
5
Biomarkers of Acute Idiosyncratic Hepatocellular Injury in Clinical Trials1
Hepatotoxicity is the adverse event that most frequently leads to regulatory action on drugs, including failure to approve, postmarketing warnings added to the label, and withdrawal from the market (Temple, 2001). Among research priorities in adverse drug events, hepatotoxicity was ranked first in a 2006 survey of pharmaceutical companies (Holden, 2008). Because most events are inaccurately classified (Aithal et al., 1999), the population incidence of drug-induced liver injury is unknown. Yet drugs are the most frequent cause of acute liver failure among those under consideration for liver transplantation in the United States (Lee, 2003).
Animal studies in rodents, dogs, and monkeys detect approximately half of compounds exhibiting hepatotoxicity in humans (Olson et al., 2000). In vitro human hepatocyte testing similarly detects 50–60 percent of drugs that can cause severe liver injury in humans, including some not detected in animal testing (Xu et al., 2008). However, no currently available preclinical tests detect the potential for serious human hepatotoxicity with both high sensitivity and high specificity.
A recent example reveals some of the issues involved in drug safety testing. A major pharmaceutical company submitted a new drug application for treatment of a chronic disease. The FDA agreed with the sponsor’s effi-
cacy data. However, it was noted that among approximately 4,000 treated patients in clinical trials, two developed elevations in both serum alanine aminotransferase (ALT) and bilirubin. As a prerequisite for approval, the company was told to conduct a new safety study of 10,000 patients treated with the drug for 1 year, and to include an additional 10,000 subjects receiving comparator treatment for 1 year to exclude an unacceptable level of risk for clinically serious acute idiosyncratic hepatocellular injury (AIHI).2 The delay and additional investment required to bring such drugs to market can be detrimental not only to their manufacturers, but also to patients with unmet medical needs.
This chapter begins with an overview of AIHI. It then describes the current state of biomarkers for AIHI and reviews potential new biomarkers now emerging from various lines of investigation. The chapter ends with highlights of the breakout session on liver safety biomarkers.
ACUTE IDIOSYNCRATIC HEPATOCELLULAR INJURY (AIHI)
The clinical and histologic presentation of drug-induced liver injury can take many forms, mimicking most types of liver disease. AIHI is of greatest concern in drug development because of its potential rapidity of development and high morbidity and mortality (Andrade et al., 2005; Bjornsson and Olsson, 2005). Table 5-1 lists marketed drugs that have been subject to regulatory actions since 1995 because of liver safety concerns. All of the drugs listed can cause AIHI, with the exception of terbenafine (mixed hepatocellular/cholestatic injury), valproate (microvesicular steatosis), and acetaminophen (hepatocellular injury, but without the characteristics of AIHI discussed below). The discussion at the workshop focused exclusively on AIHI and not on other forms of drug-induced liver injury.
Figure 5-1 shows a typical presentation of AIHI. The patient exhibited normal liver chemistries at baseline and for several weeks while receiving treatment, but then developed serious liver injury with loss of overall liver function, manifested as a rise in serum bilirubin and ultimately death.
During AIHI, if treatment is not withdrawn promptly, and in some cases even with prompt discontinuation, the progressive loss of hepatocytes leads to liver dysfunction and ultimately death (absent liver transplant). The event is frequently termed “idiosyncratic” because the majority of treated patients are able to take the drug safely at the recommended dose range; the affected individuals are different from the majority in ways that make them susceptible to injury or less able to recover from injury. With most of the drugs listed in Table 5-1, fatal AIHI typically occurs in 1 in every 10,000
TABLE 5-1 Regulatory Actions on Approved Drugs Due to Hepatotoxicity, 1995–2008
|
Withdrawals |
Second Line |
Warnings |
|
|
|
|
SOURCE: Guo et al., 2008. |
||
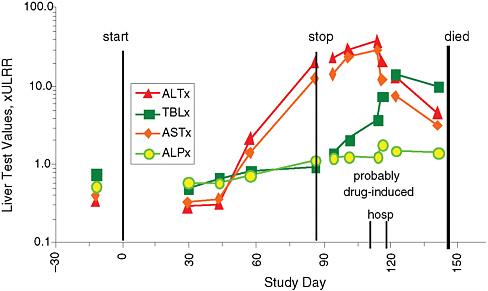
FIGURE 5-1 Acute idiosyncratic hepatocellular injury. An 80-year-old man who experienced acute idiosyncratic hepatocellular injury exhibited marked increases in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) starting about 45 days after treatment began. Total bilirubin (TLB) rose dramatically before death, while alkaline phosphatase (ALP) increased less markedly. Measures are compared with the upper limit of the reference range (ULRR).
