
3
Taking Advantage of New Tools and Techniques
INTRODUCTION
As with virtually every scientific endeavor, clinical effectiveness research can be improved and expedited through innovation. In this case, innovation means the better use of existing tools and techniques as well as the development of entirely new methods and approaches. Understanding these emerging tools and techniques is critical to the discussion of improvements to the clinical effectiveness research paradigm. Better tools and enhanced techniques are fundamental building blocks in redesigning the clinical effectiveness paradigm, and new methods and strategies for evidence development are needed to use these tools to capture and analyze the increasingly complex information and data generated. In turn, better evidence will lead to stronger clinical and policy decisions and set the stage for further research.
Opportunities provided by developments in health information technology are reviewed in Chapter 4. In this chapter we review innovative uses of existing research tools as well as emerging methods and techniques. Part of the reform needed to enhance clinical effectiveness research is a more widespread understanding of different research tools and techniques, including greater clarity about what each can offer the overall research enterprise, both alone and in synergy with other approaches. A further need is broad, substantive support for ongoing development of new approaches and applications of existing tools and techniques that researchers believe may offer more benefits. As noted in Chapter 1, greater attention is needed to understand which approach is best suited for which situation and under what circumstances.
The papers included in this chapter offer observations on improvements needed in the design and interpretation of intervention trials; methods that take better advantage of system-level data; possible improvements in analytic tools, sample size, data quality, organization, and processing; and novel techniques that researchers are beginning to use in conjunction with new information, models, and tools.
Citing models from Duke University, The Society of Thoracic Surgeons (STS), and the Food and Drug Administration’s (FDA’s) Critical Path Clinical Trials Transformation Initiative, Robert M. Califf from Duke University discusses opportunities to improve the efficiency of clinical trials and to reduce their exorbitant costs. Innovations in the structure, strategy, conduct, analysis, and reporting of trials promise to make them less expensive, faster, more inclusive, and more responsive to important questions. Particular attention is needed to identify regulations that improve clinical trial quality and eliminate practices that increase costs without an equal return in value. Finally, establishing “envelopes of creativity” in which innovation is encouraged and supported is essential to maximizing the appropriate use of this methodology.
Confounding is often the biggest issue in effectiveness analyses of large databases. Innovative analytic tools are needed to make the best use of large clinical and administrative databases. Sebastian Schneeweiss from Harvard Medical School observes that instrumental variable analysis is an underused, but promising, approach for effectiveness analyses. Recent developments of note include approaches that exploit the concepts of proxy variables using high-dimensional propensity scores and provider variation in prescribing preference using instrumental variable analysis.
Rejecting any suggestion that “one trial = all trials,” Donald A. Berry from the University of Texas M.D. Anderson Cancer Center makes the case that adaptive and, particularly, Bayesian approaches lend themselves well to synthesizing and combining sources of information, such as meta-analyses, and provide means of modeling and assessing sources of uncertainty appropriately. Therefore, Berry asserts, they are ideally suited for experimental trial design.
Mark S. Roberts of the University of Pittsburgh, representing Archimedes Inc. at the workshop, suggests that physiology-based simulation and predictive models, such as an eponymous model developed at Archimedes, have the potential to augment and enhance knowledge gained from randomized controlled trials (RCTs) and can be used to fill “gaps” that are difficult or impractical to answer using clinical trial methods. Of particular relevance is the potential for these models to perform virtual comparative effectiveness trials.
This chapter concludes with a discussion of the dramatic expansion of information on genetic variation related to common, complex disease and
the potential of these insights to improve clinical care. Teri A. Manolio of the National Human Genome Research Institute reviews recent findings from genomewide association studies that will enable examination of inherited genetic variability at an unprecedented level of resolution. She proposes opportunities to better capture and use these data to understand clinical effectiveness.
INNOVATIVE APPROACHES TO CLINICAL TRIALS
Robert M. Califf, M.D.
Vice Chancellor for Clinical Research
Duke University
As we enter the era in which we hope that “learning health systems” (IOM, 2001) will be the norm, the evolution of randomized controlled trials required to meet the tremendous need for high-quality knowledge about diagnostic and therapeutic interventions has emerged as a critical issue. All too often, discussion about medical evidence gravitates toward a comparison of randomized controlled trials and studies based on observational data, rather than toward a serious examination of ways to improve the operational methods of both approaches. My own experience in assessing the relative merits of RCTs versus observational studies dates back more than 25 years (Califf and Rosati, 1981), and recent discussions on this topic remind me of conversations I had as a medical student in 1977 with Eugene Anson Stead, Jr., M.D., the former chair of the Department of Medicine at Duke University. Dr. Stead founded the Duke Cardiovascular Disease Database, which eventually evolved into the Duke Clinical Research Institute; he is credited with helping change cardiovascular medicine from a discipline largely based on anecdotal observation to one based on clinical evidence. Dr. Stead, who was significantly ahead of his time, introduced us to a device not yet in common use—the computer—and urged us to record outcomes data on all of our patients. Further, he stressed that simply collecting information on acute, hospital-based practice was not sufficient; instead, we should add to this computerized collection throughout our patients’ lives.
I firmly believe that this approach—building human systems that take advantage of the power of modern informatics—is the key to improving both RCTs and observational studies. Within the domain of clinical trials, an informatics-based approach holds promise both for pragmatic trials in broad populations, as well as in proof-of-concept (POC) trials intended to elucidate complex biological effects in small groups of people.
In 1988, our research group published a paper in which we concluded that well-designed and carefully executed observational studies could provide research data that were comparable in quality to those provided by
RCTs (Hlatky et al., 1988). We have learned much since then, a point recently driven home during rounds in the Duke Coronary Care Unit (CCU). Time after time, we were faced with decisions that, had there had been a trial with an inception time for enrollment that coincided with the time point when we needed to make that clinical decision, the trial would likely have provided invaluable information for our CCU deliberations.
While observational studies can provide useful knowledge, they are inadequate for detecting modest differences in effects between treatments (Peto et al., 1995), because without a common inception point and randomization to equally distribute known and unknown confounding factors, the risk of an invalid answer is substantial (DeMets and Califf, 2002a, 2002b). Innovation in clinical trials, in my view, is mostly concerned with performing them in optimal fashion, so that more knowledge is created more efficiently.
How Can We Foster Quality in Clinical Trials?
The most urgently needed innovation in implementing clinical trials is a more intelligent approach to defining and producing quality. Since randomization is such a powerful tool for creating a basis to compare alternatives from a common inception point, we should abandon the assumption that the common critiques of RCTs stem from unalterable rules governing the conduct of such trials. Clinical trials are not required of their nature to be expensive, slow, noninclusive, and irrelevant to measurement of outcomes that matter to patients and medical decision makers. While innovative statistical methods have provided exciting additions to our capabilities, the main source of innovation in trials must be a focus on the fundamental “blocking and tackling” of clinical trials.
A Structural Framework for Clinical Trials
We have published a model, shown in simplified form in Figure 3-1, which integrates quantitative measurements of quality and performance into the development cycle of existing and future therapeutics (Califf et al., 2002). Such a model can serve as a basic approach to the development of reliable knowledge about medical care that is necessary but not sufficient for those wishing to provide the best possible care for their patients. Currently, it takes too long to complete this cycle, but if we had continuous, practice-based registries and the ability to randomize within those registries, we could see in real time which patients were included and excluded from trials. Further, upon completing the study, we could then measure the uptake of the results of the trial in practice. Such an approach provides a
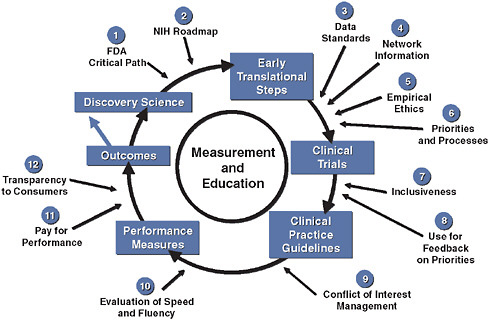
FIGURE 3-1 Innovation in clinical trials: relevance of evidence system.
SOURCE: Copyrighted and published by Project HOPE/Health Affairs as Califf, R. M., R. A. Harrington, L. K. Madre, E. D. Peterson, D. Roth, and K. A. Schulman. 2007. Curbing the cardiovascular disease epidemic: Aligning industry, government, payers, and academics. Health Affairs (Millwood) 26(1):62-74. The published article is archived and available online at www.healthaffairs.org.
system wherein everyone contributes to the registry and the results of trials are fed back into the registry in a rapid cycle.
We have invested considerable efforts in evaluating the details of the system for generating clinical evidence from the perspective of cardiovascular medicine, where there is a long history of applying scientific discoveries to large clinical trials, which in turn inform clinical practice. Figure 3-1 summarizes the complex interplay of relevant factors. If we assume that scientific discoveries are evaluated through proper clinical trials, clinical practice guidelines and performance indicators can be devised and continuous evaluation through registries can measure improved outcomes as the system itself improves. In this context, there are at least a dozen major factors that must be iteratively improved in order for this system to work more efficiently and at lower cost (Califf et al., 2007).
A specific model of this approach has been implemented by STS (Ferguson et al., 2000). Over time, STS has developed a clinical practice
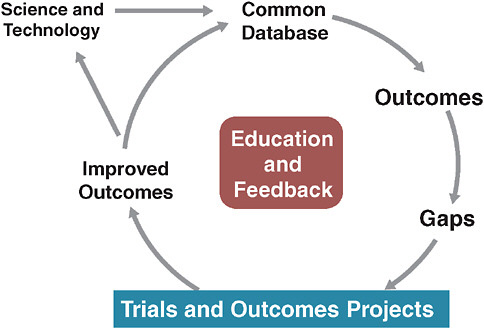
FIGURE 3-2 The Society of Thoracic Surgeons evidence system model.
SOURCE: Derived from Ferguson, T. B., et al. 2000. The STS national database: Current changes and challenges for the new millennium. Committee to establish a national database in cardiothoracic surgery, The Society of Thoracic Surgeons. The Annals of Thoracic Surgery 69(3):680-691.
database that is used for quality reporting, and, increasingly, for continuously analyzing operative issues and techniques (Figure 3-2). The STS model also allows randomized trials to be conducted within the database.
The most significant aspects of this model lie in its constantly evolving, continuously updated information base and its methods of engaging practitioners in this system by providing continuous education and feedback. Many have assumed that we must wait on fully functional electronic health records (EHRs) for such a system to work. However, we need not wait for some putatively ideal EHR to emerge. Current EHRs have serious shortcomings from the perspective of clinical researchers, since these records must be optimized for individual provider–patient transactions. Consequently, they are significantly suboptimal with respect to coded data with common vocabulary—an essential feature for the kind of clinical research enterprise we envision. This deficit severely hobbles researchers seeking to evaluate aggregated patient information in order to draw inferential conclusions about treatment effects or quality of care. While we await the
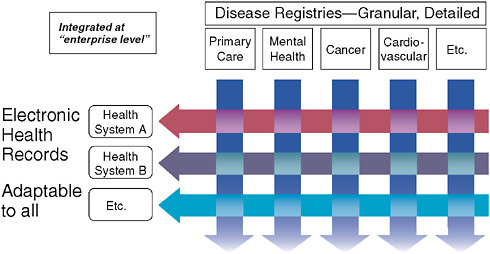
FIGURE 3-3 Fundamental informatics infrastructure—matrix organizational structure.
resolution of issues regarding EHR functionality, the best approach will be to construct a matrix between the EHR and continuous professional-based registries (disease registries) that measure clinical interactions in a much more refined and structured fashion (Figure 3-3). Such a system would allow us to perform five or six times as many trials as can now be done for the same amount of money; even better, such trials would be more relevant to clinical practice. As part of our Clinical and Translational Sciences Award (CTSA) cooperative agreement with the National Institutes of Health (NIH), we are presently working on such a county-wide matrix in Durham County, North Carolina (Michener et al., 2008).
New Strategies for Incorporating Scientific Evidence into Clinical Practice
New efficiencies can be gained through applying innovative informatics-based approaches to the broad pragmatic trials discussed above; however, we also must develop more creative methods of rapidly translating new scientific findings into early human studies. The basis for such POC clinical trials lies in applying an intervention to elucidate whether an intended biological pathway is affected, while simultaneously monitoring for unanticipated effects on unintended biological pathways (“off-target effects”). This process includes acquiring a preliminary indication of dose–response relationships and of whether unintended pathways are also being perturbed (again, while providing a basic understanding of dose–response relationships). POC studies are performed to advance purely scientific understand-
ing or to inform a decision about whether to proceed to the next stage of clinical investigation. We used to limit ourselves by thinking that we could only perform POC studies in one institution at a time, but we now know that we can perform exactly the same trials, with the same standard operating procedures and the same information systems in India and Singapore, as well as in North Carolina. The basis for this broadened capability, as in pragmatic clinical trials, is the building of clinical research networks that enable common protocols, data structures, and sharing of information across institutions. This broadening of scope affords the ability to rethink the scale, both physical and temporal, for POC clinical trials. The wide variation in costs in these different environments also deserves careful consideration by U.S. researchers.
New Approaches to Old Problems: Conducting Pragmatic Clinical Trials
When considering strategies for fostering innovation in clinical trials, several key points must be borne in mind. The most important is that there exists, particularly in the United States, an entrenched notion that each clinical trial, regardless of circumstances or aims, must be done under precisely the same set of rules, usually codified in the form of standard operating procedures (SOPs). Upon reflection, it is patently obvious that this is not (or should not be) the case; further, acting on this false assumption is impairing the overall efficiency of clinical trials. Instead, the conduct of trials should be tailored to the type of question asked by the trial, and to the circumstances of practice and patient enrollment for which the trial will best be able to answer that question. We need to cultivate environments where creative thought about the pragmatic implementation of clinical trials is encouraged and rewarded (“envelopes of innovation”), and given the existing barriers to changes in trial conduct, financial incentives may be required in order to encourage researchers and clinicians to “break the mold” of entrenched attitudes and practices.
What is the definition of a high-quality clinical trial? It is one that provides a reliable answer to the question that the trial intended to answer. Seeking “perfection” in excess of this goal creates enormous costs while at the same time paradoxically reducing the actual quality of the trial by distracting research staff from their primary mission. Obviously, in the context of a trial evaluating a new molecular entity or device for the first time in humans, there are compelling reasons to measure as much as possible about the subjects and their response to the intervention, account for all details, and ensure that the intensity of data collection is at a very high level. Pragmatic clinical trials, however, require focused data collection in large numbers of subjects; they also take place in the clinical setting where their usual medical interactions are occurring, thereby limiting the scope of detail
for the data that can be collected on each subject. To cite a modified Institute of Medicine definition of quality, “high quality with regard to procedural, recording and analytic errors is reached when the conclusion is no different than if all of these elements had been without error” (Davis, 1999).
Efficacy trials are designed to determine whether a technology (a drug, device, biologic, well-defined behavioral intervention, or decision support algorithm) has a beneficial effect in a specific clinical context. Such investigation requires carefully controlled entry criteria and precise protocols for intervention. Comparisons are often made with a placebo or a less relevant comparator (these types of studies are not sufficiently informative for clinical decision making because they do not measure the balance of risk and benefit over a clinically relevant period of time). Efficacy trials—which speak to the fundamental question, “can the treatment work?”—still require a relatively high level of rigor, because they are intended to establish the effect of an intervention on a specific end-point in a carefully selected population.
In contrast, pragmatic clinical trials determine the balance of risk and benefit in “real world” practice; i.e., “Should this intervention be used in practice compared with relevant alternatives?” (Tunis et al., 2003). The population of such a study is allowed to be “messy” in order to simulate the actual conditions of clinical practice; operational procedures for the trial are designed with these decisions in mind. The comparator is pertinent to choices that patients, doctors, and health systems will face, and outcomes typically are death, clinical events, or quality of life. Relative cost is important and the duration of follow-up must be relevant to the duration that will be recommended for the intervention in practice.
When considering pragmatic clinical trials, I would argue we actually do not want professional clinical trialists or outstanding practitioners in the field to dominate our pool of investigators. Rather, we want to incorporate real-world conditions by recruiting typical practitioners who practice the way they usually do, with an element of randomization added to the system to provide, at minimum, an inception time and a decision point from which to begin the comparison. A series of papers recently have been published that present a detailed summary of the principles of pragmatic clinical trials (Armitage et al., 2008; Baigent et al., 2008; Cook et al., 2008; Duley et al., 2008; Eisenstein et al., 2008; Granger et al., 2008; Yusuf et al., 2008).
The Importance of Finding Balance in Assessing Data Quality
If we examine the quality of clinical trials from an evidence-based perspective we might emerge with a very different system (Yusuf, 2004). We know, for example, that an on-site monitor almost never detects fraud, largely because if someone is clever enough to think they can get away with
TABLE 3-1 Taxonomy of Clinical Errors
|
Error Type |
Monitoring Method |
|
Design error |
Peer review, regulatory review, trial committee oversight |
|
Procedural error |
Training and mentoring during site visits; simulation technology |
|
Recording error |
|
|
Random |
Central statistical monitoring; focused site monitoring based on performance metrics |
|
Fraud |
Central statistical monitoring; focused site monitoring based on unusual data patterns |
|
Analytical error |
Peer review, trial committees, independent analysis |
fraud, that person is likely to be adroit at hiding the signs of their deception from inspectors. A better way to detect fraud is through statistical process control, performed from a central location. For example, a common indicator of fraudulent data is that the data appear to be “too perfect.” If data appear ideal in a clinical trial, they are unlikely to be valid: That is not the way that human beings behave. Table 3-1 summarizes monitoring methods to find error in clinical trials that take advantage of a complete perspective on the design, conduct, and analysis of trials.
Recent work sheds light on how to take advantage of natural units of practice (Mazor et al., 2007). It makes sense, for example, to randomize clusters of practices rather than individuals when a policy is being evaluated (versus treating an individual). Several studies that have followed this approach were conducted as embedded experiments within ongoing registries; the capacity to feed information back immediately within the registry resulted in improvements in practice. Although the system is not perfect, there is no question that it makes possible the rapid improvement of practice and allows us to perform trials and answer questions with randomization in that setting.
Disruptive Technologies and Resistance to Change
All this, however, suggests the question: If we are identifying more efficient ways to do clinical trials, why are they not being implemented? The problem is embedded in the issue of disruptive technology—initiating a new way of doing a clinical trial is disruptive to the old way. Such disruption upsets an industry that has become oriented, both financially and philosophically, toward doing things in the accustomed manner. In less highly regulated areas of society, technologies develop in parallel and the “winners” are chosen by the marketplace. Such economic Darwinian
selection causes companies that remain wedded to old methods to go out of business when their market is captured by an innovator who offers a disruptive technology that works better. In most markets, technology and organizational innovation drive cost and quality improvement. Providing protection for innovation that will allow those factors to play out naturally in the context of medical research might lead to improved research practices, thereby generating more high-quality evidence and, eventually, improving outcomes.
In our strictly regulated industry, however, regulators bear the mantle of authority, and the risk that applying new methods will result in lower quality is not easily tolerated. This in turn creates a decided barrier to innovation, given the extraordinarily high stakes. There is a question that is always raised in such discussions: If you do human trials less expensively and more efficiently, can you prove that you are not hurting patient safety?
What effect is all of this having? A major impact is cost: Many recent cardiovascular clinical outcomes trials have cost more than $350 million dollars to perform. In large part this expense reflects procedures and protocols that are essentially unnecessary and unproductive, but required nonetheless according to the prevailing interpretation of regulations governing clinical trials by the pharmaceutical and device companies and the global regulatory community.
