
2
Addressing the FDA’s Resource Challenges
|
Recommendation on Resources from the IOM Report The Future of Drug Safety: Promoting and Protecting the Health of the Public Recommendation 7.1 To support improvements in drug safety and efficacy activities over a product’s lifecycle, the committee recommends that the Administration should request and Congress should approve substantially increased resources in both funds and personnel for the Food and Drug Administration. |
The FDA today faces resource challenges in carrying out an expanding set of responsibilities. A central theme of the symposium was that these resource challenges must be addressed if the agency is to implement the recommendations of the IOM report.
HISTORICAL OVERVIEW1
At the beginning of the twentieth century, there were few regulations to safeguard foods, and no safety or efficacy standards for drugs or concoctions purported to be drugs. In 1906, public disclosure of unsanitary conditions in meat-packing plants as documented in Upton Sinclair’s The Jungle, the use of poisonous preservatives and dyes in foods, and cure-all claims for worthless and dangerous patent medicines led to the enactment of the Federal Pure Food and Drug Act. This act authorized regulation
of product labeling, prohibited the adulteration or misbranding of both foods and drugs, and ensured that drugs were consistent with national formulary standards.
During its 100-year history, food and drug regulation in the United States has evolved in response to a series of significant public health events. For example, the deaths of more than 100 people who had taken a new but untested sulfanilamide elixir led to the 1938 Food, Drug and Cosmetic Act, which established a requirement for premarket safety testing. When thalidomide (a sedative taken by pregnant woman to relieve morning sickness and aid sleep) caused thousands of European babies to be born with birth defects, Congress recognized the need for premarket safety and efficacy testing and passed the 1962 Kefauver-Harris Amendments. These amendments also called for the retroactive examination of all drugs introduced since 1938. Investigation of the nearly 3,500 drugs introduced between 1938 and 1962, as mandated by the Kefauver-Harris Amendments, was among the first of many increases in the scope of the FDA’s regulatory responsibility that were unfunded by Congress. More recently, the Hatch-Waxman Act, enacted in 1984 to ease the entry of generic drugs into the market and respond to concerns about drug pricing, led to an increase in the number of applications the agency had to review.
Dr. Henney commented that while necessary for preserving and advancing public health, this continual expansion of the FDA’s mandate has created financial challenges for the agency in the absence of corresponding funding. By the early 1990s, there was a substantial backlog of New Drug Applications (NDAs), and review times had increased. To quell growing industry frustration with the unpredictability of the FDA review process, as well as to meet the desperate need of AIDS patients for access to new therapies, Congress passed the Prescription Drug User Fee Act of 1992 (PDUFA).2
As noted in Chapter 1, recent public health events, such as the withdrawal of Vioxx and the association between the use of selective serotonin reuptake inhibitors (SSRIs) and increased risk of suicidal ideation in children, have renewed public concern about drug safety and the FDA’s ability to regulate it. Public outcry resulting from these events has led FDA officials and lawmakers to reexamine the agency and the current legislation that governs its role in regulating drug safety. Currently, the 110th Congress is considering the reauthorization of PDUFA, as well as several legislative proposals to improve the U.S. drug safety system.
CURRENT FDA FUNDING3
According to Mr. Thompson, the FDA has been chronically underfunded in carrying out its responsibilities for ensuring the safety of drugs, medical devices, and the nation’s food supply. While the FDA is commonly viewed as the global gold standard for consumer protection, it faces stiff competition for scarce resources and over the past 20 years has been tasked to do far more with its limited resources. As noted in Chapter 1, for example, the Coalition for a Stronger FDA (www.fdacoalition.org), comprising consumer, patient, industry, and nonprofit groups, reports that the FDA regulates products representing roughly 25 percent of all consumer spending, yet it is expected to meet its expanding mandate with only a fraction of the budget of its sister public health agencies—the Centers for Disease Control and Prevention (CDC) and the National Institutes of Health (NIH). In 1986, the FDA’s budget was 97 percent of CDC’s budget; by 1996, this figure was just 39 percent and by 2006, just 28 percent. Likewise, in 1986 and 1996, the FDA’s budget was merely 8 percent of the NIH budget and by 2006, only 5 percent (Figure 2-1). Mr. Thompson noted that not only is the budget gap between the FDA and its sister agencies increasing, but the recent doubling of NIH’s budget is likely to stimulate a flood of new drug discoveries and new development technologies that the agency will not be able to handle, given the difficulty of dealing with its current workload. He expressed concern that the agency’s limited resources could slow the development and availability of new therapies for major diseases.
