
The Evolutionary Design of Proteins
RAMA RANGANATHAN
Green Center for Systems Biology
University of Texas Southwestern Medical Center
Evolution builds proteins that display structural and functional properties that are beautifully suited for their biological role. They can fold spontaneously under physiological conditions into a compact and well-packed three-dimensional structure, and often display complex functional properties, such as signal transmission, efficient catalysis of chemical reactions, specificity in molecular recognition, and allosteric conformational change. Because these properties require great precision in the positioning and coupling of amino acids, the current view is to regard proteins as finely tuned machines that are exactly arranged for mediating their selected function. However, other aspects of proteins seem less consistent with this view and demand further delineation of the underlying design principles. For example, proteins are well known to be robust to random perturbation; that is, they display tolerance to mutagenesis at many amino acid positions. In addition, they are readily evolvable; that is, they display the capacity for rapid adaptation to changing selection pressures by allowing specific sequence variation at a few sites to profoundly alter function. This curious mixture of robustness at many sites and fragility at a few sites is interesting since it suggests that despite homogeneously good atomic packing, strong heterogeneity exists in the energetic interactions between amino acids that underlie structural stability and function. A major advance in our understanding of proteins would come with the development of methods to systematically map and then mechanistically understand the global architecture of amino acid interactions. In principle, such understanding would (1) provide a new basis for the interpretation of protein sequence and structure,
(2) provide powerful practical rules for the rational engineering of protein structure and function, (3) help predict and better understand the molecular basis for disease-causing mutations, and (4) provide the basis for beginning to understand how proteins are even possible through the random, algorithmic process of mutation and selection that we call evolution.
However, the problem is truly complex; amino acids make unequal and cooperative contributions to protein structure and function, and these contributions are generally not obvious in even high-resolution atomic structures. For example, studies of the interaction between the human growth hormone and its receptor show that the binding interface contains “hot spots” of favorable energetic interactions embedded within an overall environment of neutral interactions (Clarkson and Wells, 1995). Similarly, catalytic specificity in proteases (Hedstrom, 1996), signal transmission within G protein coupled receptors (GPCRs) (Gether, 2000) and the cooperative binding of oxygen molecules in hemoglobin (Perutz et al., 1998), catalysis in the metabolic enzyme dihydrofolate reductase (Benkovic and Hammes-Schiffer, 2003), and antigen recognition by antibody molecules (Midelfort and Wittrup, 2006; Patten et al., 1996) all depend on the concerted action of a specific set of amino acids that are distributed both near and far from the active site. Why are these energetic phenomena not obvious in atomic structures? The main problem is easily stated: we do not “see” energy in protein structures. We might observe an interaction in a crystal structure, but we do not know the net free energy value of that interaction given only the mean atomic positions. Since the native state of a protein represents a fine balance of opposing forces that operate with steep distance dependencies to produce marginally stable structures, complex and nonintuitive arrangements of amino acid interactions are possible.
These observations permit a clear statement of the goals, and that will constitute this lecture. The essence of understanding the evolutionary design of protein structure and function is globally assessing the energetic value of all amino acid interactions. Since the value of interactions is not a simple function of distance, and complex spatial arrangements of interactions between amino acids are possible, we must be open to novel strategies that go beyond structure-based inferences or high-throughput mutagenesis. In this regard we have reported a novel statistical approach (now termed “statistical coupling analysis,” or SCA) for globally estimating amino acid interactions (Lockless and Ranganathan, 1999). Treating evolution as a large-scale experiment in mutation allows this method to make the simple proposition that the energetic coupling of residues in a protein (whether for structural or functional reasons) should force the mutual evolution of those sites. That is, the conserved cooperative interactions between amino acids might be exposed through analysis of the higher order statistics of sequence variation between positions in a large and diverse multiple sequence alignment of a protein family. Application of this method in several different protein families—PDZ (Post-synaptic density 95, Discs Large, Zona Occludens 1) domains (Lockless and Ranganathan, 1999), GPCRs (Suel et al., 2003), serine proteases
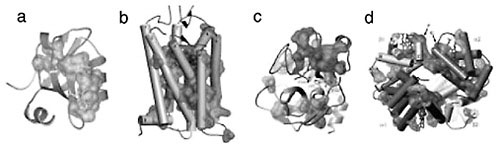
FIGURE 1 Networks of evolutionarily coupled residues in the PDZ (a), GPCR (b), serine protease (c), and hemoglobin (d) protein families. A representative member of each family is shown with a van der Waals surface drawn around the residues that emerge as strongly evolutionarily coupled in the SCA.
(Suel at al., 2003), hemoglobins (Suel et al., 2003), guanine nucleotide binding (G) proteins (Hatley et al., 2003), and the nuclear hormone receptors (Shulman et al., 2004)—suggests two general conclusions (Figure 1): (1) the global pattern of amino acid interactions is sparse, so that a small set of positions mutually co-evolves among a majority that are largely decoupled, and (2) the strongly co-evolving residues are spatially organized into physically connected networks linking distant functional sites in the structure through packing interactions. Importantly, mutagenesis experiments directed by the SCA mapping show strong correlations between the predictions and experimental measurements, implicating the co-evolving networks as hot spots for functional mechanism. Taken together these studies suggest a new model for the architecture of amino acid interactions in natural proteins: most residues interact minimally, acting as if nearly independent or locally coupled, while a few (~10-15 percent) comprise strongly interacting, sparse, and interconnected networks of co-evolving residues that define core aspects of protein function.