Costing out the consequences of the current regulatory regime can yield staggering results. As one small example, a drug already on the market evidenced a side effect that is commonly seen in the disease for which it is prescribed. The manufacturer believed that it was required to ship by overnight express the adverse event report to all 2,000 investigators, with instructions that the investigators review it carefully, classify it, and send it to their respective IRBs for further review and classification. The cost of that exercise for a single event that contributed no new knowledge about the risk and benefit balance of the drug was estimated at $450,000.
Starting a trial in the United States can cost $14,000 per site before the first patient is enrolled simply because of current regulations and procedures governing trial initiation, including IRB evaluation and contracting. A Cooperative Study Group funded by the National Cancer Institute recently published an analysis demonstrating that a minimum of more than 481 discrete processing steps are required for an average Phase II or Phase III cancer protocol to be developed and shepherded through various approval processes (Dilts et al., 2008). This results in a delay of more than 2 years from the time a protocol is developed until patient enrollment can begin, and means that “the steps required to develop and activate a clinical trial may require as much or more time than the actual completion of a trial.”
We must ask: Do the benefits conferred by documenting pre-study evaluation visits or pill counts really outweigh the costs of collecting such data, for example? Do we need 800 different IRBs reviewing protocols for large multicenter trials, or could we enact studies using central IRBs or collaborative agreements among institutional IRBs? Is all the monitoring and safety reporting that we do really necessary (or even helpful)?
Transforming Clinical Trials
All is not dire, however. One promising new initiative is the FDA Critical Path Initiative (public/private partnership [PPP]): the Clinical Trials Transformation Initiative (CTTI), which is intended to map ways to better trials (www.trialstransformation.org). A collaboration among the FDA, industry, academia, patient advocates, and nonacademic clinical researchers, CTTI is designed to conduct empirical studies that will provide evidence to support redesign of the overall framework of clinical trials and to eliminate practices that increase costs but provide no additional value. The explicit mission of CTTI is to identify practices that through adoption will increase the quality and efficiency of clinical trials.
Another model that we could adapt from the business world is the concept of establishing “envelopes of creativity.” In short, we need to create spaces within organizations where people can innovate with a certain degree of creative freedom, and where financial incentives reward this creativity. Pediatric clinical trials offer a good example of this approach. Twenty years ago, clinical trials were rarely undertaken in children; many companies argued that they simply could not be done. Pediatricians led the charge to point out that the end result of such an attitude was a shocking lack of knowledge about the risks and benefits of drugs and devices in children. Congress was persuaded to require pediatric clinical trials and grant patent extensions for companies that performed appropriate trials in children (Benjamin et al., 2006). The result was a significant increase in the number of pediatric trials and a corresponding growth in knowledge about the effects of therapeutics in children (Li et al., 2007).
Conclusions
If we all agree that clinical research must be improved in order to provide society with answers to critical questions about medical technologies and best practices, a significant transformation is needed in the way we conduct the clinical trials that provide us with the most reliable medical evidence. We need not assume that trials must be expensive, slow, noninclusive, and irrelevant to the measurement of important outcomes that matter most to patients and clinicians. Instead, smarter trials will
become an integral part of practice in learning health systems as they are embedded into the information systems that form the basis for clinical practice; over time, these trials will increasingly provide the foundation for integrating modern genomics and molecular medicine into the framework of clinical care.
INNOVATIVE ANALYTIC TOOLS FOR LARGE CLINICAL AND ADMINISTRATIVE DATABASES
Sebastian Schneeweiss, M.D., Sc.D.
Harvard Medical School
BWH DEcIDE Research Center on Comparative Effectiveness Research
Instrumental Variable Analyses for Comparative Effectiveness Research Using Clinical and Administrative Databases
Physicians and insurers need to weigh the effectiveness of new drugs against existing therapeutics in routine care to make decisions about treatment and formularies. Because FDA approval of most new drugs requires demonstrating efficacy and safety against placebo, there is limited interest by manufacturers in conducting such head-to-head trials. Comparative effectiveness research seeks to provide head-to-head comparisons of treatment outcomes in routine care. Because healthcare utilization databases record drug use and selected health outcomes for large populations in a timely way and reflect routine care, they may be the preferred data source for comparative effectiveness research.
Confounding caused by selective prescribing based on indication, severity, and prognosis threatens validity of nonrandomized database studies that often have limited details on clinical information. Several recent developments may bring the field closer to acceptable validity, including approaches that exploit the concepts of proxy variables using high-dimensional propensity scores and exploiting provider variation in prescribing preference using instrumental variable analysis. The paper provides a brief overview of those two approaches and discusses their strengths, weaknesses, and future developments.
Very briefly, what is confounding? Patient factors become confounders (“C” in Figure 3-4) if they are associated with treatment choice and are also independent predictors of the outcome. When researchers are interested in the causal effect of a treatment on an outcome, factors that are independently predicting the study outcome, such as severity of the underlying condition, prognosis, co-morbidity, are at the same time also driving the treatment decision. Once these two conditions are fulfilled, you have a confounding situation and you get biased results. In large-claims database
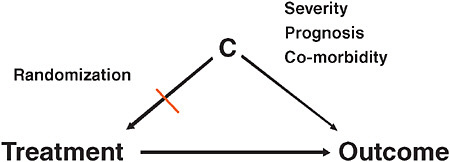
FIGURE 3-4 Explanation of confounding factors in comparative effectiveness research.
analyses, confounding is one of the biggest issues in comparative effectiveness research. Randomization breaks this association between patient factors and treatment assignment. In Figure 3-1, once you break one of the two arms of the tent, then you no longer have confounding.
We have a large continuum of comparative effectiveness research, within which some questions are heavily confounded by design while others are not; the separation is usually by unintended treatment effects and intended treatment effects. An example is in the use of selective Cox-2 inhibitors (coxibs) and cardiac events. In 1999 and 2000 when coxibs were first marketed, nobody was thinking that independent cardiovascular risk factors would influence the decision of whether to treat with the coxibs or nonselective nonsteroidal anti-inflammatory drugs (nsNSAIDs), so there was no association. Consequently there is very little potential for confounding studying unintended cardiovascular outcomes. However, when we studied coxib use and the reduction in gastric toxicity, a heavily marketed advantage of coxibs, risk factors for future gastroinestinal (GI) events drive the decision to use coxibs; consideration of GI symptoms, although often quite subtle and likely not recorded in databases, are nevertheless driving the treatment decision and may therefore cause confounding.
As Figure 3-5 suggests, epidemiologists have a whole toolbox of techniques to control confounding by measured factors (Schneeweiss, 2006). But what about the unmeasured confounders, such as the subtle GI symptoms that are not recorded in claims data, but nevertheless are driving the treatment decision?
We can sample a subpopulation and collect more detailed data there, but what options are there when such a subsampling to measure clinical details is not a possible or practical? One of the strategies is to use instrumental variables. An instrumental variable (IV) is an unconfounded substitute for the actual treatment. In this approach, instead of model-
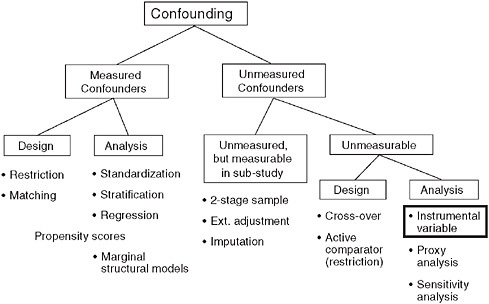
FIGURE 3-5 Dealing with unmeasured confounding factors in claims data analyses.
SOURCE: Schneeweiss, S. 2006. Sensitivity analysis and external adjustment for unmeasured confounders in epidemiologic database studies of therapeutics. Pharmacoepidemiology and Drug Safety 15:291-303. Reprinted with permission from Wiley-Blackwell, Copyright © 2006.
ing treatment and outcome, researchers model the instrument—which is unrelated to patient characteristics and therefore unconfounded—and then rescale the estimate for the correlation between the instrumental variable and the actual treatment.
One of the key assumptions is that the instrumental variable is not associated with either the measured or unmeasured confounders and is not related to the outcome directly other than through the actual treatment. This is necessary for instrumental variables to produce valid results. Consequently, in working with such instruments, researchers have to identify a sort of quasi-random treatment assignment in the real world. For the sake of this paper, two are readily identifiable:
Interruption in Medical Practice
This quasi-random treatment assignment can be caused by sudden and massive interruptions of treatment patterns, for example by regulatory changes. An example might be the FDA aprotinin advisory that reduced the medication’s use by 50 percent—a massive shift. For the same patient candi-
dates for aprotinin, a cardiac surgeon would likely choose a different course of treatment before and after the advisory. A similar example is found in the evolution of the coronary stents; a patient coming for a percutaneous procedure on one day might be treated with a bare metal stent but a year later, after the rapid adoption of drug-eluting stents that same patient might be given a drug-coated stent.
Strong Treatment Preference
Several papers have contributed to our understanding of this valuable instrument for evaluating the comparative effectiveness of therapeutics, which considers such instruments as the distance to specialist, geographic area, physician prescribing preference, and hospital formularies (Brookhart et al., 2006; McClellan et al., 1994; Stukel et al., 2007). A valid preference-based instrument would be the observation of a quasi-random treatment choice mechanism, for example, some hospitals have certain drugs on formulary and others don’t, but patients do not elect to go to one hospital versus another based on whether or not a particular medication is on formulary.
Figure 3-6 presents an example focused on the use of coxibs and nsNSAIDs, with GI complications as the causal relationship, and physician preference to prescribed coxibs versus nonselective NSAIDs (Schneeweiss et al., 2006). This nightmare for everyone writing treatment guidelines might be the dream of an epidemiologist: The same patients get treated differently by different physicians; some physicians always prescribe coxibs and some physicians never prescribe coxibs to patients that need pain therapy (Schneeweiss et al., 2005; Solomon et al., 2003).
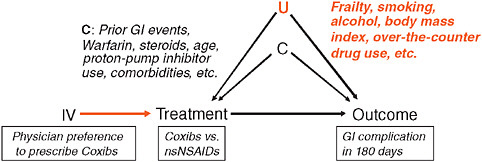
FIGURE 3-6 IV estimation of the association between NSAIDs and GI complication.
SOURCE: Adapted by permission from Macmillan Publishers, Ltd. Clinical Pharmacology & Therapeutics 82:143-156, Copyright © 2007.
Some confounders such as the use of steroids and other medications can be measured with information that we can draw from claims data. However, there will remain unmeasured confounders—for example, body mass index, and the use of over-the-counter drugs. Such information is usually not available in claims data, leading one to ask what happens when one compares the conventional multivariate-adjusted analysis to an instrumental variable based on physician preference. Data not shown here indicate that the risk difference estimates for GI complications for coxibs in a conventional multivariate analysis is around 0, meaning “no association.” What we would expect, of course, is a protective effect. When we did the instrumental variable analysis on coxibs and reduced GI toxicity (not shown), we see a negative risk difference, indicating a protective effect of the coxibs as compared to nsNSAIDs. This is an example where the confounding is strong and the confounding factor is either not measured in claims data or is measured only to a small extent.
Let us consider three core assumptions about instrumental variables (Angrist, 1996). One assumption is that the instrument is related to the actual exposure—otherwise it can’t be an instrument—and is a strong predictor of treatment. The assumption is that physician prescribing preference strongly predicts future choices of treatments. This assumption is empirically testable. In comparison with IV analyses from economics, the strength of the physician prescribing preference IV is greater than most but not all published examples (Rassen, 2008).
A second assumption is that the instrument should not be associated with any measured or unmeasured patient care characteristics. To prove such an assumption—a more difficult exercise than proving the first assumption—one must consider the extent to which one achieves balance in the measured covariates between the two treatment groups. This involves summarizing all of the measurable individual covariants into a summary metric called the Mahalanobis distance that considers the covariance between individual patient factors. In this case the physician preference for a variety of instrument definitions has led to substantial reduction in imbalance among observed patient characteristics (Rassen, 2008). The hope is that when improvement in balance in the measured covariates can be achieved by the instrument, there will be a corresponding improvement in the unmeasured covariates. This is different from the balance achieved by propensity score matching that is limited to the measured patient characteristics and their correlates (Seeger et al., 2005).
A third assumption is that there should be no direct relationship with outcome other than through actual treatment. It can be attempted to empirically test this assumption in the case of the treatment preference instrument through what is colloquially called the “good doc/bad doc” model, which suggests that treatment preference may be correlated with other physician
characteristics that relate to better outcomes. For example, some physicians who generally practice medicine better might have a preference for coxibs versus other NSAIDs. This creates a physician-level correlation and therefore introduces confounding. To test this assumption, other quality of care measures, such as prescribing long-acting benzodiazepines, or problematic tricyclic antidepressant prescribing could be assessed in a study of the effectiveness of antipsychotics. The result was that among general practitioners there was no quality of prescribing and thus a reduced chance of violations of this third assumption (Brookhart et al., 2007).
Another example used regional variation in heart catheterization rates in patients with acute myocardial infarctions as an instrument (Stukel et al., 2007). As seen in Figure 3-7, patients in this study were arranged by quintile of regional cardiac catheterization rates. In the first quintile, 43 percent of patients received a heart catheter; in the highest quintile group, 65 percent received heart catheterization.
One could argue that there shouldn’t be anything different between these populations because patients did not select their residence according to whether their regional cardiac catheterization rate is high. If this argument holds than there are some patients not receiving catheterization who would receive catheterization if they happened to live in another region. Thus there is quasi-random treatment assignment for these patients.
Looking at the effect estimates in Figure 3-7 we find that the protective effect of heart catheterization in patients with acute myocardial infarction
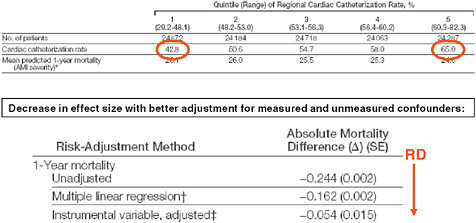
FIGURE 3-7 Regional variation in cardiac catheterization and risk of death.
SOURCE: Journal of the American Medical Association 297(3):278-285. Copyright © 2007 American Medical Association. All rights reserved.
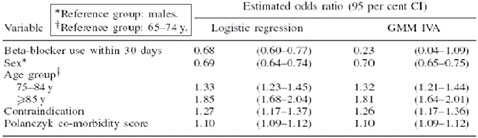
FIGURE 3-8 Time as an instrumental variable.
SOURCE: Johnston, K. M., P. Gustafson, A. R. Levy, and P. Grootendorst. 2008. Use of instrumental variables in the analysis of generalized linear models in the presence of unmeasured confounding with applications to epidemiological research. Statistics in Medicine 27(9):1539-1556. Reproduced with permission of John Wiley & Sons, Ltd.
in an unadjusted analysis of 24 less deaths per year per 100 patients reduces in the multivariate-adjusted regression to only 16 deaths prevented; and with the instrumental variable regression, only 5.
One final example (Figure 3-8) uses time as an instrumental variable. The question here concerns the use of beta-blocker after heart failure hospitalization and 1-year mortality, and whether beta-blocker use is correlated with reduced mortality. After some landmark trials had been published, beta-blocker use in patients with heart failure increased substantially. The investigators defined the binary instrument either before or after this increased use of beta-blocker. As the figure shows, the estimated odds ratio using standard logistic regression was 0.68, whereas the instrumental variable ratio was 0.23—without suggesting which is “right,” we see that there is a considerable difference between the two estimates.
The most frequently mentioned limitation of instrumental variables is that two critical assumptions are not testable but assumptions must be argued using context knowledge. Several empirical tests were suggested to partially evaluate IV assumptions using empirical data, but ultimately we cannot fully prove that assumptions are fully valid. However, readers may be reminded that conventional regression analyses are based on assumptions, including that the model is specified correctly, i.e., that all confounders are measured and included in the model, an assumption that is inherently untestable. The lower statistical efficiency as a consequence of the two-stage estimation process is another limitations. In large databases with tens of thousands of people exposed to drug therapy that is usually a minor issue.
Comparative effectiveness research should routinely explore whether
a valid instrument variable is identifiable in settings where important confounders remain unmeasured. One should search for random components in the treatment choice process, which will sometimes lead to a valid instrument. We have found that the physician prescribing preference instrument is worth considering in many situations of drug effectiveness research. We have further recommended that instrumental variable analyses should be secondary to conventional regression modeling until we better understand the qualities of preference-based instruments and how to best empirically test IV assumptions. We further suggest to perform sensitivity analyses to assess how much violation of IV assumptions may change the primary effect estimate (Brookhart, 2007).
In conclusion, instrumental variable analyses are currently underutilized but very promising approaches for comparative effectiveness research using nonrandomized data. Instrumental variable analyses can lead to substantial improvements, particularly in situations with strong unmeasured confounding. The prospect of reducing residual confounding comes at the price of somewhat untestable assumptions for valid estimation. Plenty of research is ahead, particularly developing better methods to empirically assess the validity of IV assumptions and systematic screens for instrument candidates.
ADAPTIVE AND BAYESIAN APPROACHES TO STUDY DESIGN
Donald A. Berry, Ph.D.
Head, Division of Quantitative Sciences
Professor and Frank T. McGraw Memorial Chair for Cancer Research
Chairman, Department of Biostatistics The University of Texas M.D. Anderson Cancer Center
Modern clinical studies are subject to the most rigorous of scientific standards. In particular, modern research relies heavily on the randomized clinical study that was introduced by A. Bradford Hill in the 1940s (MRC Streptomycin in Tuberculosis Studies Committee, 1948). Applying randomization in a clinical research setting was an enormous advance and it revolutionized the notion of treatment comparisons. For a variety of reasons, mostly coincidence, the RCT became tied to the frequentist approach to statistical inference. In this approach the inferential unit is the study itself, and the conventional measure of inference is the level of statistical significance. In the early days of the RCT the sample size was fixed in advance. Over time, preplanned interim analyses were incorporated to allow for stopping the study early for sufficiently conclusive results.
Randomization will continue to be important in clinical research. However, randomization is difficult and expensive to effect, and there are legiti-
mate ways of learning without randomizing. Moreover, learning can take place at any time during a study and not just when accrual is stopped and sufficient follow-up information obtained. The goal of this chapter is to describe an approach to clinical study design that improves on randomization in two ways. One way is to make RCTs more flexible, with data accrues during the study used to guide the study’s course. The other improvement is incorporating different sources of information to enable better conclusions about comparative effectiveness. Both use the Bayesian approach to statistics (Berry, 1996, 2006). This approach is ideal for both purposes. As regards the first, Bayes rule provides a formalism for updating knowledge with each new piece of information that is obtained, with updates occurring at any time. As regards the second, the Bayesian approach is inherently synthetic. Its principal measures of inference are the probabilities of hypotheses based on the totality of information available at the time.
Précis for Frequentist Statistics
Historically, the standard statistical measures used in clinical research have been frequentist. Frequentist conclusions are tailored to and driven by the study’s design. Probability calculations are restricted to the so-called “sample space,” the set of outcomes possible for the design used. To make these calculations requires the assumption that a particular mechanism that produces the observation. An especially important assumption is that the experimental treatment being evaluated is ineffective, the “null hypothesis.” Other hypotheses can be assumed as well, including that the experimental treatment has a particular specified advantage.
The most familiar frequentist inferential measure is the “p-value,” or observed statistical significance level. This is the probability of observations in the sample space as extreme or more extreme than the results actually observed, calculated assuming the null hypothesis. To make this calculation requires finding the probabilities (under the null hypothesis) of results that are potentially observable. It also requires ordering the possible results of the experiment so that “more extreme results” can be identified to enable adding probabilities over these results.
An important frequentist calculation made in advance of a study is its statistical power. This is the probability of achieving statistical significance in the study (defined as having a p-value of 0.05 or smaller) when the truth is that the experimental treatment has some particular benefit.
