
Page 563
21—
Obesity and Eating Disorders
For more than 50 years, the life insurance industry has pointed out that increased body weight is associated with excess mortality. This has been one stimulus for including measures of body weight, stature, and occasionally skinfolds in epidemiologic studies on the factors associated with the development of cardiovascular diseases and cancer. In recent years, fat distribution has also been included. It is now clear that two important factors associated with the risk of developing several chronic diseases are total body fat (most often estimated from ratios of body weight to height) and the distribution of that fat on the abdomen and trunk or peripherally on the arms and legs.
Overweight, obesity, and adiposity are commonly used terms. Overweight can be expressed as relative weight or ratios of weight to height. Relative weight is the ratio of actual to standard weight as determined from a table of reference body weights expressed relative to height, frequently as a percentage. Weight-to-height ratios can be expressed in several ways. The most widely used is the body mass index (BMI) or Quetelet index (QI), which is body weight (in kilograms) divided by the square of the height (in meters), i.e., weight/(height)2. The BMI is more highly correlated with body fat than with other indices of height and weight (Benn, 1971). The nomogram in Figure 21-1 allows rapid determination of BMI for given levels of height and weight.
Obesity refers to an excess of total body fat, which can be assessed by a variety of techniques (see Chapter 6).
Adiposity refers to both the distribution and the size of the adipose tissue depots. Since half or more of the body fat is subcutaneous, measurement of skinfold thickness has been used frequently to estimate fat and its distribution. Other techniques involve the use of soft tissue x-rays, ultrasound, electrical conductivity, electrical impedance, computed tomographic scans, and magnetic resonance imaging scans. From a practical point of view, both the ratio of waist-to-hip (WHR) circumference and the ratio of triceps-to-subscapular skinfolds have proven useful.
Fat cells in specific depots can be measured by needle biopsies of adipose tissue followed by osmium fixation of the fat cell (Hirsch and Gallian, 1968) or separation of isolated fat cells and then measurement of their size under a microscope (Lavau et al., 1977).
Weight Standards
Body Weight
The generation of national weight standards requires information on a large group of subjects. For most of the twentieth century, the life insurance industry has provided the data base for the
Page 564
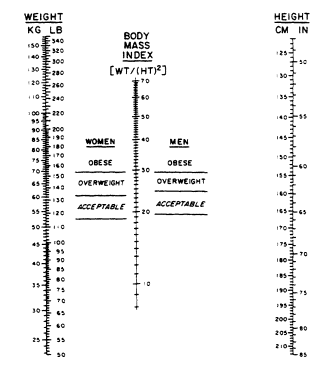
FIGURE 21-1
Nomogram for determining BMI. To use this nomogram, place
a ruler or other straight edge between the body weight in
kilograms or pounds (without clothes) located on the left-hand column
and the height in centimeters or in inches (without shoes)
located on the right-hand column. The BMI is read from the middle of
the scale and is in metric units. Copyright 1978, George A. Bray. Used by permission.
most widely used tables of desirable body weight. Both the 1959 and the 1983 Metropolitan Life Insurance tables were based on data obtained from the pooled experience of the life insurance industry in the United States (Society of Actuaries, 1959, 1980a,b). Although these surveys of weight and stature among insured individuals provide data on nearly 5 million people, they suffer from a self-selection bias, i.e., they provide data only on people who choose to take out life insurance. The insured tend to have a longer life expectancy, to be healthier, and, on average, to weigh less than the general population.
A second data base has been generated by the National Center for Health Statistics (NCHS), which in the past 20 years performed five surveys, including measurements of weight and stature of a representative sample of Americans from census tracts in the United States (Abraham et al., 1983). These surveys include approximately 20,000 people.
Appropriate weight standards can be determined in two ways. First, the normal distribution of
| TABLE 21-1 Desirable Body Mass Index in Relation to Age | |
| Age Group | BMI |
| (years) | (kg/m2) |
| 19-24 | 19-24 |
| 25-34 | 20-25 |
| 35-44 | 21-26 |
| 45-54 | 22-27 |
| 55-65 | 23-28 |
| >65 | 24-29 |
weight in relation to height can be arbitrarily divided into overweight and severely overweight groups. This approach has been used by the NCHS, which defines overweight as those in the 85th percentile of weight for height using as reference the weights of 20- to 29-year-olds. With this technique, a BMI higher than 27.8 kg/m2 for men and above 27.3 kg/m2 for women is considered overweight, and the top 5th percentile is severely overweight. This approach was used in the Surgeon General's Report on Nutrition and Health (DHHS, 1988) but not by the National Institute on Aging. There are several drawbacks with this approach. First, the standards change as the weight distribution of the population changes. Second, the 85th percentile values of the BMI, 27.8 kg/m2 and 27.3 kg/m2 for men and women, respectively, will be very difficult for health professionals and the public to remember or understand. Third, and more important, is the underlying assumption that average weight is a healthy or preferred weight. Lastly, it is assumed in this approach that optimal weights remain constant at different ages—an assumption that may not be justified (Andres, 1985).
Weight standards can also be based on the BMI associated with the lowest overall risk to health. The minimal death rate in several prospective studies is associated with a BMI of 22 to 25 kg/m2. Andres (1985) reanalyzed the Build and Blood Pressure Study of 1979 (Society of Actuaries, 1980a,b) and showed that the BMI associated with the lowest mortality increased with age. A similar increase in the BMI distribution curve with age is evident from a study conducted in Norway (Waaler, 1984). On the basis of these collated data, the ranges for BMI in relation to age proposed in Table 21-1 seem reasonable. Although the BMI is adjusted for age, the range overlaps. For example, the highest BMI for 19- to 24-year-olds is 24 kg/m2, which is the lowest for those over 65. A BMI above 25 kg/m2 was associated with increased
Page 565
risk of death in 78,612 young men followed for 32 years (Hoffmans et al., 1988). Alternatively, one might adopt the system used by the British and Australians, who define a normal BMI range as 20 to 25 kg/m2, overweight as a BMI of 25 to 30 kg/ m2, severe overweight as a BMI above 30 kg/m2, and massive overweight as a BMI above 40 kg/m2.
Data from several longitudinal surveys in the United States might also provide sufficient information for preparing weight tables. Longitudinal data bases are also available from foreign countries. For example, Waaler (1984) reported the relationship of weight and height to morbidity and mortality for all 1.7 million Norwegians, except those living in Oslo and Bergen. The size of this sample and the 5-year follow-up provide a useful basis on which to determine relationships between weight and risks for cardiovascular diseases, hypertension, and diabetes (Waaler, 1984).
Fat Distribution
Fat distribution can be estimated by skinfolds, by waist-to-hip circumference ratios, or by such sophisticated techniques as ultrasound, computed tomography, or magnetic resonance imaging. The ratio of central (abdominal) to peripheral (gluteal) fat distribution can also be estimated from subscapular skinfold thickness (Donahue et al., 1987; Stokes et al., 1985). Skinfold measurements on the trunk and extremities can be used for principal component analysis (Ducimetiere et al., 1986; Mueller, 1983).
Data bases for circumferences may be developed from Swedish studies in Göteborg (Lapidus et al., 1984; Larsson et al., 1984), from studies in Milwaukee, Wisconsin (Hartz et al., 1984; Kissebah et al., 1982), and from the Canadian Fitness Survey (Fitness and Amateur Sport, 1986). A nomogram for determining the abdominal-to-gluteal circumference ratio (AGR), or WHR, is shown in Figure 21-2. The percentile distribution of these values for men and women in relation to age is plotted in Figure 21-3 from data obtained in the Canadian Fitness Surveys (Fitness and Amateur Sport, 1986).
Adiposity Standards
Height and weight data have been accumulated on millions of people. In contrast, quantitative estimates of total fat have been determined only from much smaller samples (Cheek, 1968; Cohn et al., 1984; Garrow, 1978; Segal et al.,
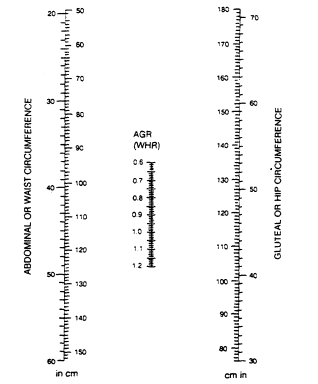
FIGURE 21-2
Nomogram for determining abdominal-to-gluteal circumference ratio (waist-to-hip ratio).
Place a straight edge between the column for waist circumference
and the column for hip circumference and read the ratio from the point
where this straight edge crosses the AGR or WHR line. The waist, or abdominal,
circumference is the smallest circumference below the rib cage and above
the umbilicus, and the hip, or gluteal, circumference is the largest circumference
at the posterior extension of the buttocks. From Bray and Gray (1988).
1985). Durnin and Womersley (1974) provided tables for estimating body fat from skinfolds measured at four different sites. No one as yet has tried to apply these Scottish standards to other populations. Steinkamp and colleagues (1965a,b) and several other groups have also provided useful equations for estimating body fat for men and women of various ages, using skinfolds from selected sites (Lohman, 1981; Lukaski, 1987; Steinkamp et al., 1965a,b). Triceps and subscapular skinfolds were measured in the surveys conducted by the NCHS (Abraham et al., 1979); however, these measurements cannot be used to establish standards for determining fatness because no data were collected on the relationship of these skinfolds to other measures of body fat. It is nonetheless possible to divide the population into percentiles of body fat by determining skinfold measurements from triceps and the subscapular region.
Page 566
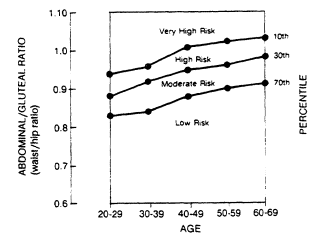
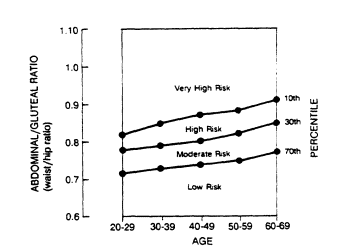
FIGURE 21-3
Percentiles of fat distribution. The percentiles of the ratio of
abdominal-to-gluteal circumference (waist-to-hip ratio) are depicted for
men and women by age groups along with relative risk. From Bray and Gray
(1988). Plotted from tabular data in the Canadian Standardized Test of Fitness, 1986.
Fat Cell Size and Number
The number of fat cells can be estimated from measurements of total body fat and the average size of a fat cell. A reliable estimate of the total number of fat cells should be based on the average size of fat cells from more than one location, because fat cells differ in size from one region to another. Normally, there are no more than 60 billion fat cells. In general, cells proliferate most rapidly from birth to 2 years of age and during late childhood and puberty. In some types of obesity, they can increase 3 to 5 times more than the normal number. In hypercellular obesity, the number of fat cells are increased. This type of obesity usually occurs in early or middle childhood but may also occur in adult life.
A higher-than-normal number of fat cells is usually present in people more than 75% above their desirable weight (Björntorp, 1985; Hirsch and Batchelor, 1976). When obesity begins during adult life, it often involves enlargement of adipose tissue cells. Hypertrophic obesity tends to correlate with an android or truncal fat distribution and is often associated with metabolic disorders such as glucose intolerance, hyperlipidemia, hypertension, and coronary artery disease (Feldman et al., 1969; Kissebah et al., 1982; Krotkiewski et al., 1983; Vague, 1956).
Stability of Total and Regional Fat
In several epidemiologic studies, investigators have examined the correlation between weights at two different ages (Borkan et al., 1986; Clarke et al., 1986; Noppa and Hällström, 1981; Zack et al., 1979). People in the lowest quintile for body weights at the end of childhood or adolescence may only have a small subsequent variation in body weight. For people in the middle or upper quintiles, however, there is considerable variability in weight; many individuals shift their weight one quintile up or down over a 5-year period. Mean body weights may show only a small upward trend during adult life, which camouflages considerable year-to-year fluctuations. In 1,302 women from Göteborg, Sweden, mean weight gain was 1.4 kg ± 5.1 kg SD over a 6-year period; 28 of the women lost more than 10 kg and 59 women gained more than 10 kg (Noppa and Hällström, 1981). In the normative aging study of 1,396 men, the weight of 168 of the subjects increased more than 10% and 75 men lost more than 10%. The baseline weight of those who lost weight was higher (84.1 kg) than that of those who gained weight (77.3 kg) (Borkan et al., 1986).
A prospective follow-up study over 36 years points to the variability in body weight with age (Bradden et al., 1986). At age 36, 3,322 people born in 1946 were divided into weight categories, based on BMI. In this cohort, 5.3% of the men and 8.4% of the women were severely overweight (BMI >30 kg/m2) and 38.0% of the men and 24.2% of women were overweight (BMI of 25 to 29.9 kg/m2). The correlation between BMI at ages 26 and 36 was R = .64 for men and R = .66 for women. The authors drew the following conclusions: First, approximately 25% of the men and
Page 567
women in the cohort were obese both as children and as adults. Second, the remaining 75% of this cohort became obese adults—an event that could not be predicted from weights before age 20. Those who became obese between the ages of 11 and 36 were often not the heaviest during childhood. On the basis of all socioeconomic, demographic, and weight data, only 50 to 60% of the men and women in the top decile for weight at age 36 could be correctly predicted at age 26 to attain that weight.
Causes of Obesity: Increased Energy Intake or Decreased Energy Expenditure?
Energy Intake
There is little question that extreme changes in food intake produce corresponding changes in body weight (Bray, 1976; Forbes, 1987; Garrow, 1978). Short-term studies in men and one in women demonstrate that normal or overweight people who overeat on a metabolic ward gain considerable amounts of body fat. However, this is influenced by the genetic makeup of individuals (Poehlman et al., 1986) (see below). Total weight gain can be predicted for groups from the degree of excess caloric ingestion (Forbes, 1987). Early in this century, Gulick (1922) and Neumann (1902) suggested that overeating might not produce obesity. In a reanalysis of these two studies, Forbes (1984) showed a linear relationship for each subject between degree of increased food intake and change in body weight. On the other hand, previously obese individuals at normal or near-normal weight may require fewer calories to maintain this weight (Geissler et al., 1987; Leibel and Hirsch, 1984).
Keys and colleagues (1950), in their classic study of starvation in humans, demonstrated that reduced-calorie diets were associated with weight loss in normal, healthy volunteers. Kinsell et al. (1964) and others (e.g., Bray, 1976; Forbes, 1987; Garrow, 1978) reported that calorie restriction in obese subjects reduced body weight and fat stores. A vast literature collected primarily in developing countries also demonstrates a correlation between calorie restriction, body weight, and other indices of malnutrition.
In animals, there is also little question that increasing total caloric intake causes them to increase body weight. There are several ways of increasing voluntary food intake in animals, including the provision, of more palatable diets (Cruce et al., 1974; Sclafani and Springer, 1976), the administration of calorically dense drinking solutions (Faust et al., 1978), switching diets frequently (Collier, 1985), and increasing the fat content of the diet (Schemmel et al., 1970). In most of these cases, the ability to voluntarily increase intake is complicated by dietary composition. Thus, it is frequently difficult to separate the effects of caloric intake per se from the specific effects of dietary macronutrients, i.e., fats, carbohydrates, and proteins. In addition, such dietary manipulations have sometimes been confounded by the ingestion of diets whose protein-to-energy ratio is too low to support normal growth and maintenance.
There is no epidemologic evidence indicating that total fat intake per se, independent of total caloric intake, is associated with increased adiposity in the population. Obesity itself has not been found to be associated with dietary fat in either inter- or intrapopulation studies. A few clinical studies suggest that a high saturated fatty acid intake may be positively associated with obesity (see Chapter 7). Animal studies suggest that both the type and the amount of dietary fat, independent of total caloric intake, may contribute to obesity, possibly through more efficient metabolism of this nutrient relative to other nutrients (see Chapters 6 and 7).
Female rats fed isocaloric high-fat and high-carbohydrate diets spontaneously overingest the high-fat diet, compared to male rats, and become fatter (Hoyenga and Hoyenga, 1982; Sclafani and Gorman, 1977). In general, reduction in dietary fat leads to weight reduction, but this effect may be secondary to concomitant reductions in caloric intake. A more detailed discussion of the association of dietary fat with obesity can be found in Chapter 7.
Energy Intake and Energy Expenditure
As discussed in Chapter 6, epidemiologic studies of the relationship between body weight and energy intake have shown either no correlation or an inverse correlation (Baecke et al., 1983; Braitman et al., 1985; Keen et al., 1979; Kromhout, 1983; Lapidus et al., 1986; Romieu et al., 1988). In contrast, the direct measurements of energy expenditure for periods ranging from a few hours to 72 hours by indirect or direct calorimeters show a direct positive correlation between energy expenditure and body weight, body fat, fat-free mass,
Page 568
and body surface area (de Boer et al., 1986; Garrow, 1978; Jequier, 1984; Owen et al., 1986, 1987; Ravussin et al., 1986). This direct positive correlation between energy expenditure and fat-free mass has an important genetic component, since the variance among siblings is smaller than the variance among families (Bogardus et al., 1986; Bouchard et al., 1986). Thus, increases in adiposity in some cases must reflect decreases in energy expenditure or changing metabolic efficiency.