SOURCE: Watkins slide presentation.
to 100,000 treated patients. It is rare for fatal AIHI to occur in preapproval clinical trials, in part because most such trials involve insufficient numbers of patients treated for long enough to have a high likelihood of identifying such rare individuals.
CURRENT STATE OF BIOMARKERS FOR AIHI
The primary biomarkers discussed at the workshop were those that detect a drug’s potential to cause AIHI in a preapproval clinical trial. Biomarkers to identify individual susceptibility to a drug with established AIHI potential were discussed to the extent that they were relevant in this context. Other types of biomarkers, such as those that may aid in causality assessment, were not discussed.
Serum ALT activity is the biomarker used most frequently to detect hepatocellular injury in clinical trials and is more liver-specific than aspartate aminotransferase (AST) (Green and Flamm, 2002). Serum ALT can increase, even markedly (for example, to levels exceeding 20 times the upper limits of normal [ULN]), as a result of events other than hepatocyte necrosis, including hepatocyte autophagy in anorexia nervosa (Rautou et al., 2008) or hepatic glycogen accumulation in uncontrolled type 1 diabetes (Olsson et al., 1989; Sayuk et al., 2007). Lesser ALT elevations are observed with hepatic steatosis (Browning et al., 2004). Activation of ALT gene transcription can occur in response to Peroxisome proliferators activated receptors (PPARs) agonists in cell culture (Thulin et al., 2008). In addition, it is theoretically possible that drugs could interfere with ALT degradation or blood clearance. However, no published data support transcriptional or clearance-related ALT increases due to these events occurring in humans.
Drugs recognized to cause AIHI have demonstrated an increased incidence of ALT elevations of more than three times ULN relative to placebo or controls in preapproval clinical trials (Temple, 2001). However, ALT elevations have a limited specificity to predict AIHI. Even frequent and fairly high ALT elevations do not reliably predict the potential to cause AIHI in clinical trials, as evidenced by tacrine. In clinical trials, about 25 percent of Alzheimer’s disease patients receiving tacrine developed ALT elevations of greater than three times ULN, and 2 percent exhibited ALT elevations of greater than 20 times ULN (Watkins et al., 1994). However, tacrine exhibits a very low risk of causing AIHI. Similarly, although statins have demonstrated up to a 3 percent incidence of ALT of greater than three times ULN in clinical trials relative to placebo or controls, statin use has not been associated with an increased risk of acute liver failure (Kaplowitz, 2005). Heparins also can cause ALT elevations but pose a very low or zero risk of causing AIHI. Drugs such as tacrine, heparins, and statins generally exhibit transient, self-limited liver injury that resolves with continued
treatment in a process termed “adaptation.” Adaptation is observed not only with drugs that pose a low risk of causing AIHI but also in many or most patients who experience ALT elevations while receiving drugs that can cause AIHI, such as troglitazone (Watkins, 1998) and isoniazid (Mitchell et al., 1975).
Common pathways likely underlie initial injury, regardless of whether the injury progresses or resolves with continued treatment. These pathways include those that determine the intracellular “dose” of hepatotoxic metabolites or bile acids (for example, cellular transporters, Phase 1 and 2 drug metabolism, and concomitant medications) and the production of hepatocyte injury (for example, oxidative stress or mitochondrial impairment), as well as regenerative or hepatoprotective abilities (including hepatic glutathione redox status, Nrf2 activation of cell defense systems, and liver regeneration). All of these pathways may be influenced by genetic (Larrey, 2002; Daly et al., 2007; Kindmark et al., 2007; Wilke et al., 2007), epigenetic (Murata et al., 2007), demographic (Kaplowitz, 2005; Uetrecht, 2007), infectious/inflammatory (Roth et al., 2003), and environmental factors (Larrey, 2002; Kaplowitz, 2005).
A popular theory is that progressive liver injury occurs in those individuals who fail to adapt to the initial insult. However, data to support this theory are sparse. It has been claimed that with drugs capable of causing AIHI, such as isoniazid, troglitazone, and ximelagatran, the AIHI events typically occur with a latency similar to that of the more frequent ALT elevations observed in clinical trials (personal communication, D. Larrey, J. Uetrecht, P. Watkins), a view consistent with a mechanistic link between isolated ALT elevations and AIHI events. The temporal relationship between ALT elevations observed in clinical trials and postmarketing AIHI events has not been systematically examined.
