would have been rolled out only to those patients with arthritis who were at risk of GI bleeding. It would not have been until prospective Phase IV studies were initiated that we would have begun to look at the potential benefit in other patients. That is where we would have seen the safety signal—while the drugs were still restricted with respect to being prescribed only to patients for whom the benefit– risk ratio was known to be favorable. He argued that had this route been taken, Vioxx would still be on the market and available for those patients. For many other drugs as well, the majority of side effects are not going to show a signal during Phase I, II, or III.
When asked to comment on Dr. Leiden’s proposal for change, Dr. Throckmorton noted that some components of Dr. Leiden’s proposal are things that the FDA is already doing but perhaps could be doing more consistently or better. Other components would require changes in regulatory law. We need to determine which parts of the process would add the most value if improved—and which parts are not being addressed—in order to decide how to move forward.
Dr. Goldman3 said that she was intrigued by Dr. Leiden’s proposal and noted that although the EPA created a fast-track process for reduced-
risk pesticides, it was not easy and it required a lot of work with companies and other stakeholders. Congress eventually adopted it and put legislation in place that strengthened it considerably. She suggested that the FDA could pursue a similar path in a step-by-step fashion.
LESSONS FROM OTHER INDUSTRIES4
Two of the workshop sessions focused on lessons to be learned from other industries. The first session, “Assessing the Effectiveness of Risk– Benefit Algorithms from Other Industries,” focused on the methodologies used to evaluate benefit–risk ratios of various nonpharmaceutical products. The content of that session is summarized here.
Lessons Learned from Chemical Risk Assessment
Dr. Paustenbach highlighted key developments in the history of chemical risk assessment, including the 1983 publication of the “Red Book” (National Research Council 1983). The anticipated role of the Red Book was that it would provide a framework for a well-integrated risk-assessment process that could quantitatively characterize chemical hazards in a way that was objective and “separate and distinct” from decision making. While the field has realized some of that early optimism and risk assessment has become integrated into most regulatory guidance and policy involving chemicals, there have been some shortcomings. (For example, Dr. Jasanoff5 argued that experience and social science research have shown that risk assessment is limited by uncertainty and ignorance, and the boundary line between where science [risk assessment] ends and policy [risk management] begins is not as clear-cut as we sometimes believe.)
Dr. Paustenbach identified 10 lessons from chemical risk assessment that may be relevant to the pharmaceutical industry:
-
Humility about the limits of science is critical to enjoy the trust and respect of the public. Scientific analyses are not often trusted by the public. They often wonder at any given dose, why they should have to tolerate any risk.
-
Transparency is critical to maintaining the trust of the public as well as satisfying the expectations of trial attorneys. He predicted that the
-
pharmaceutical industry will see an avalanche of suits in the future, as the chemical industry has for the past 25 years. He observed that every single award of any magnitude that he has seen over the past 10 years (in chemical industry law suits) has involved lack of disclosure, not actual outcome. Transparency provides the means to reconstruct scientific analyses to determine where objective quantitative methods end and professional judgment begins, and it allows for a better understanding of the uncertainty in analyses and conclusions.
-
Use quantitative techniques to describe uncertainty in risk estimates. He noted that there are methods than can be used to do this.
-
Acknowledge that genetic polymorphisms exist and that most have not been characterized. Genetic polymorphisms are responsible for multiple dose-response curves for some chemicals. The same is true of drugs.
-
Unlike chemical risk assessment, where safe doses can be estimated with confidence (because exposures are usually quite low, on the order of 1 in 100,000 or less), pharmaceutical agents often involve relatively high doses, making it difficult to pinpoint human safety. Some pharmaceuticals carry risks as low as 1 in 10 or 1 in 20, again pointing to the importance of transparency.
-
Clearly describe the benefits of taking the drug and compare this with the possible risks. Most Americans are taking a more active role in their medical decisions than in the past, and they want to know the benefit–risk relationship of the drugs they are considering. The challenge is in communicating that information.
-
Discuss with patients the risks of not taking a drug. Both patients and trial lawyers need to know this as much as they need to know the risks of taking a drug.