Metabolic Efficiency
Animal studies provide clear evidence that increased energy intake is not required to induce obesity (Bray and York, 1971, 1979). The most clear-cut examples are animals with recessively inherited forms of obesity, which, despite precise paired feeding, had greater weight gain and fat deposition than their lean littermates (Cleary et al., 1980; Coleman, 1979; Cox and Powley, 1981). These differences cannot be explained by differences in work-related energy expenditure.
Similar studies have been conducted in animals with hyperphagia and obesity resulting from damage to the hypothalamus (Bray and York, 1979; Cox and Powley, 1981). When such animals are pair-fed with lean animals, the animals with lesions become obese but the lean animals do not. These studies imply differences in metabolic efficiency.
Studies in animals indicate that increased energy intake usually results in obesity (Bray, 1987a) but is not essential for the development of modest levels of obesity. If this phenomenon exists among heterogeneous groups of obese humans, then some forms of obesity are probably associated with enhanced metabolic efficiency, whereas others may require increased energy intake or decreased energy expenditure. Recent prospective studies of Pima Indians (Ravussin et al., 1988) and infants born to overweight mothers (Roberts et al., 1988) further support the possibility that lower energy expenditure can predict the subsequent development of obesity.
Genetic Factors in Obesity
It has been known for decades that obesity is a familial trait (Bray, 1976). Since members of a kindred share many dietary and other environmental exposures, the identification of genetic factors is complex. As discussed in Chapter 4, the most compelling evidence for a genetic component in obesity comes from examinations of monozygotic and dizygotic twins (e.g., Börjeson, 1976; Bouchard, 1988; Fabsitz et al., 1980; Feinleib et al., 1977; Medlund et al., 1976; Stunkard et al., 1986). Börjeson (1976) studied 40 monozygotic and 61 same-sex dizygotic twin pairs and estimated heritability of obesity to be 88%. (Heritability may range from 0, i.e., no genetic factors, to 100%, indicating that a trait is entirely genetically determined.) A similar conclusion by Brook et al. (1975) was based on studies of monozygotic and dizygotic twins. Two large studies of twins confirmed the importance of genetic factors in obesity and the distribution of body fat (Bouchard, 1988; Stunkard et al., 1986). A strong interaction between genetic predisposition and environmental stimuli is suggested by the findings of Poehlman et al. (1986), who reported differential responses of twin pairs to overfeeding. Although weight gain was closely correlated within a pair of twins, there was wide scatter in values among pairs of twins. Studies of twins suggest that there is an important genetic component in the etiology of human obesity.
In addition to obesity per se, there is increasing evidence that patterns of fat distribution are inherited. This was indicated in a study of twins (Bouchard, 1988) and in several studies of ethnic groups (Mueller, 1988). These patterns may be apparent among children. For example, Mexican-American children have a pronounced upper-body distribution of fat, which is sometimes independent of adiposity per se (Mueller, 1988), whereas children of European origin seem to have a more peripheral fat distribution (Mueller, 1988).
The specific genes involved in obesity are still unknown. Such genes may have played an important evolutionary role by improving the survival of people who could store fat more efficiently during periods of prosperity and use it during caloric deprivation. If this is true, then one might expect to find some people who develop obesity more readily than others.
Obesity in mice and rats can be caused by several different single gene mutations (Bray and York, 1979). Some strains of pigs (Steele et al., 1974) and rats become obese, whereas control animals fed the same diet do not (Schemmel et al., 1970). The exact biochemical or metabolic defects leading to such observations have not yet been discovered. Intensive research is under way to clone relevant candidate genes.
Monogenic animal models of obesity will most probably not fully explain the genetic factors op-
Page 569
erative in human obesity, which do not appear to be monogenic; however, some genes involved in monogenic animal obesity may play some role in human obesity. Information on the genes involved in animal obesity will provide a basis for the direct testing of similar genes in humans with the techniques of molecular genetics.
Evidence Associating Overweight or Obesity with Chronic Diseases and Physiologic Function
Epidemiologic data on the relationship between body weight and the risk of chronic diseases, or between body weight and mortality, indicate that the relationship is curvilinear and often J- or U-shaped. That is, mortality increases as body weight increases or decreases away from the mean. This J-shaped relationship applies to overall mortality as well as to cardiovascular diseases, cancer, diabetes, hypertension, gallbladder disease, and osteoporosis (Bray, 1985). For example, data from the Pooling Project show that the underweight group has the highest mortality (McGee and Gordon, 1976).
Cardiovascular Diseases
Epidemiologic Studies
Many epidemiologic studies have contributed to our current perspective on the relationship of overweight and adiposity to atherosclerotic cardiovascular diseases. These include retrospective as well as prospective studies.
Cardiovascular diseases are the major cause of death in the United States. This fact has provided the stimulus for many epidemiologic studies, which have attempted to identify the risk factors associated with these multifactorial diseases. In a review of many such studies, Manson et al. (1987) concluded that overweight generally increases the risk of death, especially sudden death, although it is not always an independent risk factor. Manson et al. (1987) also pointed out in a review of mortality statistics related to body weight indices that there are three major drawbacks to most studies in this area. First, the authors frequently do not separate smokers, who tend to have lower weights and higher mortality, from nonsmokers, who have lower mortality. Second, they fail to take into account the impact of early mortality on longer-range mortality trends. Third, they focus on the identification of obesity as an independent risk factor rather than on its relationship to intermediary complications such as diabetes, hypertension, or hyperlipidemia through which the effects of increased body weight on chronic diseases are likely to become manifest. Obesity must modify some intermediate mechanism, such as cardiac function or the metabolism of lipid or glucose, to produce its effects on risks that have not otherwise been controlled for or identified. Overweight and obesity are more likely to be identified as significant risk factors in studies with large numbers of subjects of all ages, with a large proportion of younger people, and with a follow-up longer than 15 years.
Obesity itself is not independently associated with the severity of atherosclerosis (Solberg and Strong, 1983), and the precise role of obesity in the etiology of atherosclerosis and CHD is unclear. However, some studies show that there is an association between obesity and risk of coronary death at the upper range of body weight, e.g. > 140% of ideal body weight or a BMI >30 (Donahue et al., 1987). Furthermore, the pattern of fat distribution, as discussed below, may be an important risk factor for CHD. In general, dietary composition (e.g., type of fat) and lifestyle are more closely associated with risk of CHD than is overweight, except for severe overweight.
Applicants for individual life insurance policies usually undergo a medical examination. In the Build and Blood Pressure Study of 1979 (Society of Actuaries, 1980a,b), weights were measured for approximately 87% of the almost 4 million men and 600,000 women on whom policies were written (Society of Actuaries, 1980a,b).
The lowest death rate occurred at a body weight slightly lower than the average weight for the entire population. As body weight increased, there was a progressive increase in mortality. Life insurance data show that mortality is increased to 125% of expected levels in individuals who are 5 to 15% overweight and rises to more than 500% of the expected level in people who are 25% or more overweight. There was also an increase in excess mortality at very low body weights, but the causes were different from the causes of death for people with a high BMI.
The curvilinear relationship of excess mortality to excess body weight is evident for all age groups in the life insurance studies published in 1959 and 1980 (Society of Actuaries, 1959, 1980a,b). The lowest mortality occurred at high BMIs in this
Page 570
study as well as in a study conducted by Waaler (1984) in Norway.
Data on the association between BMI and mortality in the elderly are limited. Mattila et al. (1986) reported that survival over a 5-year observation period was related to the BMI of 674 people 65 years of age or older. Those with a BMI of 22 kg/m2 or less had a lower survival rate compared to people with a BMI between 22 and 28 kg/m2. These data are consistent with most population data, which show that increased risk is associated with underweight and that it also increases gradually with BMI above 25 kg/m2.
Few morbidly obese people (greater than twice the ideal body weight) receive life insurance and, thus, are not represented in life insurance statistics. Drenick et al. (1980) have provided some insight into the effects of gross obesity on life expectancy. They reviewed medical records of 200 morbidly obese men (average weight, 143.5 kg), who were admitted to a Veterans Administration hospital for a weight control program and then followed for an average of 7.5 years. One hundred eighty-five of these men were followed until death or termination of the study. The mortality rate for these subjects was higher at all ages when compared with the mortality expected for the general population of U.S. males. In men ages 25 to 34, mortality increased 12-fold. In those ages 35 to 44, mortality increased fivefold. For the 55- to 64-year-old group, however, the mortality rate was only double that of the average U.S. population.
In the Pooling Project, data from five prospective studies of factors involved in the development of coronary heart disease (CHD) were pooled for analytical purposes. These studies included three occupation-based studies (Albany Civil Servants, Chicago Gas Company, Chicago Western Electric Company) and two community-based studies (Framingham, Massachusetts, and Tecumseh, Michigan). The subjects were 8,422 white men ages 40 to 64 years. The mean length of follow-up was 8.6 years. A high relative weight was associated with an increased risk of a first major coronary event only for men in their forties. For the older groups, there was no effect of age on the risk of developing CHD. Using quintiles of body weight at 5-year age intervals, the investigators found that the relative risk of developing heart disease was 2.1 among 40- to 44-year-old men and that the risk fell to 1.6 for men ages 45 to 49. This analysis means that men ages 40 to 44 in the highest quintile for relative weight were 2.1 times as likely to develop a first coronary event within an average of 3.6 years than men in the lowest quintile. In the men over 55, the gradual increase in risk with increasing weight was negligible.
The Framingham Study included 2,252 men and 2,818 women living in Framingham, Massachusetts. Participants were initially examined between 1948 and 1950 and reexamined at 2-year intervals thereafter. The median body weights of these subjects were almost identical to the upper limits for large-frame people in life insurance tables published in 1959 by the Metropolitan Life Insurance Company. On the basis of these tables, 15% of the men and 19% of the women were 20% overweight, and 3% of the men and 9% of the women were more than 50% overweight. Weight gain was associated with an increase in serum cholesterol, blood pressure, uric acid, and blood glucose (Ashley and Kannel, 1974). Overweight was also associated with an increased risk of sudden death, presumably from CHD; however, the exact cause is unknown.
After 26 years, there were 870 deaths among the men and 688 among the women. Relative weight at entry into the study was an independent predictor for development of cardiovascular diseases, particularly in women (Hubert et al., 1983). The 26-year incidence of CHD (including angina pectoris), death from CHD, and the likelihood of developing congestive heart failure in men was predicted from the initial degree of overweight using multiple logistic regression analysis. The predictive power for the relative degree of overweight was independent of age, cholesterol levels, systolic blood pressure, cigarette smoking, or glucose intolerance.
Relative body weight of the women was also positively and independently predictive of the likelihood for developing cardiovascular diseases, stroke, congestive heart failure, and death from CHD. Weight gain after the young adult years increased the risk of cardiovascular diseases in both sexes. There was, however, either no effect or a small inverse relationship of body weight to the frequency of intermittent claudication (Kannel and McGee, 1985).
Although excess weight was shown to be predictive of developing CHD in the Framingham study, it is not predictive of the outcome. Survival and functional status after a myocardial infarct were not affected by weight status (Kannel and Schatzkin, 1984) (see Chapter 19).
The Seven Countries Study involved an inter-
Page 571
national collaborative examination of risk factors for development of CHD in 16 cohorts of men in the United States, Europe, and Japan. Men at entry into this prospective study were between 40 and 59 years old. Overweight was defined as a BMI greater than 27 kg/m2, and obesity as the sum of triceps and subscapular skinfolds greater than 37 mm. On the basis of these criteria, more than half (52%) of the U.S. participants were obese. This was a substantially higher prevalence of obesity compared to that of the northern European group, but it was comparable to the prevalence in the southern European group.
Among the men from the United States and southern Europe, few, if any, significant relationships were found between body weight, the risk of myocardial infarction, and CHD death, but overweight was found to be correlated with angina pectoris. Among the men from northern Europe, the findings were nearly reversed. There were statistically significant correlations between all measures of body weight and ''hard" criteria (an actual occurrence) for coronary events, but no significant association was found between body weight and "soft" diagnostic criteria (e.g., elevated blood lipids) for CHD.
In the 15-year follow-up of these groups, there were 2,289 deaths—618 of which were from CHD. Relative body weight did not predict the risks of death from all causes or from CHD alone. These findings are contrary to the conclusions based on life insurance statistics.
The American Cancer Society studied the association of mortality with body weight in more than 750,000 people followed prospectively between 1959 and 1972 (Lew and Garfinkel, 1979). Relative death rates among subgroups that deviated above or below the average body weight were compared to the death rate for the group with weights that were 90 to 109% of the group mean. The overall mortality rate increased with increasing body weight.
This study also indicated that mortality for a smoker with a normal body weight (a BMI of 25 kg/m2) is comparable to that of a nonsmoker with a BMI of 30 to 35 kg/m2. Cardiovascular disease was a major factor in this increased mortality. No increase in mortality was observed until the BMI exceeded 25 kg/m2, at which point the increase in relative mortality was almost linear for both sexes. These findings are similar to the increases observed both in the Build and Blood Pressure Study of 1979 (Society of Actuaries, 1980a,b) and in the Framingham study (Kannel and Schatzkin, 1984).
In the Norwegian prospective study, height and weight measurements were obtained for most of that country's population during the course of mass x-ray screenings conducted between 1963 and 1975 (Waaler, 1984). For more than 95% of the subjects, BMIs were calculated on the basis of body weights and heights measured without shoes and with the upper body undressed in preparation for the x-rays. Of the 1.7 million men and women ages 15 to 90 followed in this study, 176,574 deaths occurred from 1963 to 1979. A J-shaped relationship was found between mortality and BMI, with the lowest mortality rates for males and females occurring at a BMI of 23 kg/m2, and increased rates observed for BMIs of less than 23 kg/m2. Relative mortality increased only slightly when at BMIs between 23 and 27 kg/m2. As the BMI increased above 27 kg/m2, however, there was a curvilinear increase in excess mortality.
There was also a strong inverse association between mortality and height. That is, short people had a higher death rate. The principal causes of excess mortality in the shorter people were tuberculosis, obstructive lung diseases, and cancer of the stomach and lung. Among the overweight subjects, the principal causes of death were cerebrovascular diseases, cardiovascular diseases, diabetes mellitus, and cancer of the colon. From his analysis, Waaler (1984) concluded that at the optimal BMI, the total mortality would be reduced by an additional 15%.
Clinical Studies
Associations of obesity with alterations in lipoprotein metabolism may be related to the risk of developing coronary atherosclerosis (Egusa et al., 1985; Grundy et al., 1979) (see Chapter 19). First, high-density-lipoprotein (HDL2) cholesterol decreases in obese males and females (Gordon et al., 1977b). Second, serum total cholesterol in obesity is normal or only slightly elevated, although the transport of low-density-lipoprotein (LDL) cholesterol through the plasma compartment increases. This increased transport is consistent with the correlation between increased cholesterol production and obesity, which amounts to approximately 20 more milligrams of cholesterol for each extra kilogram of body fat (Miettinen, 1971; Nestel et al., 1973). Third, the production of very-low-density-lipoprotein (VLDL) triglyceride and the corresponding apoprotein (VLDL-B) tends to increase in relation to the degree of obesity in
Page 572
Caucasians and Pima Indians (Bennion and Grundy, 1978). Whether or not triglyceride levels increase, as reported in many studies, depends on the rate at which triglycerides are cleared from the plasma. Fourth, the high rate of apoprotein B synthesis in LDL-B is probably related to the high rate of synthesis of apoprotein B for incorporation into VLDL. Fifth, lipoprotein lipase, the clearing-factor enzyme for lipoproteins, increases in obesity and may help prevent elevated triglyceride levels in some subjects. Finally, free fatty acid concentrations frequently increase in obesity, reflecting their higher rate of turnover.
Hypertension
It is widely believed that the indirect auscultatory method of obtaining blood pressure with an inflatable cuff produces higher readings in obese individuals than does direct intraarterial measurement. This may not occur, however, if the blood pressure cuff bladder is sufficiently long. With short cuffs, greater differences have been observed between systolic and diastolic pressures measured by direct intraarterial methods and indirect measurements. Despite this potential problem, there is clear evidence that increased body weight is associated with hypertension (Chaing et al., 1969; Havlik et al., 1983).
The increased blood pressure probably results from increased peripheral arteriolar resistance (Messerli, 1982), but the etiology of the increase in peripheral resistance is unknown. Tuck et al. (1981) suggested that it results from the increased secretion of catecholamines due to a hyperactive sympathetic nervous system.