A logical conclusion would be that drugs that cause ALT elevations can be placed along a spectrum. On one end are drugs causing ALT elevations that may reflect liver injury but never cause AIHI. On the other end are drugs that cause liver injury that progresses to AIHI with relatively high frequency (perhaps 1 in 10 subjects with ALT elevations who are continued on treatment). While ALT is a sensitive biomarker for liver injury, it alone cannot differentiate between drugs at opposite ends of this spectrum.
Drug-Induced Liver Injury with Jaundice: The Current Gold Standard Biomarker for AIHI Potential
Hy Zimmerman (1968) first noted that a patient who presented with drug-induced hepatocellular injury with jaundice had at least a 10 percent chance of dying from liver failure (before liver transplantation was available). In hepatocellular injury, a rise in total and direct bilirubin reflects a
substantial risk because it indicates a major loss of functioning hepatocytes (when other causes for increased bilirubin are excluded). The approximately 10 percent mortality or transplant rate for drug-induced hepatocellular jaundice—known as “Hy’s Law”—has been confirmed in recent reports from Sweden and Spain (Andrade et al., 2005; Bjornsson and Olsson, 2005).
FDA draft guidance for evaluating liver safety in a clinical trial defines “Hy’s Law cases” as subjects in a clinical trial who experience ALT elevations of more than three times ULN and total bilirubin of more than two times ULN and satisfy the following three criteria: (1) the liver injury should be hepatocellular in nature, and there should not be a prominent cholestatic component (e.g., serum alkaline phosphatase of more than two times ULN); (2) there should be no more likely alternative cause than drug-induced liver injury, such as acute viral hepatitis A or B or C, preexisting or other acute liver disease, or another drug capable of causing the observed injury; and (3) there should be evidence that the drug causes more frequent but less severe hepatocellular injury as shown by more frequent ALT elevations of greater than three times ULN in the treated group relative to the control group (FDA, 2007). The FDA has placed great confidence in the specificity of Hy’s Law cases as a biomarker for identifying drugs capable of inducing serious AIHI, reporting, “We are not aware of false positive Hy’s Law findings” (FDA, 2007).
Hy’s Law cases are, however, a specific but imperfect biomarker for drugs capable of causing AIHI. In an FDA review of 26 new drug applications (13 of the drugs were known to be “hepatotoxic”), Hy’s Law events were seldom observed in the clinical trials, even with compounds later demonstrated to be capable of causing severe AIHI (Pauls, 2004). This is not surprising since the rarity of susceptible individuals and the delayed appearance of the event generally would require very large and prolonged clinical trials for detection. Also, clinical protocols usually mandate frequent monitoring of and strict stopping rules based on serum ALT, especially once a liver safety issue has been established for a drug in development. Stopping treatment at a low level of liver injury may allow a patient susceptible to AIHI to recover without demonstrating a rise in serum bilirubin. The only way to determine whether a patient with ALT elevation will adapt or progress is to continue treatment and observe the patient closely, with frequent monitoring of liver chemistries. The draft FDA guidance on liver safety (FDA, 2007) suggests that continued treatment may be considered in subjects with asymptomatic ALT elevations exceeding three times ULN. This practice may place these study subjects at greater health risk than subjects without ALT elevations, which raises ethical concerns.
Do Better Biomarkers Than Hy’s Law Cases Exist?
The ideal biomarker would not require placing patients at significant risk in the course of distinguishing between drugs capable of causing AIHI and drugs, such as heparin and statins, that do not appear to cause AIHI. The ideal biomarker also would be able to make this distinction in a relatively small clinical trial of short duration. The plausibility of such a biomarker rests on the mechanistic differences between drugs that have the potential to cause AIHI and those that cause only reversible ALT elevations. At least two possibilities exist:
-
The mechanisms that distinguish AIHI are many, complex, and agent-specific, to the extent that identifying a manageable number of predictive markers applicable to most drugs capable of causing severe AIHI is impractical or impossible.
-
Common mechanisms for AIHI exist and can be translated to a manageable number of validated biomarkers that could be applied to better understand the hepatic safety of candidate drugs in both the preregistration and postmarketing settings.
The first of these possibilities suggests the need for drug-specific biomarkers for those agents whose risk/benefit balance warrants continued marketing (as when agent-specific markers for patients at risk are available) when the incidence of liver safety events is high enough to characterize pre-marketing development. The existence of a small group of markers that, in the aggregate, have predictive value for AIHI in many or most cases relies on the second possibility being correct. What follows are three lines of thought regarding the pathogenesis of AIHI that can be used to examine possible biomarkers.