-
Remind the public about its role and responsibilities in minimizing the disease process. Be clear about the risks and benefits of taking a drug with other pharmaceutical or recreational drugs and the roles of diet, exercise, and other factors in the “total approach” to dealing with illness. This kind of information not only contributes to the patient’s complete understanding but also impacts litigation.
-
Strong, credible, science-based regulators that perform with integrity and diligence protect industry from public suspicion—and tort litigation—and play an important role in building trust relationships between the public and industry.
-
Don’t try to hide risks. In recent years, Americans have insisted that they be informed of all possible risks to which they are exposed. If the risk is clearly discussed, rarely will the public become angry with a manufacturer.
In conclusion, Dr. Paustenbach stated that after 30 years, chemical risk assessors have learned that conducting good scientific analyses is not enough. One has to be transparent, direct, forthcoming, and willing to acknowledge uncertainties in medical or scientific understanding.
Need for a Framework
Dr. Samet reemphasized the long history of chemical risk assessment and how the Red Book provided a much-needed formal framework for addressing environmental health risk questions. He discussed what is widely considered one of the best human data-based quantitative chemical risk assessments conducted thus far: radon exposure. He described the history of the EPA’s awareness of the problem and the subsequent risk assessment process conducted by a National Research Council committee, of which Dr. Samet was the chair (National Research Council 1999).
He described how models derived from a quantitative risk assessment can be used to answer questions about risk at both the population level (e.g., what is the population risk?) and the individual level (e.g., if I have been living in a home with radon levels 10 times above the EPA’s action guidelines, has my family sustained increased risk?). He noted that one of the key challenges of risk assessment is the reality that there are often multiple dose-response curves. He commented on the importance of using pooled epidemiological data and mechanistic knowledge to guide more certain risk models. His committee was able to derive a reasonably precise description of how risk varies with exposure because it had 100 years of epidemiological data on the relationship between exposure and cancer risk and a good mechanistic understanding of how radon might damage a cell and cause cancer. He also noted that uncertainties can be characterized and that understanding how uncertainty estimates change the behavior of decision makers is a relevant topic that has not received much attention.
Dr. Samet emphasized that pharmaceutical risk assessment needs a framework—its own Red Book—so that questions about risk and benefit can be asked and answered as precisely as possible. A framework provides a common understanding of concepts and terminology and serves as a foundation for readily identifying what information is needed in any given situation to accurately assess benefit–risk.
Lessons Learned from Pesticide and Mercury Risk Assessments
Dr. Cohen agreed with Dr. Samet that having a framework is critical. He noted that unlike the EPA framework, which is focused on reducing risk to acceptable levels and for the most part does not really con-
sider benefit (except when considering economic cost), pharmaceutical benefit–risk assessment (and management) demands a more comprehensive framework based on the recognition that risks do not occur in a vacuum and must be weighed with benefits.
Dr. Cohen’s talk revolved around two case studies, the first involving a pesticide ban (Gray and Hammitt 2000) and the second mercury in fish (Cohen et al. 2005). He used both to demonstrate difficulties encountered when conducting benefit–risk trade-off analyses. He used the second study to elaborate on how some of those difficulties can be overcome. Specifically, Dr. Cohen and his colleagues used an alternative, QALY-based risk-assessment approach to evaluate the benefit–risk trade-offs associated with shifts in fish consumption. Their more comprehensive analysis and presentation of the data provided the public (and the professional community) with more meaningful information than a single reference data point.
Dr. Cohen concluded by noting that it is important to compare pharmaceuticals to realistic alternatives, not just to nothing (that is, placebos). While comparing to a placebo is a good starting point, accurate risk assessment ultimately requires a realistic comparison. He noted that outcome probabilities need to be quantified, particularly if they vary among individuals, which is the case with pharmaceuticals (e.g., people have different underlying risk factors and take different doses of medication) and that outcome severity needs to be quantified using clinical outcomes, not intermediary measures such as enzyme biomarkers. Finally, both the “natural unit” and common metric estimates should be reported. While these demands complicate pharmaceutical drug efficacy assessments and increase uncertainty, he said that there are ways to deal with the uncertainty.