Several associations emerge from a review of the relationship between body weight and hypertension. First, hypertension has a striking correlation not only with body weight but also with lateral body build. People with a large chest circumference relative to their height and weight have higher blood pressure than do slender individuals. In one study, hypertension was present in 37% of the broad-chested men but in only 3% of the narrow-chested men (Bray 1976). Body build was associated with an almost proportional effect on systolic and diastolic blood pressure. Second, when blood pressure was compared in groups with constant body build, there was no significant correlation between obesity and hypertension. The greatest correlation of blood pressure with obesity was observed in men with a slender build. A much smaller correlation was found in the broad-chested men. From these data, it appears that body build may be more important than obesity per se in the positive correlation between blood pressure and body weight (Weinsier et al., 1985). Two recent epidemiologic studies suggest that the effect of hypertension may be less severe in obese subjects than in normal-weight subjects. Two different groups found that the risk of cardiovascular mortality associated with hypertension and obesity is less than the risk of hypertension in normal-weight people (Barrett-Connor and Khaw, 1985; Cambien et al., 1985).
Messerli (1982) compared the cardiovascular consequences of hypertension in subjects with and without obesity. Cardiac output is increased in obese people, but not in nonobese, hypertensive people. The preload (volume inside the heart before it contracts), which is increased in obese people with or without hypertension, is normal in nonobese, hypertensive individuals. The presence of hypertension and obesity increases abnormalities of most other cardiac parameters, including afterload (resistance against which heart has to beat), stroke work (volume that the heart ejects in one heart beat against resistant pressure), and left ventricular mass. On the other hand, the nonobese hypertensive subject has a smaller chamber volume but a larger relative thickness of the left ventricular wall. Total peripheral resistance is sharply elevated in nonobese hypertensives but may decrease in obese people and become only modestly elevated in those who are obese and hypertensive. These data suggest an interaction between obesity and hypertension with reduced morbidity in obese hypertensives. However, this does not imply that becoming obese is an effective approach to treatment of hypertension, but may suggest that type of obesity, distribution of body fat (Hartz et al., 1984), weight history, or other lifestyle factors may be more important.
A reduction in blood pressure usually follows weight loss. During periods of caloric deprivation, such as World War I or World War II, hypertension was almost nonexistent. A number of clinical studies correlating changes in blood pressure with weight reduction have shown that blood pressure drops in 50 to 70% of those who lose weight. One explanation might be the reduced intake of salt that is associated with reduced caloric intake. However, Reisin et al. (1978) and Tuck et al. (1981) showed that blood pressure decreased even when salt intake was not reduced. Weight reduction is more effective in lowering systolic than diastolic pressure. Because reduction in blood pressure takes place as soon as food restriction begins,
Page 573
and then levels off, it has been suggested that the lowering of blood pressure may be a direct result of caloric deprivation rather than the result of eliminating obesity (Ernsberger and Nelson, 1988; Nelson and Ernsberger, 1984). Whether the therapeutic effect of weight reduction is related to the magnitude of the decline in body weight or to other environmental factors is still not clear. However, weight reduction was observed to produce a significant reduction in blood pressure in more than half of the hypertensive patients studied by MacMahon et al. (1986).
Diabetes Mellitus
Epidemiologic Studies
A variety of epidemiologic data indicate that adiposity increases the risk of noninsulin-dependent (Type II) diabetes mellitus (NIDDM). The U.S. National Diabetes Commission (DHEW, 1976) reported that the chance of becoming diabetic more than doubles with every 20% excess in body weight. In an epidemiologic study of 10,000 Israeli civil servants, those destined to develop diabetes were found to be considerably fatter. In a study of Pima Indians, the incidence of NIDDM was strongly related to preexisting obesity. When BMI was below 20 kg/m2, the incidence of diabetes was 0.8 cases per 1,000 person-years. The rate increased steadily as BMI increased, peaking at 72.2 cases per 1,000 person-years at a BMI over 40 kg/m2. This effect remained when subjects were classified according to the diabetic status of their parents (Knowler et al., 1978, 1981).
Rimm et al. (1975) investigated the relationship between diabetes and obesity in a retrospective analysis of more than 73,000 responses to a questionnaire submitted to members of a voluntary weight-loss group. Increases in body weight and aging were associated with increases in the frequency of NIDDM. Less than 1% of normal-weight women ages 25 to 44 reported NIDDM, whereas 7% of those of the same age who were 100% overweight (twice the ideal weight) reported this disease. Data from the Framingham study (Gordon et al., 1977a) also show that obesity is significantly associated with diabetes. Only plasma glucose was a better predictor than body weight for the risk of developing glucose intolerance over a 14-year period. Moreover, fasting glucose level changed in the same direction as body weight (Ashley and Kannel, 1974).
The increase in percentage of body weight for males and females between the ages of 25 and 60 in four different countries is also related to the frequency of diabetes. The mortality from NIDDM was highest among females who had the greatest gain in weight between ages 25 and 60. Canadian and U.S. populations had the greatest percentage increases in body weight between these ages compared to other populations of countries and thus had the highest mortality from diabetes.
The epidemiologic data are buttressed by the cross-cultural studies of West and Kalbfeisch (1971), who examined the prevalence of diabetes in 12 age-matched populations from 11 countries. There was a positive correlation (R = .89) between the prevalence of diabetes and standard weight in these populations. Larsson et al. (1981) also demonstrated a positive correlation between obesity and the development of diabetes over a 10-year period in a longitudinal study of nearly 900 middle-aged men in Göteborg.
Clinical Studies
In a retrospective analysis of patient charts, Joslin et al. (1935) found that 51% of diabetic males and 59% of diabetic females were at least 20% overweight and that 17% of the males and 26% of the females were actually 40% or more above average weight. Patients who developed diabetes between the ages of 20 and 35—presumably a mixture of insulin-dependent diabetes mellitus (IDDM) and NIDDM—had appreciably less excess body weight compared to those in older age groups.
Pyke and Please (1957) extended these studies by comparing the body weights of 946 patients. Below age 30, there was little difference in weight distribution between subjects with and without diabetes. Among 30-year-olds, the percentage of overweight diabetics exceeded that of nondiabetics. Among the older diabetics, 43 to 55% of the men and 51 to 55% of the women exceeded 110% of normal weight. Among nondiabetics, only 21 to 27% were >110% of normal weight.
Early studies demonstrated that impaired glucose tolerance is related to the duration of obesity. The prevalence of glucose intolerance in grossly obese subjects has been repeatedly found to be around 50%, varying somewhat with age, sex, and genetics. Lillioja et al. (1987) demonstrated that obese Pima Indians are at high risk of developing NIDDM and that this phenomenon aggregates in families. The risk of developing diabetes was highest among those with the most resistance to the effects of insulin (lowest rate of glucose disposal), and this resistance ran in families. Why only half
Page 574
the obese develop glucose intolerance remains an unresolved question. Its answer may be related to the impact of regional fat distribution.
Vague (1956) suggested that increased central body fat is more likely to be associated with the onset of diabetes mellitus. The female (or gynoid) type of body fat distribution (i.e., fat deposited primarily on the hips and thighs) was found to have a lower association with diabetes than the male (or android) type of obesity (i.e., fat deposited predominantly on the abdomen and upper body). Subsequently, Feldman et al. (1969), Hartz et al. (1984), and Ohlson et al. (1985) confirmed this association by demonstrating that diabetic subjects showed a significant shift toward an android or abdominal distribution of fat after onset of diabetes. Kissebah and colleagues (1982), who were pioneers in this area of research, conducted glucose tolerance tests and found that glucose and insulin levels increased more in subjects with upper-body obesity (high WHR) than in those with lower-body obesity when both groups had comparable amounts of total body fat. Krotkiewski et al. (1983), who also conducted a glucose tolerance test, found that both high total body fat and upper-body fat distribution were associated with a greater rise in glucose and insulin.
Recent data from the San Antonio Heart Study have extended these observations to 2,217 randomly selected Mexican-American individuals ages 25 to 60 (Hafner et al., 1987). A high ratio of subscapular to tricep skinfold or high WHR in a medically examined subset of 736 subjects was associated with high rates of NIDDM. BMI, a high WHR, and the ratio of subscapular tricep skinfold measurements all were independent predictors for the risk of NIDDM onset in females but not in males.
Beginning with the work of Himsworth (1935) and Newburgh and Conn (1939), studies have consistently shown that weight loss in obese subjects could improve glucose tolerance and that weight gain could worsen it. Glucose levels rise when normal-weight subjects gain weight and decline when they lose weight (Ashley and Kannel, 1974). This relationship was demonstrated most elegantly by Drenick et al. (1972), who showed the marked amelioration in glucose tolerance and insulin secretion after weight loss. In five obese men with normal glucose tolerance, insulin levels rose less during a glucose tolerance test when weight fell from 270 to 196 lb (112.5 to 81.6 kg) than before weight loss. After a 10- to 20-pound (4.5- to 9.0-kg) weight gain, glucose tolerance deteriorated substantially, and insulin secretion was inadequate. In another group of six men with abnormal glucose tolerance, there was improvement in insulin secretion and glucose tolerance after weight fell from 132.3 kg to 94.1 kg. After a modest regain of weight to 103.2 kg, glucose tolerance deteriorated significantly.
When normal-weight subjects voluntarily overeat to gain weight, not only is there a small but significant increase in plasma glucose but there is also a rise in the fasting insulin level (Sims et al., 1973). Basal levels of insulin increase linearly with the degree of overweight. This primarily represents increased insulin secretion associated with insulin resistance. Obese people also have a reduced response to the infusion of exogenous insulin, which indicates insulin resistance. This has been shown when the human forearm is perfused in vivo, when total glucose disposal is measured during the infusion of insulin, and when various tissues are studied in vitro (Olefsky et al., 1982; Rabinowitz, 1970). One mechanism for the resistance to insulin may be a reduction in the number of receptor sites on fat cells and other tissue cells (Olefsky et al., 1982). A second mechanism in some obese people is a postreceptor disturbance, in which genetic factors play an important role (Lillioja et al., 1987).
Animal Studies
Almost all forms of experimental obesity in animals are associated with hyperinsulinemia, and most of the animals have abnormalities in glucose tolerance. This is most prominent in obese (ob/ob) and diabetic (db/db) mice and obese (fa/fa) rats. This impairment in glucose tolerance can be mild, as in the fatty rat or the rat with hypothalamic obesity, or can be pronounced with associated ketoacidosis, as observed in the diabetic mouse (Bray and York, 1979) and the Wistar fatty rat (Ikeda et al., 1981; Kava et al., 1989). The impairment in glucose tolerance and the diabetic features of these animals have been shown to be improved by adrenalectomy (Bray, 1987a).
The mechanism for the hyperinsulinemia in obesity is only partly understood. Both humoral and neural mechanisms may play a role. The raised levels of several amino acids could act synergistically with glucose to enhance the secretion of insulin. Increased vagal tone or reduced sympathetic tone could also augment the release of insulin. The increased secretion of b-endorphin by the pituitary may stimulate insulin secretion, or alteration in fat storage enzymes such as lipoprotein lipase may lead to increased fat cell size and insulin resistance.
Page 575
Gallbladder Disease
Epidemiologic Studies
According to the American Cancer Society study by Lew and Garfinkel (1979), digestive diseases, primarily gallbladder disease, are next to diabetes in demonstrating the detrimental curvilinear effect of excess and substandard body weight. In a cross-sectional study of 62,739 respondents to a questionnaire developed by a self-help group, Bernstein et al. (1977) found that the prevalence of gallbladder disease increases with age, body weight, and parity. Eighteen percent of women between 25 and 34 years of age whose weight was twice or more higher than the ideal weight had gallbladder disease compared to nearly 35% of the women at ages 45 to 55 who were in the same weight category. In this study, 88% of the variation in frequency of gallbladder disease was accounted for by weight, age, and parity; weight was the most important variable. Obese women between 20 and 30 years of age had a sixfold increase in the risk of developing gallbladder disease compared to normal-weight women. By age 60, nearly one-third of obese women can expect to develop gallbladder disease. In the Framingham study (Friedman et al., 1966), people at least 20% above the median weight for their height had about twice the risk of developing gallbladder disease as those who were less than 90% of the median weight for height.
In a case-control autopsy study, Sturdevant et al. (1973) found that the body weight of men without gallstones was considerably less than that of men with gallstones. The incidence of gallstones at autopsy was 16% (25/156) in men who were more than 9.1 kg overweight.
Clinical Studies
Increased cholesterol production and secretion provide one explanation for the increased risk of gallbladder disease in overweight people. Nestel et al. (1973) showed that cholesterol production rate was correlated with body weight and the number of fat cells. For each kilogram of excess body weight, cholesterol production increased 22 mg/day. Miettinen (1971) estimated that an additional 20 mg of cholesterol per day was produced for each additional kilogram of adipose tissue. Bennion and Grundy (1978) found that bile was more highly saturated with cholesterol in 23 obese subjects than in the 23 nonobese controls. The hepatic secretion of cholesterol was higher in 11 subjects before weight loss than afterward, but neither phospholipids nor bile salt secretion changed.
Cancers
Epidemiologic Studies
In the American Cancer Society cohort study conducted between 1959 and 1972, Lew and Garfinkel (1979) reported positive associations between excess weight and cancers of the gallbladder, biliary duct, endometrium, ovary, breast, and cervix in women and cancers of the colon and prostate in men. The finding of an increase in risk for endometrial cancer with increasing weight has been a consistent finding in the majority of case-control studies in northern Italy (La Vecchia et al., 1986), Denmark (Jensen, 1985), and the United States (Henderson et al., 1983), including the Framingham cohort study.
Studies of breast cancer have also provided evidence for an association between obesity and increased risk. The effect has primarily been observed in postmenopausal women in studies in the Netherlands (de Waard and Baanders-van Halewijn, 1975), northern Italy (Talamini et al., 1984), and Israel (Lubin et al., 1985). Associations have also been reported in premenopausal women (Kelsey et al., 1981), although less consistently. On the other hand, Willett et al. (1985) used BMI as a basis for examining the relationship between weight status and the development of premenopausal breast cancer among 121,964 U.S. women ages 30 to 55 who were enrolled in the Nurses Health Study. There was a significant inverse relationship between the BMI and age-adjusted relative risks for breast cancer. In the Israeli case-control study (Lubin et al., 1985), risk decreased in postmenopausal women past the age of 60 who had lost weight during their adult life. Several studies suggest that height rather than weight may be the better predictor of breast cancer risk (de Waard and Baanders-van Halewijn, 1974; Tulinius et al., 1985). If this is the case, nutritional status during adolescence may be a determinant of breast cancer risk.
In one case-control study of ovarian cancer, there was a weakly positive association with weight (Tzonou et al., 1984). In another, there was a weakly negative association (Byers et al., 1983). Several, but not all, studies have suggested that weight gain is associated with increased risk of prostate cancer (Kolonel et al., 1988; Snowdon et al., 1984; Talamini et al., 1986).
Page 576
There have been isolated reports of increased risk of various cancers associated with decreases in weight, for example, cancers of the esophagus (Ziegler et al., 1981) and lung (Tulinius et al., 1985). It seems likely, however, that these are indicators of less-than-optimal nutritional status, which may reflect social factors or the use of tobacco or alcohol.
Clinical Studies
The incidence of endometrial cancer has been shown to increase in obese women, especially in postmenopausal obese women when endogenous estrogen levels fall. One explanation for this increased risk might be the increased production of estrogen in fat tissue. Urinary estrone production rates are increased in obese postmenopausal women; 50 to 120 µg/day are produced in obese women in contrast to 20 to 40 µg/day in nonobese women. In postmenopausal women, the increased estrogen appears to be produced by the conversion of the adrenal steroid androstenedione to estrogen by the stromal cells in adipose tissue.
Animal Studies
Most animal studies of diet and cancer have been designed to assess the effects of total caloric intake or the effects of specific nutrients on the induction or prevention of various cancers. Some of these are reviewed under the sections on total energy intake in this chapter and in more detail in Chapter 22. Thus, body weight gain or adiposity may affect cancer incidence, but most probably because of the effect of specific macronutrients rather than because of obesity per se.
Effects of Fluctuations in Body Weight
Epidemiologic Studies
Weight gain in adults (Ashley and Kannel, 1974; Borkan et al., 1986; Noppa, 1980) and in children (Clarke et al., 1986) has been associated with increased blood pressure and blood lipids, suggesting an increased risk of heart disease. Borkan et al. (1986) found that changes in weight status were long-term predictors of changes in blood pressure, triglycerides, serum cholesterol, glucose, and uric acid. Clarke et al. (1986) reported that changes in adiposity among children were also directly related to changes in blood pressure.
Dieting to reduce body weight has become a major phenomenon due to health and cosmetic concerns in the United States and many other industrialized societies. The Health Promotion and Disease Prevention Questionnaire, a part of the National Health Interview Survey (1985), indicated that the prevalence of dieting in 1974 and 1975 was 47% among 18- to 44-year-old women, 45% among 45- to 64-year-old women, and 24% and 30%, respectively, among men in the same age groups. A 1985 Gallup poll revealed that almost 90% of Americans believed that they weighed too much, 16% of the women questioned considered themselves perpetual dieters, and 31% reported dieting at least once monthly.
Most dieters eventually regain the weight they lose. Recidivism rates after weight loss have been estimated to range between 60 and 90%. Most people engage in many dieting attempts in their pursuit of thinness (Brownell, 1982; Jeffery et al., 1984; Stunkard, 1980).