Cumulative Injury Theory
Cumulative injury theory maintains that drugs capable of causing AIHI induce progressive impairment of critical functions of hepatocytes that may start soon after the initiation of treatment but is not detected by elevation in serum ALT. An example is progressive mitochondrial injury (for example, as demonstrated for fialuridine and in cell culture for other drugs, such as nefazadone and troglitazone) where adenosine triphosphate (ATP) generation is progressively compromised over a period of weeks or months during treatment (McKenzie et al., 1995; Dykens et al., 2008; Xu et al., 2008). When mitochondrial function deteriorates to a critical level, hepatocellular necrosis may ensue, releasing or recruiting injury-propagating factors and/
or increasing metabolic demands on neighboring hepatocytes, which may progress to liver failure.
Some recent data suggest that a small number of critical pathways may compromise hepatocyte function to produce AIHI. In a recent study, fluorescent imaging of human hepatocytes was used to examine the effects of 300 hepatotoxins and nonhepatotoxins on mitochondrial damage, oxidative stress, and intracellular glutathione. These in vitro studies predicted approximately 60 percent of drugs capable of causing AIHI (many of which had not been detected in preclinical testing) with a high specificity (a false positive rate of 0–5 percent) (Xu et al., 2008). Because mitochondrial damage, oxidative stress, or depletion of intracellular glutathione may be downstream of molecule-specific events, such as reactive metabolite accumulation, a drug capable of causing severe AIHI could induce characteristic changes in the serum proteome or metabolome or in the urinary metabolome that would not be present in patients treated with drugs incapable of causing AIHI.
Immune Response Theory
Another mechanism proposed to account for the temporal delay in the onset and progression of liver injury is the production of reactive metabolites resulting in immune activation (Uetrecht, 2007). Within the hepatocyte, a drug is bioactivated to a reactive metabolite that binds to and modifies hepatocellular proteins. When this modified protein or hapten is presented by antigen-presenting cells to T cells, they transform to cytotoxic T cells and antibody-producing B cells (Kaplowitz, 2005; Park et al., 2005). Such drug-induced immune reactions typically occur within the first month of treatment and more rapidly with rechallenge (Kaplowitz, 2005), as seen with halothane (Mushin et al., 1971), and may be accompanied by clinical signs of hypersensitivity, such as fever, rash, and eosinophilia. The role of a specific hepatotoxin/metabolite in this immune response can be assessed in some cases by the lymphocyte stimulation test (Kaplowitz, 2005; Sanderson et al., 2006). These immune responses may be enhanced by acute inflammation or circulating lipopolysaccharide in rodents (Roth et al., 2003), and may explain why immunoallergic hepatotoxicity is more common in AIDS patients (Kaplowitz, 2005). It is quite probable that many AIHI cases result from episodic environmental/infectious/inflammatory changes that occur during drug therapy and that affect susceptibility or directly trigger a toxic interaction with a drug.
A variety of data suggest that immune mechanisms may underlie AIHI even when there are no clinical signs of hypersensitivity; an example is a report of human leukocyte antigen (HLA) associations with zimelagatran
hepatotoxicity (Kindmark et al., 2007). It is possible that biomarkers of immune activation could be useful in distinguishing benign ALT elevations from those that can portend AIHI. In support of this concept, ALT elevations accompanied by hepatitis symptoms (fatigue, nausea, right upper quadrant pain) appear to be more predictive of AIHI potential than are asymptomatic ALT elevations (Nolan et al., 1999). These symptoms may be mediated by cytokines or other endogenous proteins, which may be detectable long before symptoms appear.
Failure of Adaptation
If the critical issue in AIHI is failure to adapt to the initial injury, there may be biomarkers that could identify patients likely to adapt—and, conversely, those likely to progress to severe liver injury—at a very early stage in the injury process.
POTENTIAL NEW BIOMARKERS FOR AIHI
Sources of Candidate Biomarkers
Candidate biomarkers for AIHI are emerging from many lines of investigation, including extensive transcriptomic profiling of rats treated with a variety of hepatotoxic drugs. In the Liver Toxicity Biomarker Study (LTBS), pangenomic approaches are being used in rats to identify biomarkers capable of distinguishing pairs of drugs that are structurally and pharmacologically similar but differ in that one is capable of causing AIHI and the other is not. The Predictive Safety Testing Consortium has been identifying potential liver safety biomarkers but has not yet focused on detecting those for AIHI.