In all of the above calculations the design must be completely described in advance for otherwise the probabilities in the sample space and even the sample space itself will be unknown. And the study must be complete, having followed the design as specified in advance. The mathematics are easiest when the sample size is fixed and treatment assignments do not depend on
the interim results. But frequentist measures can be calculated (perhaps only via simulation) for any prospective design, however complicated. One potential stumbling block in a complicated study is identifying an ordering of the study results. There is no natural way of ordering study results in the frequentist approach when the study has a complicated design. For example, there is no good frequentist approach to answer questions such as, “Given the current results of the study, how much credibility should I place in the null hypothesis as opposed to competing hypotheses?” That makes it difficult to alter the course of the study on the basis of those results.
Précis for Bayesian Statistics
There are many publications describing the Bayesian approach—for example, Berry (2006) and Spiegelhlater (2004). I will give a brief description here, highlighting some points of special importance in clinical study design. In the Bayesian approach, anything which is unknown—including hypotheses—has a probability. So the null hypothesis has a probability. And this probability can be calculated at any time: at the end of the study, during the study, and at the beginning of the study. The last of these is called a “prior probability.” Probabilities calculated during or after a study are based on whatever results are available at the time and are called “posterior probabilities.” For example, a Bayesian can always answer the question in the previous paragraph by giving the current (posterior) probability of the null hypothesis.
The Bayesian approach has a characteristic that is very important in designing clinical studies: It enables calculating probabilities of future observations based on previous observations. Frequentists can calculate probabilities of future observations only by assuming particular hypotheses. In the Bayesian approach predictive probabilities do not require assuming a particular hypothesis because these probabilities are averages with respect to the current posterior probabilities of the various hypotheses.
The online learning aspect of the Bayesian approach makes it ideal for building adaptive designs. If a study’s design is developed as the study is being conducted, which is possible in the Bayesian approach, it is impossible to calculate the study’s false-positive rate. This is why I insist on building designs prospectively. It is more work because one must consider many possibilities that will not arise in the actual trial: “What would I want to do if the data after 40 patients are as follows: …?” The various “operating characteristics” of any prospective study design, including its false-positive rate, can be calculated. Except in the simplest of adaptive designs, such calculation will require simulation.
Clinical Studies with Adaptive Designs
Clinical studies, including RCTs, are usually static in the sense that sample size and treatment assignment are fixed in advance. Results observed during the study are not used to guide the study’s course. There are exceptions. One is a two-stage Phase II cancer trial in which stopping is possible after the first stage if the results are either very promising or very discouraging. Also, Phase III and Phase IV trial designs usually prescribe interim analyses for early stopping in case one treatment arm is performing much better than the other. However, these methods are crude and they are limited in the design modifications that are possible. In particular, interim analyses are allowed at only a small number of epochs, limiting ability to adjust course in mid-study. In addition, traditional early stopping criteria in late phase studies are so conservative that few of them stop early in practice.
The simplicity of studies that have static designs makes them appealing inferential tools. But such studies are costly, in both time and resources. Late-phase clinical trials tend to be large. Large clinical trials are expensive, which increases the cost of health care. And large studies use patient resources that might be used more effectively for other investigations. Moreover, large sample size means exposing many patients to a treatment that may be ineffective and perhaps even harmful. Despite being large, static studies too often reach their full accrual goal and prescribed patient followup time only to conclude that the scientific goal was not achieved.
A more flexible approach is to use the information that accrues in a study to modify its subsequent course. Such designs are adaptive in that modifications depend on the interim results. Among the modifications possible are stopping the study early, changing eligibility criteria, expanding accrual (by adding additional clinical sites), extending accrual beyond the study’s original sample size if its conclusion is still not clear, dropping or adding arms (including combinations of other arms) or doses, switching from one clinical phase to another, and shifting focus to subsets of the patient populations (such as responders). Combinations of these are possible. For example, one might learn that an arm performs poorly in one subset of patients and so that arm is dropped within that subset but it continues otherwise. Adaptive designs also include unbalanced randomization (more patients assigned to some of the treatment arms than others based on interim results of the study) where the degree of imbalance depends on the accumulating data. For example, arms that will provide more information or that are performing better than other arms can be weighted more heavily in the randomization. Adaptations are considered in the light of accumulating information concerning the hypotheses in question.
Consider two examples. First is a circumstance that occurs commonly in drug studies. Patient accrual and follow-up end without a clear
conclusion—the results are neither clearly positive nor clearly negative. For example, the statistical significance level for the primary end-point may be slightly larger than the targeted 5 percent. The company has to carry out another study. A flexible approach in the original study would include the possibility of continuing to accrue patients depending on the results available at the time of the targeted end of accrual. (The overall false-positive rate is affected by such analyses but the final significance levels can be adjusted accordingly.) Allowing for the possibility of extending accrual may increase the study’s sample size. A modest increase in average sample size buys a substantial increase in statistical power. This favorable trade-off is because accrual is extended only when the available information indicates that such an extension is worthwhile. Most importantly, the possibility of extending accrual minimizes the chance of having to carry out an additional study when the drug is in fact effective. Moreover, any increase in average sample size can be more than compensated by incorporating frequent interim analyses with the possibility of stopping for futility (that is, if the results on the experimental agent are not sufficiently promising).
A more extreme example of flexibility has the explicit goal of treating patients in the study as effectively as possible, while learning rapidly about relative therapeutic benefits. Patients are assigned with higher probabilities to therapies that are performing better. Such designs are attractive to patients and so can lead to increased participation in clinical studies. And they lead to rapid learning about better performing therapies. Inferior treatments are dropped from consideration early (Giles et al., 2003). Logistics are more complicated because study databases must be updated as soon as results become available; such updating includes information about early end-points that may be related to the primary long-term end-points.
Adaptations are not limited to the data accumulating in the study in question. Information that is reported from other studies also may be used in affecting a study’s course.
Using Multifarious Sources of Information
The Bayesian approach is inherently synthetic. Inferences use all available sources of information. Appropriately combining these sources is seldom easy. Populations may be different. Protocols may be different. Some sources may be clinical trials while others are databases accumulated in clinical practice.
Because the Bayesian approach is tailored to combining information, it is increasingly used in meta-analyses (Stangl, 2000). But it can be used in much more complicated settings as well. One of the most complicated is the following. Breast cancer mortality in the United States started to drop in about 1990, decreasing by about 24 percent over the decade 1990–2000.
Possible explanations included mammographic screening and adjuvant treatment with tamoxifen and chemotherapy. The National Cancer Institute funded seven groups to sort out the issue, with the goal of proportionally attributing the decrease to these explanations (Berry et al., 2005).
One of the seven groups took a simulation-based Bayesian approach (Berry et al., 2006). We used relevant empirical information from 1975 to 2000, including the use of screening mammography (schedules such as annual, biennial, haphazard) by the woman’s age and year, the characteristics of tumors detected by screening (and which screen) and symptomatically (including interval cancers), the use of tamoxifen by disease stage and the woman’s age (and the tumor’s hormone-receptor status), the use of polychemotherapy by disease stage and age, and the survival benefits of tamoxifen and chemotherapy by disease stage, age, and hormone-receptor status. We did not have longitudinal information on any set of women and so we had to piece together the effects of the various factors.
As in Bayesian modeling more generally, the important unknown parameters (benefits of treatment, survival after breast cancer depending on method of detection, background incidence of cancer [no screening] over time) had prior probability distributions. For example, for the survival benefit of tamoxifen for women with hormone-receptor positive tumors we based the prior distribution on the Oxford Overview of randomized trials, but with much greater standard deviation than that from the Overview to account for the possibility that tamoxifen used in clinical practice might not have the same benefit as in clinical trials. We generated many thousands of cohorts of 2 million U.S. women having the age distribution of U.S. women in 1975. We accounted for emigration and immigration. For each simulation we selected a particular value from each of the various prior distributions. For example, for one cohort we might have chosen a 20 percent reduction in the risk of breast cancer death when using tamoxifen. We assigned non-breast-cancer survival times to each woman consistent with the overall survival pattern of the actual U.S. population. Women in each simulation got breast cancer with probabilities according to their ages and their use of screening, again consistent with the actual U.S. population. Their cancers had characteristics depending on age and method of detection. Their treatment depended on their tumors’ characteristics and was consistent with the mores of the day. We generated breast cancer survival ages for women who were diagnosed with the disease, and these women were recorded as dying of breast cancer if these ages were younger than their non-breast-cancer survival.
For each simulation we tabulated over 1975–2000 the incidence of breast cancer by stage and breast cancer mortality. If these matched the actual U.S. population statistics sufficiently well then we “accepted” the values of the parameters for that simulation into the posterior distribution
of the parameters. Most simulations did not match actual mortality. But some did. We simulated enough cohorts to form reasonable conclusions about the posterior distributions.
One set of conclusions in this example was the relative contributions of screening and treatment to the observed decrease in mortality. Another was that despite having access to the various sources of data, our conclusions about the relative contributions of screening and treatment were uncertain. The Bayesian approach allowed for quantifying this uncertainty. The six non-Bayesian models provided point estimates of the relative contributions. Interestingly, these point estimates were consistent with the uncertainty concluded by the Bayesian model.
Still another conclusion from the Bayesian model was that the benefits of tamoxifen and chemotherapy in clinical practice are similar to the benefits seen in the clinical trials. Again, there is some uncertainty in this statement. Although the means of the posterior distributions of these parameters were very similar to the means of the corresponding prior distributions, the posterior standard deviations were not much smaller than the prior distributions.
Conclusion
Statistical philosophy and methodology has contributed in important ways to medical research. The standard approaches are rigorous and not very flexible. Such a tack has been critical to establishing medicine as a science. But having achieved a high plateau, we must move even higher. In this chapter I have suggested some ways that medical research can be more flexible and yet maintain scientific rigor. Bayesian thinking and methodology can help in synthesizing information from various sources and in building more efficient designs. Efficiencies include smaller sample sizes, usually, but also greater accuracy in comparing treatment effectiveness.
SIMULATION AND PREDICTIVE MODELING
Mark S. Roberts, M.D., M.P.P.,
University of Pittsburgh
David M. Eddy, M.D., Ph.D.,
Archimedes, Inc.
Randomized clinical trials have substantial advantages in isolating and testing the effect of an intervention. However, RCTs have weaknesses and limitations, including problems with generalizability, duration, and costs. Physiology-based models, such as the Archimedes model, have the potential to augment and enhance knowledge gained from clinical trials and can be
used to fill in “gaps” that are difficult or impractical to answer using clinical trial methods.
Physiology-based models are mechanistic in nature and model disease processes at a biological level rather than through statistical relationships between observed data and outcomes. When properly constructed, they replicate the results of the studies used to build them, not only in terms of outcomes but also in terms of the changes in biomarkers and clinical findings as well. A unique characteristic of a properly constructed physiology-based model is its ability to predict the results of studies and trials that have not been used in the model’s construction, a process that provides very strong validation of its predictions.
This paper will describe the Archimedes model as an example of a physiology-based model and will propose uses for such models. The methods for representing and calibrating the mechanistic processes will be described, and comparisons of simulated trials to actual clinical trials as a method of validation will be presented. Multiple uses of the Archimedes model to enhance and extend existing clinical trials as well as to conduct virtual comparative effectiveness trials also will be discussed.
Strengths and Weakness of Randomized Controlled Trials
The main strength of randomized controlled trials is that the random assignment to treatment and control group renders those groups equivalent and eliminates bias by indication, resulting in intervention and control groups that are balanced in known and unknown parameters. At the same time, strictly controlled protocols isolate the specific effect of the intervention.
The weaknesses of RCTs are well known. They often represent a narrow spectrum of disease, are conducted in specialized, highly controlled environments, and are expensive. Patients and physicians must agree to participate, which produces a selection bias that limits generalizability to other populations. They often require a large number of patients and follow-up times so long that the trial results might be eclipsed by the pace of technologic change. This is true, for example, in HIV disease, in which antiretroviral resistance patterns are rapidly and constantly changing, and the number of HIV drugs is rapidly expanding. Finally, RCTs usually represent efficacy, not effectiveness, as they are typically conducted in tightly controlled settings in which care processes have high levels of compliance and protocol adherence.
Physiology-Based Models
The use of physiology-based or mechanistic models as an adjunct or alternative to RCTs has been increasing in several different fields. Although
only recently used in medicine, there are some interesting examples of this in sepsis (Day et al., 2006; Reynolds et al., 2006; Vodovotz et al., 2004), in critical care and injury (Clermont et al., 2004b; Saka et al., 2007), in the acquisition of antiretroviral resistance in HIV disease (Braithwaite et al., 2006, 2008), and in the Archimedes model, which currently includes cardiovascular and metabolic diseases (Eddy and Schlessinger, 2003a; Heikes et al., 2007; Sherwin et al., 2004).
Physiology-based models seek to represent the underlying biology of the disease. They are continuous in time and generally model the physiological processes that create the data observed in the world: They do not simply model the relationship between observed variables and outcomes statistically. Physiology-based models can represent many different levels of detail, from physiologic variables and biomarkers that create disease through anatomy, symptoms, behaviors, all the way up through interactions with health systems, utilization, and costs.
The Archimedes model is designed to represent actual biological relationships and is best illustrated visually in a similar manner to how these relationships are presented in a standard textbook of physiology, with physiological parameters and their relationships described with influence-diagrams at multiple levels of detail from whole organ relationships to processes that occur within organs to those within cells, etc. Similarly, every virtual individual in the Archimedes model has a virtual heart with four virtual chambers, a virtual circulatory system that has a virtual blood pressure and responds to virtual changes in cardiovascular dynamics. The virtual individual has a virtual liver that produces virtual glucose, a virtual gut that absorbs virtual nutrients, a virtual pancreas with virtual beta cells that make virtual insulin, and virtual muscle mass and virtual fat cell mass that utilizes glucose as a function of the amount of virtual insulin available.
Figure 3-9 shows a small portion of the model, but illustrates the types of variables and relationships that are in the Archimedes model. The figure resembles the “bubble diagrams” from physiology texts, and in this particular example, represents some of the factors that affect diabetes and other metabolic conditions. In the figure, every oval represents a characteristic, biological parameter, condition, test, intervention, symptom, or other type of clinically important variable. Some of the relationships are trivial and obvious as, for example, is the relationship between height and weight that defines the body mass index (BMI) with a simple functional form. Most of the functions are substantially more complicated and are typically represented as differential equations that relate the instantaneous change in a particular physiological parameter to the level and change of many other variables. The equations that are contained in the Archimedes model relate the various physiological variables to each other and to specific outcomes, such as the development of diabetes and heart disease. The functional
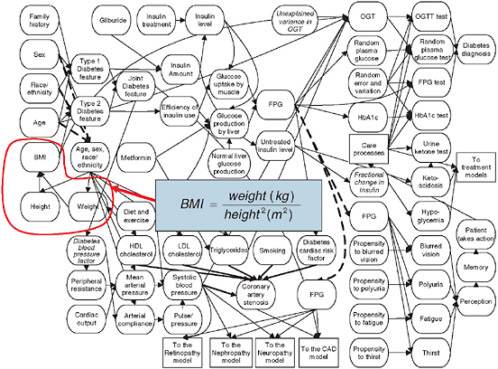
FIGURE 3-9 Physiological factors affecting development of diabetes. BMI is shown as one such variable, composed of the components height and weight through the indicated equation.
SOURCE: Copyright © 2003 American Diabetes Association. From Diabetes Care, Vol. 26, 2003; 3093-3101. Modified with permission from the American Diabetes Association.
form of the equations and the coefficients on the terms of the equations are derived from and calibrated with data from a wide variety of empirical sources, ranging from studies of basic biology to large longitudinal trials and datasets. A more complete description of the Archimedes model and its development is available elsewhere (Eddy and Schlessinger, 2003a; Schlessinger and Eddy, 2002).
Validation of a Physiology-Based Model
One of the most important steps in the building and use of a model is validation. Confidence in a model’s predictions is necessary if models are to be used for clinical and health policy decisions. In general, model validation starts with demonstrating that the model can replicate the results of the trials and studies that were used to develop and calibrate the model. This is called a “dependent” validation. This method of validation is used in both biological and statistical models. However, perhaps the most appropriate “gold standard” of validation is the ability to replicate the results of multiple actual clinical trials that have not been used to build or modify the model. This is called an “independent” validation. A clinical trial enrolls real people, administers real treatments (usually by randomizing them to specific therapies), and records real outcomes a specified time later. The Archimedes model can replicate that process by enrolling virtual people with the exact characteristics of their counterparts in real clinical trials and randomly assign them to virtual treatments that represent the real treatments used in the trial, record virtual outcomes using the same definitions and methods used in the trials, and then compare the results of the virtual trial to those of the real trial. Data available from separate Phase I or Phase II trials can be used to estimate the effects of the intervention on the relevant biomarkers. The Archimedes model has been validated by successfully replicating more than 50 major clinical trials. About half of these validations have been independent.
An example of a dependent validation is provided in Figure 3-10, which compares the actual results from the UK Prospective Diabetes Study (UKPDS) to the simulated results calculated by replicating the trial in Archimedes. Although technically a dependent validation, it is important to note that the models results shown in Figure 3-10 were not “fitted” to the results of the trial. Rather, data from the trial were used to fit only two equations: the rate of progression of insulin resistance in untreated diabetes and the effect of insulin resistance on progression of plaque in coronary arteries. Simulation of the trial involved scores of other equations that were not touched by any data from the trial. Thus even though dependent, this validation tests large parts of the model.
Prospective and independent validations also have been conducted.
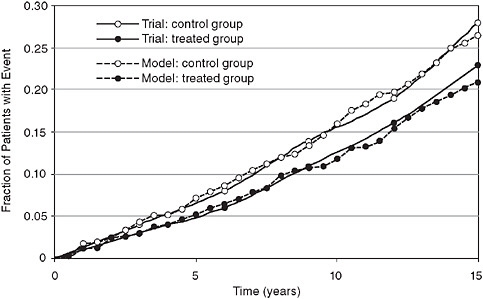
FIGURE 3-10 Retrospective (dependent) validation: Simulated UKPDS trial comparing real trial results (fatal and nonfatal myocardial infarction) to a simulated version of the trial using the Archimedes model.
SOURCE: Copyright © 2003 American Diabetes Association. From Diabetes Care, Vol. 26, 2003; 3102-3110. Modified with permission from the American Diabetes Association.
Figure 3-11 shows the results of a validation that was both prospective and independent. It predicted the results of the Collaborative Atorvastatin Diabetes Study (CARDS), which tested the ability of a lipid-lowering medication to reduce cardiovascular events in patients with diabetes. The figure shows the actual trial result for both the intervention and control arm (solid lines) and the predictions of the Archimedes model (dotted lines). In this validation, the model’s results were sent in sealed envelopes to the ADA and the study investigators prior to the release of the study’s results.
The results for 18 clinical trials have been published. Figure 3-12 compares the results of 74 simulated trials in diabetes, lipid control, and cardiovascular disease, and graphs the actual relative risk found from a trial and the results calculated by the Archimedes model. Because the ability to replicate the results from each arm is considered a validation of the model, this graph represents many more validations than the simple number of clinical trials. The correlation coefficient of the actual and predicted results is r = 0.99.
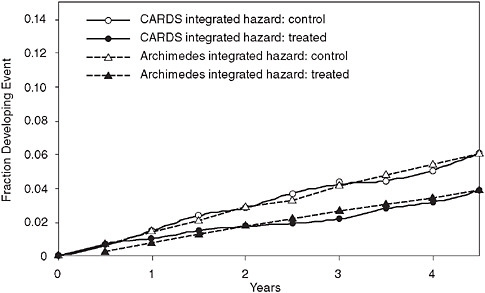
FIGURE 3-11 Prospective and independent validation of the CARDS trial comparing real trial results to results predicted by the Archimedes model.