Although adherence to weight reduction diets is traditionally viewed as a health-enhancing behavior, it is clear that some dieting practices, such as those associated with eating disorders, can seriously damage health. Furthermore, there is evidence that repeated cycles of weight loss and regain alone may enhance metabolic efficiency in animals (Brownell et al., 1986). Given the high prevalence of dieting, it is surprising that the long-term consequences of weight loss and weight gain are not well understood, especially in relation to the normal aging process, and that even less is known about the health consequences of repeated cycles of dieting. The few studies that have addressed these issues in humans are summarized below.
In the Framingham study, weight gain was shown to be a risk factor for atherogenesis and total mortality (Ashley and Kannel, 1974; Hubert et al., 1983). These analyses indicate that even small weight gains carry incremental health risks. In contrast, an analysis of life insurance data suggests that the optimal BMI, or that associated with minimal mortality, increases slightly with age (Table 21-1) (Andres, 1985). This observation is consistent with the results of two longitudinal studies, which also suggest that moderate increments in body weight over a lifetime may be beneficial (Avons et al., 1983; Rhoads and Kagan, 1983). Some investigators have found positive associations between weight gain and cardiovascular diseases (Abraham et al., 1971; Noppa, 1980; Shapiro et al., 1969), whereas others have found
Page 577
no association (Barrett-Connor, 1985; Heyden et al., 1971; Hsu et al., 1977).
Weight loss was associated with decreased relative risk of CHD in the Framingham study, which is consistent with the negative consequences of weight gain observed in the same group (Ashley and Kannel, 1974). In contrast, results of a study of Parisian civil servants associated weight loss with elevated mortality from all causes and from cancer, although the possibility that occult disease was present during the periods of weight loss makes it difficult to ascribe causality with certainty (Avons et al., 1983). Paradoxically, data from the Honolulu Heart Study indicate that weight loss is associated with a decrease in CHD risk but with an increase in CHD mortality (Bloom et al., 1987). One possible interpretation of this finding is that weight loss in the absence of disease is beneficial but that weight loss is also an indicator of antecedent disease.
In the Framingham study, a 30% increase in CHD was associated with a 10% weight gain, and a 20% decrease in CHD was associated with a 10% weight loss (Ashley and Kannel, 1974). Repeated cycles of weight loss and gain might exert a net negative effect; however, relatively few attempts have been made to address this question directly by studying subjects who lose and subsequently regain weight.
Data from the Chicago Gas and Electric Study indicate that one cycle of gain and loss is a risk factor for death from CHD independent of high BMI (Hamm et al., 1989). In that study, Hamm et al. (1989) examined the self-reported weights of Western Electric workers at ages 20, 25, 30, 35, and 40. They divided them into three groups: those who had a large gain in weight and no loss, those who gained little or no weight, and those who had both a large weight gain and a large weight loss. The gain-and-loss group had a significantly higher risk of CHD mortality when compared to the no-change group. The adjusted relative risk was 1.8 for the gain-and-loss group compared to the no-change group. No significant effects were observed in the gain-and-no-loss group.
The study of the temporal sequence of weight change in relation to chronic diseases is particularly challenging in humans. It is critical that epidemiologic studies of weight change and chronic diseases be long term and prospective in nature. Body weight should be measured repeatedly before the onset of disease. People who lose large amounts of weight immediately before diagnosis of CHD should be excluded from the analysis. In such studies, systematic trends in body weight need to be distinguished from random or periodic fluctuations. Such data are accumulating in several ongoing longitudinal studies. Thus, it may soon be possible to address this issue more thoroughly.
Clinical Studies
Keys et al. (1950) observed in clinical studies that healthy male volunteers who lost weight by food restriction and then regained it were fatter after regain than at the beginning of their weight reduction. Over time, the body weight and composition returned to preweight reduction levels. During studies of overfeeding, Sims and colleagues (1973) observed an increase in circulating cholesterol and triglycerides as well as a small increase in glucose and insulin. Weight loss has been associated with considerable reductions in blood pressure and levels of triglycerides, insulin, and glucose (Henry et al., 1985; Tuck et al., 1981). In another study of the loss-regain cycle, Drenick et al. (1964) and Johnson and Drenick (1977) placed approximately 200 obese men on total fasts, some for more than 2 months and the rest for 3 to 8 weeks. After refeeding, the men regained their lost weight, often exceeding their original weight. Eighty percent developed diabetes; one-half of these cases were severe.
Weight loss may be accompained by changes in several chronic disease risk factors, e.g., lowered blood pressure or lowered triglycerides. If weight is regained, however, levels of these parameters return to preweight-loss level. (See section on Clinical Studies under Evidence Relating Total Energy Intake to Chronic Diseases: Athersclerotic Cardiovascular Disease, Chapter 6.)
Some animal studies of cycles of weight gain and weight loss and regain suggest that repeated cycles are associated with increases in metabolic efficiency, i.e., greater energy storage at a fixed energy intake (Brownell et al., 1986; Reed et al., 1988) and increases in abdominal fat depots (Reed et al., 1988), but others have not found an increase in energy efficiency (Gray et al., 1988; Hill et al., 1984).
Page 578
Effects of Regional Fat Distribution
Prospective Epidemiologic Studies
In a prospective study of men living in Göteborg, Sweden, Larsson et al. (1984) showed that BMI and WHR (AGR) were positively correlated in those who developed strokes. That is, over a 13-year period, men in the highest tertile, both for WHR ratio and BMI, had 20.8% of the total number of strokes compared to 5.6% for men in the lowest tertile of BMI and WHRs. For cardiovascular diseases, there was a much different relationship. The highest risk was observed in the group with the highest tertile of WHR and the lowest tertile of BMI. Thus, carrying extra fat around the waist poses a particular risk, regardless of BMI.
A similar set of relationships apply to women (Lapidus et al., 1986). Among 14,462 women between 38 and 60 years of age, the 12-year age-specific incidence rates for myocardial infarction, stroke, and overall death were correlated with WHR. Among the highest quintile, the relative risk of myocardial infarction was 8.2 times higher than the risk for those in the lowest quintile. For stroke and overall death rate, the relative risk was increased 3.8 and 2.0 times, respectively, for those in the highest quintile for WHR compared to those in the lowest quintile. When women in the top 5% were compared to women in the lowest quintile, the risk of myocardial infarction was increased 14.8 times, the risk of having a stroke was increased 11.0 times, and the risk of death from all causes was increased 4.8 times. Increasing BMI along with a higher WHR enhanced the risk of developing CHD in women.
At the fourth biennial examination in the Framingham study, several measures of regional fat distribution were taken, including waist, but not hip, circumference, plus measurements of subscapular, tricep, abdominal, and quadricep skinfolds. The 22-year incidence of CHD was significantly related to thickness of the subscapular skinfold. For men, only the relationship to CHD with serum total cholesterol was stronger than the relationship to subscapular skinfold thickness. For women, cholesterol and blood pressure coefficients were higher (.222 and .293, respectively) (Stokes et al., 1985).
This study of 8,006 men of Japanese ancestry living in Hawaii provided data on the 12-year incidence of CHD in relation to BMI and subscapular skinfold thickness. The men in the highest tertile for subscapular skinfold experienced a more than twofold excess relative risk compared to those in the lowest tertile. There was a direct and significant relationship between the rate of CHD and subscapular skinfold within each tertile of BMI. Men in the middle tertile of subscapular skinfold thickness experienced a 70% excess of definite CHD. After adjustment for concomitant risk factors, those in the middle tertile still had a 40% excess risk of CHD and those in the highest tertile of subscapular skinfold thickness still had a 50% excess risk (Donahue et al., 1987).
In a prospective study conducted on 7,535 civil servants in Paris, investigators examined the relationship between body weight and fat distribution based on a principal-component analysis of several skinfolds in men ages 42 to 53. This analysis provides three parameters that correspond to total fatness, trunk versus extremity fatness, and upper extremity versus lower extremity fatness. The parameter corresponding to the trunk/extremity estimate of fatness had a linear relationship to the annual incidence of CHD, for which there was a nearly threefold increase in risk ratio between the lowest and highest quintiles (Ducimetiere et al., 1986).
All these studies indicate that regional fat distribution is a more important variable than BMI for predicting risks associated with adiposity.
Cardiac Function
Heart mass increases as body weight rises, whether assessed by postmortem examination or by echocardiographic measurements of the posterior wall and interventricular septal dimensions (Bray, 1987b). This increase involves left and right ventricles and is related not only to the degree of obesity but also to its duration.
The increased cardiac mass observed in obese people is associated with a number of functional changes. Blood volume increases along with intracellular and extracellular fluid volumes. Cardiac output and stroke volume are elevated and positively correlated with body weight and the degree of overweight. Left and right ventricular end-
Page 579
diastolic pressures are high as are the pulmonary artery and pulmonary capillary wedge pressures. In the most obese subjects, cardiac catheterization studies have revealed the presence of impaired left ventricular function, and a cardiomyopathy of obesity has been clearly identified (Bray, 1987b).
In contrast to most other cardiac indices, heart rate does not increase in very obese subjects. However, abnormalities can be detected on the electrocardiogram. Data from the second National Health and Nutrition Examination Survey (NHANES II) show that there is a leftward shift of the mean QRS complex with increasing fatness in both men and women (Zack et al., 1984). This effect is independent of age and blood pressure. However, this shift in the QRS complex is confined to the normal range, since left-axis deviation (QRS axis of -30° or less) is not associated with body fatness. In a study of more than 1,000 electrocardiograms from obese people, the PR interval, QRS duration, QTc interval, and voltage increased with increasing obesity. ST and T wave abnormalities were present in 11% of the subjects and correlated better with increasing age and blood pressure than with the magnitude of the obesity. A prolonged QTc interval was present in 28.3% of those tested (Frank et al., 1986). Changes in the QTc interval have been considered a harbinger of potential cardiac dysrhythmia in obese individuals during prolonged starvation or when eating very low-calorie diets.
Pulmonary Function
Abnormalities of pulmonary function have been observed in obese subjects (Ray et al., 1983). At one extreme are patients with the Pickwickian syndrome, named after Joe, the overweight boy in Dickens's Pickwick Papers. This syndrome, also called the obesity-hypoventilation syndrome, is characterized by somnolence, obesity, and alveolar hypoventilation. At the other extreme are impairments in work capacity and pulmonary function that are due to obesity itself. In obese subjects, there is a fairly uniform decrease in expiratory reserve volume (i.e., the volume of air that can be exhaled after normal respiration) (Ray et al., 1983). There is also a low maximum rate of voluntary respiration as well as a tendency toward a general reduction in lung volumes. Lung compliance appears to be normal, but studies on the mechanics of breathing show increased oxygen consumption associated with breathing, since more work is required to move the mass of the obese chest (Sharp et al., 1980). Finally, there appears to be some element of venous admixture: segments of the lung that are not well perfused but are ventilated and other regions that are perfused but not adequately ventilated, leading to the consistent but modest decrease in arterial oxygenation without a corresponding increase in arterial carbon dioxide content.
Extensive alterations in pulmonary function are observed primarily in massively obese subjects or in obese people with some other respiratory or cardiovascular problem. In a careful study of 29 obese women and 14 obese men, there was a progressive decrease in expiratory reserve volume as the weight-to-height (kg/cm) ratio increased. On the other hand, vital capacity, inspiratory capacity, residual volume, and diffusion capacity remained fairly constant over a range of weights, except in subjects who were massively obese (i.e., with a WHR above 1.0 kg/cm) (Ray et al., 1983).
In a study by Sharp et al. (1980), the higher metabolic rate of obese subjects at rest and during exercise resulted in an increased oxygen uptake and carbon dioxide output. This demand is met by increased minute ventilation. The extra weight on the chest and abdomen of subjects weighing 114 kg on average was associated with a two- to fourfold increase in the mechanical work required to passively ventilate the lungs. The compliance of the chest wall, however, was not related to BMI (Suratt et al., 1984).
Hematologic and Immunologic Consequences
Epidemiologic Studies
An increase in the hemoglobin levels in obese subjects was reported by Garn and Ryan (1982). Data from the NCHS show a difference of approximately 0.2 g/dl between the obese (the top 15% of the sample) and the lean (the lowest 15% by triceps skinfold). This increase in hemoglobin concentration in obese subjects was not associated with race.
Clinical Studies
Nutritional factors influence the immune response under many conditions, including obesity. For example, Krishnan et al. (1982) showed that when monocytes were incubated in vitro, the number that matured into macrophages was significantly less in obese people than in lean subjects. The generation of migration-inhibiting factor by lymphocytes from obese subjects with normal glu-
Page 580
cose levels was also significantly less in the presence of purified protein derivative than in normal controls. In obese and lean children, the levels of serum immunoglobulins, the complement components C3 and C4, as well as the numbers of T and B lymphocytes were similar. The obese children and adolescents had a variably impaired cell-mediated immune response and reduced intracellular bacterial killing by polymorphonuclear leukocytes. In the search for an explanation, Krishnan et al. (1982) examined serum copper and zinc levels, since it is well known that zinc deficiency can impair immune functions. They found a subclinical deficiency of both micronutrients. To test this further, the authors treated patients with zinc and copper for 4 weeks and found that immunologic function improved. These data suggest that immunologic responses may be impaired in obese subjects, in part because of other nutritional deficiencies.
Bone, Joint, and Skin Disorders
Epidemiologic Studies
Added body weight might be expected to increase trauma to the weight-carrying joints and thus accelerate the development of osteoarthritis. The literature on this issue is contradictory (Bray, 1985). The distribution of body weight in people with primary and secondary osteoarthritis of the hip is similar to that of people with normal hip joints. Surprisingly, one study of people with more than twice ideal body weight indicated that the incidence of osteoarthritis, determined by x-ray, was only 12%. Several authors reported an increased prevalence of osteoarthritis in obese subjects, and an increased mean weight for people with osteoarthritis has been reported in other studies. The knee joint seems to be the most frequently involved. The prevalence of osteoarthritis of the hands and the ankles increased with age among 2,548 people divided into four age groups in NHANES II (Engel, 1968). Within each age group, however, there was a clear increase in the prevalence of osteoarthritis in relation to body weight for all groups of women over 35 years of age. The slope of the increase with weight was sharpest below 90 kg (200 lbs), suggesting that body weight is only one factor.
Obesity is also associated with an increased risk of gout. Rimm et al. (1975), in a study of 73,532 weight-conscious women, found that the crude relative risks for gout and arthritis were 2.56 and 1.55, respectively. In women whose weights were 85% higher than desirable, the frequency of gout was 1.56 times that of women who were less than 10% overweight. There was also a significant correlation between serum uric acid levels and body weight (Ashley and Kannel, 1974). This effect was particularly marked in the 35- to 44-year age group; somewhat lower correlation coefficients were found in the older age groups. The risk of osteoporosis is reduced in the obese (Dalen et al., 1975), possibly because of the increased bone mass accrued during the early years of bone formation.
Clinical Studies
In a study of obese and normal subjects, Bell et al. (1985) found that obese white people had an increase in serum immunoreactive parathyroid hormone, serum 1,25-dihydroxy vitamin D, and urinary cyclic adenosine monophosphate. There was a corresponding decrease in urinary calcium excretion comparable to that observed in obese menopausal women who were also found to have a lower ratio of urinary calcium to creatinine compared to nonobese postmenopausal controls.
Complications of Pregnancy
Body weight before pregnancy and weight gain during pregnancy both influence the course of labor and its outcome. Among heavy women (the top 10% in weight of 3,939 women), the frequency of toxemia and hypertension was greatly increased, and the duration of labor was longer (Peckham and Christianson, 1971). In more than 7% of the heavy women, labor lasted more than 24 hours. This occurred in only 0.8% of the light-weight women (the lowest 10%). Caesarean section was performed in 5.5% of the overweight patients but in only 0.7% of the light-weight ones, indicating that obese patients had more obstetrical complications than the nonobese ones. Gross et al. (1980) reviewed 2,746 consecutive deliveries, which included 279 obese women weighing more than 90 kg. The obese women were older, had a higher parity, and had an increased incidence of hypertension, diabetes mellitus, and twin gestation. The occurrence of abnormal labor, including oxytocin infusion and caesarean section, was also higher among the obese women.
Infants born to heavy women weigh more than the offspring of light women. There is also a direct relationship between placental weight and prepregnancy body weight. At the age of 7, approximately 50% of the incremental weight gain can be accounted for by the differences in placental weight at birth and the remaining 50% is attribut-
Page 581
able to postnatal environment. Naeye (1979) found that the fewest fetal and neonatal deaths occurred when mothers who were overweight at the beginning of the pregnancy gained an average of 7.3 kg (16 lb) or less. The optimal weight gain during pregnancy was 9.1 kg (20 lb) for normal-weight women and 13.6 kg (30 lb) for those under weight.