Another path to identifying potential biomarkers for AIHI is the ongoing effort to study patients who have actually experienced the condition. The Severe Adverse Event Consortium (SAEC) (Holden, 2008) has begun whole-genome single nucleotide polymorphism (SNP) analysis on germ-line DNA from patients who have experienced varying degrees of drug-induced liver injury, including AIHI. The expanding U.S.-based Drug Induced Liver Injury Network (DILIN) (Hoofnagle, 2004) will begin genetic analysis on a similar cohort and has the advantage of maintaining identity links to the participants so that additional phenotyping studies can be performed. Because subjects are enrolled in these registries only after a diagnosis of drug-induced liver injury has been made, it is generally not possible to obtain blood or urine early in the course of or prior to the injury.
One research priority will be to generate hypotheses that can be tested in gene banks and in the DILIN subjects themselves. International drug-
induced liver injury registries in the United States, the United Kingdom, Japan, Spain, Sweden, and Denmark now contain thousands of expert-adjudicated cases, which can be combined and analyzed for risk factors predicting progression to AIHI. Mining of large postmarketing adverse event databases also may suggest drug–environment susceptibility factors that could lead to testable hypotheses or be used to provide supportive data for genetic associations observed in these networks. In addition, analysis of blood/urine samples obtained in clinical trials of drugs known to cause AIHI may be useful in identifying biomarkers, especially when compared with blood/urine samples obtained in clinical trials of drugs that cause ALT elevations but do not have the potential to cause AIHI. A large prospective trial in isoniazid-treated patients has been proposed for this purpose (Watkins et al., 2008). Studying differences in susceptibility to hepatotoxicity across panels of inbred strains of mice and performing quantitative trait loci mapping may be a promising approach to generating hypotheses that would be testable in relatively small numbers of human subjects. Recent models have emerged in which drugs that cause AIHI in humans also cause liver injury in animals. It may be productive to explore biomarkers in these animal models, especially for biomarkers that are related to injury progression and adaptation and that predict serious downstream injury. In choosing drugs for study, consideration should be given to AIHI-associated drugs having a negative comparator in the same pharmacologic/structural class that is devoid of an equivalent degree of AIHI liability (e.g., trovafloxacin-levofloxacin).
Finally, the FDA has sponsored a cooperative research and development agreement to develop a computer-based model for understanding and predicting drugs capable of causing AIHI.3 The goal of this effort is to incorporate current mechanistic knowledge, as well as data and insights gained from ongoing efforts such as the SAEC and DILIN analyses. This evolving model could suggest novel biomarkers and provide a biological rationale for biomarkers discovered by other means.
Validation of Candidate Biomarkers
Biomarkers that are predictive in small clinical trials of short duration would be extremely useful. Potential biomarkers could be tested by administering examples of drugs both with and without AIHI liability to small groups of closely monitored patients or healthy volunteers and analyzing prospectively collected blood/urine samples. For example, the first pair of drugs studied in the LTBS were tolcapone (whose use is restricted because of
|
3 |
More information about this agreement can be found at http://www.entelos.com/newsReleases.php?ID=press101. |
liver toxicity) and entacapone. Since both drugs are in clinical use, it should be possible to test candidate biomarkers that emerge from the LTBS effort in patients or possibly healthy volunteers treated with these drugs. Short-term studies of this design with healthy volunteers may be ethical since the onset of liver injury is typically delayed weeks or months with tolcapone (Olanow and Watkins, 2007). However, it is possible that drugs capable of causing AIHI will be distinguishable only once liver injury has begun as signaled by ALT elevations, so that longer-term treatment would be required to evoke the phenotype. In this case, blood/urine samples would have to be obtained from patients with ALT elevations induced by drugs capable of causing AIHI and then compared with blood/urine samples obtained from patients with ALT elevations induced by drugs that do not cause AIHI. If (for at least some drugs) human AIHI occurs via interaction with an acute inflammatory stress, then plasma biomarkers based on this mode of action can be examined. For example, prolonged elevation of plasma cytokines, hemostatic biomarkers, and/or markers of neutrophil activation, when used in conjunction with traditional biomarkers such as ALT, might prove predictive. Generally, biomarkers with mechanistic/mode-of-action underpinnings are likely to be the most consistent predictors of AIHI.
True validation of biomarkers will ultimately require large numbers of samples obtained from individuals with well-known phenotypes, including both healthy and diseased populations, as well as populations treated with many different drugs. One path would be to institute protocols for standard data and blood/urine collection once ALT elevations have been observed in a clinical trial. An example of a liver safety data management system is eDish (Guo et al., 2008). This or a similar format could be directly linked to the sample bank, and would allow immediate identification of individuals of interest and immediate access to all pertinent clinical and laboratory data for those patients for detailed evaluation. Because the true potential of a drug to cause AIHI may not be evident preapproval, the blood/urine samples and data bank would need to be maintained for some time postmarketing. It would obviously be ideal if scientists had access to samples and clinical data from the clinical trials of many of the drugs listed in Table 5-1.