Parallels Between Food and Drugs
Dr. Hall remarked that even though the U.S. Congress has combined foods with drugs in the same legislative act for the past 100 years, the differences between the regulation of foods and drugs are much more apparent than the similarities. He discussed the limited regulatory authority of the FDA and the greater complexity and uncertainty of measuring food-related risks (compared to measuring drug-related risks). He noted that, in contrast to drugs (with the exception of Olestra), there have been no clinical studies for safety or observation of possible adverse effects, and postmarket surveillance is uncommon (exceptions include aspartame and sterol and stanol esters in bread spreads).
Dr. Hall noted that while public perception of acceptable risk in food is zero, the unacceptability of risk is perception only. Obviously we do
accept risks, and food-borne illness is second only to the common cold as a cause of lost time from work, and obesity—a nutritional food-related risk—is widespread. The discrepancy between zero-risk perception and the acceptance of food risks exists, Dr. Hall argued, because the latter are regarded as voluntary. For example, weight gain is something that is within our control, so we don’t think of it as a risk.
Dr. Hall concluded by remarking that dietary supplements reflect a trend toward the “medicalization” of food. If that trend continues, then perhaps the regulatory challenges associated with food will become somewhat more similar to those of drugs. For now, however, the risks and benefits of foods and the public perception of them do not offer many parallels to drugs.
This session ended with a brief discussion of risk communication and whether the discrepancy between public perception of risk and actual risk, in any of these situations, may stem from the fact that the public seems to be hearing exaggerated claims, not nuanced messages, about risks and benefits. Dr. Cohen remarked that yes, risk communication must be improved. In the case of mercury in fish, he argued that the FDA and EPA have worked very hard on improving their risk communication but that it still poses a problem. Regulators need to anticipate how people are really going to react to something and realize that they are not going to follow the recommendations exactly as advised.
Dr. Paustenbach emphasized the importance of clearly expressing what we have learned from quantitative risk analysis, including uncertainty analysis. He suggested that communicators conduct dry runs by communicating such information to stakeholders, for example in an afternoon session, and then immediately testing the communications package by asking the stakeholders what they heard. Dr. Hall agreed that conducting a communications dry run provides important information about what the listeners bring with them in terms of preconceived perceptions.
The discussion ended with a question about whether there are any circumstances in which it does not matter if information about risk is available. Dr. Paustenbach argued that from a legal perspective, there is no exception to complete transparency, disclosure, and communication of all information.
CRISIS IN CREDIBILITY6
A recurring topic of discussion over the course of the two-day workshop was loss of public trust in the U.S. drug safety system and wide-
spread misunderstanding about the meaning of drug safety and the scientific process that moves drug approvals forward. Highlights are summarized here.
Dr. Strom identified three major sets of limitations associated with analyzing benefit–risk ratios early in the drug’s regulatory life cycle. The first limitation is the experiential difference between premarketing and postmarketing drug use and the fact that efficacy, a measure of how well a drug works in an experimental setting, is very different from effectiveness, which is how well an intervention works in the real world. Second is the growing cost of drug development, which has led to an increased need for immediate blockbuster sales and aggressive marketing even though knowledge of adverse events is inherently incomplete prior to marketing. Third, premarketing studies are of short duration, which means that only the short-term effects are known at the time of marketing.
Dr. Strom commented on other limitations of the current system, including lack of incentives (e.g., to complete promised postmarketing surveillance studies) and the “historic” lack of commercial and regulatory interest in adverse drug events, both of which feed into public misunderstanding that drugs have zero risks at launch. He stated that direct-to-consumer advertising exacerbates the situation, leading to overuse of drugs by patients for whom use of the drug is not compelling and for whom there may be substantial risk of unknown adverse reactions.
The effect of these limitations is that the public misunderstands drug safety, believing that postmarketing discovery of adverse drug reactions means that “somebody messed up.” In reality, almost all postmarketing safety issues involve rare adverse events that could not have been detected prior to marketing. This misunderstanding, coupled with growing concern about drug safety has led to overreaction, increased premarketing requirements, and delayed access to new drugs.