Endocrine Disorders
Epidemiologic Studies
There is a reduction in the concentration of total serum testosterone and sex-hormone binding globulin (SHBG) in obese males. The decline in testosterone is directly related to the degree of overweight. There is also a weight-related increase in estradiol and estrone in males. The low testosterone level sometimes found in obese men probably results from the reduced concentrations of SHBG, which transports testosterone in the serum. Although the mechanism for the SHBG reduction is presently unknown, the concentration of free testosterone remains essentially normal, except in massively obese men, in whom free testosterone is often low. Testicular size and the basal concentration of pituitary gonadotropins, follicle-stimulating hormones (FSH), and luteinizing hormones (LH), are normal. Similarly, the pituitary release of LH and FSH in response to an injection of luteinizing hormone-releasing hormone (LHRH) is normal, as is the concentration of these pituitary peptides during treatment with clomiphene.
Menarche occurs at younger ages among heavy women and is somewhat delayed among light women (Bray, 1976). Obese females often experience irregular menstrual cycles and an increased frequency of menstrual abnormalities. But obesity frequently occurs secondary to menstrual and other hormonal disorders, for example, abnormal steroid hormone metabolism. In these disorders, weight loss does not correct the hormonal imbalance. In one study (Bray, 1976), 43% of the 100 women with menstrual disorders, including amenorrhea, functional uterine bleeding, premature menopause, and infertility, were 20% above standard weight. In contrast, only 13% of 201 women with no menstrual abnormalities were overweight. Forty-eight percent of the women with amenorrhea were overweight, and 58% of the 19 patients with functional uterine bleeding were obese. Women with hirsutism and anovulatory cycles (i.e., irregular cycles of greater than 36 days) were on average 14 kg heavier than women with no menstrual abnormalities. There was also an increasing percentage of women with anovulatory cycles as the degree of excess weight increased. Among women less than 20% overweight, only 2.6% had anovulatory cycles compared to 8.4% of the women who were more than 74% overweight. Facial hair was associated with a longer duration of obesity.
Teenage-onset obesity was associated with a greater number of never-pregnant married women and with a higher likelihood of surgery for polycystic ovaries (Hartz et al., 1979). (The syndrome of polycystic ovaries, however, is associated with obesity, as discussed in the next section, and is not corrected by weight loss.) Thus, it appears that alterations in body weight can influence both the onset of menstruation as well as the subsequent initiation of menstruation in women who have developed secondary amenorrhea. Just as menarche occurs earlier in obese women, ovarian failure in menopause with its increased FSH production begins on average 4 years earlier in obese than in normal-weight women.
Clinical Studies
There are two abnormalities in the menstrual cycle of obese women (Sherman and Korenman, 1974). First, the rise in FSH production in the first half of the cycle is lower than in normal-weight women. Second, progesterone fails to rise normally in the second half of the menstrual cycle. The mechanism leading to these abnormalities in obese women is unknown, but it is clear that they are reversible with weight loss.
Function of the reproductive system in women is complicated by the metabolism of steroids in nonendocrine organs. Androstenedione, a product of the adrenal cortex, can be converted to estrone by nonsteroid-producing tissues. This conversion probably takes place in the stromal cells of adipose tissue. Muscle can also convert estrone to estradiol. Despite this enhanced rate of conversion of androstenedione to estrone, most studies do not demonstrate an increase in the circulating concentration of estrogens in premenopausal females (Zumoff, 1982); however, in the obese postmenopausal woman, the concentration of estrogen appears to be increased (Lobo et al., 1982). When bilaterally oophorectomized obese and nonobese women with estradiol implants were compared, the obese women had higher levels of androstenediol, androstenedione, and unbound testosterone.
The polycystic ovary syndrome is associated with obesity. This syndrome is defined clinically by
Page 582
the presence of oligomenorrhea, or amenorrhea, and obesity in 16 to 49% of the cases reported in various studies. Hirsutism, hyperandrogenism, elevated LH-to-FSH ratio, and polycystic ovaries are also present. In one study, SHBG was reduced in obese women with or without this syndrome, but levels of testosterone, albumin-bound testosterone, and androstenedione were higher than normal in those with polycystic ovaries (Pasquali et al., 1982). These alterations suggest that women with the polycystic ovary syndrome may have a defective control of hormone secretion.
Animal Studies
Many endocrine abnormalities accompany the obese condition in animals. Much of the pathology associated with obesity may in fact result from the accompanying endocrine dysfunction. Hyperinsulinemia is a prominent feature of virtually all experimentally induced or genetically determined obesities (Bray et al., in press). The hyperinsulinemia may precede, coincide with, or follow the development of hyperphagia in obese rodents (Berthaud and Jeanrenaud, 1979; Turkenkopf et al., 1982). Hyperinsulinemia contributes to the hypertriglyceridemia and glucose intolerance, further enhances adipose tissue lipoprotein lipase (LPL), and contributes to blood flow alterations in obese animals. In addition, the prevention of hyperinsulinemia in some experimentally, but not genetically, obese rodents can retard the development of obesity.
The role of sex hormones in the control of food intake has been extensively described (Wade and Gray, 1979). There is little question that sex hormones regulate certain aspects of food ingestion, fat distribution, and the redistribution of fat during pregnancy and lactation. In animals, much of the effect of the sex hormones on the distribution of fat is believed to be mediated through the action of the hormones on adipose tissue LPL activity.
There have been numerous reports of hypothalamus-pituitary dysfunction in obese rodents. These include altered levels of endorphin, adrenocorticotropic hormone (ACTH), LH, FSH, and somatomedin. Growth hormone abnormalities have also been observed in several strains and in experimentally produced obese animals. The removal of the pituitary reduces linear growth but does not prevent, and may enhance, the development of obesity (Han, 1967) in normally lean animals. In genetically obese rodents, hypophysectomy normalizes the growth curve but not the body composition of treated animals (Cox and Powley, 1977); however, treatment with growth hormone does not reverse the development of the obesity. The role of glucocorticoids in the development of experimental and genetic obesities is somewhat unclear. There are few clear-cut examples of altered plasma glucocorticoids in experimental animals when conditions are carefully controlled. Nonetheless, the removal of the adrenal gland prevents the further development of obesity in genetically obese strains of rodents and reverses much of the associated pathology (Bray, 1987a). Overall, many of the endocrine abnormalities associated with obesity may cause certain aspects of the associated morbidity, but no single endocrine abnormality has been shown to be responsible for experimentally produced or genetically determined obesity.
Misconceptions
Do Fat Children Become Fat Adults?
It is widely held that fat children become fat adults. Longitudinal studies show that the risk of a fat child becoming a fat adult is far less than many think. When a large sample of U.S. children were examined after an average of 3.5 years following the initial measurement of weight and height, considerable redistribution in relative weights was observed (Zack et al., 1984). Many children who are at the extreme end of the spectrum for weight relative to height at one age in childhood do not remain there in subsequent years. Of individuals who were overweight at age 26, less than 7% were overweight at age 7 (Bradden et al., 1986). The principal concern about obesity in childhood and adolescence should be focused on those whose proportion of body weight to height continues to deviate from the norm, especially in those with a family history of marked obesity.
For those with so-called progressive obesity, the risks of remaining obese in adult life appear to be substantially higher than for those in the upper 20% of weight for height but who remain in that category for several years. Current estimates suggest that more than 80% of obese adolescents remain obese into adulthood. The likelihood of remission decreases as severity increases. For example, 54% of 7-year-olds who are 130 to 145% of ideal body weight [50th percentile of the NHANES] will remit, whereas remission occurs in only 18% of 7-year-olds who are 157 to 165% over ideal body weight and in 0% among those who are
Page 583
>165% of ideal body weight (Börjesson, 1962). Two factors influence whether or not an obese child becomes an obese adult: the time of onset and the degree of obesity. Obese adolescents who become obese adults may be fatter than obese adults whose obesity began in adulthood.
A variety of family variables are also associated with childhood and adolescent obesity. The association of obesity in children with parental obesity has been well described. This can be readily explained by environmental factors or family practices, although genetic factors may affect susceptibility (Dietz, 1987).
Do All Fat People Overeat?
Overweight individuals are popularly believed to be gluttons. Carefully controlled studies in animals indicate, however, that overeating is not a necessary requirement for the development of obesity (Bray and York, 1979; Bray et al., in press). The precise measurement of food intake by humans is difficult, but it is clear from several studies that normal as well as overweight individuals tend to underreport their dietary intake. Most studies comparing normal and overweight people suggest that those who are overweight eat fewer calories than those of normal weight (see Chapter 6). Yet, in-patient studies show that the energy required to maintain body weight in heavy people is greater than that required for lean subjects. In formerly obese people who have lost weight, energy requirements are lower than expected for their age (Leibel and Hirsch, 1984). If food intake, corrected for errors in reporting, is lower in overweight than in normal-weight people, this suggests that their energy expenditure may be proportionally lower or their metabolic efficiency may be greater. In the absence of techniques for modifying metabolic efficiency, people should be encouraged to increase their overall level of exercise and to moderate their food intake.
Do Fat Cells Increase in Number and Size During Adult Life?
Early studies on measurements of fat cell size and number indicated that most laboratory animals that became obese early in life had more fat cells than did lean controls. Development of obesity in older animals was initially believed to result entirely from an increase in the size of fat cells—not by an increase in the number. It has recently become clear, however, that the number of fat cells in laboratory animals eating a high-fat diet can increase at any age, although the increase is less than that seen in genetically obese animals (Faust et al., 1978; Lemonnier, 1972). Recent clinical data also suggest that the number of fat cells in the human body may increase in adult life (Sjöstrom and William-Olsson, 1981). When the number of fat cells in a group of 19 Swedish women were compared over 7 to 9 years, Sjöstrom and William-Olsson (1981) observed that the changes in the amount of body fat in women who gained weight or lost weight were related to the total number—not the size—of their fat cells. Data consistently indicate that very obese humans have an increased fat cell number and that this is not reversible.
Does Low Caloric Intake Increase Life Span?
For more than 40 years, studies in mice and rats have shown that reducing caloric intake below baseline levels prolongs life (Masoro, 1985; McCay et al., 1935). This reduction is also associated with slower rates of growth and smaller body sizes. The mechanism for these observations is not clear. In contrast to the substantial amount of data on animals, there are no convincing data that restricting energy intake in humans has any effect on life expectancy. Furthermore, there are no epidemiologic data regarding the timing of the imposition of the dietary restriction and its effect on life expectancy. Data from several natural famines indicate that the incidence of certain diseases was lowered following caloric restriction, but its effect on longevity is not known since caloric intake subsequently returned to normal.
Eating Disorders
Two major eating disorders, anorexia nervosa and bulimia, are becoming increasingly recognized as serious psychiatric disorders with physiological sequelae, especially among young white women in the middle and upper socioeconomic classes (Fadiman, 1982; Golden and Sacker, 1984; Levin, 1983). Anorexia nervosa is characterized by extreme weight loss, body-image disturbance, and an intense fear of becoming obese. Bulimia is characterized by binge-eating episodes in private followed by self-induced vomiting, fasting, or use of laxatives or diuretics (Herzog and Copeland, 1985). On college campuses, these disorders may affect as many as 20% of female students (Boskind-Lodahl
Page 584
and White, 1978; Cooper and Fairburn, 1983; Halmi et al., 1981; Kretchmar, 1984; Pyle et al., 1983). The term epidemic has been used in conjunction with eating disorders; however, there is disagreement over its applicability. For example, Williams and King (1987) suggest that this term is inappropriate because the statistics simply reflect a high hospital readmission rate and not necessarily new cases. They do note, however, that in the population they studied (patients admitted to psychiatric facilities in England between 1972 and 1981), although there was no increase in risk, there was an increase in the size of the population at risk for anorexia nervosa.
Anorexia Nervosa
Prevalence
Anorexia nervosa is found primarily among adolescent girls and young women; approximately 4 to 6% of the cases are male (Halmi, 1985). A recent British study estimates the incidence of anorexia nervosa at 3.82 per 100,000 per annum (Szmukler et al., 1986), while a Dutch study puts this figure at 5 per 100,000 per annum (Hoek and Brook, 1985). In a population-based study in Rochester, Minnesota, A. Lucas (Mayo Clinic, personal communication, 1989) reported that over a 45-year period (19351979) the overall rate for males and females was 7.3 per 100,000. Relentless pursuit of a thin body size despite emaciation is the central phenomenon that must always be present for a diagnosis of anorexia nervosa. Central to this weight phobia is a preoccupation with maintaining a low subpubertal body weight and avoiding any weight gain. Low body weight control may be sustained mainly by carbohydrate and fat avoidance—a form of starvation unique to anorexia nervosa (Crisp, 1977; Garfinkel and Garner, 1982).
Clinical Diagnosis
Several objective diagnostic clinical criteria have been developed (Feighner et al., 1972). According to the Diagnostic and Statistical Manual DSM-III (APA, 1987), onset usually occurs before 25 years of age. To be classified as anorectic, the subject must have lost at least 25% of original body weight or, if under 18 years of age, 9.25% of original body weight plus the amount of weight that would have been gained based on growth charts. Furthermore, there must be no other known medical or psychiatric illness accounting for the weight loss. The diagnostic criteria usually require evidence of a psychological disturbance in addition to a morbid fear of becoming fat, e.g., a disturbance (misperception) of body image and a claim to feel fat even when emaciated (Dally, 1969; Morgan and Russell, 1975). The patient may resort to a variety of devices to lose weight (e.g., starvation, vomiting, laxatives). Patients with anorexia nervosa show a desperate need to control and manipulate their environments, and there is a refusal to maintain body weight over a minimal normal weight for age and height range. Despite severe weight loss, these people exhibit excessive activity. In the natural course of events, approximately 40% of anorectics recover within 6 years after initial diagnosis and about 5% die (Crisp, 1983).
A variety of endocrine abnormalities have been observed in anorectic patients. Among them are abnormal responses to exposure to heat and cold (Burman et al., 1977; Frankel and Jenkins, 1975), which suggest hypothalamic dysfunction. Amenorrhea, dry skin, constipation, bradycardia, and low basal metabolic rate have also been reported. Thyroid-stimulating hormone appears to be normal, but serum triiodothyronine (T3) levels have been found to be low (Bhat and Cama, 1978; Burman et al., 1977; Curran-Celentano et al., 1985). Thyroxine (T4) levels have also been found to be in the low-normal range, but not in all patients (Curran-Celentano et al., 1985; Schwabe et al., 1981). Since similar thyroid test results are found in other malnourished states, these findings cannot be considered specific to anorexia nervosa. All the endocrine abnormalities mentioned above are observed in other forms of starvation and are reversible with adequate refeeding and return of the resting metabolic rate to normal.
Hypercarotenemia is also associated with anorexia nervosa, but the mechanism for this has not been established (Curran-Celentano et al., 1985; Robboy et al., 1974). Other endocrine abnormalities include low basal plasma levels of FSH, LH, and estradiol (Russell, 1965; Vigersky et al., 1976). Resting levels of growth hormone can be greatly elevated (Frankel and Jenkins, 1975; Vigersky and Loriaux, 1977) as can basal plasma cortisol levels (Boyar et al., 1977).
Bulimia
Prevalence and Clinical Features
Bulimia is much more common than anorexia nervosa, especially during late adolescence and young adulthood (Halmi et al., 1981; Herzog, 1982). Halmi et al. (1981) found a prevalence of
Page 585
13% in a population survey of 355 college students and a 7-to-1 ratio of females to males. Other studies show that the prevalence of bulimia may be as low as 3.2% among college women (Pyle et al., 1986). The wide range of results may be due to differences in methods of assessment and in the stringency with which the diagnostic criteria are applied. In addition to women of college age, other groups identified as being vulnerable to this disorder include jockeys, wrestlers, gymnasts, and ballet dancers.
Bulimia may exist concomitantly with anorexia nervosa or as an entirely separate disorder (Palmer and Guay, 1985). Bulimic patients are usually less cachectic than anorectics (Pyle et al., 1981) but are difficult to treat, since the pattern of binge eating and vomiting, or purging, is usually extremely difficult to interrupt (Smith, 1984).
Diagnostic criteria for bulimia include such behaviors as recurrent rapid consumption of a large amount of high-calorie, easily ingested food, such as carbohydrates, in a discrete period of time, usually less than 2 hours, and frequent weight fluctuations greater than 10 pounds due to alternating binges and fasts (APA, 1987). Body weight may, however, hover around normal or even be slightly higher. Other symptoms that may accompany bulimia include an increase in dental caries (possibly related to recurrent contact with acidic vomitus or excessive carbohydrate consumption during binging), hypoglycemia, parotid gland enlargement, and, in extreme cases, acute gastric dilation and rupture (Pyle et al., 1981).
Summary
Overweight as defined by a BMI of 27 to 30 kg/m2 afflicts more than 25% of the females and 31% of the males over 18 years of age in the U.S. population. Severe overweight—a BMI higher than 30 kg/m—is present in 12%. For most adults, overweight results from the expenditure of fewer calories than are ingested. This may result from a sedentary lifestyle or increased metabolic efficiency. As BMI increases, there is an increased risk of chronic disease.
Obesity is an independent risk factor that modifies some intermediate mechanism, such as cardiac function or the metabolism of lipids or glucose, to produce its effects on risks that have not otherwise been identified or controlled for. For example, a high relative weight has been associated with an increased risk of a first major coronary event for men in their forties. As age increases, the relationship between weight and CHD decreases. In the Framingham study, relative body weight of women was positively and independently associated with developing CHD. In other studies, however, few significant relationships were found between body weight and the risk of myocardial infarction and CHD death. Associations of obesity with alterations in lipoprotein metabolism may be related to the risk of developing coronary atherosclerosis.