HIGHLIGHTS OF THE BREAKOUT DISCUSSION
In plenary session, John Bloom of Eli Lilly presented the main conclusions of participants in the breakout session on biomarkers for liver toxicity. Discussants in the breakout session observed that candidate AIHI biomarkers are best identified and validated in three relevant human populations: Hy’s Law cases; subjects in prospective, controlled clinical trials with established and well-characterized AIHI agents, including isoniazid;
and subjects in clinical trials who receive a drug known to cause ALT elevations but not yet known to cause AIHI. Some data suggest that subpopulations of these groups may exhibit changes that share common mechanisms with those associated with AIHI. Discussants identified the following six priority research efforts.
Accessing and Characterizing Hy’s Law Cases
The first priority is to develop methods for overcoming key barriers to accessing clinical information and biospecimens from Hy’s Law cases, which arguably constitute the most relevant population to study. This priority need raises several important questions. How can these rare cases be better accessed and characterized? How can well-annotated specimens be obtained, including specimens from matched controls? Can ongoing initiatives such as those of SAEC, DILIN, and other partnerships be integrated more effectively? How can electronic health records and large databases, such as those from the U.S. Department of Veterans Affairs or private insurers, be better leveraged? Can a warehouse for data be established? Could such an effort be integrated into the FDA’s Sentinel Initiative?
Developing and Implementing Protocols for Specimen and Data Collection in Clinical Trials of Specific Marketed Drugs Known to Either Cause or Not Cause AIHI
A second priority is to develop and implement protocols for specimen and data collection in prospective clinical trials of isoniazid and other drugs known to cause AIHI or known to cause ALT elevations but not AIHI. A number of key questions need to be addressed. What would a protocol look like in terms of the subjects who are enrolled and controls for concomitant treatments and diseases? Are there markers that can be used to enrich this patient population? Can signals or markers for adaptation and severity of liver injury be differentiated and stratified? Are there markers that can predict, rather than simply demonstrate, the effect of the disorder? What other agents should be considered for prospective trials? To what extent will the identified markers be agent-specific and therefore not more broadly applicable? How can this kind of study be sponsored or funded, and can it be coupled with ongoing studies?
Investigating ALT Signals in Clinical Trials During Drug Development
A third need is to develop and implement protocols for standardized data and biospecimen collection in clinical trials when an ALT signal is identified. Important questions include the following: What should be the
trigger for collection, and which specimens should be collected? How can standardization of specimens and data be achieved, including ascertainment as well as phenotyping? What should be the role of regulators? Should access to specimens and data be restricted? How can the risk for the sponsor be managed in such a situation?
Making Use of Existing Databases
A fourth priority is to conduct a thorough examination of existing FDA liver safety databases from Phase III clinical trials, and perhaps from the Adverse Event Reporting System database, to test hypotheses for the more frequent benign ALT elevations. An important hypothesis is that such elevations in this population, or more likely a subset of these patients, are mechanistically linked to AIHI, and that this link could be validated or at least corroborated. How can these data be mined? How can privacy issues be addressed? How can alignment among regulatory agencies and private companies be achieved? What are the incentives for alignment? What resources and oversight are needed? Can this research be decoupled from regulatory decision making?
Prioritizing Biomarker Discovery
A fifth research need is to prioritize biomarker discovery options using the data and biospecimens from the three populations described above. This work will require answers to a number of questions. How should the right specimens be collected when candidate markers are not known? How should options be kept open? Should candidate biomarker domains be prioritized according to established and emerging hypotheses? For instance, if a toxic metabolite or other form of injury leads to subsequent immune responses, there are pathways from which one can derive biomarkers for these responses. Should biomarker discovery searches be prioritized along the lines of these hypotheses or enabling technology platforms? Should this guide which specimens are collected?
Identifying and Prioritizing Nonclinical Research Options
Finally, there is a need to identify and prioritize nonclinical research options for generating biomarker hypotheses for testing in clinical specimen banks. Can animal models enable method development? If relevant models are identified, can they provide information on progression factors, reversibility, the kinetics of biomarker changes, and other questions enabled by tightly controlled conditions? Can nonclinical studies be linked to clinical studies to inform biomarker identification? Are there surrogate
in vivo or in silico models that can suggest new candidate biomarkers for human studies?