SOURCE: Derived from Mount Hood Modeling Group. 2007. Computer modeling of diabetes and its complications: A report on the fourth mount hood challenge meeting. Diabetes Care 30(6):1638-1646. Modified with permission from the American Diabetes Association.
Applications of Physiology-Based Models
There are several ways that physiology-based prediction models can be used to enhance clinical trials. One is to help identify and set priorities for new trials. Another is to facilitate the design of new trials. For example, as the validations described above have shown the Archimedes model can be used to estimate the rates of outcomes in control groups and the expected magnitude of the effects of treatments. This information can then be used to help calculate sample sizes, and the durations required to detect outcomes with specified powers. Another use of physiology-based models is to extend clinical trials to estimate long-term outcomes. If a model has successfully calculated the outcomes in the trial of interest over the duration of the trial, and if it has successfully calculated the important biomarkers and clinical outcomes in a variety of other trials that involve similar populations and interventions, then there is good reason to believe its projections for the outcomes of trial over a longer follow-up period will be accurate. At the least, such a trial-validated application is the best available method for estimating longer term outcomes. Related roles of well-validated physiology-
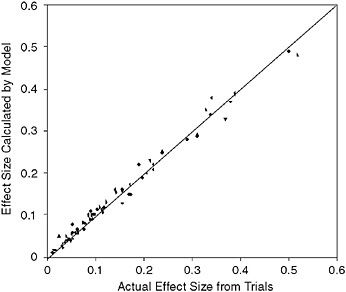
FIGURE 3-12 Comparison of Archimedes model and multiple trials. The x-axis represents the size of the effect measured in the actual trial; the y-axis is the size of the effect in the simulated version of the trial in Archimedes.
SOURCE: Copyright © 2003 American Diabetes Association. From Diabetes Care, Vol. 26, 2003; 3102-3110. Modified with permission from the American Diabetes Association. Modified from Eddy and Schlessinger, 2003a.
based models are to extend a trial’s results to other outcomes that were not examined in the original trial, such as logistic or economic outcomes, and to examine the results for subpopulations.
Physiology-based models can also be used to customize the results of atrial to different settings. For example, a model that has been demonstrated to be accurate in predicting the results of the original trial and related trials can be used to address such issues as settings that have different levels of performance and compliance, and settings that have different background protocols and/or cost structures. For example, a common complaint of clinical trials is that they represent efficacy, the effect of a medication or intervention in tightly controlled, highly specialized environments. However, the effectiveness of these therapies in real-world conditions may be quite different, because of different levels of adherence to the intervention or differences in the quality of baseline care. The model also can study variations in the background rates of healthcare practices seen in different settings. For example, if we are testing a medication for decreasing cardiovascular risk in diabetic patients, but happen to be con-
cerned about a setting in which patients seen in emergency rooms have a very small chance of being treated with thrombolytic, the overall effect on cardiovascular outcomes will be different than would be seen in a setting in which the use of thrombolytics is very high. These types of processes can be included large-scale physiologic-based models but are virtually impossible to incorporate in regression based and Markov models.
Physiology-based models also can be used for analyzing the comparative effectiveness of different treatments for a condition. Suppose there are trials of Medication A versus placebo and of Medication B versus placebo but no trials directly comparing Medication A versus Medication B. Rather than conduct a new trial that compares A versus B, which could be extremely expensive and take years (by which time new medications will invariably have been introduced), physiology-based models that have successfully predicted the two original trials can provide the best currently available estimate of what a real trial of A versus B would be likely to show. This information can then be used to understand the potential value of a new trial of A versus B, to plan a new trial if it is deemed to be desirable, and to recommend what practices should be followed while waiting for the trials results.
The development and calibration of physiology-based models require good data for each of the elements it includes. A model like Archimedes would not have been possible without large-scale cross-sectional and longitudinal datasets such as National Health and Nutrition Examiniation Survey, Framingham, and Atherosclerosis Risk in Communities. A model like Archimedes also rests on clinical trials for understanding the natural progression of diseases and the effects of treatments, for both dependent and independent validations. Data for physiology-based models are most useful if they contain data on demographics (e.g., age, gender, ethnicity); past medical history, family history, physical findings; biomarkers; signs and symptoms; and outcomes. The volume and quality of data of these types can be expected to increase as the use of electronic medical records spreads.
The key to all of these applications is that if a model is to be used to predict, plan, extend, or help fill the gaps between clinical trials, it must prove its ability to reproduce and predict the results of many real clinical trials, using only data available at the start of the trial, and not using any results from the trial to build or modify the model to fit the results of new trials. It is very easy to build models that fit the results of any particular trial, using regression models, Markov models, or other non-physiology-based approaches. It also is easy to build simple models that fit data from multiple disparate sources if each of the sources addresses a different part of the model (e.g., one study of incidence, another of progression, a third of the effect of a treatment on one outcome, and a fourth of the effect of a
treatment on a different outcome). This type of validation by itself provides little evidence about the model’s ability to predict the results of a new trial. For the latter problem, which is our main interest in this paper, it is important that there be multiple validations, involving overlapping populations, treatments, and outcomes, and that the model accurately predicts the results of all of the trials without using the results of any of them.
In this paper we have used the Archimedes model to illustrate these types of validations and the types of applications to which such a model can be put. However, it is important to note that over the past few years some other physiology-based models also have succeeded in predicting the results of some trials. For example, a physiology-based model of HIV resistance predicted the actual resistance rates seen in two independent trials not used to develop the model (Braithwaite et al., 2006). Similarly, physiology-based models for sepsis have been able to prospectively predict outcomes and cytokine patterns (in animals) after acute injury by applying large systems of differential equations that relate the insult to the cytokine production (Clermont et al., 2004a; Reynolds et al., 2006; Vodovotz et al., 2004).
In conclusion, the strengths and limitations of clinical trials are well known. Physiology-based models have substantial promise to, and a growing track record of, addressing many of these limitations. If carefully built and rigorously validated they can be used to enhance and extend the knowledge gained from trials.
EMERGING GENOMIC INFORMATION
Teri A. Manolio, M.D., Ph.D.
U.S. Department of Health and Human Services
National Institutes of Health
National Human Genome Research Institute
The recent advent of high-density, cost-effective, genomewide genotyping technologies as led to a virtual explosion of information on genetic variants related to common, complex diseases (Pearson and Manolio, 2008). In just the past 3 years, over 100 genetic variants associated with nearly 40 complex diseases and traits have been reliably identified and replicated using this revolutionary technology. Several of these findings have sufficient supporting evidence for functional significance or biologic plausibility, and many are sufficiently common that they provide real potential for translation into diagnostic, preventive, or therapeutic interventions. In this new era of genomic discovery, one of the most pressing questions for clinical effectiveness research is thus: What is needed to facilitate the reliable and timely introduction of emerging genetic information into research and clinical databases?
Genomewide Association Studies
The identification and mapping of the most common form of genetic variation, the single nucleotide polymorphism (SNP), has permitted the development of cost-effective genotyping platforms that utilize the patterns of association, or co-inheritance, among SNPs to assay the majority of common variants in the human population (Frazer et al., 2007; A haplotype map of the human genome, 2005; The International HapMap Project, 2003). Upward of 80–90 percent of common variants (those present at an allele frequency of 5 percent or more) can now be assayed by typing 500,000–1,000,000 carefully selected SNPs (Manolio et al., 2007). This allows a systematic approach to association testing that frees genomic investigation from dependence on what is as yet an imperfect understanding of genomic function, or on strongly supported prior hypotheses (Chanock et al., 2007; Frazer et al., 2007).
Success to Date of Genomewide Association Studies
The genomewide association (GWA) approach has been enormously successful in identifying genetic variants related to complex diseases, or diseases likely influenced by multiple genes and environmental factors. The first notable success of this method came in March 2005, with the identification of a variant in the gene for complement factor H (CFH) associated with age-related macular degeneration (Klein et al., 2005). This strong and highly significant relationship was simultaneously reported using two other study designs, and subsequently replicated many times (Edwards et al., 2005; Haines et al., 2005; Magnusson et al., 2006; Sepp et al., 2006; Zareparsi et al., 2005). Two additional GWA studies were published within that year, of Parkinson’s disease and obesity (Herbert et al., 2006; Maraganore et al., 2005), but efforts at replicating these findings have been inconsistent (Hall et al., 2006; Lyon et al., 2007; Myers, 2006). Later in 2006, strong, robust associations with electrocardiographic QT interval prolongation (Arking et al., 2006), neovascular macular degeneration (Dewan et al., 2006), and inflammatory bowel disease (Duerr et al., 2006) were identified and have since been the subjects of a substantial body of follow-up research to determine gene function and population impact.
This pace of genomic discovery increased dramatically in 2007, following the increased availability of high-density genotyping platforms and experience in interpreting the results. Simultaneous publication of coordinated efforts in prostate cancer (Gudmundsson et al., 2007; Yeager et al., 2007), diabetes (Saxena et al., 2007; Scott et al., 2007; Zeggini et al., 2007), and breast cancer (Easton et al., 2007; Hunter et al., 2007; Stacey
et al., 2007) demonstrated the power and value of collaborative projects involving multiple investigative efforts, often involving tens of thousands of study subjects. These were soon followed by publication of the landmark Wellcome Trust Case Control Consortium of 2,000 cases each of 7 common diseases and 3,000 shared controls (Wellcome Trust Case Control Consortium, 2007). Rapid progress has continued into 2008 with investigation of a wide variety of diseases and traits, though not all have produced definitive results (Table 3-2). Indeed, as Hunter and Kraft have noted, “There have been few, if any, similar bursts of discovery in the history of medical research” (Hunter and Kraft, 2007).
Recombination Rate
Unique aspects of the GWA methodology that have made these discoveries possible include its potential for examining inherited genetic variability at an unprecedented level of resolution. GWA studies allow the investigator to narrow an association region to a 5–10 kilobase length of DNA, in contrast to the 5–10 megabases usually detected in familial linkage studies. Because GWA regions typically contain only a few genes, rather than the dozens or hundreds implicated in linkage regions, potentially causative variants can be examined much more rapidly and in greater depth. As noted above, systematic interrogation of the entire genome frees the investigator from reliance on inaccurate prior hypotheses based on incomplete understanding of genome structure and function. The critical importance of this is illustrated by the fact that many of the associations identified to date, such as complement factor H in macular degeneration (Klein et al., 2005) and TCF7L2 in Type 2 diabetes (Grant et al., 2006; Sladek et al., 2007) have not been with genes previously suspected of being related to the disease. Some, such as the strong associations of prostate cancer with SNPs in the 8q24 region (Scott et al., 2007), and Crohn’s disease with the 5p13 region (Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls, 2007), have been in genomic regions containing no known genes at all. And because current genotyping assays capture the vast majority of human variation genomewide, rather than being focused on particular regions or pathways, once a GWA scan is completed it can be applied to any condition or trait measured in that same individual and consistent with his or her informed consent.
The potential for harnessing these data to examine additional traits has been amply demonstrated in GWA studies of anthropometric traits (such as obesity and height) and laboratory measures (such as serum urate and lipoproteins) performed in cohorts with a primary focus on diabetes
TABLE 3-2 Diseases and Traits Studied Using Genomewide Association Testing Assaying 100,000 Variants or More, March 2005–March 2008
or hypertension (Frayling et al., 2007; Wallace et al., 2008; Weedon et al., 2007; Willer et al., 2008). In addition, application of GWA genotyping to long-standing, extensively characterized cohorts such as the Framingham Heart Study and Women’s Health Study (Cupples et al., 2007; Ridker et al., 2008) opens the door to investigation of the genetics of every disease and trait measured in these extensive studies and consistent with participants’ informed consent, adding substantially to their research value both now and for the future.
Challenges of GWA Studies
Against the context of this remarkable flow of findings, however, lies a fundamental challenge of GWA studies: With hundreds of thousands of comparisons performed per study, the potential for spurious associations is unprecedented (Hunter and Kraft, 2007). This was widely recognized as a major shortcoming of candidate gene association studies, in which small sample sizes, publication bias, and the play of chance led to a rash of irreproducible results early on (Colhoun et al., 2003; Ioannidis et al., 2001). The problem was illustrated by Hirschhorn et al. in a seminal paper in 2002, who demonstrated that of 600 genetic associations reviewed, only 6 could be reliably reproduced (Hirschhorn et al., 2002). A variety of statistical approaches has been proposed for dealing with this problem in GWA studies, including the use of a standard Bonferroni correction, by dividing the conventional p-value (typically 0.05) by the number of tests performed (often 106 or more) (Yang et al., 2005). Other approaches include calculation of the false discovery rate or the false-positive report probability to estimate the proportion of significant associations that are actually false positives (Pearson and Manolio, 2008). But the approach most widely accepted is replication of findings (Todd, 2006), often in a staged design expanding from an initial set of 500 or 1,000 cases and similar number of matched controls to studies involving as many as 40,000 or 50,000 participants (Chanock et al., 2007; Hoover, 2007). These large numbers are necessitated by the very stringent p-values demanded by the hundreds of thousands of comparisons performed, and by the relatively modest effect sizes of the variants detected, typically carrying odds ratios of 1.5 or less (Pearson and Manolio, 2008). Such numbers have generally been achieved by combining many smaller studies (Easton et al., 2007; Frayling, 2007), but the potential for conducting this research in large healthcare systems involving hundreds of thousands or millions of participants should not be overlooked.
Use of GWA Information in Research and Clinical Databases
One way of using this emerging genomic technology in research and clinical databases is to perform GWA genotyping in patients with comprehensive (and typically, electronic) medical records and suitable consent to investigate a wide variety of past and current diagnoses or traits. Record linkage may also permit subsequent follow-up for development and progression of new clinical diagnoses or characteristics. Such studies are designed primarily for genomic discovery, to identify additional variants, genes, or regions associated with disease, which then require extensive additional investigation to identify causal variants, biologic mechanisms, and potential
interventions. This approach is being used in large-scale biorepositories such as those organized by Kaiser Permanente (Research, 2008) and the Children’s Hospital of Philadelphia in the United States (Philadelphia, 2008), deCODE Genetics in Iceland (Gulcher and Stefansson, 1998), and the UK Biobank in Britain (Palmer, 2007). GWA genotyping is also being applied to a more limited degree (that is, only to subsets of participants selected for presence or absence of disease in case control studies) in a number of biorepositories with electronic health records (EHRs), such as those participating in the National Human Genome Research Institute’s eMERGE network (The eMERGE Network, 2008). Substantial efforts will be needed to examine the reliability and standardization of phenotypic measures derived from EHRs for genomic research, as well as the adequacy of participants’ consent for the extensive investigation and widespread data sharing common in these studies.
A simpler and more immediate way of using emerging genomic information in research and clinical databases is to test only the variants that have been strongly implicated in disease causation or progression from GWA and other genomic discovery studies. This is particularly suited to clinical settings without the extensive research infrastructures needed for gene discovery (such as standardized phenotype and exposure measures, flexible informatics systems, biospecimen repositories, and consent for broad research uses), where real-world implications of these discoveries are best assessed. Limited genotyping for specific variants of interest in specific conditions can often be conducted more readily than GWA genotyping, assuming consent is adequate and phenotypic measures are reliable, allowing evaluation of the clinical and public health impact of these variants on a very large scale.
Genomic Information Suitable for Clinical Effectiveness Research
Assays of genetic variants related to two traits—Type 2 diabetes risk and warfarin dosing requirements—have sufficient scientific foundation and clinical availability to serve as prototypes for applying genomic information emerging from GWA studies to clinical effectiveness research. In a longer paper we also might have considered CFH and age-related macular degeneration (Klein et al., 2005), IL23R and inflammatory bowel disease (Duerr et al., 2006), or chromosome 8q24 variants and prostate cancer (Gudmundsson et al., 2007; Yeager et al., 2007), all of which would also lend themselves well to investigating questions of clinical effectiveness.
Type 2 Diabetes and TCF7L2
GWA studies have identified a number of variants associated with risk of diabetes to a modest degree, but the one first implicated by this approach, TCF7L2, clearly carries the greatest increased risk (Weedon,
2007). TCF7L2 is a transcription factor that is part of the Wnt signaling pathway, a pathway critical for cell proliferation, motility, and development, particularly of the pancreas (Weedon, 2007). It is an excellent example of the power of the hypothesis-free approach exemplified by GWA studies, since this gene was not previously suspected of playing any role in diabetes. The variant was initially identified in a linkage study of diabetes in Icelanders by deCODE Genetics, Inc., and was shown to be present in 36 percent of patients with diabetes but only 28 percent of unaffected individuals (Grant et al., 2006). An estimated 38 percent of the Icelandic population was heterozygous for the risk allele, and 7 percent were homozygous. Each copy of the risk allele increased the odds of diabetes 1.56-fold with a p-value of 10–18 when the Icelandic study was combined with similar studies from Denmark and the United States (Grant et al., 2006).
When this finding was first published in January 2006, it evoked surprise and a certain degree of perplexity, since there was no a priori biologic information supporting such an association. The data presented, however, were quite robust and convincing, and the finding was subsequently replicated in a GWA study of French cases and controls in February 2007 (Sladek et al., 2007). Three additional GWA studies in British and Scandinavian participants published in April 2007 all found TCF7L2 to be their strongest association signal (Saxena et al., 2007; Scott et al., 2007; Zeggini et al., 2007). These and subsequent studies have suggested a slightly lower odds ratio, closer to 1.4, but the association has been replicated in every population in which it has been examined (Frayling, 2007; Weedon, 2007).
Clinical testing for TCF7L2 variants is currently offered by DNA Direct (DNA Direct, 2008) and deCODE Diagnostics (Genetics, 2008), the corporate home of the team that published the original paper (Grant et al., 2006). deCODE Diagnostics also offers DNA-based tests for assessing risk of atrial fibrillation, myocardial infarction, glaucoma, and prostate cancer, all conditions for which deCODE Genetics published the first or one of the first GWA studies (Gudbjartsson et al., 2007; Gudmundsson et al., 2007; Helgadottir et al., 2007; Thorleifsson et al., 2007). Information about TCF7L2 testing (provided in a 4-gene panel referred to as deCODE T2TM) is provided for physicians and patients on the company’s website and describes the research conducted at deCODE and elsewhere demonstrating the TCF7L2–diabetes association (Genetics, 2008). Data from the NIH-sponsored clinical trial of diabetes prevention, the Diabetes Prevention Program (DPP), are cited showing that prediabetics homozygous for the risk allele were at 1.8-fold increased risk of developing diabetes in the next 4 years compared to heterozygotes or those without a risk allele (Florez et al., 2006). Evidence from the DPP on the effectiveness of weight loss and metformin treatment in reducing the risk of diabetes is also sum-
marized, demonstrating the availability of clinical-trial proven interventions to reduce diabetes incidence in persons at risk for the disease. The website notes that deCODE offers a Clinical Laboratory Improvement Amendments (CLIA)-certified testing facility, that the test is not FDA-approved, and that information from the test may “offer a new means to help physicians decide which prediabetic patients they wish to treat more aggressively either through lifestyle modification or drug treatment.” It also includes an important caveat: “Information gained from a genetic test does not itself prevent the development of disease, but can be used in formulating better preventive strategies. A positive genetic test result can emphasize the increased importance of using available and appropriate means in that regard” (deCODE, 2008).