Although measuring blood pressure in obese patients is difficult, there is evidence that increased body weight is associated with hypertension. Hypertension also has a striking correlation with lateral body build, which may be more important than obesity in the positive correlation between blood pressure and body weight. Other researchers have reported that hypertension may be less severe in obese subjects than in normal-weight subjects. The relationship between hypertension and body size needs further exploration.
As adiposity increases, the risk of NIDDM also increases. Fasting glucose levels have also been reported to change in the same direction as body weight. It has been postulated that there is a genetic component that contributes to the risk of developing insulin resistance. In general, weight loss in obese subjects could improve glucose tolerance and weight gain could worsen it.
Certain cancers, such as cancers of the gallbladder, biliary duct, endometrium, ovary, breast, and cervix in women, and cancers of the colon and prostate in men, have been associated with excess weight. In particular, the incidence of endometrial cancer has been shown to increase among obese, postmenopausal women. The association of obesity with cancer is discussed further in Chapters 6 and 22.
Obesity is also associated with increased risk of gallbladder disease. Cholesterol production rate has been reported to correlate with body weight and the number of fat cells. Increased abdominal circumference relative to hip circumference is associated more strongly than BMI per se with early death and with the risk of developing heart disease, hypertension, stroke, diabetes mellitus, and in women, endometrial carcinoma.
Childhood obesity increases the likelihood of obesity in adult life, but only a minority of the overweight adults were overweight as children or adolescents. The degree and timing of obesity in childhood are the two most important factors that influence whether or not the obesity will carry over into adulthood. Weight gain in adult life usually results from a relative decrease in energy expendi-
Page 586
ture, which is on average greater than the decline in food intake. This conclusion arises from an epidemiologic study showing that energy intake by men decreased over 10 years during which body weight rose. Weight gain or gain-and-loss cycles in adults may carry greater risks for chronic disease than does stable body weight.
Directions for Research
· It is now recognized that fat distribution, particularly the fat deposition in the abdominal and probably the intraabdominal region, is an important determinant of health risk; however, several important questions remain to be answered: Are intra- and extraabdominal subcutaneous fat equally important? Why does fat accumulate in one region as opposed to another? What are the relative risks of regional fat distribution? What are the feedback signals for regulation of fat stores? One possible clue to this last question is the observation that testosterone, estrogen, and adrenal steroids have powerful influences on the sites of fat deposition. The mechanisms by which these steroid hormones produce their effects on fat, and how this can be influenced, deserve high priority.
· Understanding food intake is a second area of importance. In most adults, body fat increases with age, even though average food intake declines (see Chapter 6). Research is needed to study why food intake does not decrease further to keep the stores of fat in balance, and why some humans and some animals are more susceptible to increasing body fat stores when eating a high-fat diet, whereas others are not responsive to this dietary shift. This implies that there are genetic or individual differences that are not understood and that deserve more research.
· Big eaters who are not obese tend to be more active. Research is needed to elucidate the relationship between physical activity and the ingestion and storage of fat and to understand the environmental and genetic interactions that regulate food intake as a function of physical activity. Better understanding of the mechanisms by which food intake and physical activity are related will provide new insights into the prevention and management of obesity.
References
Abraham, S., G. Collins, and M. Nordsieck. 1971. Relationship of childhood weight status to morbidity in adults. HSMHA Health Rep. 86:273-284.
Abraham, S., C.L. Johnson, and M.F. Najjar. 1979. Weight and Height of Adults 18-74 Years of Age: United States, 1971-74. Vital and Health Statistics, Ser. 11, No. 211. DHEW Publ. No. PHS-79-1659. National Center for Health Statistics, Public Health Service, U.S. Department of Health, Education, and Welfare, Hyattsville, Md. 49 pp.
Abraham, S., M.D. Carroll, M.F. Najjar, and R. Fulwood. 1983. Obese and overweight adults in the United States. Vital and Health Statistics, Ser. 11, No. 230. DHHS Publ. No. (PHS) 83-1680. National Center for Health Statistics, Public Health Service, U.S. Department of Health and Human Services, Hyattsville, Md. 93 pp.
Andres, R. 1985. Mortality and obesity: the rationale for age-specific height-weight tables. Pp. 311-318 inR. Andres, E.L. Bierman, and W.R. Hazzard, eds. Principles of Geriatric Medicine. McGraw-Hill, New York.
APA (American Psychiatric Association). 1987. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. American Psychiatric Association, Washington, D.C. 567 pp.
Ashley, F.W., Jr., and W.B. Kannel. 1974. Relation of weight change to changes in atherogenic traits: the Framingham Study. J. Chronic Dis. 27:103-114.
Avons, P., P. Ducimetiere, and R. Rakotovao. 1983. Weight and mortality. Lancet 1:1104.
Baecke, J.A., W.A. van Staveren, and J. Burema. 1983. Food consumption, habitual physical activity, and body fatness in young Dutch adults. Am. J. Clin. Nutr. 37:278-286.
Barrett-Connor, E.L. 1985. Obesity, atherosclerosis, and coronary artery disease. Ann. Intern. Med. 103:1010-1019.
Barrett-Connor, E., and K.T. Khaw. 1985. Is hypertension more benign when associated with obesity? Circulation 72: 53-60.
Bell, N.H., S. Epstein, A. Greene, J. Shary, M.J. Oexmann, and S. Shaw. 1985. Evidence for alteration of the vitamin D-endocrine system in obese subjects. J. Clin. Invest. 76: 370-373.
Benn, R.T. 1971. Some mathematical properties of weight-for-height indices used as measures of adiposity. Br. J. Prev. Soc. Med. 25:42-50.
Bennion, L.J., and S.M. Grundy. 1978. Risk factors for the development of cholelithiasis in man (part 2). N. Engl. J. Med. 299:1221-1227.
Bernstein, R.A., E.E. Geifer, J.J. Vieira, L.H. Werner, and A.A. Rimm. 1977. Gallbladder disease. II. Utilization of the life table method in obtaining clinically useful information. A study of 62,739 weight-conscious women. J. Chronic Dis. 30:529-541.
Berthaud, H.R., and B. Jeanrenaud. 1979. Changes of insulinemia, glycemia and feeding behavior induced by VMH-procainization in the rat. Brain Res. 174:184-187.
Bhat, M.K., and H.R. Cama. 1978. Thyroidal control of hepatic release and metabolism of vitamin A. Biochim. Biophys. Acta 541:211-222.
Björntorp, P. 1985. Adipose tissue in obesity (Willendorf lecture). Pp. 163-170 in J. Hirsch and T.B. Van Itallie, eds. Recent Advances in Obesity Research, Vol. IV. John Libbey, London.
Bloom, E., K. Yano, J.D. Curb, D.M. Reed, and C.J. MacLean. 1987. Smoking cessation and incidence of coronary heart disease. CVD Epidemiol. Newsletter (AHA) 41: 36.
Bogardus, C., S. Lillioja, E. Ravussin, W. Abbott, J.K. Zawadzki, A. Young, W.C. Knowler, R. Jacobowitz, and P.P. Moll. 1986. Familial dependence on the resting metabolic rate. N. Engl. J. Med. 315:96-100.
Page 587
Börjeson, M. 1962. Overweight children. Acta Paediatr. 51 suppl. 132:1-76.
Börjeson, M. 1976. The aetiology of obesity in children. A study of 101 twin pairs. Acta Paediatr. Scand. 65:279-287.
Borkan, G.A., D. Sparrow, C. Wisniewski, and-P.S. Vokonas. 1986. Body weight and coronary disease risk: patterns of risk factor change associated with long-term weight change. Am. J. Epidemiol. 124:410-419.
Boskind-Lodahl, M., and W.C. White, Jr. 1978. The definition and treatment of bulimarexia in college women—a pilot study. J. Am. Coll. Health Assoc. 27:84-86.
Bouchard, C. 1988. Inheritance of human fat distribution. Pp. 103-125 in C. Bouchard and F.E. Johnston, eds. Fat Distribution During Growth and Later Health Outcomes: Current Topics in Nutrition and Disease, Vol. 17. Alan R. Liss, New York.
Bouchard, C., R. Lesage, G. Lortie, J.A. Simoneau, P. Hamel, M.R. Boulay, L. Perusse, G. Theriault, and C. Leblanc. 1986. Aerobic performance in brothers, dizygotic and monozygotic twins. Med. Sci. Sports Exercise 18:639-646.
Boyar, R.M., L.D. Hellman, H. Roffwarg, J. Katz, B. Zumoff, J. O'Connor, H.L. Bradlow, and D.K. Fukushima. 1977. Cortisol secretion and metabolism in anorexia nervosa. N. Engl. J. Med. 296:190-193.
Bradden, F.E., B. Rodgers, M.E. Wadsworth, and J.M. Davies. 1986. Onset of obesity in a 36-year birth cohort study. Br. Med. J. 293:299-303.
Braitman, L.E., E.V. Adlin, and J.L. Stanton, Jr. 1985. Obesity and caloric intake: the National Health and Nutrition Examination Survey of 1971-1975 (HANES I). J. Chronic Dis. 38:727-732.
Bray, G.A. 1976. The Obese Patient: Major Problems in Internal Medicine, Vol. IX. W.B. Saunders, Philadelphia. 450 pp.
Bray, G.A. 1978. Definition, measurement, and classification of the syndromes of obesity. Int. J. Obes. 2:99-112.
Bray, G.A. 1985. Complications of obesity. Ann. Intern. Med. 103:1052-1062.
Bray, G.A. 1987a. Obesity—a disease of nutrient or energy balance? Nurr. Rev. 45:33-43.
Bray, G.A. 1987b. Obesity and the heart. Mod. Concepts Cardiovasc. Dis. 56:67-71.
Bray, G.A., and D.S. Gray. 1988. Obesity: Part I—Pathogenesis. West. J. Med. 149:429-441.
Bray, G.A., and D.A. York. 1971. Genetically transmitted obesity in rodents. Physiol. Rev. 51:598-646.
Bray, G.A., and D.A. York. 1979. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol. Rev. 59:719-809.
Bray, G.A., D.A. York, and J.S. Fissler. In press. Experimental obesity: a homeostatic failure due to defective nutrient stimulation of the sympathetic nervous system. Vitam. Horm.
Brook, C.G., R.M. Huntley, and J. Slack. 1975. Influence of heredity and environment in determination of skinfold thickness in children. Br. Med. J. 2:719-721.
Brownell, K.D. 1982. Obesity: understanding and treating a serious, prevalent, and refractory disorder. J. Consult. Clin. Psychol. 50:820-824.
Brownell, K.D., M.R. Greenwood, E. Stellar, and E.E. Shrager. 1986. The effects of repeated cycles of weight loss and regain in rats. Physiol. Behav. 38:459-464.
Burman K.D., R.A. Vigersky, D.L. Loriaux, D. Strum, Y.Y. Djuh, F.D. Wright, and L. Wartofsky. 1977. Investigations concerning thyroxine deiodinative pathways in patients with anorexia nervosa. Pp. 255-261 in R.A. Vigersky, ed. Anorexia Nervosa. Raven Press, New York.
Byers, T., J. Marshall, S. Graham, C. Mettlin, and M. Swanson. 1983. A case-control study of dietary and nondietary factors in ovarian cancer. J. Natl. Cancer Inst. 71:681-686.
Cambien, F., J.M. Chretien, P. Ducimetiere, L. Guize, and J.L. Richard. 1985. Is the relationship between blood pressure and cardiovascular risk dependent on body mass index? Am. J. Epidemiol. 122:434-442.
Chaing, B.N., L.V. Perlman, and F.H. Epstein. 1969. Overweight and hypertension: a review. Circulation 39:403-421.
Cheek, D.B. 1968. Human Growth. Lea & Febiger, Philadelphia. 781 pp.
Clarke, W.R., R.F. Woolson, and R.M. Lauer. 1986. Changes in ponderosity and blood pressure in children: the Muscatine Study. Am. J. Epidemiol. 124:195-206.
Cleary, M.P., J.R. Vasselli, and M.R. Greenwood. 1980. Development of obesity in Zucker obese (fafa) rat in absence of hyperphagia. Am. J. Physiol. 238:E284-E292.
Cohn, S.H.. A.N. Vaswani, S. Yasumura, K. Yuen, and K.J. Ellis. 1984. Improved models for determination of body fat by in vivo neutron activation. Am. J. Clin. Nutr. 40:255-259.
Coleman, D.L. 1979. Obesity genes: beneficial effects in heterozygous mice. Science 203:663-665.
Collier, G.H. 1985. Satiety: an ecological perspective. Brain Res. Bull. 14:693-700.
Cooper, P.J., and C.G. Fairburn. 1983. Binge-eating and self-induced vomiting in the community: a preliminary study. Br. J. Psychiatry 142:139-144.
Cox, J.E., and T.L. Powley. 1977. Development of obesity in diabetic mice pair-fed with lean siblings. J. Comp. Physiol. Psychol. 91:347-358.
Cox, J.E., and T.L. Powley. 1981. Prior vagotomy blocks VMH obesity in pair-fed rats. Am. J. Physiol. 240:E573-E583.
Crisp, A.H. 1977. Some psychobiological aspects of adolescent growth and their relevance for the fat/thin syndrome (anorexia nervosa). Int. J. Obes. 1:231-238.
Crisp, A.H. 1983. Treatment and outcome in anorexia nervosa. Pp. 91-104 in R.K. Goodstein, ed. Eating and Weight Disorders: Advances in Treatment and Research. Springer Publishing Co., New York.
Cruce, J.A., M.R. Greenwood, P.R Johnson, and D. Quartermain. 1974. Genetic versus hypothalamic obesity: studies of intake and dietary manipulations in rats. J. Comp. Physiol. Psychol. 87:295-301.
Curran-Celentano, J., J.W. Erdman, Jr., R.A. Nelson, and S.J. Grater. 1985. Alterations in vitamin A and thyroid hormone status in anorexia nervosa and associated disorders. Am. J. Clin. Nutr. 42:1183-1191.
Dalen, N., D. Hallberg, and B. Lamke. 1975. Bone mass in obese subjects. Acta Med. Scand. 197:353-355.
Dally, P. 1969. Anorexia Nervosa. Grune & Stratton, New York. 137 pp.
de Boer, J.O., A.J. van Es, L.C. Roovers, J.M. van Raaij, and J.G. Hautvast. 1986. Adaptation of energy metabolism of overweight women to low-energy intake, studied with whole-body calorimeters. Am. J. Clin. Nutr. 44:585-595.
de Waard, F., and E.A. Baanders-van Halewijn. 1974. A prospective study in general practice on breast-cancer risk in postmenopausal women. Int. J. Cancer 14:153-160.
Page 588
DHEW (Department of Health, Education, and Welfare). 1976. Report of the National Commission on Diabetes to the Congress of the United States, Vol. 1: The Long Range Plan to Combat Diabetes. DHEW Publ. No. (NIH) 76-1018. National Institutes of Health, Public Health Service, U.S. Department of Health, Education, and Welfare, Washington, D.C. 97 pp.
DHHS (U.S. Department of Health and Human Services). 1988. The Surgeon General's Report on Nutrition and Health. DHHS (PHS) Publ. No. 88-50210. Public Health Service, U.S. Department of Health and Human Services. U.S. Government Printing Office, Washington, D.C. 712 pp.
Dietz, W.H. 1987. Pediatric obesity and the risk of obesity in adulthood. Presented at a workshop in La Jolla, California, on January 21, 1987, held by the Committee on Diet and Health, Food and Nutrition Board, Commission on Life Sciences, National Research Council, Washington, D.C. 7 pp.
Donahue, R.P., R.D. Abbott, E. Bloom, D.M. Reed, and K. Yano. 1987. Central obesity and coronary heart disease in men. Lancet 1:821-824.
Drenick, E.J., M.E. Swendseid, W.H. Blahd, and S.G. Tuttle. 1964. Prolonged starvation as treatment for severe obesity. J. Am. Med. Assoc. 187:100-105.
Drenick, E.J., A.S. Brickman, and E.M. Gold. 1972. Dissociation of the obesity-hyperinsulinism relationship following dietary restriction and hyperalimentation. Am. J. Clin. Nutr. 25:746-755.
Drenick, E.J., G.S. Bale, F. Seltzer, and D.G. Johnson. 1980. Excessive mortality and causes of death in morbidly obese men. J. Am. Med. Assoc. 243:443-445.
Ducimetiere, P., J. Richard, and F. Cambien. 1986. The pattern of subcutaneous fat distribution in middle-aged men and the risk of coronary heart disease: the Paris Prospective Study. Int. J. Obes. 10:229-240.
Durnin, J.V., and J. Womersley. 1974. Body fat assessed from total body density and its estimation from skinfold thickness: measurements on 481 men and women aged from 16 to 72 years. Br. J. Nutr. 32:77-97.