Next Steps
The questions identified by the discussants in each of the above six areas attest to the complexity and challenges of implementing such a multidisciplinary and interinstitutional endeavor. Many additional questions remain regarding coordination, oversight, and sponsorship of efforts in these areas. Breakout session participants discussed potential roles for the IOM in facilitating efforts of the research community in addressing these questions. One suggested approach would be for the IOM to convene and facilitate working groups in each of the six priority research areas.
REFERENCES
Aithal, G. P., M. J. Rawlins, and C. P. Day. 1999. Accuracy of hepatic adverse drug reaction reporting in one English health region. British Medical Journal 319:1541.
Andrade, R. J., M. I. Lucena, M. C. Fernández, G. Pelaez, K. Pachkoria, E. García-Ruiz, B. García-Muñoz, R. González-Grande, A. Pizarro, J. A. Durán, M. Jiménez, L. Rodrigo, M. Romero-Gomez, J. M. Navarro, R. Planas, J. Costa, A. Borras, A. Soler, J. Salmerón, R. Martin-Vivaldi, and Spanish Group for the Study of Drug-Induced Liver Disease. 2005. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology 129:512–521.
Bjornsson, E., and R. Olsson. 2005. Outcome and prognostic markers in severe drug-induced liver disease. Hepatology 42(2):481–489.
Browning, J. D., L. S. Szczepaniak, and R. Dobbins. 2004. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 40:1387–1395.
Daly, A. K., G. P. Aithal, J. B. S. Leathart, R. A. Swainsbury, T. S. Dang, and C. P. Day. 2007. Genetic susceptibility to diclofenac-induced hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 132(1):272–281.
Dykens, J. A., J. D. Jamieson, L. D. Marroquin, S. Nadanaciva, J. J. Xu, M. C. Dunn, A. R. Smith, and Y. Will. 2008. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone and buspirone. Toxicological Sciences 103(2):335–345.
FDA (Food and Drug Administration). 2007. Guidance for industry (draft) drug-induced liver injury: Premarketing clinical evaluation. www.fda.gov/cder/guidance/7507dft.htm (accessed October 17, 2008).
Green, R. M., and S. Flamm. 2002. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology 123:1367–1384.
Guo, T., K. Gelperin, and J. Senior. 2008. A tool to help you decide: Detect potentially serious liver injury. AASLD-FDA-NIH-PhRMA-Hepatotoxicity Special Interest Group meeting. http://www.fda.gov/cder/livertox/presentations2008/D-GelperinGuo2.pdf (accessed October 17, 2008).
Holden, A. 2008. Detecting and investigating drug-induced adverse events: The international serious adverse event consortium’s experience. www.fda.gov/cder/livertox/presentations2008/T-Holden2.pdf (accessed October 17, 2008).
Hoofnagle, J. H. 2004. Drug-Induced Liver Injury Network (DILIN). Hepatology 40(4):773.
Kaplowitz, N. 2005. Drug hepatotoxicity. Nature Reviews. Drug Discovery 4:489–499.
Kindmark, A., A. Jawaid, C. G. Harbron, B. J. Barratt, O. F. Bengtsson, T. B. Andersson, S. Carlsson, K. E. Cederbrant, N. J. Gibson, M. Armstrong, M. E. Lagerström-Fermér, A. Dellsén, E. M. Brown, M. Thornton, C. Dukes, S. C. Jenkins, M. A. Firth, G. O. Harrod, T. H. Pinel, S. M. Billing-Clason, L. R. Cardon, and R. E. March. 2007. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. The Pharmacogenomics Journal 8(3)186–195.
Larrey, D. 2002. Epidemiology and individual susceptibility to adverse drug reactions affecting the liver. Seminars in Liver Disease 22:145–155.
Lee, W. M. 2003. Drug-induced hepatotoxicity. New England Journal of Medicine 349(5):474–485.
McKenzie, R., M. W. Fried, R. Sallie, H. Conjeevaram, A. M. Di Bisceglie, Y. Park, B. Savarese, D. Kleiner, M. Tsokos, and C. Luciano. 1995. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. New England Journal of Medicine 333:1099–1105.
Mitchell, J. R., M. W. Long, U. P. Thorgeirsson, and D. J. Jollow. 1975. Acetylation rates and monthly liver function tests during one year of isoniazid preventive therapy. Chest 68(2):181–190.
Murata, K., M. Hamada, K. Sugimoto, and T. Nakano. 2007. A novel mechanism for drug-induced liver failure: inhibition of histone acetylation by hydralazine derivatives. Journal of Hepatology 46:322–329.
Mushin, W. W., M. Rosen, and E. V. Jones. 1971. Post-halothane jaundice in relation to previous administration of halothane. British Medical Journal 3(5765):18–22.