Marketing or application of diagnostic genetic testing in this way has raised some anxieties, primarily due to the lack of evidence that genetic testing improves outcome or adds significantly to readily available clinical information (Haga et al., 2003; Janssens et al., 2006). Such evidence should be derivable, however, by linking genotypic data on these variants to phenotypic characteristics (such as presence of diagnosed diabetes or intermediate traits) and environmental exposures (such as lifestyle factors or medication use) in real-world clinical databases or ongoing research studies. If possible, it would be best to demonstrate that patients and/or their clinicians understood and retained the information and implemented appropriate interventions if improved outcomes are to be correctly attributed to the testing (Feero et al., 2008; Hunter et al., 2008).
Warfarin
Warfarin is a commonly used anticoagulant for prevention of pulmonary embolism in venous thromboembolic disease and of stroke in atrial fibrillation. Dosage must be maintained within a narrow range specific to each patient to prevent over-anticoagulation, and subsequent hemorrhage, or inadequate anticoagulation, and subsequent thrombosis. Dose requirements vary widely between individuals and are influenced by age, sex, body size, diet, medication use, and presence of other medical conditions (Kimmel, 2008). Variants in the cytochrome P450 system, specifically the *2 and *3 alleles of CYP2C9, have been shown to be associated with significantly lower dose requirements (Higashi et al., 2002). More recently, variants in the gene encoding vitamin K epoxide reductase complex 1 (VKORC1) have been shown to have similar effects (Figure 3-13 [Rieder et al., 2005]). A number of dosing algorithms have been proposed to reduce time to achieve therapeutic levels and avoid over-anticoagulation, incorporating a variety of clinical and genetic information (Kimmel, 2008).
Like TCF7L2 and diabetes, the effectiveness of including pharmaco-
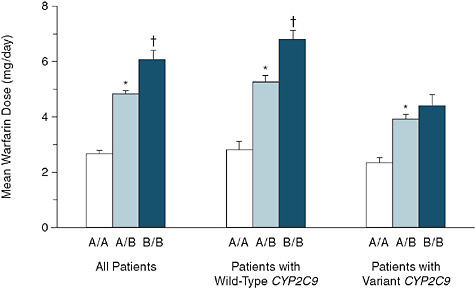
FIGURE 3-13 Patients were genotyped and assigned a VKORC1 haplotype combination (A/A, A/B, or B/B). The patients were further classified according to CYP2C9 genotype (the wild type or either the *2 or *3 variant). The total numbers of patients having a group A combination, a group B combination, or both were 182 (all patients), 124 (wild-type CYP2C9), and 58 (variant CYP2C9). The asterisks denote P < 0.05 for the comparison with combination A/A and the daggers P < 0.05 for the comparison with combination A/B. The T bars represent standard errors. SOURCE: Rieder, M. J., A. P. Reiner, B. F. Gage, D. A. Nickerson, C. S. Eby, H. L. McLeod, D. K. Blough, K. E. Thummel, D. L. Veenstra, and A. E. Rettie. 2005. Effect of vkorc1 haplotypes on transcriptional regulation and warfarin dose. New England Journal of Medicine 352(22):2285-2293. Copyright © 2005 Massachusetts Medical Society. All rights reserved.
genetic information in warfarin-dosing algorithms has yet to be demonstrated, despite clear evidence of dose dependence on a number of variants (Figure 3-14) (Anderson et al., 2007; Shurin and Nabel, 2008). Fortunately, a large, NIH-sponsored, randomized trial of genotype-guided warfarin therapy, designed to provide definitive answers to questions of clinical effectiveness, is about to get underway (Shurin and Nabel, 2008).
Incorporating Genomic Information into Clinical Effectiveness Research
With the examples of TCF7L2 and warfarin-dosing-related variants in hand, we can return to our original question of what is needed to facilitate the reliable and timely introduction of emerging genomic information into
![FIGURE 3-14 Average stable maintenance warfarin doses (mg/wk) by number of variant alleles (reproduced with permission from [Anderson et al., 2007]). Numbers of patients in each group: wild type (no variants), 56 (30%); 1 variant, 75 (43%); 2 variants, 36 (21%); 3 variants, 7 (4%); and 4 variants, 1 (0.6%). SEM is 2.0 for wild type and 1.4 for 1-, 2-, and 3-variant groups. Dose differences across groups are highly significant (P << 0.001).](/openbook/12197/xhtml/images/p2001bb37g198001.jpg)
FIGURE 3-14 Average stable maintenance warfarin doses (mg/wk) by number of variant alleles (reproduced with permission from [Anderson et al., 2007]). Numbers of patients in each group: wild type (no variants), 56 (30%); 1 variant, 75 (43%); 2 variants, 36 (21%); 3 variants, 7 (4%); and 4 variants, 1 (0.6%). SEM is 2.0 for wild type and 1.4 for 1-, 2-, and 3-variant groups. Dose differences across groups are highly significant (P << 0.001).
SOURCE: Anderson, J. L., B. D. Horne, S. M. Stevens, A. S. Grove, S. Barton, Z. P. Nicholas, S. F. Kahn, H. T. May, K. M. Samuelson, J. B. Muhlestein, J. F. Carlquist; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard war-farin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563-2570.
research and clinical databases. Needs can be identified in several areas, including the information needed for rational clinical decision making likely to affect outcomes; the laboratory and clinical infrastructure needed to utilize this information in clinical practice; and the policy and educational infrastructures needed to facilitate this research.
Epidemiologic Information Needed
Much of the basic information needed for informed decision making about newly identified genetic variants relates to fundamental epidemiologic questions such as prevalence, risk, and potential for risk reduction. Genetic variants such as those in TCF7L2, CYP2C9, and VKORC1 are essentially risk factors for complex diseases, similar in many ways to non-genetic risk factors such as obesity, smoking, or hypertension. Before recommending screening of TCF7L2 variants (or any risk factor) in persons at risk for diabetes to increase the intensity of efforts to prevent diabetes, for example, or of CYP2C9 or VKORC1 variants to guide warfarin dosing,
several pieces of information would be useful (Box 3-1). The prevalence of the variant is an important factor—testing for common variants is likely to be more useful and cost-effective than testing for rare ones. It may still be important to measure rare variants if their effects are devastating and essentially avoidable (such as TPMT variants in 6-mercaptopurine dosing [Wang and Weinshilboum, 2006] or phenylalanine hydroxylase deficiency in phenylketonuria [Scriver, 2007]), but even there information on prevalence is useful in estimating the costs of screening or the magnitude of the population likely to need intervention, if interventions are available.
Magnitude of increased risk associated with the variant is also important—variants of large effect would likely have more impact than those of small effect. Differences in the presence or magnitude of associated risk across different demographic groups, such as those defined by age, sex, or race/ethnicity, or in persons with particular medical conditions, lifestyle factors, or medications, would be useful in targeting testing or interpreting results. Independence of the association from other known risk factors, such as body mass, family history, or age, also would be important—it would seem unwise at present to substitute a genetic test for other information that is readily available clinically. Association of the variant with earlier onset or more severe course, or with response to treatment, might suggest targeted interventions or time-sensitive ones, or provide clues to pathogenesis. Evidence that knowledge of the variant would improve patients’ adherence to effective interventions also would be useful.
Much of this epidemiologic information about genetic variants identified as potentially causally associated with complex diseases can be readily obtained by assaying them in well-characterized population studies.
|
BOX 3-1 Epidemiologic Information Needed to Assess the Usefulness of Genetic Variants in Clinical Practice
|
Genomewide genotyping in cohorts such as the Framingham Heart Study or the Women’s Health Study (Cupples et al., 2007; Ridker et al., 2008), will provide this type of information, as will typing of specific variants in large numbers of extensively phenotyped participants in other cross-sectional studies, prospective cohorts, and clinical trials. Such efforts have been quite valuable in understanding the epidemiology of diabetes risk variants as studied in the Diabetes Prevention Program (Florez et al., 2006, 2007), particularly when the resulting prevalence and association data are made widely available. The National Human Genome Research Institute (NHGRI) is initiating a program to expand genotyping of putative causal variants in large-scale population studies and disseminate the results, as permitted by participants’ informed consent, for application in research and clinical settings (Services, 2007).
Genetic Information Needed
Detailed information about the genomic region proposed for testing is also needed (Box 3-2). The TCF7L2 gene, for example, is large and complex, with 17 exons and 4 alternate splice sites yielding multiple isoforms (John Hopkins University, 2008). The markers first identified as associated with diabetes lay in the third intron of the gene, in a well-defined linkage disequilibrium (LD) block spanning 92 kb (Figure 3-15 [Grant et al., 2006]). Conceivably, any of the variants in this block, which encompasses all of exon 4 and parts of introns 3 and 4, could be responsible for the observed association, since they would all tend to be inherited together. This would include variants that were not assayed on the original genotyping platform or, possibly, not even yet known to exist. Substantial investigative effort was needed to narrow this down; in this instance, the deCODE investigators identified another SNP, rs7903146, that carried a higher relative risk at a
|
BOX 3-2 Genetic Information Needed to Select Genetic Variants for Use in Clinical Effectiveness Research
|
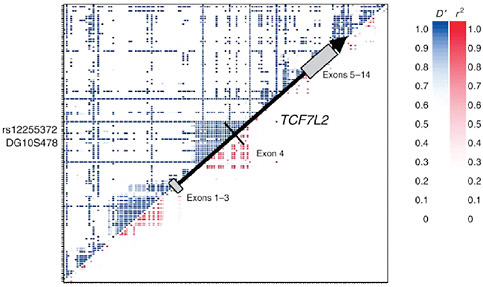
FIGURE 3-15 Region of interest in TCF7L2 and associated linkage disequilibrium (LD) pattern. LD between pairs of SNPs is shown by standard D′ (upper left) or r2 measures (lower right); the 216-kb gene spans several LD blocks as shown by the black arrow, indicating the direction of transcription and position of exons. Relative location of two of the most strongly associated markers, rs12255372 and DG10S478, are also shown.
SOURCE: Reprinted by permission from Macmillan Publishers, Ltd. Nature Genetics 38(3):320-323. Copyright © 2006.
much stronger significance level (Grant et al., 2006). It is this variant that is reported from deCODE T2TM. One might want to demonstrate, however, that assaying this variant is sufficient to measure all of the relevant variation in the region, as LD blocks may contain multiple independent signals (Haiman et al., 2007).
Similarly for CYP2C9 and VKORC1, it would be important to determine exactly which variants to test from the frequency and association patterns of SNPs in the genetic region surrounding the association signal. These patterns may not be known, but they may be possible to infer from available LD patterns in well-characterized samples such as those included in the HapMap (A haplotype map of the human genome, 2005). The relationship of LD patterns in these defined populations, however, to those in the clinical populations to be tested may be unclear, and this should be recognized as a limitation of proposed testing strategies.
Information on rarer SNPs and structural variants such as insertions, deletions, and duplications not captured through the HapMap also is needed, as these may be causing an underlying association signal but may
not be identified by existing genotyping platforms (Pearson and Manolio, 2008). Sequencing of association regions in hundreds or thousands of people may be needed to identify these rarer variants, and such efforts are ongoing on a small scale in follow-up to GWA findings. In-depth sequencing of the entire human genome is the best way to develop a comprehensive catalog of rarer variants, and an international effort to do just this has recently gotten underway (Genomes, 2008). Efforts to identify functional variants and their phenotypic effects also are continuing with projects such as ENCODE and the Knockout Mouse Project (Austin et al., 2004; Birney et al., 2007) which should help to extend and refine testing and interpretation of genomic regions associated with complex diseases and traits. Approaches are needed for updating clinical testing strategies and intervention approaches with this emerging information.
Laboratory and Clinical Infrastructure
Incorporation of genomic information into clinical effectiveness research also requires broad capacity to conduct testing and interpret the results (Box 3-3). First, we would need a laboratory infrastructure that could support such research, including a test that is valid, readily available, affordable, and (preferably) FDA-certified, and that is conducted under the auspices of a CLIA-certified laboratory. It would be important to have available a test for whatever genetic variant(s) one is interested in. Such a test should be eligible, or preferably approved, for reimbursement by insurers, though this raises the conundrum that even provisional approval requires a sufficient evidence base to support use of the test. It also would need to meet estab-
|
BOX 3-3 Laboratory and Clinical Infrastructure Needed to Conduct Clinical Effectiveness Research on Genetic Variants
|
lished guidelines for analytic validity, clinical validity, clinical utility, and public health utility (Grosse and Khoury, 2006; National Human Genome Research Institute, 2008b).
Flexible, comprehensive, accessible, user-friendly EHRs and personal health records (PHRs) also would be important for receiving and providing the results of genetic testing and related phenotypic or other measures. Such systems would need adequate privacy protections, as does all protected health information (U.S. Department of Health and Human Services, 1996) and ideally should provide point of care performance feedback so that clinicians or patients who receive results of genetic testing will know what actions they should then take. Automated prompts and patient management tools recommending genetic testing for patients in whom it has been demonstrated to improve outcomes, particularly in response to changing clinical parameters also recorded in the EHR, would also be useful. Testing might be recommended, for example, in persons who cross a certain threshold of increased risk for diabetes based on obesity or fasting glucose, or those who need to initiate warfarin therapy. To facilitate research as well as practice, a secure network of interoperable EHR and PHR systems (Murphy et al., 2006; Murray et al., 2003) would make possible rapid, large-scale, multicenter clinical studies to assess the effectiveness of testing for specific genetic variants and the interventions that follow. Additional standardized, structured nomenclatures for genomic applications in such systems also may be needed.
Accessible but secure large-scale databases to receive, archive, and distribute results of studies of genotype–phenotype associations are needed, such as the database of Genotype and Phenotype (dbGaP) (Mailman et al., 2007). Developed and maintained by the National Center for Biotechnology Information of the NIH, dbGaP includes genotype and phenotype data from genomewide association studies, medical sequencing, and molecular diagnostic assays, as well as phenotypic and clinical characteristics and environmental and lifestyle exposures. dbGaP provides access to data at two levels—open and controlled—allowing for both the broad release of summary data on allele frequencies and associations while also restricting access and ensuring investigator accountability for sensitive datasets involving individual-level genotype and phenotype data. By NIH policy, data in dbGaP are assumed to be pre-competitive and are expected to remain unencumbered by premature intellectual property claims (U.S. Department of Health and Human Services, 2007). On the open access public site, dbGaP supports searches for studies, protocols, and questionnaires. Visitors to dbGaP can view phenotype summary data, genotype summary data, and pre-computed or published genetic associations. As such it provides a powerful tool for identifying emerging genomic information that may potentially be applied to clinical effectiveness research, but as yet the association
information is not readily searchable and may be difficult to extract and distill. Published genomewide association studies also are catalogued by the National Human Genome Research Institute (2008a) and the Centers for Disease Control and Prevention (Centers for Disease Control and Prevention, 2008). Such catalogues provide information on genetic associations that may be closer to practical application than those in dbGaP, but as noted above, much additional investigation is needed following an initial genomewide association report before one can contemplate applying them to clinical research.
Policy and Educational Infrastructure
The need to ensure confidentiality and privacy is vitally important to databases containing individual-level genotype or phenotype information, whether they be research databases such as dbGaP or clinical databases derived from EHRs used in clinical care. Major, justified, and unresolved concerns about the potential for persons with genetic variants putting them at increased disease risk becoming the object of discrimination by employers or insurers must be addressed. Clinical effectiveness research on genomic information will be difficult, if not impossible, to conduct on a large scale without the formal legal protection against discrimination provided by the Genetic Information Non-Discrimination Act (GINA). The Act would protect individuals against discrimination based on their genetic information in health insurance and employment. Enactment would prohibit insurers from denying health insurance coverage or charging higher premiums or employers from using genetic information in hiring or firing decisions. These protections are intended to encourage Americans to take advantage of genetic testing as part of their medical care and to participate in genetic research. Originally introduced over a decade ago, genetic non-discrimination legislation was passed unanimously by the Senate in both the 108th and 109th Congresses, but, until this year had never been passed by the House. The Genetic Nondiscrimination Act of 2007 was passed in the House by a vote of 420-3, and has the support of the current administration, but as of this writing has yet to be voted on in the Senate. This kind of policy infrastructure is absolutely crucial not only to clinical effectiveness research but also to any genomic research and its incorporation in clinical care.
Additional needs include consensus on reporting of variants or abnormalities to patients, consent requirements for research, approaches to counseling, and education of clinicians and patients (Box 3-4). Debate continues on whether, when, and how results of genetic tests, especially for common, complex diseases for which genetic variants are not deterministic, should be reported to patients, particularly in research settings (Bookman et al., 2006). This is a legacy in part of the era of Mendelian genetics when
|
BOX 3-4 Policy and Educational Infrastructure
|
identification of at-risk variants such as those for Huntington’s disease carried grave implications, often with little in the way of effective intervention (Manolio, 2006). Many research studies have taken the approach of informing patients in the consent form that under no circumstances will any results of genetic testing be provided to them, their physicians, or anyone outside the research setting, but this also precludes informing patients of the presence of potentially modifiable genetic risk that this research is specifically intended to identify (Bookman et al., 2006).
Research to develop valid, feasible approaches to informing patients of the modest increased risk conferred by many variants associated with complex diseases, and the avenues open to them for reducing that risk, is needed if this work is to be conducted in a way that maximizes benefits and minimizes risks to research participants. To that end, consistent and agreed-upon consent policies and procedures also are needed, as well as consistent policies by Institutional Review Boards (IRBs), particularly for multicenter research and practice. Flexible approaches to genetic counseling also are needed, including approaches for providing adequate counseling, where appropriate, through means other than one-on-one counseling with a certified genetic counselor. There are simply not enough certified genetic counselors available, nor is that level of counseling necessary for variants of modest effect, to provide it for every genetic test performed in the course of clinical care or effectiveness research. Less cumbersome alternatives that still bring qualified expertise to the discussion are needed. Finally, a better educational infrastructure is needed to improve “genomic literacy” for clinicians, research participants, and patients. Importantly, education should extend to family members who may be in a decision-making or advisory role and who may well be personally affected by genetic information provided to a blood relative with whom they share many of the same genetic variants. Rapid and reliable systems also are needed for updating clinicians
and the public on emerging genomic information, without the hyperbole that can sometimes accompany initial media reports.
As genomic information evolves and clinical effectiveness research progresses, the lines between research and patient care will blur and the two purposes may well merge. A number of other issues will need to be addressed, such as whose responsibility it will be to contact patients who have been genotyped for markers of unknown or questionable significance, in either the research or clinical setting, once actionable information on them becomes available. Whose responsibility will it be to store and maintain the data long term? What if a clinician acts on a marker and later the association is shown to be of lesser importance or have different implications than initially thought? Will clinicians be criticized or held accountable for inaction on risk variants of questionable clinical but very strong statistical significance? The agenda for clinical effectiveness research on emerging genomic information is clearly substantial and will likely continue for some time after initial identification of a clinically important variant.
Conclusion
Advances in genotyping technology coupled with expanding knowledge of genome structure and function have fueled a virtual explosion of genomic information on common, complex diseases. New insights into disease mechanisms, and new avenues to diagnosis, prevention, and treatment, are opening more rapidly than we can keep pace with them, but most remain many steps away from actual clinical application. Incorporation of this information into research and clinical databases, and ultimately into effective clinical practice, will require substantial additional epidemiologic and genetic information. It also will require a substantial infrastructure, including a valid, certified, reimbursable, available test; secure, interoperable, and transportable electronic medical records; genetic information, non-discrimination legislation; guidelines for reporting results to patients; guidelines for obtaining consent and institutional review; practical approaches to counseling; and genomically literate clinicians and patients. Although this is a tall order, it is one we must fill if we are to capitalize on the enormous investment, and the enormous promise, of genomic research in common, complex diseases.
REFERENCES
Anderson, J. L., B. D. Horne, S. M. Stevens, A. S. Grove, S. Barton, Z. P. Nicholas, S. F. Kahn, H. T. May, K. M. Samuelson, J. B. Muhlestein, and J. F. Carlquist. 2007. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 116(22):2563-2570.