Egusa, G., W.F. Beltz, S.M. Grundy, and B.V. Howard. 1985. Influence of obesity on the metabolism of apolipoprotein B in humans. J. Clin. Invest. 76:596-603.
Engel, A. 1968. Osteoarthritis and body measurements. Vital Health Stat. 11:1-37.
Ernsberger, P., and D.O. Nelson. 1988. Refeeding hypertension in dietary obesity. Am. J. Physiol. 254:R47-R55.
Fabsitz, R., M. Feinleib, and Z. Hrubec. 1980. Weight changes in adult twins. Acta Genet. Med. Gemellol. 29: 273-279.
Fadiman, A. 1982. The skeleton at the feast: a case study of anorexia nervosa. Life 5:62-76.
Faust, I.M., P.R. Johnson, J.S. Stem, and J. Hirsch. 1978. Diet-induced adipocyte number increase in adult rats: a new model of obesity. Am. J. Physiol. 235:E279-E286.
Feighner, J.P., E. Robins, S.B. Guze, R.A. Woodruff, Jr., G. Winokur, and R. Munoz. 1972. Diagnostic criteria for use in psychiatric research. Arch. Gen. Psychiatry 26:57-63.
Feinleib, M., R.J. Garrison, R. Fabsitz, J.C. Christian, Z. Hrubec, N.O. Borhani, W.B. Kannel, R. Rosenman, J.T. Schwartz, and J.O. Wagner. 1977. The NHLBI twin study of cardiovascular disease risk factors: methodology and summary of results. Am. J. Epidemiol. 106:284-285.
Feldman, R., A.J. Sender, and A.B. Sieglaub. 1969. Difference in diabetic and non diabetic fat distribution patterns by skin fold measurements. Diabetics 18:478-486.
Fitness and Amateur Sport. 1986. Canadian Standardized Test of Fitness (for 15 to 69 Years of Age), 3rd ed. Operations Manual. FAS 7378. Minister of State, Fitness and Amateur Sport, Government of Canada, Ottawa, Ontario, Canada. 41 pp.
Forbes, G.B. 1984. Energy intake and body weight: a reexamination of two ''classic" studies. Am. J. Clin. Nutr. 39:349-350.
Forbes, G.B. 1987. Lean body mass-body fat interrelationships in humans. Nutr. Rev. 45:225-231.
Frank, S., J.A. Colliver, and A. Frank. 1986. The electrocardiogram in obesity. Statistical analysis of 1,129 patients. J. Am. Coll. Cardiol. 7:295-299.
Frankel, R.J., and J.S. Jenkins. 1975. Hypothalamic-pituitary function in anorexia nervosa. Acta Endocrinol. 78:209-221.
Friedman, G.D., W.B. Kannel, and T.R. Dawber. 1966. The epidemiology of gallbladder disease: observations in the Framingham Study. J. Chronic Dis. 19:273-292.
Garfinkel, P.E., and D.M. Garner. 1982. Anorexia Nervosa: A Multidimensional Perspective. Brunner/Mazel, New York. 379 pp.
Garn, S.M., and A.S. Ryan. 1982. The effect of fatness on hemoglobin levels. Am. J. Clin. Nutr. 36:189-192.
Garrow, J.S. 1978. Energy Balance and Obesity in Man, 2nd ed. Elsevier/North Holland, New York. 243 pp.
Geissler, C.A., D.S. Miller, and M. Shah. 1987. The daily metabolic rate of the post-obese and the lean. Am. J. Clin. Nutr. 45:914-920.
Golden, N., and I.M. Sacker. 1984. An overview of the etiology, diagnosis, and management of anorexia nervosa. Clin. Pediatr. 4:209-214.
Gordon, T., W.B. Kannel, W.P. Castelli, M.C. Hjortland, W.B. Kannel, and T.R. Dawber. 1977a. Diabetes, blood lipids, and the role of obesity in coronary heart disease risk for women. The Framingham study. Ann. Intern. Med. 87: 393-397.
Gordon, T., W.P. Castelli, M.C. Hjortland, W.B. Kannel, and T.R. Dawber. 1977b. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 62:707-714.
Gray, D.S., J.S. Fisler, and G.A. Bray. 1988. Effects of repeated weight loss and regain on body composition in obese rats. Am. J. Clin. Nutr. 47:393-399.
Gross, T., R.J. Sokol, and K.C. King. 1980. Obesity in pregnancy: risks and outcome. Obstet. Gynecol. 56:446-450.
Grundy, S.M., H.Y. Mok, L. Zech, D. Steinberg, and M. Berman. 1979. Transport of very low density lipoprotein triglycerides in varying degrees of obesity and hypertriglyceridemia. J. Clin. Invest. 63:1274-1283.
Gulick, A. 1922. A study of weight regulation in the adult human body during over-nutrition. Am. J. Physiol. 60:371-395.
Haffner, S.M., M.P. Stern, H.P. Hazuda, J. Pugh, and J.K. Patterson. 1987. Do upper-body and centralized adiposity measure different aspects of regional body-fat distribution? Relationship to non-insulin-dependent diabetes mellitus, lipids, and lipoproteins. Diabetes 36:43-51.
Halmi, K.A. 1985. Eating disorders. Pp. 1731-1736 in H.I. Kaplan and B.J. Sadock, eds. Comprehensive Textbook of Psychiatry IV, Vol. 2. Williams and Wilkins, Baltimore.
Page 589
Halmi, K.A., J.R. Falk, and E. Schwartz. 1981. Binge-eating and vomiting: a survey of a college population. Psychol. Med. 11:697-706.
Hamm, P., R.B. Shekelle, and J. Stamler. 1989. Large fluctuations in body weight during young adulthood and twenty-five-year risk of coronary death in men. Am. J. Epidemiol. 129:312-318.
Han, P.W. 1967. Hypothalamic obesity in rats without hyperphagia. Trans. N.Y. Acad. Sci. 30:229-242.
Hartz, A.J., P.N. Barboriak, A. Wong, K.P. Katayama, and A.A. Rimm. 1979. The association of obesity with infertility and related menstrual abnormalities in women. Int. J. Obes. 3:57-73.
Hartz, A.J., D.C. Rupley, and A.A. Rimm. 1984. The association of girth measurements with disease in 32,856 women. Am. J. Epidemiol. 119:71-80.
Havlik, R.J., H.B. Hubert, R.R. Fabsitz, and M. Feinleib. 1983. Weight and hypertension. Ann. Intern. Med. 98: 855-859.
Henderson, B.E., J.T. Casagrande, M.C. Pike, T. Mack, and I. Rosario. 1983. The epidemiology of endometrial cancer in young women. Br. J. Cancer 47:749-756.
Henry, R.R., L. Scheaffer, and J.M. Olefsky. 1985. Glycemic effects of intensive caloric restriction and isocaloric refeeding in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 61:917-925.
Herzog, D.B. 1982. Bulimia in the adolescent. Am. J. Dis. Child. 136:985-989.
Herzog, D.B., and P.M. Copeland. 1985. Eating disorders. N. Engl. J. Med. 313:295-303.
Heyden, S., C.G. Hames, A. Bartel, J.C. Cassel, H.A. Tyroler, and J.C. Cornoni. 1971. Weight and weight history in relation to cerebrovascular and ischemic heart disease. Arch. Intern. Med. 128:956-960.
Hill, J.O., S.K. Fried, and M. DiGirolamo. 1984. Effects of fasting and restricted refeeding on utilization of ingested energy in rats. Am. J. Physiol. 247:R318-R327.
Himsworth, H.P. 1935. Diet and incidence of diabetes mellitus. Clin. Sci. 2:117-148.
Hirsch, J., and B. Batchelor. 1976. Adipose tissue cellularity in human obesity. Clin. Endocrinol. Metab. 5:299-311.
Hirsch, J., and E. Gallian. 1968. Methods for the determination of adipose cell size in man and animals. J. Lipid Res. 9:110-119.
Hoek, W.H., and F.G. Brook. 1985. Patterns of care of anorexia nervosa. J. Psychiatr. Res. 19:155-160.
Hoffmans, M.D., D. Kromhout, and C. de Lezenne Coulander. 1988. The impact of body mass index of 78,612 18-year-old Dutch men on 32-year mortality from all causes. J. Clin. Epidemiol. 41:749-756.
Hoyenga, K.B., and K.T. Hoyenga. 1982. Gender and energy balance: sex differences in adaptations for feast and famine. Physiol. Behav. 28:545-563.
Hsu, P.H., F.A. Mathewson, H.A.Abu-Zeid, and S.W. Rabkin. 1977. Change in risk factor and the development of chronic disease. A methodological illustration. J. Chronic Dis. 30:567-584.
Hubert, H.B., M. Feinleib, P.M. McNamara, and W.P. Castelli. 1983. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 67:968-977.
Ikeda, H., A. Shino, T. Matsuo, H. Iwatsuka, and Z. Suzuoki. 1981. A new genetically obese-hyperglycemic rat (Wistar fatty). Diabetes 30:1045-1050.
Jeffery, R.W., A.R. Folsom, R.V. Luepker, D.R. Jacobs, Jr., R.F. Gillum, H.L. Taylor, and H. Blackburn. 1984. Prevalence of overweight and weight loss behavior in a metropolitan adult population: the Minnesota Heart Survey experience. Am. J. Public Health 74:349-352.
Jensen, H. 1985. Endometrial carcinoma. A retrospective, epidemiological study. Dan. Med. Bull. 32:219-228.
Jequier, E. 1984. Energy expenditure in obesity. Clin. Endocrinol. Metab. 13:563-580.
Johnson, D., and E.J. Drenick. 1977. Therapeutic fasting in morbid obesity. Arch. Intern. Med. 137:1381-1382.
Joslin, E.P., L.I. Dublin, and H.H. Marks. 1935. Studies in diabetes mellitus; interpretations of variations in diabetes incidence. Am. J. Med. Sci. 189:163-192.
Kannel, W.B., and D.L McGee. 1985. Update on some epidemiologic features of intermittent claudication: the Framingham Study. J. Am. Geriatr. Soc. 33:13-18.
Kannel, W.B., and A. Schatzkin. 1984. Risk factor analysis. Prog. Cardiovasc. Dis. 26:309-332.
Kava, R.A., D.B. West, V.A. Lukasik, and M.R.C. Greenwood. 1989. Sexual dimorphism of hyperglycemia and glucose tolerance in Wistar fatty rat. Diabetes 38:159-163.
Keen, H., B.J. Thomas, R.J. Jarrett, and J.H. Fuller. 1979. Nutrient intake, adiposity, and diabetes. Br. Med. J. 1:655-658.
Kelsey, J.L., D.B. Fischer, T.R. Holford, V.A. LiVoisi, E.D. Mostow, I.S. Goldenberg, and C. White. 1981. Exogenous estrogens and other factors in the epidemiology of breast cancer. J. Natl. Cancer Inst. 67:327-333.
Keys, A., J. Brozek, A. Henschel, O. Mickelsen, and H.L. Taylor. 1950. The Biology of Human Starvation, Vols. I and 2. University of Minnesota Press, Minneapolis. 1385 pp.
Kinsell, L.W., B. Gunning, G.D. Michaels, J. Richardson, S.E. Cox, and C. Lemon. 1964. Calories do count. Metabolism 13:195-204.
Kissebah, A.H., N. Vydelingum, R. Murray, D.J. Evans, A.J. Hartz, R.K. Kalkhoff, and P.W. Adams. 1982. Relation of body fat distribution to metabolic complications of obesity. J. Clin. Endocrinol. Metab. 54:254-260.
Knowler, W.C., P.H. Bennett, R.F. Hamman, and M. Miller. 1978. Diabetes incidence and prevalence in Pima Indians: a 19-fold greater incidence than in Rochester, Minnesota. Am. J. Epidemiol. 108:497-505.
Knowler, W.C., D.J. Pettitt, P.J. Savage, and P.H. Bennett. 1981. Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes. Am. J. Epidemiol. 113:144-156.
Kolonel, LN., C.N. Yoshizawa, and J.H. Hankin. 1988. Diet and prostatic cancer: a case-control study in Hawaii. Am. J. Epidemiol. 127:999-1012.
Kretchmar, L. 1984. A look at eating disorders on campus: feast and famine. Dartmouth Alumni Magazine 76:39-46.
Krishnan, E.C., L. Trost, S. Aarons, and W.R. Jewell. 1982. Study of function and maturation of monocytes in morbidly obese individuals. J. Surg. Res. 33:89-97.
Kromhout, D. 1983. Energy and macronutrient intake in lean and obese middle-aged men (the Zutphen study). Am. J. Clin. Nutr. 37:295-299.
Krotkiewski, M., P. Björntorp, L. Sjöström, and U. Smith. 1983. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J. Clin. Invest. 72:1150-1162.
Lapidus, L., C. Bengtsson, B. Larsson, K. Pennert, E. Rybo,
Page 590
and L. Sjöström. 1984. Distribution of adipose tissue and risk of cardiovascular disease and death: a 12 year follow-up of participants in the population study of women in Gothenburg, Sweden. Br. Med. J. 289:1257-1261.
Lapidus, L., H. Andersson, C. Bengtsson, and I. Bosaeus. 1986. Dietary habits in relation to incidence of cardiovascular disease and death in women: a 12-year follow-up of participants in the population study of women in Gothenburg, Sweden. Am. J. Clin. Nutr. 44:444-448.
Larsson, B., P. Björntorp, and G. Tibblin. 1981. The health consequences of moderate obesity. Int. J. Obesity 5:97-116.
Larsson, B., K. Svärdsudd, L. Welin, L. Wilhelmsen, P. Björntorp, and G. Tibblin. 1984. Abdominal adipose tissue distribution, obesity, and risk of cardiovascular disease and death: 13 year follow-up of participants in the study of men born in 1913. Br. Med. J. 288:1401-1404.
Lavau, M., C. Susini, J. Knittle, S. Blanchet-Hirst, and M.R.C. Greenwood. 1977. A reliable photomicrographic method to determining fat cell size and number: application to dietary obesity. Proc. Soc. Exp. Biol. Med. 156:251-256.
La Vecchia, C., A. Decarli, M. Fasoli, and A. Gentile. 1986. Nutrition and diet in the etiology of endometrial cancer. Cancer 57:1248-1253.
Leibel, R.L., and J. Hirsch. 1984. Diminished energy requirements in reduced obese patients. Metabolism 33:164-170.
Lemonnier, D. 1972. Effect of age, sex, and site on the cellularity of the adipose tissue in mice and rats rendered obese by a high-fat diet. J. Clin. Inves. 51:2907-2915.
Levin, E. 1983. A sweet surface hid a troubled soul in the late Karen Carpenter, a victim of anorexia nervosa. People Weekly 19:52-59.
Lew, E.A., and L. Garfinkel. 1979. Variations in mortality by weight among 750,000 men and women. J. Chronic Dis. 32: 563-576.
Lillioja, S., D.M. Mott, J.K. Zawadzki, A.A. Young, W.G. Abbott, W.C. Knowler, P.H. Bennett, P. Moll, and C. Bogardus. 1987. In vivo insulin action is familial characteristic in nondiabetic Pima Indians. Diabetes 36:1329-1335.
Lobo, R.A., C.M. March, U. Goebelsmann, and D.R. Mishell, Jr. 1982. The modulating role of obesity and 17 beta-estradiol (E2) on bound and unbound E2 and adrenal androgens in oophorectomized women. J. Clin. Endocrinol. Metab. 54:320-324.
Lohman, T.G. 1981. Skinfolds and body density and their relation to body fatness: a review. Hum. Biol. 53:181-225.
Lubin, F., A.M. Ruder, Y. Wax, and B. Modan. 1985. Overweight and changes in weight throughout adult life in breast cancer etiology. A case-control study. Am. J. Epidemiol. 122:579-588.
Lukaski, H.C. 1987. Methods for the assessment of human body composition: traditional and new. Am. J. Clin. Nutr. 46:537-556.
MacMahon, S.W., D.E. Wilcken, and G.J. Macdonald. 1986. The effect of weight reduction on left ventricular mass. A randomized controlled trial in young, overweight hypertensive patients. N. Engl. J. Med. 314:334-339.
Manson, J.E., M.J. Stampfer, C.H. Hennekens, and W.C. Willett. 1987. Body weight and longevity. A reassessment. J. Am. Med. Assoc. 257:353-358.
Masoro, E.J. 1985. Aging and nutrition—can diet affect life span? Trans. Assoc. Life Insur. Med. Dir. Am. 67:30-44.
Mattila, K., M. Haavisto, and S. Rajala. 1986. Body mass index and mortality in the elderly. Br. Med. J. 292:867-868.
McCay, C.M., M.F. Crowell, and L.A. Maynard. 1935. Effect of retarded growth upon length of life span and upon ultimate body size. J. Nutr. 10:63-79.
McGee, D., and T. Gordon. 1976. The results of the Framingham Study applied to four other U.S. based epidemiological studies of cardiovascular disease. Section 31 in W. B. Kannel and T. Gordon, eds. The Framingham Study: An Epidemiological Investigation of Cardiovascular Disease. DHEW Publ. No. (NIH) 76-1083. National Heart and Lung Institute, National Institutes of Health, Public Health Service, U.S. Department of Health, Education, and Welfare, Bethesda, Md.