The finding that the global pattern of energetic interactions between residues in many protein families is so sparse suggested an interesting and testable hypothesis: proteins may be far simpler in their energetic architecture than we think. Indeed, the SCA suggests the possibility that all the information required for specifying the fold and characteristic function of a protein family may be sufficiently encoded in the matrix of amino acid interactions revealed by the global co-evolution analysis. If so, the proof lies in building artificial members of a protein family using only information extracted by the SCA. We developed a computational method for creation of artificial amino acid sequences according to the statistical rules revealed by the SCA and built large libraries of designed sequences in the laboratory (Russ et al., 2005; Socolich et al., 2005). We find that for the WW (Trp-Trp) domain, a small β-structured protein fold containing
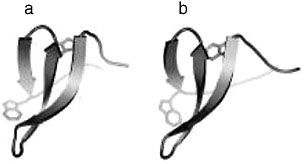
FIGURE 2 A comparison of a 1.45Å crystal structure of the human Pin-1 WW domain (a) and a NMR structure of an artificial WW domain, cc45 (b). The root mean squared deviation of backbone atoms between cc45 and all natural WW domains is within that of natural domains to each other. CC45 shows 35 percent mean identity and 61 percent top-hit identity with natural WW domains.
two conserved tryptophan residues, the computational method produces artificial sequences that in fact efficiently fold into the characteristic WW domain structure (Figure 2). In contrast, randomly designed sequences, or even sequences preserving the conservation pattern of the WW domain but excluding the co-evolution of residues, failed to show native folding. Remarkably, phage display and peptide binding studies indicate that the artificial sequences not only fold but also exhibit binding specificity that quantitatively mirrors that of natural WW domains (Russ et al., 2005). More recently these findings have been extended to the successful design of functional PDZ domains, a roughly 90 amino acid protein interaction module with a mixed α/β fold. These results demonstrate that the information extracted from the SCA is necessary and sufficient to specify the WW and PDZ folds and their characteristic function. The very few numbers we imposed in the computational process in building these artificial sequences suggests that evolution’s rules for building proteins could be vastly less complex than theoretically possible. These results suggest a new reparameterization of proteins guided by the higher order statistics of conservation rather than by traditional principles of primary, secondary, or tertiary structure. Recent work on developing and testing this parameterization will be presented.
To date, our work has been focused on mapping and experimentally verifying the architecture of amino acid interactions implied by the evolutionary analysis—essentially, a mechanism-free description of what is built through the evolutionary process. The results have been unexpected and suggest potential practical avenues for directed mutagenesis and design of proteins. However, this
work primarily has set the stage for two critical further problems that limit our understanding of the evolutionary design of proteins: (1) how does the SCA-predicted architecture of amino acid interactions physically operate in proteins and (2) why is the SCA-predicted architecture a good solution for specifying protein structure and function through evolution? In essence the goal is to ultimately connect the mechanism-free description of statistical interactions between amino acids with a mechanistic model for the physics of these interactions and with an underlying evolutionary theory. It will be interesting to see what experiments can be designed to test the theory that emerges from this work for the dynamics of the evolutionary process.
REFERENCES
Benkovic, S. J., and S. Hammes-Schiffer. 2003. A perspective on enzyme catalysis. Science 301(5637): 1196-1202.
Clackson, T., and J. A. Wells. 1995. A hot spot of binding energy in a hormone-receptor interface. Science 267(5196):383-386.
Gether, U. 2000. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocrine Reviews 21(1):90-113.
Hatley, M. E., S. W. Lockless, S. K. Gibson, A. G. Gilman, and R. Ranganathan. 2003. Allosteric determinants in guanine nucleotide-binding proteins. Proceedings of the National Academy of Sciences 100(suppl. 2):14445-14450.
Hedstrom, L. 1996. Trypsin: A case study in the structural determinants of enzyme specificity. Biological Chemistry 377(7-8):465-470.
Lockless, S. W., and R. Ranganathan. 1999. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286(5438):295-299.
Midelfort, K. S., and K. D. Wittrup. 2006. Context-dependent mutations predominate in an engineered high-affinity single chain antibody fragment. Protein Science 15(2):324-334.
Patten, P. A., N. S. Gray, P. L. Yang, C. B. Marks, G. J. Wedemayer, J. J. Boniface, and P. G. Schutz. 1996. The immunological evolution of catalysis. Science 271:1086-1091.
Perutz, M. F., A. J. Wilkinson, M. Paoli, and G. G. Dodson. 1998. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annual Review of Biophysics and Bimolecular Structure 27:1-34.
Russ, W. P., D. M. Lowery, P. Mishra, M. B. Yaffe, and R. Ranganathan. 2005. Natural-like function in artificial WW domains. Nature 437(7058):579-583.
Shulman, A. I., C. Larson, D. J. Mangelsdorf, and R. Ranganathan. 2004. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell 116(3):417-429.
Socolich, M., S. W. Lockless, W. P. Russ, H. Lee, K. H. Gardner, and R. Ranganatahan. 2005. Evolutionary information for specifying a protein fold. Nature 437(7058):512-518.
Suel, G. M., S. W. Lockless, M. A. Wall, and R. Ranganathan. 2003. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature Structural Biology 10(1):59-69.