Angrist, J. D. 1996. Identification of casual effects using instrumental variables Journal of the American Statistical Association (91):444-455.
Arking, D. E., A. Pfeufer, W. Post, W. H. Kao, C. Newton-Cheh, M. Ikeda, K. West, C. Kashuk, M. Akyol, S. Perz, S. Jalilzadeh, T. Illig, C. Gieger, C. Y. Guo, M. G. Larson, H. E. Wichmann, E. Marban, C. J. O’Donnell, J. N. Hirschhorn, S. Kaab, P. M. Spooner, T. Meitinger, and A. Chakravarti. 2006. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nature Genetics 38(6):644-651.
Armitage, J., R. Souhami, L. Friedman, L. Hilbrich, J. Holland, L. H. Muhlbaier, J. Shannon, and A. Van Nie. 2008. The impact of privacy and confidentiality laws on the conduct of clinical trials. Clinical Trials 5(1):70-74.
Austin, C. P., J. F. Battey, A. Bradley, M. Bucan, M. Capecchi, F. S. Collins, W. F. Dove, G. Duyk, S. Dymecki, J. T. Eppig, F. B. Grieder, N. Heintz, G. Hicks, T. R. Insel, A. Joyner, B. H. Koller, K. C. Lloyd, T. Magnuson, M. W. Moore, A. Nagy, J. D. Pollock, A. D. Roses, A. T. Sands, B. Seed, W. C. Skarnes, J. Snoddy, P. Soriano, D. J. Stewart, F. Stewart, B. Stillman, H. Varmus, L. Varticovski, I. M. Verma, T. F. Vogt, H. von Melchner, J. Witkowski, R. P. Woychik, W. Wurst, G. D. Yancopoulos, S. G. Young, and B. Zambrowicz. 2004. The knockout mouse project. Nature Genetics 36(9):921-924.
Baigent, C., F. E. Harrell, M. Buyse, J. R. Emberson, and D. G. Altman. 2008. Ensuring trial validity by data quality assurance and diversification of monitoring methods. Clinical Trials 5(1):49-55.
Benjamin, D. K., Jr., P. B. Smith, M. D. Murphy, R. Roberts, L. Mathis, D. Avant, R. M. Califf, and J. S. Li. 2006. Peer-reviewed publication of clinical trials completed for pediatric exclusivity. Journal of the American Medical Association 296(10):1266-1273.
Berry, D. A. 1996. Statistics: A Bayesian Perspective. Belmont, CA: Duxbury Press.
———. 2006. Bayesian clinical studies. Nature Reviews and Drug Discovery (5):27-36.
Berry, D. A., K. A. Cronin, S. K. Plevritis, D. G. Fryback, L. Clarke, M. Zelen, J. S. Mandelblatt, A. Y. Yakovlev, J. D. Habbema, and E. J. Feuer. 2005. Effect of screening and adjuvant therapy on mortality from breast cancer. New England Journal of Medicine 353(17):1784-1792.
Berry, D. A., L. Inoue, Y. Shen, J. Venier, D. Cohen, M. Bondy, R. Theriault, and M. F. Munsell. 2006. Modeling the impact of treatment and screening on U.S. breast cancer mortality: A Bayesian approach. Journal of the National Cancer Institute Monographs (36):30-36.
Birney, E., J. A. Stamatoyannopoulos, A. Dutta, R. Guigo, T. R. Gingeras, E. H. Margulies, Z. Weng, M. Snyder, E. T. Dermitzakis, R. E. Thurman, M. S. Kuehn, C. M. Taylor, S. Neph, C. M. Koch, S. Asthana, A. Malhotra, I. Adzhubei, J. A. Greenbaum, R. M. Andrews, P. Flicek, P. J. Boyle, H. Cao, N. P. Carter, G. K. Clelland, S. Davis, N. Day, P. Dhami, S. C. Dillon, M. O. Dorschner, H. Fiegler, P. G. Giresi, J. Goldy, M. Hawrylycz, A. Haydock, R. Humbert, K. D. James, B. E. Johnson, E. M. Johnson, T. T. Frum, E. R. Rosenzweig, N. Karnani, K. Lee, G. C. Lefebvre, P. A. Navas, F. Neri, S. C. Parker, P. J. Sabo, R. Sandstrom, A. Shafer, D. Vetrie, M. Weaver, S. Wilcox, M. Yu, F. S. Collins, J. Dekker, J. D. Lieb, T. D. Tullius, G. E. Crawford, S. Sunyaev, W. S. Noble, I. Dunham, F. Denoeud, A. Reymond, P. Kapranov, J. Rozowsky, D. Zheng, R. Castelo, A. Frankish, J. Harrow, S. Ghosh, A. Sandelin, I. L. Hofacker, R. Baertsch, D. Keefe, S. Dike, J. Cheng, H. A. Hirsch, E. A. Sekinger, J. Lagarde, J. F. Abril, A. Shahab, C. Flamm, C. Fried, J. Hackermuller, J. Hertel, M. Lindemeyer, K. Missal, A. Tanzer, S. Washietl, J. Korbel, O. Emanuelsson, J. S. Pedersen, N. Holroyd, R. Taylor, D. Swarbreck, N. Matthews, M. C. Dickson, D. J. Thomas, M. T. Weirauch, J. Gilbert, J. Drenkow, I. Bell, X. Zhao, K. G. Srinivasan, W. K. Sung, H. S. Ooi, K. P. Chiu, S. Foissac, T. Alioto, M. Brent, L. Pachter, M. L. Tress, A. Valencia, S. W. Choo, C. Y. Choo, C. Ucla, C. Manzano, C. Wyss, E. Cheung, T. G. Clark, J. B. Brown, M. Ganesh, S. Patel, H. Tammana, J. Chrast, C. N.
Henrichsen, C. Kai, J. Kawai, U. Nagalakshmi, J. Wu, Z. Lian, J. Lian, P. Newburger, X. Zhang, P. Bickel, J. S. Mattick, P. Carninci, Y. Hayashizaki, S. Weissman, T. Hubbard, R. M. Myers, J. Rogers, P. F. Stadler, T. M. Lowe, C. L. Wei, Y. Ruan, K. Struhl, M. Gerstein, S. E. Antonarakis, Y. Fu, E. D. Green, U. Karaoz, A. Siepel, J. Taylor, L. A. Liefer, K. A. Wetterstrand, P. J. Good, E. A. Feingold, M. S. Guyer, G. M. Cooper, G. Asimenos, C. N. Dewey, M. Hou, S. Nikolaev, J. I. Montoya-Burgos, A. Loytynoja, S. Whelan, F. Pardi, T. Massingham, H. Huang, N. R. Zhang, I. Holmes, J. C. Mullikin, A. Ureta-Vidal, B. Paten, M. Seringhaus, D. Church, K. Rosenbloom, W. J. Kent, E. A. Stone, S. Batzoglou, N. Goldman, R. C. Hardison, D. Haussler, W. Miller, A. Sidow, N. D. Trinklein, Z. D. Zhang, L. Barrera, R. Stuart, D. C. King, A. Ameur, S. Enroth, M. C. Bieda, J. Kim, A. A. Bhinge, N. Jiang, J. Liu, F. Yao, V. B. Vega, C. W. Lee, P. Ng, A. Yang, Z. Moqtaderi, Z. Zhu, X. Xu, S. Squazzo, M. J. Oberley, D. Inman, M. A. Singer, T. A. Richmond, K. J. Munn, A. Rada-Iglesias, O. Wallerman, J. Komorowski, J. C. Fowler, P. Couttet, A. W. Bruce, O. M. Dovey, P. D. Ellis, C. F. Langford, D. A. Nix, G. Euskirchen, S. Hartman, A. E. Urban, P. Kraus, S. Van Calcar, N. Heintzman, T. H. Kim, K. Wang, C. Qu, G. Hon, R. Luna, C. K. Glass, M. G. Rosenfeld, S. F. Aldred, S. J. Cooper, A. Halees, J. M. Lin, H. P. Shulha, M. Xu, J. N. Haidar, Y. Yu, V. R. Iyer, R. D. Green, C. Wadelius, P. J. Farnham, B. Ren, R. A. Harte, A. S. Hinrichs, H. Trumbower, H. Clawson, J. Hillman-Jackson, A. S. Zweig, K. Smith, A. Thakkapallayil, G. Barber, R. M. Kuhn, D. Karolchik, L. Armengol, C. P. Bird, P. I. de Bakker, A. D. Kern, N. Lopez-Bigas, J. D. Martin, B. E. Stranger, A. Woodroffe, E. Davydov, A. Dimas, E. Eyras, I. B. Hallgrimsdottir, J. Huppert, M. C. Zody, G. R. Abecasis, X. Estivill, G. G. Bouffard, X. Guan, N. F. Hansen, J. R. Idol, V. V. Maduro, B. Maskeri, J. C. McDowell, M. Park, P. J. Thomas, A. C. Young, R. W. Blakesley, D. M. Muzny, E. Sodergren, D. A. Wheeler, K. C. Worley, H. Jiang, G. M. Weinstock, R. A. Gibbs, T. Graves, R. Fulton, E. R. Mardis, R. K. Wilson, M. Clamp, J. Cuff, S. Gnerre, D. B. Jaffe, J. L. Chang, K. Lindblad-Toh, E. S. Lander, M. Koriabine, M. Nefedov, K. Osoegawa, Y. Yoshinaga, B. Zhu, and P. J. de Jong. 2007. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature 447(7146):799-816.
Bookman, E. B., A. A. Langehorne, J. H. Eckfeldt, K. C. Glass, G. P. Jarvik, M. Klag, G. Koski, A. Motulsky, B. Wilfond, T. A. Manolio, R. R. Fabsitz, and R. V. Luepker. 2006. Reporting genetic results in research studies: Summary and recommendations of an NHLBI working group. American Journal of Medical Genetics Part A 140(10):1033-1040.
Braithwaite, R. S., S. Shechter, M. S. Roberts, A. Schaefer, D. R. Bangsberg, P. R. Harrigan, and A. C. Justice. 2006. Explaining variability in the relationship between antiretroviral adherence and HIV mutation accumulation. Journal of Antimicrobial Chemotherapy 58(5):1036-1043.
Braithwaite, R. S., M. S. Roberts, C. C. Chang, M. B. Goetz, C. L. Gibert, M. C. Rodriguez-Barradas, S. Shechter, A. Schaefer, K. Nucifora, R. Koppenhaver, and A. C. Justice. 2008. Influence of alternative thresholds for initiating HIV treatment on quality-adjusted life expectancy: A decision model. Annals of Internal Medicine 148(3):178-185.
Brookhart, M. A. 2007. Evaluating the validity of an instrumental variable study of nueroleptics: Can between-physician differences in prescribing patterns be used to estimate treatment effects? Medical Care 45(10):116-122.
Brookhart, M. A., S. Schneeweiss, K. J. Rothman, R. J. Glynn, J. Avorn, and T. Sturmer. 2006. Variable selection for propensity score models. American Journal of Epidemiology 163(12):1149-1156.
Brookhart, M. A., J. A. Rassen, P. S. Wang, C. Dormuth, H. Mogun, and S. Schneeweiss. 2007. Evaluating the validity of an instrumental variable study of neuroleptics: Can between-physician differences in prescribing patterns be used to estimate treatment effects? Medical Care 45(10 Supl 2):S116-S122.
Califf, R. M., and R. A. Rosati. 1981. The doctor and the computer. Western Journal of Medicine 135(4):321-323.
Califf, R. M., E. D. Peterson, R. J. Gibbons, A. Garson, Jr., R. G. Brindis, G. A. Beller, and S. C. Smith, Jr. 2002. Integrating quality into the cycle of therapeutic development. Journal of the American College of Cardiology 40(11):1895-1901.
Califf, R. M., R. A. Harrington, L. K. Madre, E. D. Peterson, D. Roth, and K. A. Schulman. 2007. Curbing the cardiovascular disease epidemic: Aligning industry, government, payers, and academics. Health Affairs (Millwood) 26(1):62-74.
Centers for Disease Control and Prevention. 2008. HuGE Literature Finder. http://www. hugenavigator.net/ (accessed July 9, 2008).
Chanock, S. J., T. Manolio, M. Boehnke, E. Boerwinkle, D. J. Hunter, G. Thomas, J. N. Hirschhorn, G. Abecasis, D. Altshuler, J. E. Bailey-Wilson, L. D. Brooks, L. R. Cardon, M. Daly, P. Donnelly, J. F. Fraumeni, Jr., N. B. Freimer, D. S. Gerhard, C. Gunter, A. E. Guttmacher, M. S. Guyer, E. L. Harris, J. Hoh, R. Hoover, C. A. Kong, K. R. Merikangas, C. C. Morton, L. J. Palmer, E. G. Phimister, J. P. Rice, J. Roberts, C. Rotimi, M. A. Tucker, K. J. Vogan, S. Wacholder, E. M. Wijsman, D. M. Winn, and F. S. Collins. 2007. Replicating genotype-phenotype associations. Nature 447(7145):655-660.
Children’s Hospital of Philadelphia. 2008. Center for Applied Genomics. http://www.chop.edu/consumer/jsp/division/generic.jsp?id=84930 (accessed July 9, 2008).
Clermont, G., J. Bartels, R. Kumar, G. Constantine, Y. Vodovotz, and C. Chow. 2004a. In silico design of clinical trials: A method coming of age. Critical Care Medicine 32(10):2061-2070.
Clermont, G., V. Kaplan, R. Moreno, J. L. Vincent, W. T. Linde-Zwirble, B. V. Hout, and D. C. Angus. 2004b. Dynamic microsimulation to model multiple outcomes in cohorts of critically ill patients. Intensive Care Medicine 30(12):2237-2244.
Colhoun, H. M., P. M. McKeigue, and G. Davey Smith. 2003. Problems of reporting genetic associations with complex outcomes. Lancet 361(9360):865-872.
Cook, D., A. Moore-Cox, D. Xavier, F. Lauzier, and I. Roberts. 2008. Randomized trials in vulnerable populations. Clinical Trials 5(1):61-69.
Cupples, L. A., H. T. Arruda, E. J. Benjamin, R. B. D’Agostino, Sr., S. Demissie, A. L. DeStefano, J. Dupuis, K. M. Falls, C. S. Fox, D. J. Gottlieb, D. R. Govindaraju, C. Y. Guo, N. L. Heard-Costa, S. J. Hwang, S. Kathiresan, D. P. Kiel, J. M. Laramie, M. G. Larson, D. Levy, C. Y. Liu, K. L. Lunetta, M. D. Mailman, A. K. Manning, J. B. Meigs, J. M. Murabito, C. Newton-Cheh, G. T. O’Connor, C. J. O’Donnell, M. Pandey, S. Seshadri, R. S. Vasan, Z. Y. Wang, J. B. Wilk, P. A. Wolf, Q. Yang, and L. D. Atwood. 2007. The Framingham Heart Study 100k SNP genome-wide association study resource: Overview of 17 phenotype working group reports. BMC Medical Genetics 8(Supl 1):S1.
Davis, J. R. 1999. Assuring data quality and validiy in clinical trials for regulatory descision making. Washington, D.C.
Day, J., J. Rubin, Y. Vodovotz, C. C. Chow, A. Reynolds, and G. Clermont. 2006. A reduced mathematical model of the acute inflammatory response II. Capturing scenarios of repeated endotoxin administration. Journal of Theoretical Biology 242(1):237-256.
deCODE. 2008. deCODE Diagnostics. http://www.decodediagnostics.com (accessed June 21, 2010).
DeMets, D. L., and R. M. Califf. 2002a. Lessons learned from recent cardiovascular clinical trials: Part I. Circulation 106(6):746-751.
———. 2002b. Lessons learned from recent cardiovascular clinical trials: Part II. Circulation 106(7):880-886.
Department of Health and Human Services. 2007. Epidemiologic Investigation of Putative Causal Genetic Variants—Study Investigators (u01). http://grants.nih.gov/grants/guide/rfa-files/RFA-HG-07-014.html (accessed July 9, 2008).
Dewan, A., M. Liu, S. Hartman, S. S. Zhang, D. T. Liu, C. Zhao, P. O. Tam, W. M. Chan, D. S. Lam, M. Snyder, C. Barnstable, C. P. Pang, and J. Hoh. 2006. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 314(5801):989-992.
Dilts, D. M., A. Sandler, S. Cheng, J. Crites, L. Ferranti, A. Wu, R. Gray, J. Macdonald, D. Marinucci, and R. Comis. 2008. Development of clinical trials in a cooperative group setting: The Eastern Cooperative Oncology Group. Clinical Cancer Research 14(11):3427-3433.
DNA Direct. 2008. DeCODE T2™—Diabetes Risk Marke. http://www.dnadirect.com/patients/tests/decode_diabetes/decode_t2.jsp (accessed July 9, 2008).
Duerr, R. H., K. D. Taylor, S. R. Brant, J. D. Rioux, M. S. Silverberg, M. J. Daly, A. H. Steinhart, C. Abraham, M. Regueiro, A. Griffiths, T. Dassopoulos, A. Bitton, H. Yang, S. Targan, L. W. Datta, E. O. Kistner, L. P. Schumm, A. T. Lee, P. K. Gregersen, M. M. Barmada, J. I. Rotter, D. L. Nicolae, and J. H. Cho. 2006. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314(5804):1461-1463.
Duley, L., K. Antman, J. Arena, A. Avezum, M. Blumenthal, J. Bosch, S. Chrolavicius, T. Li, S. Ounpuu, A. C. Perez, P. Sleight, R. Svard, R. Temple, Y. Tsouderous, C. Yunis, and S. Yusuf. 2008. Specific barriers to the conduct of randomized trials. Clinical Trials 5(1):40-48.
Easton, D. F., K. A. Pooley, A. M. Dunning, P. D. Pharoah, D. Thompson, D. G. Ballinger, J. P. Struewing, J. Morrison, H. Field, R. Luben, N. Wareham, S. Ahmed, C. S. Healey, R. Bowman, K. B. Meyer, C. A. Haiman, L. K. Kolonel, B. E. Henderson, L. Le Marchand, P. Brennan, S. Sangrajrang, V. Gaborieau, F. Odefrey, C. Y. Shen, P. E. Wu, H. C. Wang, D. Eccles, D. G. Evans, J. Peto, O. Fletcher, N. Johnson, S. Seal, M. R. Stratton, N. Rahman, G. Chenevix-Trench, S. E. Bojesen, B. G. Nordestgaard, C. K. Axelsson, M. Garcia-Closas, L. Brinton, S. Chanock, J. Lissowska, B. Peplonska, H. Nevanlinna, R. Fagerholm, H. Eerola, D. Kang, K. Y. Yoo, D. Y. Noh, S. H. Ahn, D. J. Hunter, S. E. Hankinson, D. G. Cox, P. Hall, S. Wedren, J. Liu, Y. L. Low, N. Bogdanova, P. Schurmann, T. Dork, R. A. Tollenaar, C. E. Jacobi, P. Devilee, J. G. Klijn, A. J. Sigurdson, M. M. Doody, B. H. Alexander, J. Zhang, A. Cox, I. W. Brock, G. MacPherson, M. W. Reed, F. J. Couch, E. L. Goode, J. E. Olson, H. Meijers-Heijboer, A. van den Ouweland, A. Uitterlinden, F. Rivadeneira, R. L. Milne, G. Ribas, A. Gonzalez-Neira, J. Benitez, J. L. Hopper, M. McCredie, M. Southey, G. G. Giles, C. Schroen, C. Justenhoven, H. Brauch, U. Hamann, Y. D. Ko, A. B. Spurdle, J. Beesley, X. Chen, A. Mannermaa, V. M. Kosma, V. Kataja, J. Hartikainen, N. E. Day, D. R. Cox, and B. A. Ponder. 2007. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447(7148):1087-1093.