Medlund, P., R. Cederlöf, B. Floderus-Myrhed, L. Friberg, and S. Sörensen. 1976. A new Swedish twin registry. Acta Med. Scand., Suppl. 600:1-111.
Messerli, F.H. 1982. Cardiovascular effects of obesity and hypertension. Lancet 1:1165-1168.
Miettinen, T.A. 1971. Cholesterol production in obesity. Circulation 44:842-850.
Morgan, H.G., and G.F. Russell. 1975. Value of family background and clinical features as predictors of long-term outcome in anorexia nervosa: four-year follow-up study of 41 patients. Psychol. Med. 5:355-371.
Mueller, W.H. 1983. The genetics of human fatness. Yearb. Phys. Anthropol. 26:215-230.
Mueller, W.H. 1988. Ethnic differences in fat distribution during growth. Pp. 127-145 in C. Bouchard and F.E. Johnston, eds. Current Topics in Nutrition and Disease, Vol. 17: Fat Distribution During Growth and Later Health Outcomes. Alan R. Liss, New York.
Naeye, R.L. 1979. Weight gain and the outcome of pregnancy. Am. J. Obstet. Gynecol. 135:3-9.
Nelson, D.O., and P. Ernsberger. 1984. Feed-starve cycling in dietary obesity induces moderate hypertension via alterations in the autonomic regulation of cardiovascular function. Soc. Neurosci. 10:716.
Nestel, P.J., P.H. Schreibman, and E.H. Ahrens, Jr. 1973. Cholesterol metabolism in human obesity. J. Clin. Invest. 52:2389-2397.
Neumann, R.O. 1902. Experimentelle Beiträge zur Lehre von dem täglichen Nahrungsbedarf des Menschen unter besondeer Berücksichtigung der notwendigen Eiweissmenge. Arch. Hyg. 45:1-87.
Newburgh, L.H., and J.W. Conn. 1939. New interpretation of hypergylcemia in obese middle-aged persons. J. Am. Med. Assoc. 112:7-11.
Noppa, H. 1980. Body weight change in relation to incidence of ischemic heart disease and change in risk factors for ischemic heart disease. Am. J. Epidemiol. 111:693-704.
Noppa, H., and T. Hällström. 1981. Weight gain in adulthood in relation to socioeconomic factors, mental illness and personality traits: a prospective study of middle-aged women. J. Psychosom. Res. 25:83-89.
Noppa, H., C. Bengtsson, H. Wedel, and L. Wilhelmsen. 1980. Obesity in relation to morbidity and morality from cardiovascular disease. Am. J. Epidemiol. 111:682-692.
Ohlson, L.O., B. Larsson, K. Svärdsudd, L. Welin, H. Eriksson, L. Wilhelmsen, P. Björntorp, and G. Tibblin. 1985. The influence of body fat distribution on the incidence of diabetes mellitus. 13.5 years of follow-up of the participants in the study of men born in 1913. Diabetes 34:1055-1058.
Olefsky, J.M., O.G. Kolterman, and J.A. Scarlett. 1982. Insulin action and resistance in obesity and non insulin-
Page 591
dependent type II diabetes mellitus. Am. J. Physiol. 243:E15-E30.
Owen, O.E., E. Kavle, R.S. Owen, M. Polansky, S. Caprio, M.A. Mozzoli, Z.V. Kendrick, M.C. Bushman, and G. Boden. 1986. A reappraisal of caloric requirements in healthy women. Am. J. Clin. Nutr. 44:1-19.
Owen, O.E., J.L. Holup, D.A. D'Alessio, E.S. Craig, M. Polansky, K.J. Smalley, E.C. Kavle, M.C. Bushman, L.R. Owen, M.A. Mozzoli, Z.V. Kendrick, and G.H. Boden. 1987. A reappraisal of the caloric requirements of men. Am. J. Clin. Nutr. 46:875-885.
Palmer, E.P., and A.T. Guay. 1985. Reversible myopathy secondary to abuse of ipecac in patients with major eating disorders. N. Engl. J. Med. 313:1457-1459.
Pasquali, R., S. Venturoli, R. Paradis, M. Capelli, M. Parenti, and N. Melchionda. 1982. Insulin and C-peptide levels in obese patients with polycystic ovaries. Horm. Metab. Res. 14:284-287.
Peckham, C.H., and R.E. Christianson. 1971. The relationship between pre-pregnancy weight and certain obstetric factors. Am. J. Obstet. Gynecol. 111:1-7.
Poehlman, E.T., A. Tremblay, J.P. Deprés, E. Fontaine, L. Pérusse, G. Thériault, and C. Bouchard. 1986. Genotype-controlled changes in body composition and fat morphology following overfeeding in twins. Am. J. Clin. Nutr. 43:723-731.
Pyke, D.A., and N.W. Please. 1957. Obesity, parity and diabetes. J. Endocrinol. 15:xxvi-xxxiii.
Pyle, R.L., J.E. Mitchell, and E.D. Eckert. 1981. Bulimia: a report of 34 cases. J. Clin. Psychiatry 42:60-64.
Pyle, R.L, J.E. Mitchell, E. Eckert, P.A. Halvorson, P.A. Neuman, and G.M. Goff. 1983. The incidence of bulimia in freshman college students. Int. J. Eating Disorders 2:75-85.
Pyle, R.L, P.A. Halvorson, P.A. Neuman, and J.E. Mitchell. 1986. The increasing prevalence of bulimia in freshman college students. Int. J. Eating Disorders 5:631-636.
Rabinowitz, D. 1970. Some endocrine and metabolic aspects of obesity. Annu. Rev. Med. 21:241-258.
Ravussin, E., S. Lillioja, T.E. Anderson, L. Christin, and C. Bogardus. 1986. Determinants of 24-hour energy expenditure in man. Methods and results using a respiratory chamber. J. Clin. Invest. 78:1568-1578.
Ravussin, E., S. Lillioja, W.C. Knowler, L. Christin, D. Freymond, W.G. Abbott, V. Boyce, B.V. Howard, and C. Bogardus. 1988. Reduced rate of energy expenditure as a risk factor for body-weight gain. N. Engl. J. Med. 318:467-472.
Ray, C.S., D.Y. Sue, G. Bray, J.E. Hansen, and K. Wasserman. 1983. Effects of obesity on respiratory function. Am. Rev. Respir. Dis. 128:501-506.
Reed, D.R., R.J. Contreras, C. Maggio, and M.R. Greenwood, and J. Rodin. 1988. Weight cycling in female rats increases dietary fat selection and adiposity. Physiol. Behav. 42:389-395.
Reisin, E., R. Abel, M. Modan, D.S. Silverberg, H.E. Eliahou, and B. Modan. 1978. Effect of weight loss without salt restriction on the reduction of blood pressure in overweight hypertensive patients. N. Engl. J. Med. 298:1-6.
Rhoads, G.G., and A. Kagan. 1983. The relation of coronary disease, stroke, and mortality to weight in youth and in middle age. Lancet 1:492-495.
Rimm, A.A., L.H. Werner, B.V. Yserloo, and R.A. Bernstein. 1975. Relationship of obesity and disease in 73,532 weight-conscious women. Public Health Rep. 90:44-54.
Robboy, M.S., A.S. Sato, and A.D. Schwabe. 1974. The hypercarotenemia in anorexia nervosa: a comparison of vitamin A and carotene levels in various forms of menstrual dysfunction and cachexia. Am. J. Clin. Nutr. 27:362-367.
Roberts, S.B., J. Savage, W.A. Coward, B. Chew, and A. Lucas. 1988. Energy expenditure and intake in infants born to lean and overweight mothers. N. Engl. J. Med. 318:461466.
Romieu, I., W.C. Willett, M.J. Stampfer, G.A. Colditz, L. Sampson, B. Rosner, C.H. Hennekens, and F.E. Speizer. 1988. Energy intake and other determinants of relative weight. Am. J. Clin. Nutr. 47:406-412.
Russell, G.F. 1965. Metabolic aspects of anorexia nervosa. Proc. R. Soc. Med. 58:811-814.
Schemmel, R., O. Mickelsen, and J.L. Gill. 1970. Dietary obesity in rats: body weight and body fat accretion in seven strains of rats. J. Nutr. 100:1041-1048.
Schwabe, A.D., B.M. Lippe, R.J. Chang, M.A. Pops, and J. Yager. 1981. Anorexia nervosa. Ann. Intern. Med. 94:371-381.
Sclafani, A., and A.N. Gorman. 1977. Effects of age, sex, and prior body weight on the development of dietary obesity in adult rats. Physiol. Behav. 18:1021-1026.
Sclafani, A., and D. Springer. 1976. Dietary obesity in adult rats: similarities to hypothalamic and human obesity syndromes. Physiol. Behav. 17:461-471.
Segal, K.R., B. Gutin, E. Presta, J. Wang, and T.B. Van Itallie. 1985. Estimation of human body composition by electrical impedance methods: a comparative study. J. Appl. Physiol. 58:1565-1571.
Shapiro, S., E. Weinblatt, C.W Frank, and R.V. Sager. 1969. Incidence of coronary heart disease in a population insured for medical care (HIP): myocardial infarction, angina pectoris, and possible myocardial infarction. Am. J. Public Health 59:1-101.
Sharp, J.T., M. Barrocas, and S. Chokroverty. 1980. The cardiorespiratory effects of obesity. Clin. Chest Med. 1:103-118.
Sherman, B.M., and S.G. Korenman. 1974. Measurement of serum LH, FSH, estradiol and progesterone in disorders of the human menstrual cycle: the inadequate luteal phase. J. Clin. Endocrinol. Metab. 39:145-149.
Sims, E.A., E. Danforth, Jr., E.S. Horton, G.A. Bray, J.A. Glennon, and LB. Salans. 1973. Endocrine and metabolic effects of experimental obesity in man. Recent Prog. Horm. Res. 29:457-496.
Sjöström, L, and T. William-Olsson. 1981. Prospective studies on adipose tissue development in man. Int. J. Obes. 5: 597-604.
Smith, M.S. 1984. Anorexia nervosa and bulimia. J. Fam. Practice 18:757-766.
Snowdon, D.A., R.L. Phillips, and W. Choi. 1984. Diet, obesity, and risk of fatal prostate cancer. Am. J. Epidemiol. 120:244-250.
Society of Actuaries. 1959. Build and Blood Pressure Study, Vol. 1. Society of Actuaries, Chicago. 268 pp.
Society of Actuaries. 1980a. Blood Pressure Study of 1979. Society of Actuaries/Association of Life Insurance Medical Directors of America, Chicago. 359 pp.
Society of Actuaries. 1980b. Build Study of 1979. Society of Actuaries/Association of Life Insurance Medical Directors of America, Chicago. 255 pp.
Solberg, L.A., and J.P. Strong. 1983. Risk factors and atherosclerotic lesions. A review of autopsy studies. Arteriosclerosis 3:187-198.
Page 592
Steele, N.C., L.T. Frobish, and M. Keeney. 1974. Lipogenesis and cellularity of adipose tissue from genetically lean and obese swine. J. Anim. Sci. 39:712-719.
Steinkamp, R.C., N.L. Cohen, W.E. Siri, T.W. Sargent, and H.E. Walsh. 1965a. Measures of body fat and related factors in normal adults. I. Introduction and methodology. J. Chronic Dis. 18:1279-1291.
Steinkamp, R.C., N.L. Cohen, W.R. Gaffey, T. McKey, G. Bron, W.E. Siri, T.W. Sargent, and E. Isaacs. 1965b. Measures of body fat and related factors in normal adults. II. A simple clinical method to estimate body fat and lean body mass. J. Chronic Dis. 18:1291-1307.
Stokes, J., III, R.J. Garrison, and W.B. Kannel. 1985. The independent contributions of various indices of obesity to the 22-year incidence of coronary heart disease: the Framingham Heart Study. Pp. 49-57 in J. Vague, P. Björntorp, B. Guy-Grand, M. Rebuffé-Scrive, and P. Vague, eds. Metabolic Complications of Human Obesities. Excerpta Medica, Amsterdam.
Stunkard, A.J., ed. 1980. Obesity. W.B. Saunders, Philadelphia. 470 pp.
Stunkard, A.J., T.T. Foch, and Z. Hrubec. 1986. A twin study of human obesity. J. Am. Med. Assoc. 256:51-54.
Sturdevant, R.A., M.L. Pearce, and S. Dayton. 1973. Increased prevalence of cholelithiasis in men ingesting a serum-cholesterol-lowering diet. N. Engl. J. Med. 288:24-27.
Suratt, P.M., S.C. Wilhoit, H.S. Hsiao, R.L. Atkinson, and D.F. Rochester. 1984. Compliance of chest wall in obese subjects. J. Appl. Physiol. 57:403-407.
Szmukler, G., C. McCance, L. McCrone, and D. Hunter. 1986. Anorexia nervosia: a psychiatric case register study from Aberdeen. Psychol. Med. 16:49-58.
Talamini, R., C. La Vecchia, A. Decarli, S. Franceschi, E. Grattoni, E. Grigoletto, A. Liberati, and G. Tognoni. 1984. Social factors, diet and breast cancer in a northern Italian population. Br. J. Cancer 49:723-729.
Talamini, R., C. La Vecchia, A. Decarli, E. Negri, and S. Franceschi. 1986. Nutrition, social factors and prostatic cancer in a northern Italian population. Br. J. Cancer 53:817-821.
Tuck, M.L., J. Sowers, L Dornfeld, G. Kledzik, and M. Maxwell. 1981. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N. Engl. J. Med. 304:930-933.
Tulinius, H., N. Sigfusson, H. Sigvaldason, and N.E. Day. 1985. Relative weight and human cancer risk. Int. Cong. Ser. (685):173-189.
Turkenkopf, I., P.R. Johnson, and M.R.C. Greenwood. 1982. Development of pancreatic and plasma insulin in prenatal and suckling Zucker rats. Am. J. Physiol. 242:E220-E229.
Tzonou, A., N.E. Day, D. Trichopoulos, A. Walker, M. Saliaraki, M. Papapostolou, and A. Polychronopoulou. 1984. The epidemiology of ovarian cancer in Greece: a case-control study. Eur. J. Cancer Clin. Oncol. 20:1045-1052.
Vague, J. 1956. Degree of masculine differentiation of obesities: factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am. J. Clin. Nutr. 4:20-34.
Vigersky, R.A., and D.L. Loriaux. 1977. Anorexia nervosa as a model of hypothalamic dysfunction. Pp. 109-21 in R.A. Vigersky, ed. Anorexia Nervosa. Raven Press, New York.
Vigersky, R.A., D.L. Loriaux, A.E. Andersen, R.S. Mecklenburg, and J.L. Vaitukaitis. 1976. Delayed pituitary hormone response to LRF and TRF in patients with anorexia nervosa and with secondary amenorrhea associated with simple weight loss. J. Clin. Endocrinol. Metab. 43:893-900.
Waaler, H.T. 1984. Height, weight and mortality: the Norwegian experience. Acta Med. Scand., Suppl. 679:1-56.
Wade, G.N., and J.M. Gray. 1979. Gonadal effects on food intake and adiposity: a metabolic hypothesis. Physiol. Behav. 22:583-593.
Weinsier, R.L., D.J. Norris, R. Birch, R.S. Bernstein, J. Wang, M.U. Yang, R.N. Pierson, Jr., and T.B. Van Itallie. 1985. The relative contribution of body fat and fat pattern to blood pressure level. Hypertension 7:578-585.
West, K.M., and J.M. Kalbfleisch. 1971. Influence of nutritional factors on prevalence of diabetes. Diabetes 20:289-296.
Willett, W.C., M.L. Browne, C. Bain, R.J. Lipnick, M.J. Stampfer, B. Rosner, G.A. Colditz, C.H. Hennekens, and F.E. Speizer. 1985. Relative weight and risk of breast cancer among premenopausal women. Am. J. Epidemiol. 122:731-740.
Williams, P., and M. King. 1987. The "epidemic" of anorexia nervosa: another medical myth? Lancet 1:205-207.
Zack, P.M., W.R. Harlan, P.E. Leaverton, and J. Cornoni-Huntley. 1979. A longitudinal study of body fatness in childhood and adolescence. J. Pediatr. 95:126-130.
Zack, P.M., R.D. Wiens, and H.L. Kennedy. 1984. Left-axis deviation and adiposity: the United States Health and Nutrition Examination Survey. Am. J. Cardiol. 53:1129-1134.
Ziegler, R.G., L.E. Morris, W.J. Blot, L.M. Pottern, R. Hoover, and J.F. Fraumeni, Jr. 1981. Esophageal cancer among black men in Washington, D.C. II. Role of nutrition. J. Natl. Cancer Inst. 67:1199-1206.
Zurnoff, B. 1982. Relationship of obesity to blood estrogens. Cancer Res. 42:3289s-3292s.






