Nolan, C. M., S. V. Goldberg, and S. E. Buskin. 1999. Hepatotoxicity associated with isoniazid preventative therapy: A 7-year survey from a public health tuberculosis clinic. Journal of the American Medical Association 282(23):2207–2208.
Olanow, C. R., and P. B. Watkins. 2007. Tolcapone: An efficacy and safety review. Clinical Neuropharmacology 30(5):287–294.
Olson, H., G. Betton, D. Robinson, K. Thomas, A. Monro, G. Kolaja, P. Lilly, J. Sanders, G. Sipes, W. Bracken, M. Dorato, K. Van Deun, P. Smith, B. Berger, and A. Heller. 2000. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regulatory Toxicology and Pharmacology 23:56–57.
Olsson, R., C. Wesslau, T. W. Olsson, and L. Zettergren. 1989. Elevated aminotransferases and alkaline phosphatases in unstable diabetes mellitus without ketoacidosis or hypoglycemia. Journal of Clinical Gastroenterology 11:541–545.
Park, B. K., N. R. Kitteringham, J. L. Maggs, M. Pirmohamed, and D. P. Williams. 2005. The role of metabolic activation in drug-induced hepatotoxicity. Annual Review of Pharmacology and Toxicology 45:177–202.
Pauls, L. 2004 (February 5). Can we predict hepatotoxicity based on MO reviews of submitted data? Presentation by Center for Drug Evaluation and Research. FDA Hepatotoxicity Steering Committee. http://www.fda.gov/cder/livertox/presentations2004/LP5Feb04.ppt (accessed October 5, 2008).
Rautou, P. E., D. Cazals-Hatem, R. Moreau, C. Francoz, G. Feldmann, D. Lebrec, E. Ogier-Denis, P. Bedossa, D. Valla, and F. Durand. 2008. Acute liver cell damage in patients with anorexia nervosa: A possible role of starvation-induced hepatocyte autophagy. Gastroenterology 135:840–848.
Roth, R. A., J. P. Luyendyk, J. F. Maddox, and P. E. Ganey. 2003. Inflammation and drug idiosyncrasy—is there a connection? The Journal of Pharmacology and Experimental Therapeutics 307(1):1–8.
Sanderson, J. P., D. J. Naisbitt, and B. K. Park. 2006. Role of bioactivation in drug-induced hypersensitivity reactions. The AAPS Journal 8(1):E55–E64.
Sayuk, G. S., J. E. Elwing, and M. Lisker-Melman. 2007. Hepatic glycogenosis: An underrecognized source of abnormal liver function tests? Digestive Diseases and Sciences 52:936–938.
Temple, R. 2001. Hepatotoxicity through the years: Impact on the FDA. www.fda.gov/cder/livertox/meeting2001.htm#Drug-Induced (accessed October 17, 2008).
Thulin, P., I. Rafter, K. Stockling, C. Tomkiewicz, E. Norjavaara, M. Aggerbeck, H. Hellmold, E. Ehrenborg, U. Andresson, I. Cotgreave, and B. Glinghammar. 2008. PPARalpha regulates the hepatotoxic biomarker alanine aminotransferase (ALT1) gene expression in human hepatocytes. Toxicology and Applied Pharmacology 231(1):1–9.
Uetrecht, J. 2007. Idiosyncratic drug reactions: Current understanding. Annual Review of Pharmacology and Toxicology 47:513–539.
Watkins, P. B. 1998. Hepatic dysfunction associated with troglitazone. New England Journal of Medicine 338:916–917.
Watkins, P. B., H. J. Zimmerman, M. J. Knapp, S. I. Gracon, and K. W. Lewis. 1994. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. Journal of the American Medical Association 271(13):992–998.
Watkins, P. B., P. J. Seligman, J. S. Pears, M. I. Avigan, and J. R. Senior. 2008. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology 48(5):1680–1689.
Wilke, R. A., D. W. Lin, D. M. Roden, P. B. Watkins, D. Flockhart, I. Zineh, K. M. Giacomini, and R. M. Krauss. 2007. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nature Reviews Drug Discovery 7:904–916.
Xu, J. J., P. V. Henstock, M. C. Dunn, A. R. Smith, J. R. Chabot, and D. de Graaf. 2008. Cellular imaging predictions of clinical drug-induced liver injury. Toxicological Sciences 105:97–105.
Zimmerman, H. J. 1968. The spectrum of hepatotoxicity (Kober lecture). Perspectives in Biology and Medicine 12(1)135–161.
