Eddy, D. M., and L. Schlessinger. 2003a. Archimedes: A trial-validated model of diabetes. Diabetes Care 26(11):3093-3101.
———. 2003b. Validation of the Archimedes diabetes model. Diabetes Care 26(11):3102-3110.
Edwards, A. O., R. Ritter, 3rd, K. J. Abel, A. Manning, C. Panhuysen, and L. A. Farrer. 2005. Complement factor H polymorphism and age-related macular degeneration. Science 308(5720):421-424.
Eisenstein, E. L., R. Collins, B. S. Cracknell, O. Podesta, E. D. Reid, P. Sandercock, Y. Shakhov, M. L. Terrin, M. A. Sellers, R. M. Califf, C. B. Granger, and R. Diaz. 2008. Sensible approaches for reducing clinical trial costs. Clinical Trials 5(1):75-84.
The eMERGE Network. 2008. Electronics Medical Records and Genomics. http://www.gwas.net (accessed July 9, 2008).
Feero, W. G., A. E. Guttmacher, and F. S. Collins. 2008. The genome gets personal—almost. Journal of the American Medical Association 299(11):1351-1352.
Ferguson, T. B., Jr., S. W. Dziuban, Jr., F. H. Edwards, M. C. Eiken, A. L. Shroyer, P. C. Pairolero, R. P. Anderson, and F. L. Grover. 2000. The STS national database: Current changes and challenges for the new millennium. Committee to Establish a National Database in Cardiothoracic Surgery, The Society of Thoracic Surgeons. Annals of Thoracic Surgery 69(3):680-691.
Florez, J. C., K. A. Jablonski, N. Bayley, T. I. Pollin, P. I. de Bakker, A. R. Shuldiner, W. C. Knowler, D. M. Nathan, and D. Altshuler. 2006. Tcf7l2 polymorphisms and progression to diabetes in the diabetes prevention program. New England Journal of Medicine 355(3):241-250.
Florez, J. C., K. A. Jablonski, M. W. Sun, N. Bayley, S. E. Kahn, H. Shamoon, R. F. Hamman, W. C. Knowler, D. M. Nathan, and D. Altshuler. 2007. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. Journal of Clinical Endocrinology & Metabolism 92(4):1502-1509.
Frayling, T. M. 2007. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nature Reviews Genetics 8(9):657-662.
Frayling, T. M., N. J. Timpson, M. N. Weedon, E. Zeggini, R. M. Freathy, C. M. Lindgren, J. R. Perry, K. S. Elliott, H. Lango, N. W. Rayner, B. Shields, L. W. Harries, J. C. Barrett, S. Ellard, C. J. Groves, B. Knight, A. M. Patch, A. R. Ness, S. Ebrahim, D. A. Lawlor, S. M. Ring, Y. Ben-Shlomo, M. R. Jarvelin, U. Sovio, A. J. Bennett, D. Melzer, L. Ferrucci, R. J. Loos, I. Barroso, N. J. Wareham, F. Karpe, K. R. Owen, L. R. Cardon, M. Walker, G. A. Hitman, C. N. Palmer, A. S. Doney, A. D. Morris, G. D. Smith, A. T. Hattersley, and M. I. McCarthy. 2007. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316(5826):889-894.
Frazer, K. A., D. G. Ballinger, D. R. Cox, D. A. Hinds, L. L. Stuve, R. A. Gibbs, J. W. Belmont, A. Boudreau, P. Hardenbol, S. M. Leal, S. Pasternak, D. A. Wheeler, T. D. Willis, F. Yu, H. Yang, C. Zeng, Y. Gao, H. Hu, W. Hu, C. Li, W. Lin, S. Liu, H. Pan, X. Tang, J. Wang, W. Wang, J. Yu, B. Zhang, Q. Zhang, H. Zhao, J. Zhou, S. B. Gabriel, R. Barry, B. Blumenstiel, A. Camargo, M. Defelice, M. Faggart, M. Goyette, S. Gupta, J. Moore, H. Nguyen, R. C. Onofrio, M. Parkin, J. Roy, E. Stahl, E. Winchester, L. Ziaugra, D. Altshuler, Y. Shen, Z. Yao, W. Huang, X. Chu, Y. He, L. Jin, Y. Liu, W. Sun, H. Wang, Y. Wang, X. Xiong, L. Xu, M. M. Waye, S. K. Tsui, H. Xue, J. T. Wong, L. M. Galver, J. B. Fan, K. Gunderson, S. S. Murray, A. R. Oliphant, M. S. Chee, A. Montpetit, F. Chagnon, V. Ferretti, M. Leboeuf, J. F. Olivier, M. S. Phillips, S. Roumy, C. Sallee, A. Verner, T. J. Hudson, P. Y. Kwok, D. Cai, D. C. Koboldt, R. D. Miller, L. Pawlikowska, P. Taillon-Miller, M. Xiao, L. C. Tsui, W. Mak, Y. Q. Song, P. K. Tam, Y. Nakamura, T. Kawaguchi, T. Kitamoto, T. Morizono, A. Nagashima, Y. Ohnishi, A. Sekine, T. Tanaka, T. Tsunoda, P. Deloukas, C. P. Bird, M. Delgado, E. T. Dermitzakis, R. Gwilliam, S. Hunt, J. Morrison, D. Powell, B. E. Stranger, P. Whittaker, D. R. Bentley, M. J. Daly, P. I. de Bakker, J. Barrett, Y. R. Chretien, J. Maller, S. McCarroll, N. Patterson, I. Pe’er, A. Price, S. Purcell, D. J. Richter, P. Sabeti, R. Saxena, S. F. Schaffner, P. C. Sham, P. Varilly, L. D. Stein, L. Krishnan, A. V. Smith, M. K. Tello-Ruiz, G. A. Thorisson, A. Chakravarti, P. E. Chen, D. J. Cutler, C. S. Kashuk, S. Lin, G. R. Abecasis, W. Guan, Y. Li, H. M. Munro, Z. S. Qin, D. J. Thomas, G. McVean, A. Auton, L. Bottolo, N. Cardin, S. Eyheramendy, C. Freeman, J. Marchini, S. Myers, C. Spencer, M. Stephens, P. Donnelly, L. R. Cardon, G. Clarke, D. M. Evans, A. P. Morris, B. S. Weir, J. C. Mullikin, S. T. Sherry, M. Feolo, A. Skol, H. Zhang, I. Matsuda, Y. Fukushima, D. R. Macer, E. Suda, C. N. Rotimi, C. A. Adebamowo, I. Ajayi, T. Aniagwu, P. A. Marshall, C. Nkwodimmah, C. D. Royal, M. F. Leppert, M. Dixon, A. Peiffer, R. Qiu, A. Kent, K. Kato, N. Niikawa, I. F. Adewole, B. M. Knoppers, M. W. Foster, E. W. Clayton, J. Watkin, D. Muzny, L. Nazareth, E. Sodergren, G. M. Weinstock, I. Yakub, B. W. Birren, R. K. Wilson, L. L. Fulton, J. Rogers, J. Burton, N. P. Carter, C. M. Clee, M. Griffiths, M. C. Jones, K. McLay, R. W. Plumb, M. T. Ross,
S. K. Sims, D. L. Willey, Z. Chen, H. Han, L. Kang, M. Godbout, J. C. Wallenburg, P. L’Archeveque, G. Bellemare, K. Saeki, D. An, H. Fu, Q. Li, Z. Wang, R. Wang, A. L. Holden, L. D. Brooks, J. E. McEwen, M. S. Guyer, V. O. Wang, J. L. Peterson, M. Shi, J. Spiegel, L. M. Sung, L. F. Zacharia, F. S. Collins, K. Kennedy, R. Jamieson, and J. Stewart. 2007. A second generation human haplotype map of over 3.1 million SNPS. Nature 449(7164):851-861.
Genomes. 2008. A Deep Catalog of Human Genetic Variation. http://www.1000genomes.org/ (accessed July 9, 2008).
Giles, F. J., H. M. Kantarjian, J. E. Cortes, G. Garcia-Manero, S. Verstovsek, S. Faderl, D. A. Thomas, A. Ferrajoli, S. O’Brien, J. K. Wathen, L. C. Xiao, D. A. Berry, and E. H. Estey. 2003. Adaptive randomized study of idarubicin and cytarabine versus troxacitabine and cytarabine versus troxacitabine and idarubicin in untreated patients 50 years or older with adverse karyotype acute myeloid leukemia. Journal of Clinical Oncology 21(9):1722-1727.
Granger, C. B., V. Vogel, S. R. Cummings, P. Held, F. Fiedorek, M. Lawrence, B. Neal, H. Reidies, L. Santarelli, R. Schroyer, N. L. Stockbridge, and Z. Feng. 2008. Do we need to adjudicate major clinical events? Clinical Trials 5(1):56-60.
Grant, S. F., G. Thorleifsson, I. Reynisdottir, R. Benediktsson, A. Manolescu, J. Sainz, A. Helgason, H. Stefansson, V. Emilsson, A. Helgadottir, U. Styrkarsdottir, K. P. Magnusson, G. B. Walters, E. Palsdottir, T. Jonsdottir, T. Gudmundsdottir, A. Gylfason, J. Saemundsdottir, R. L. Wilensky, M. P. Reilly, D. J. Rader, Y. Bagger, C. Christiansen, V. Gudnason, G. Sigurdsson, U. Thorsteinsdottir, J. R. Gulcher, A. Kong, and K. Stefansson. 2006. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics 38(3):320-323.
Grosse, S. D., and M. J. Khoury. 2006. What is the clinical utility of genetic testing? Journal of Medical Genetics 8(7):448-450.
Gudbjartsson, D. F., D. O. Arnar, A. Helgadottir, S. Gretarsdottir, H. Holm, A. Sigurdsson, A. Jonasdottir, A. Baker, G. Thorleifsson, K. Kristjansson, A. Palsson, T. Blondal, P. Sulem, V. M. Backman, G. A. Hardarson, E. Palsdottir, A. Helgason, R. Sigurjonsdottir, J. T. Sverrisson, K. Kostulas, M. C. Ng, L. Baum, W. Y. So, K. S. Wong, J. C. Chan, K. L. Furie, S. M. Greenberg, M. Sale, P. Kelly, C. A. MacRae, E. E. Smith, J. Rosand, J. Hillert, R. C. Ma, P. T. Ellinor, G. Thorgeirsson, J. R. Gulcher, A. Kong, U. Thorsteinsdottir, and K. Stefansson. 2007. Variants conferring risk of atrial fibrillation on chromosome 4Q25. Nature 448(7151):353-357.
Gudmundsson, J., P. Sulem, A. Manolescu, L. T. Amundadottir, D. Gudbjartsson, A. Helgason, T. Rafnar, J. T. Bergthorsson, B. A. Agnarsson, A. Baker, A. Sigurdsson, K. R. Benediktsdottir, M. Jakobsdottir, J. Xu, T. Blondal, J. Kostic, J. Sun, S. Ghosh, S. N. Stacey, M. Mouy, J. Saemundsdottir, V. M. Backman, K. Kristjansson, A. Tres, A. W. Partin, M. T. Albers-Akkers, J. Godino-Ivan Marcos, P. C. Walsh, D. W. Swinkels, S. Navarrete, S. D. Isaacs, K. K. Aben, T. Graif, J. Cashy, M. Ruiz-Echarri, K. E. Wiley, B. K. Suarez, J. A. Witjes, M. Frigge, C. Ober, E. Jonsson, G. V. Einarsson, J. I. Mayordomo, L. A. Kiemeney, W. B. Isaacs, W. J. Catalona, R. B. Barkardottir, J. R. Gulcher, U. Thorsteinsdottir, A. Kong, and K. Stefansson. 2007. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8Q24. Nature Genetics 39(5):631-637.
Gulcher, J., and K. Stefansson. 1998. Population genomics: Laying the groundwork for genetic disease modeling and targeting. Clinical Chemistry and Laboratory Medicine 36(8):523-527.
Haga, S. B., M. J. Khoury, and W. Burke. 2003. Genomic profiling to promote a healthy lifestyle: Not ready for prime time. Nature Genetics 34(4):347-350.
Haiman, C. A., N. Patterson, M. L. Freedman, S. R. Myers, M. C. Pike, A. Waliszewska, J. Neubauer, A. Tandon, C. Schirmer, G. J. McDonald, S. C. Greenway, D. O. Stram, L. Le Marchand, L. N. Kolonel, M. Frasco, D. Wong, L. C. Pooler, K. Ardlie, I. Oakley-Girvan, A. S. Whittemore, K. A. Cooney, E. M. John, S. A. Ingles, D. Altshuler, B. E. Henderson, and D. Reich. 2007. Multiple regions within 8Q24 independently affect risk for prostate cancer. Nature Genetics 39(5):638-644.
Haines, J. L., M. A. Hauser, S. Schmidt, W. K. Scott, L. M. Olson, P. Gallins, K. L. Spencer, S. Y. Kwan, M. Noureddine, J. R. Gilbert, N. Schnetz-Boutaud, A. Agarwal, E. A. Postel, and M. A. Pericak-Vance. 2005. Complement factor H variant increases the risk of age-related macular degeneration. Science 308(5720):419-421.
Hall, D. H., T. Rahman, P. J. Avery, and B. Keavney. 2006. INSIG-2 promoter polymorphism and obesity related phenotypes: Association study in 1428 members of 248 families. BMC Medical Genetics 7:83.
Heikes, K. E., D. M. Eddy, B. Arondekar, and L. Schlessinger. 2007. Diabetes risk calculator: A simple tool for detecting undiagnosed diabetes and prediabetes. Diabetes Care 31(5):1040-1045.
Helgadottir, A., G. Thorleifsson, A. Manolescu, S. Gretarsdottir, T. Blondal, A. Jonasdottir, A. Sigurdsson, A. Baker, A. Palsson, G. Masson, D. F. Gudbjartsson, K. P. Magnusson, K. Andersen, A. I. Levey, V. M. Backman, S. Matthiasdottir, T. Jonsdottir, S. Palsson, H. Einarsdottir, S. Gunnarsdottir, A. Gylfason, V. Vaccarino, W. C. Hooper, M. P. Reilly, C. B. Granger, H. Austin, D. J. Rader, S. H. Shah, A. A. Quyyumi, J. R. Gulcher, G. Thorgeirsson, U. Thorsteinsdottir, A. Kong, and K. Stefansson. 2007. A common variant on chromosome 9P21 affects the risk of myocardial infarction. Science 316(5830):1491-1493.
Herbert, A., N. P. Gerry, M. B. McQueen, I. M. Heid, A. Pfeufer, T. Illig, H. E. Wichmann, T. Meitinger, D. Hunter, F. B. Hu, G. Colditz, A. Hinney, J. Hebebrand, K. Koberwitz, X. Zhu, R. Cooper, K. Ardlie, H. Lyon, J. N. Hirschhorn, N. M. Laird, M. E. Lenburg, C. Lange, and M. F. Christman. 2006. A common genetic variant is associated with adult and childhood obesity. Science 312(5771):279-283.
Higashi, M. K., D. L. Veenstra, L. M. Kondo, A. K. Wittkowsky, S. L. Srinouanprachanh, F. M. Farin, and A. E. Rettie. 2002. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. Journal of the American Medical Association 287(13):1690-1698.
Hirschhorn, J. N., K. Lohmueller, E. Byrne, and K. Hirschhorn. 2002. A comprehensive review of genetic association studies. Journal of Medical Genetics 4(2):45-61.
Hlatky, M. A., R. M. Califf, F. E. Harrell, Jr., K. L. Lee, D. B. Mark, and D. B. Pryor. 1988. Comparison of predictions based on observational data with the results of randomized controlled clinical trials of coronary artery bypass surgery. Journal of the American College of Cardiology 11(2):237-245.
Hoover, R. N. 2007. The evolution of epidemiologic research: From cottage industry to “big” science. Epidemiology 18(1):13-17.
Hunter, D. J., and P. Kraft. 2007. Drinking from the fire hose—statistical issues in genomewide association studies. New England Journal of Medicine 357(5):436-439.
Hunter, D. J., P. Kraft, K. B. Jacobs, D. G. Cox, M. Yeager, S. E. Hankinson, S. Wacholder, Z. Wang, R. Welch, A. Hutchinson, J. Wang, K. Yu, N. Chatterjee, N. Orr, W. C. Willett, G. A. Colditz, R. G. Ziegler, C. D. Berg, S. S. Buys, C. A. McCarty, H. S. Feigelson, E. E. Calle, M. J. Thun, R. B. Hayes, M. Tucker, D. S. Gerhard, J. F. Fraumeni, Jr., R. N. Hoover, G. Thomas, and S. J. Chanock. 2007. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nature Genetics 39(7):870-874.
Hunter, D. J., M. J. Khoury, and J. M. Drazen. 2008. Letting the genome out of the bottle—will we get our wish? New England Journal of Medicine 358(2):105-107.
The International HapMap Consortium. 2003. The International HapMap Project. Nature 426(6968):789-796.
———. 2005. Nature 437(7063):1299-1320.
Ioannidis, J. P., E. E. Ntzani, T. A. Trikalinos, and D. G. Contopoulos-Ioannidis. 2001. Replication validity of genetic association studies. Nature Genetics 29(3):306-309.
IOM (Institute of Medicine). 2001. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press.
Janssens, A. C., M. Gwinn, R. Valdez, K. M. Narayan, and M. J. Khoury. 2006. Predictive genetic testing for type 2 diabetes. British Medical Journal 333(7567):509-510. Johns Hopkins University. 2008. Online Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/omim/ (accessed July 9, 2008).
Kaiser Permanente Medical Plan of Northern California. 2008. Research Program on Genes, Environment, and Health. http://www.dor.kaiser.org/studies/rpgeh/ (accessed July 9, 2008).
Kimmel, S. E. 2008. Warfarin therapy: In need of improvement after all these years. Expert Opinion on Pharmacotherapy 9(5):677-686.
Klein, R. J., C. Zeiss, E. Y. Chew, J. Y. Tsai, R. S. Sackler, C. Haynes, A. K. Henning, J. P. SanGiovanni, S. M. Mane, S. T. Mayne, M. B. Bracken, F. L. Ferris, J. Ott, C. Barnstable, and J. Hoh. 2005. Complement factor H polymorphism in age-related macular degeneration. Science 308(5720):385-389.
Li, J. S., E. L. Eisenstein, H. G. Grabowski, E. D. Reid, B. Mangum, K. A. Schulman, J. V. Goldsmith, M. D. Murphy, R. M. Califf, and D. K. Benjamin, Jr. 2007. Economic return of clinical trials performed under the pediatric exclusivity program. Journal of the American Medical Association 297(5):480-488.
Lyon, H. N., V. Emilsson, A. Hinney, I. M. Heid, J. Lasky-Su, X. Zhu, G. Thorleifsson, S. Gunnarsdottir, G. B. Walters, U. Thorsteinsdottir, A. Kong, J. Gulcher, T. T. Nguyen, A. Scherag, A. Pfeufer, T. Meitinger, G. Bronner, W. Rief, M. E. Soto-Quiros, L. Avila, B. Klanderman, B. A. Raby, E. K. Silverman, S. T. Weiss, N. Laird, X. Ding, L. Groop, T. Tuomi, B. Isomaa, K. Bengtsson, J. L. Butler, R. S. Cooper, C. S. Fox, C. J. O’Donnell, C. Vollmert, J. C. Celedon, H. E. Wichmann, J. Hebebrand, K. Stefansson, C. Lange, and J. N. Hirschhorn. 2007. The association of a SNP upstream of INSIG2 with body mass index is reproduced in several but not all cohorts. PLoS Genetics 3(4):e61.