Dr. Slavin discussed how the regulation of science and technology has evolved from a culture of policy makers, industrialists, and scientists meeting behind closed doors, with citizen and stakeholder groups rarely consulted, to one where science is “just another stakeholder.” The public now questions scientific results, including results about drug benefit and risk. Dr. Slavin remarked that high public trust is typically associated with low perceived risk and, conversely, low public trust with high perceived risk and eventually evidence resistance. He argued that the precautionary principle and growth of risk aversion have led to widespread expectation that there is always a new scandal around the corner and that it is “better to be safe than sorry.” He disagreed with this public expectation.
Dr. Goldman discussed the results of a survey recently published in the Wall Street Journal (2006), demonstrating that while over time most people have thought highly of the FDA, the trend now shows
the public becoming increasingly dissatisfied. She argued that the biomedical research enterprise is driven increasingly by money; researchers are funded through consulting agreements with pharmaceutical companies and the medical profession is becoming less independent of the regulatory process. Those who used to be the trusted representatives of consumers (e.g., medical professionals, biomedical researchers) are no longer trusted by the public. She noted that there are few sources of funding for pharmacology research through the National Institutes of Health (NIH), FDA, or other federal sources, and funding is not allotted for independent data assessments, consumer surveys, or efforts to communicate with consumers about pharmaceutical risks. Dr. Goldman suggested that consumers be made equal partners in the process.
Dr. Goldman’s presentation raised questions about whether there is any way to “turn the system around.” The biomedical research enterprise is at a point where the best experts consult with industry and government on the same products. Dr. Goldman replied that the FDA has not created space for a consumer role in its culture and that the agency needs to encourage more dialogue among academia, industry, and especially consumer groups. Opening communication, she argued, will create a culture of collaboration and trust.
There were several comments on how the system needs to make room for relevant industry players to collaborate with the best scientific talent in order to bring good products to market. One attendee argued that by preventing an academic researcher who collaborates with industry on product development from participating in the regulatory process, one eliminates from the process those people who truly understand the intricacies and subtleties of the research and know enough to ask the right questions on behalf of patients. Another participant remarked that there are well-designed mechanisms in place at various agencies for disclosing acceptable conflict and identifying unacceptable conflict. While this is a complex issue, it can be addressed with integrity and balance.
Dr. Goldman suggested that the FDA open the door to dialogue to consumers. She relayed an experience that she had as a consumer testifying before an FDA advisory committee. She was struck by the absence of consumer input at that meeting. Consumers attending the meeting were extraordinarily disadvantaged by not having materials made available to them until immediately prior to the meeting. Making those materials available earlier during the process is an example of a small change that the FDA can make that would begin to make the agency more consumer-friendly.
Ms. Musa Mayer7 commented that the FDA very judiciously and responsibly solicits comments from patients who serve as patient rep-
resentatives or consultants in various programs. When not serving in that capacity, however, she shares the frustration of other members of the public with respect to not being able to access materials until about 24 hours before the meeting and having such a limited amount of time for preparing comments.
Dr. Goldman further suggested that there be more public funding for pharmaceutical research, so that researchers have more options in addition to working with industry. This prompted some discussion about where the funding would come from, given that within academia there is no other clear path of advancement for pharmacological researchers. (NIH funding is the main path, but pharmacological researchers generally do not pursue it.) One participant remarked that there is really no such thing as an independent source of funding and that the challenge is to create diversity in funding sources.
Dr. Leiden argued that it is precisely these complex interactions between academicians, industry and to some extent regulators that are the reason for the success of the biomedical enterprise in the United States. If we are not careful in how we handle conflict of interest, in our attempts to untangle it, we may severely damage the system. He stated that the best evidence for this is that it wasn’t until the Bayh–Dole Act of 1980 stimulated the translation of basic scientific discoveries in academia to applications in industry that the U.S. biotechnology industry emerged. Dr. Leiden mentioned a recent article reporting that there has not been a single case of research fraud caused by these financial conflicts of interest between industry and academia (Stossel 2005).






