Dr. Henney argued that the FDA’s challenge in overcoming a weakened drug safety system stems from its lack of the resources required to implement any major changes. She stressed that the FDA has been requesting funding in the form of user fees for postmarket activities since the early 1990s, yet these requests have been removed during budget negotiations; moreover, as specified in the PDUFA I and II legislation, monies derived from user fees were to fund only premarket review activities. Dr. Henney added that the FDA has consistently requested funding from Congress to implement state-of-the-art systems for monitoring the postmarket safety of drugs, biologics, and devices; however, those requests have likewise gone unmet. During authorization of PDUFA III in 2002, $71 million was earmarked to fund drug safety activities at the FDA; however, Congress rescinded much of that money and reprogrammed the remainder elsewhere in the agency. The FDA has experienced difficulty in receiving adequate funding not necessarily because appropriations
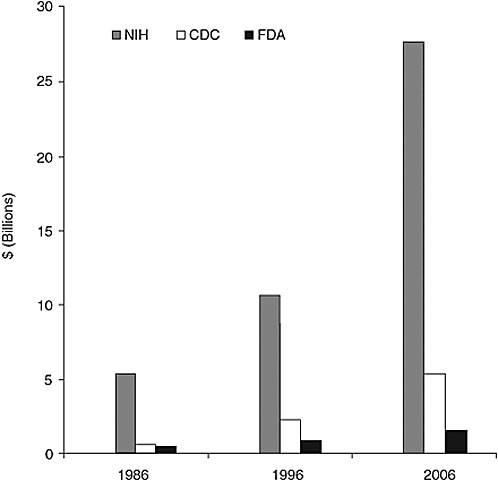
FIGURE 2-1 Comparison of NIH, CDC, and FDA budgets between 1986, 1996, and 2006. In 1986, FDA’s budget was $416.7 million, CDC’s was $429.4 million, and NIH’s was $5.1 billion. In 1996, FDA’s budget was $865 million, CDC’s was $2.2 billion, and NIH’s was $10.2 billion. In 2006, FDA’s budget was $1.5 billion, CDC’s was $5.2 billion, and NIH’s was $27.7 billion.
SOURCE: Coalition for a Stronger FDA, 2007.
committees are unsympathetic to the agency’s drug safety mission, but because the majority of resources are consumed by the agricultural sector. Dr. Henney concluded by expressing her hope that in fiscal year 2008, “both the administration and Congress will be mindful of the incredible needs of the agency.”
INDUSTRY USER FEES
Mary Pendergast, President, Pendergast Consulting, asserted that since the authorization of PDUFA in 1992, Congress has increasingly relied on user fees rather than congressional appropriations to fund the
FDA, a situation that contrasts with the funding provided to CDC and NIH. She explained that since 1992, the FDA has doubled the number of staff performing new drug reviews from approximately 1,300 to 2,600, yet congressional appropriations paid for only 11 of those 1,300 new staff. User fees, originally representing about 30 percent of the FDA budget, now make up more than 50 percent, and PDUFA IV authorization may cause that figure to rise to as high as 80 percent or more. Not only is there active public debate about the FDA’s accepting funding from the industry it regulates, but Dr. Henney asserted that the need for additional scientists to conduct the agency’s drug safety–related activities is “more than obvious.”
As discussed previously, Congress authorized PDUFA as a way to subsidize the FDA and help alleviate the backlog of NDAs. Following PDUFA, the FDA’s congressional appropriations failed to keep pace with user fees. Further, Mr. Thompson remarked that while PDUFA has paid for nearly 1,300 new drug reviewers since 1992, over the last 10 years the FDA has lost some 1,000 staffers from the food, drug, and medical device safety programs not supported by PDUFA fees. Alta Charo, Professor, University of Wisconsin–Madison, and member of the IOM Drug Safety Committee, reiterated that the FDA regulates an extraordinary proportion of the products on the American market in the form of food, drugs, biologics, and medical devices, but operates with a budget that is not commensurate with this broad regulatory authority. Indeed, the committee responsible for the IOM report concluded that the agency’s mission of promoting and protecting the health of the American public warrants more public funding in the form of general appropriations, as opposed to PDUFA funds. The committee further suggested that restrictions on the use of PDUFA funds be reduced to allow FDA management more flexibility in carrying out its mission. Echoing the IOM report, Ms. Pendergast encouraged Congress to increase appropriations for the FDA.
RESOURCE NEEDS
Dr. Henney noted that a late 1990s estimate of the resource requirements for strengthening the FDA’s postmarket review system was in excess of $100 million.4 However, this was an old estimate for a system less robust than that recommended by the IOM. Mr. Thompson commented that although the administration’s 2008 budget request for the agency provides a modest increase for improvements in the right areas—
$11 million for drug safety, $7 million for medical device safety, and $10.6 million for food safety—funding for the agency remains insufficient. He noted that these increases will barely allow the agency to operate at last year’s level, and will do little to make up for the steady loss of staff that the agency has endured for the past decade. Moreover, while the lack of national standards impedes the adoption of the information technology needed to improve the drug safety system, insufficient funding for the necessary purchases and upgrades would remain an insurmountable barrier even if such standards were in place.
As part of its call for a renewed public commitment to the FDA, the Coalition for a Stronger FDA is advocating a total of $175 million in increased appropriations for the agency for 2008 (over the fiscal year 2007 budget and over PDUFA IV increases).5 This figure includes $40 million for drug reviews, $20 million for medical device programs, and $115 million for food safety programs. Among other improvements, the $40 million increase in the drug budget would enhance the agency’s postmarket surveillance capabilities.