Magnusson, K. P., S. Duan, H. Sigurdsson, H. Petursson, Z. Yang, Y. Zhao, P. S. Bernstein, J. Ge, F. Jonasson, E. Stefansson, G. Helgadottir, N. A. Zabriskie, T. Jonsson, A. Bjornsson, T. Thorlacius, P. V. Jonsson, G. Thorleifsson, A. Kong, H. Stefansson, K. Zhang, K. Stefansson, and J. R. Gulcher. 2006. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Medicine 3(1):e5.
Mailman, M. D., M. Feolo, Y. Jin, M. Kimura, K. Tryka, R. Bagoutdinov, L. Hao, A. Kiang, J. Paschall, L. Phan, N. Popova, S. Pretel, L. Ziyabari, M. Lee, Y. Shao, Z. Y. Wang, K. Sirotkin, M. Ward, M. Kholodov, K. Zbicz, J. Beck, M. Kimelman, S. Shevelev, D. Preuss, E. Yaschenko, A. Graeff, J. Ostell, and S. T. Sherry. 2007. The NCBI dbGaP database of genotypes and phenotypes. Nature Genetics 39(10):1181-1186.
Manolio, T. A. 2006. Taking our obligations to research participants seriously: Disclosing individual results of genetic research. American Journal of Bioethics 6(6):32-34; author reply W10-W32.
Manolio, T. A., L. L. Rodriguez, L. Brooks, G. Abecasis, D. Ballinger, M. Daly, P. Donnelly, S. V. Faraone, K. Frazer, S. Gabriel, P. Gejman, A. Guttmacher, E. L. Harris, T. Insel, J. R. Kelsoe, E. Lander, N. McCowin, M. D. Mailman, E. Nabel, J. Ostell, E. Pugh, S. Sherry, P. F. Sullivan, J. F. Thompson, J. Warram, D. Wholley, P. M. Milos, and F. S. Collins. 2007. New models of collaboration in genome-wide association studies: The genetic association information network. Nature Genetics 39(9):1045-1051.
Maraganore, D. M., M. de Andrade, T. G. Lesnick, K. J. Strain, M. J. Farrer, W. A. Rocca, P. V. Pant, K. A. Frazer, D. R. Cox, and D. G. Ballinger. 2005. High-resolution whole-genome association study of Parkinson’s disease. American Journal of Human Genetics 77(5):685-693.
Mazor, K. M., J. E. Sabin, D. Boudreau, M. J. Goodman, J. H. Gurwitz, L. J. Herrinton, M. A. Raebel, D. Roblin, D. H. Smith, V. Meterko, and R. Platt. 2007. Cluster randomized trials: Opportunities and barriers identified by leaders of eight health plans. Medical Care 45(10 Supl 2):S29-S37.
McClellan, M., B. J. McNeil, and J. P. Newhouse. 1994. Does more intensive treatment of acute myocardial infarction in the elderly reduce mortality? Analysis using instrumental variables. Journal of the American Medical Association 272(11):859-866.
Michener, J. L., S. Yaggy, M. Lyn, S. Warburton, M. Champagne, M. Black, M. Cuffe, R. Califf, C. Gilliss, R. S. Williams, and V. J. Dzau. 2008. Improving the health of the community: Duke’s experience with community engagement. Academic Medicine 83(4):408-413.
Mount Hood Modeling Group. 2007. Computer modeling of diabetes and its complications: A report on the fourth Mount Hood challenge meeting. Diabetes Care 30(6):1638-1646.
MRC Streptomycin in Tuberculosis Studies Committee. 1948. Streptomycin treatment of pulmonary tuberculosis: A medical research council investigation. British Medical Journal 2(4582):769-782.
Murphy, S. N., M. E. Mendis, D. A. Berkowitz, I. Kohane, and H. C. Chueh. 2006. Integration of clinical and genetic data in the I2B2 architecture. AMIA Annual Symposium Proceedings 1040.
Murray, M. D., F. E. Smith, J. Fox, E. Y. Teal, J. G. Kesterson, T. A. Stiffler, R. J. Ambuehl, J. Wang, M. Dibble, D. O. Benge, L. J. Betley, W. M. Tierney, and C. J. McDonald. 2003. Structure, functions, and activities of a research support informatics section. Journal of the American Medical Informatics Association 10(4):389-398.
Myers, R. H. 2006. Considerations for genomewide association studies in Parkinson disease. American Journal of Human Genetics 78(6):1081-1082.
National Human Genome Research Institute. 2008a. A Catalog of Published Genome-wide Association Studies. http://www.genome.gov/GWAstudies/ (accessed July 9, 2008).
———. 2008b. Promoting Safe and Effective Genetic Testing in the United States. http://www. genome.gov/10001733 (accessed July 9, 2008).
Palmer, L. J. 2007. UK biobank: Bank on it. Lancet 369(9578):1980-1982.
Pearson, T. A., and T. A. Manolio. 2008. How to interpret a genome-wide association study. Journal of the American Medical Association 299(11):1335-1344.
Peto, R., R. Collins, and R. Gray. 1995. Large-scale randomized evidence: Large, simple trials and overviews of trials. Journal of Clinical Epidemiology 48(1):23-40.
Rassen, J. A., M. A. Brookhart, et al. 2009. Instrumental variables II: Instrumental variable application—in 25 variations, the physician prescribing preference generally was strong and reduced covariate imbalance. Journal of Clinical Epidemiology 62(12):1233-1241.
Reynolds, A., J. Rubin, G. Clermont, J. Day, Y. Vodovotz, and G. Bard Ermentrout. 2006. A reduced mathematical model of the acute inflammatory response: I. Derivation of model and analysis of anti-inflammation. Journal of Theoretical Biology 242(1):220-236.
Ridker, P. M., D. I. Chasman, R. Y. Zee, A. Parker, L. Rose, N. R. Cook, and J. E. Buring. 2008. Rationale, design, and methodology of the women’s genome health study: A genome-wide association study of more than 25,000 initially healthy American women. Clinical Chemistry 54(2):249-255.
Rieder, M. J., A. P. Reiner, B. F. Gage, D. A. Nickerson, C. S. Eby, H. L. McLeod, D. K. Blough, K. E. Thummel, D. L. Veenstra, and A. E. Rettie. 2005. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. New England Journal of Medicine 352(22):2285-2293.
Saka, G., J. E. Kreke, A. J. Schaefer, C. C. Chang, M. S. Roberts, and D. C. Angus. 2007. Use of dynamic microsimulation to predict disease progression in patients with pneumonia-related sepsis. Critical Care 11(3):R65.
Saxena, R., B. F. Voight, V. Lyssenko, N. P. Burtt, P. I. de Bakker, H. Chen, J. J. Roix, S. Kathiresan, J. N. Hirschhorn, M. J. Daly, T. E. Hughes, L. Groop, D. Altshuler, P. Almgren, J. C. Florez, J. Meyer, K. Ardlie, K. Bengtsson Bostrom, B. Isomaa, G. Lettre, U. Lindblad, H. N. Lyon, O. Melander, C. Newton-Cheh, P. Nilsson, M. Orho-Melander, L. Rastam, E. K. Speliotes, M. R. Taskinen, T. Tuomi, C. Guiducci, A. Berglund, J. Carlson, L. Gianniny, R. Hackett, L. Hall, J. Holmkvist, E. Laurila, M. Sjogren, M. Sterner, A. Surti, M. Svensson, R. Tewhey, B. Blumenstiel, M. Parkin, M. Defelice, R. Barry, W. Brodeur, J. Camarata, N. Chia, M. Fava, J. Gibbons, B. Handsaker, C. Healy, K. Nguyen, C. Gates, C. Sougnez, D. Gage, M. Nizzari, S. B. Gabriel, G. W. Chirn, Q. Ma, H. Parikh, D. Richardson, D. Ricke, and S. Purcell. 2007. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316(5829):1331-1336.
Schlessinger, L., and D. M. Eddy. 2002. Archimedes: A new model for simulating health care systems—the mathematical formulation. Journal of Biomedical Information 35(1): 37-50.
Schneeweiss, S. 2006. Sensitivity analysis and external adjustment for unmeasured confounders in epidemiologic database studies of therapeutics. Pharmacoepidemiology and Drug Safety 15(5):291-303.
Schneeweiss, S., R. J. Glynn, J. Avorn, and D. H. Solomon. 2005. A medicare database review found that physician preferences increasingly outweighed patient characteristics as determinants of first-time prescriptions for cox-2 inhibitors. Journal of Clinical Epidemiology 58(1):98-102.
Schneeweiss, S., D. H. Solomon, P. S. Wang, J. Rassen, and M. A. Brookhart. 2006. Simultaneous assessment of short-term gastrointestinal benefits and cardiovascular risks of selective cyclooxygenase 2 inhibitors and nonselective nonsteroidal antiinflammatory drugs: An instrumental variable analysis. Arthritis and Rheumatism 54(11):3390-3398.
Scott, L. J., K. L. Mohlke, L. L. Bonnycastle, C. J. Willer, Y. Li, W. L. Duren, M. R. Erdos, H. M. Stringham, P. S. Chines, A. U. Jackson, L. Prokunina-Olsson, C. J. Ding, A. J. Swift, N. Narisu, T. Hu, R. Pruim, R. Xiao, X. Y. Li, K. N. Conneely, N. L. Riebow, A. G. Sprau, M. Tong, P. P. White, K. N. Hetrick, M. W. Barnhart, C. W. Bark, J. L. Goldstein, L. Watkins, F. Xiang, J. Saramies, T. A. Buchanan, R. M. Watanabe, T. T. Valle, L. Kinnunen, G. R. Abecasis, E. W. Pugh, K. F. Doheny, R. N. Bergman, J. Tuomilehto, F. S. Collins, and M. Boehnke. 2007. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316(5829):1341-1345.
Scriver, C. R. 2007. The PAH gene, phenylketonuria, and a paradigm shift. Human Mutation 28(9):831-845.
Sepp, T., J. C. Khan, D. A. Thurlby, H. Shahid, D. G. Clayton, A. T. Moore, A. C. Bird, and J. R. Yates. 2006. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Investigative Ophthalmology Visual Science 47(2):536-540.
Sherwin, R. S., R. M. Anderson, J. B. Buse, M. H. Chin, D. Eddy, J. Fradkin, T. G. Ganiats, H. N. Ginsberg, R. Kahn, R. Nwankwo, M. Rewers, L. Schlessinger, M. Stern, F. Vinicor, and B. Zinman. 2004. Prevention or delay of type 2 diabetes. Diabetes Care 27(Supl 1): S47-S54.
Shurin, S. B., and E. G. Nabel. 2008. Pharmacogenomics—ready for prime time? New England Journal of Medicine 358(10):1061-1063.
Sladek, R., G. Rocheleau, J. Rung, C. Dina, L. Shen, D. Serre, P. Boutin, D. Vincent, A. Belisle, S. Hadjadj, B. Balkau, B. Heude, G. Charpentier, T. J. Hudson, A. Montpetit, A. V. Pshezhetsky, M. Prentki, B. I. Posner, D. J. Balding, D. Meyre, C. Polychronakos, and P. Froguel. 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445(7130):881-885.
Solomon, D. H., S. Schneeweiss, R. J. Glynn, R. Levin, and J. Avorn. 2003. Determinants of selective cyclooxygenase-2 inhibitor prescribing: Are patient or physician characteristics more important? American Journal of Medicine 115(9):715-720.
Spiegelhlater, D. A. K., Myles, J. 2004. Bayesian Approaches to Clinical Trials and Health-care Evaluation. Chichester, West Sussex, UK: John Wiley & Sons.
Stacey, S. N., A. Manolescu, P. Sulem, T. Rafnar, J. Gudmundsson, S. A. Gudjonsson, G. Masson, M. Jakobsdottir, S. Thorlacius, A. Helgason, K. K. Aben, L. J. Strobbe, M. T. Albers-Akkers, D. W. Swinkels, B. E. Henderson, L. N. Kolonel, L. Le Marchand, E. Millastre, R. Andres, J. Godino, M. D. Garcia-Prats, E. Polo, A. Tres, M. Mouy, J. Saemundsdottir, V. M. Backman, L. Gudmundsson, K. Kristjansson, J. T. Bergthorsson, J. Kostic, M. L. Frigge, F. Geller, D. Gudbjartsson, H. Sigurdsson, T. Jonsdottir, J. Hrafnkelsson, J. Johannsson, T. Sveinsson, G. Myrdal, H. N. Grimsson, T. Jonsson, S. von Holst, B. Werelius, S. Margolin, A. Lindblom, J. I. Mayordomo, C. A. Haiman, L. A. Kiemeney, O. T. Johannsson, J. R. Gulcher, U. Thorsteinsdottir, A. Kong, and K. Stefansson. 2007. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nature Genetics 39(7):865-869.
Stangl, D. K. B., D.A. 2000. Meta-analysis in Medicine and Health Policy. New York: Marcel Dekker.
Stukel, T. A., E. S. Fisher, D. E. Wennberg, D. A. Alter, D. J. Gottlieb, and M. J. Vermeulen. 2007. Analysis of observational studies in the presence of treatment selection bias: Effects of invasive cardiac management on AMI survival using propensity score and instrumental variable methods. Journal of the American Medical Association 297(3):278-285.
Thorleifsson, G., K. P. Magnusson, P. Sulem, G. B. Walters, D. F. Gudbjartsson, H. Stefansson, T. Jonsson, A. Jonasdottir, G. Stefansdottir, G. Masson, G. A. Hardarson, H. Petursson, A. Arnarsson, M. Motallebipour, O. Wallerman, C. Wadelius, J. R. Gulcher, U. Thorsteinsdottir, A. Kong, F. Jonasson, and K. Stefansson. 2007. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 317(5843):1397-1400.
Todd, J. A. 2006. Statistical false positive or true disease pathway? Nature Genetics 38(7): 731-733.
Tunis, S. R., D. B. Stryer, and C. M. Clancy. 2003. Practical clinical trials: Increasing the value of clinical research for decision making in clinical and health policy. Journal of the American Medical Association 290(12):1624-1632.
U.S. Department of Health and Human Services. 1996. Health Insurance Portability and Accountability Act of 1996. http://aspe.hhs.gov/admnsimp/pL104191.htm (accessed July 10, 2008).
———. 2007. Policy for Sharing of Data Obtained in NIH Supported or Conducted Genomewide Association Studies (GWAS). http://grants.nih.gov/grants/guide/notice-files/NOT-OD-07-088.html (accessed July 10, 2008).
Vodovotz, Y., G. Clermont, C. Chow, and G. An. 2004. Mathematical models of the acute inflammatory response. Current Opinion in Critical Care 10(5):383-390.
Wallace, C., S. J. Newhouse, P. Braund, F. Zhang, M. Tobin, M. Falchi, K. Ahmadi, R. J. Dobson, A. C. Marcano, C. Hajat, P. Burton, P. Deloukas, M. Brown, J. M. Connell, A. Dominiczak, G. M. Lathrop, J. Webster, M. Farrall, T. Spector, N. J. Samani, M. J. Caulfield, and P. B. Munroe. 2008. Genome-wide association study identifies genes for biomarkers of cardiovascular disease: Serum urate and dyslipidemia. American Journal of Human Genetics 82(1):139-149.
Wang, L., and R. Weinshilboum. 2006. Thiopurine s-methyltransferase pharmacogenetics: Insights, challenges and future directions. Oncogene 25(11):1629-1638.
Weedon, M. N. 2007. The importance of TCF7L2. Diabetes Medicine 24(10):1062-1066.
Weedon, M. N., G. Lettre, R. M. Freathy, C. M. Lindgren, B. F. Voight, J. R. Perry, K. S. Elliott, R. Hackett, C. Guiducci, B. Shields, E. Zeggini, H. Lango, V. Lyssenko, N. J. Timpson, N. P. Burtt, N. W. Rayner, R. Saxena, K. Ardlie, J. H. Tobias, A. R. Ness, S. M. Ring, C. N. Palmer, A. D. Morris, L. Peltonen, V. Salomaa, G. Davey Smith, L. C. Groop, A. T. Hattersley, M. I. McCarthy, J. N. Hirschhorn, and T. M. Frayling. 2007. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nature Genetics 39(10):1245-1250.
Wellcome Trust Case Control Consortium. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447(7145):661-678.
Willer, C. J., S. Sanna, A. U. Jackson, A. Scuteri, L. L. Bonnycastle, R. Clarke, S. C. Heath, N. J. Timpson, S. S. Najjar, H. M. Stringham, J. Strait, W. L. Duren, A. Maschio, F. Busonero, A. Mulas, G. Albai, A. J. Swift, M. A. Morken, N. Narisu, D. Bennett, S. Parish, H. Shen, P. Galan, P. Meneton, S. Hercberg, D. Zelenika, W. M. Chen, Y. Li, L. J. Scott, P. A. Scheet, J. Sundvall, R. M. Watanabe, R. Nagaraja, S. Ebrahim, D. A. Lawlor, Y. Ben-Shlomo, G. Davey-Smith, A. R. Shuldiner, R. Collins, R. N. Bergman, M. Uda, J. Tuomilehto, A. Cao, F. S. Collins, E. Lakatta, G. M. Lathrop, M. Boehnke, D. Schlessinger, K. L. Mohlke, and G. R. Abecasis. 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nature Genetics 40(2):161-169.
Yang, Q., J. Cui, I. Chazaro, L. A. Cupples, and S. Demissie. 2005. Power and type I error rate of false discovery rate approaches in genome-wide association studies. BMC Genetics 6(Supl 1):S134.
Yeager, M., N. Orr, R. B. Hayes, K. B. Jacobs, P. Kraft, S. Wacholder, M. J. Minichiello, P. Fearnhead, K. Yu, N. Chatterjee, Z. Wang, R. Welch, B. J. Staats, E. E. Calle, H. S. Feigelson, M. J. Thun, C. Rodriguez, D. Albanes, J. Virtamo, S. Weinstein, F. R. Schumacher, E. Giovannucci, W. C. Willett, G. Cancel-Tassin, O. Cussenot, A. Valeri, G. L. Andriole, E. P. Gelmann, M. Tucker, D. S. Gerhard, J. F. Fraumeni, Jr., R. Hoover, D. J. Hunter, S. J. Chanock, and G. Thomas. 2007. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nature Genetics 39(5):645-649.
Yusuf, S. 2004. Randomized clinical trials: Slow death by a thousand unnecessary policies? Canadian Medical Association Journal 171(8):889-892; discussion 892-883.
Yusuf, S., J. Bosch, P. J. Devereaux, R. Collins, C. Baigent, C. Granger, R. Califf, and R. Temple. 2008. Sensible guidelines for the conduct of large randomized trials. Clinical Trials 5(1):38-39.
Zareparsi, S., K. E. Branham, M. Li, S. Shah, R. J. Klein, J. Ott, J. Hoh, G. R. Abecasis, and A. Swaroop. 2005. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. American Journal of Human Genetics 77(1):149-153.
Zeggini, E., M. N. Weedon, C. M. Lindgren, T. M. Frayling, K. S. Elliott, H. Lango, N. J. Timpson, J. R. Perry, N. W. Rayner, R. M. Freathy, J. C. Barrett, B. Shields, A. P. Morris, S. Ellard, C. J. Groves, L. W. Harries, J. L. Marchini, K. R. Owen, B. Knight, L. R. Cardon, M. Walker, G. A. Hitman, A. D. Morris, A. S. Doney, M. I. McCarthy, and A. T. Hattersley. 2007. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316(5829):1336-1341.


































































