
Regulation of Predictive Tests
The predictive tests used in personalized medicine are overseen by two federal agencies—the FDA and the Centers for Medicare & Medicaid Services (CMS). The Medical Device Amendments of 1976 to the Federal Food, Drug, and Cosmetic Act brought the marketing of devices, including in vitro diagnostics, under FDA regulation (hereinafter, in this report, “companion diagnostic tests.”)1 The FDA has exercised regulatory discretion with regard to laboratory-developed predictive tests (hereinafter, in this report, “laboratory-developed tests”), and does not oversee the development of these tests. The laboratories that provide these tests are, however, subject to oversight by CMS under CLIA, with the goal of ensuring quality laboratory testing services.
The FDA and CMS authority for the oversight of predictive tests are described in detail below. These sections are followed by a discussion on whether the current, dichotomous system is the best approach to overseeing these types of tests.
OVERVIEW OF THE FDA’S REGULATION OF PREDICTIVE TESTS
Dr. Alberto Gutierrez of the Office of In Vitro Diagnostic Devices (OIVD), FDA, explained how the FDA regulates companion diagnostics
(predictive tests that have gone through the FDA approval process). He began his talk by pointing out that “in personalized medicine, the companion diagnostic really becomes key, because if you’re going to be given a therapeutic, or you’re going to be taking a clinical action based on the companion diagnostic, the diagnostic has to be right.” The Medical Device Amendments of 1976 gave the FDA authority to regulate devices, including companion diagnostic tests, based on the amount of risk that is linked to the use of that device. Devices are classified into one of three risk categories (Classes I, II, and III), where Class I devices have the lowest level of risk and Class III devices have the highest.
The regulatory requirements necessary for approval of a device are based on the devices classification. Manufacturers of Class I devices, such as Band-Aids or pH tests, have to register their test with the FDA and follow general controls, such as adhering to good manufacturing practices, reporting device failures, and developing and using a system for remedying such failures (FDA, 2009b). The requirements for Class II devices are more complex. This is where most companion diagnostic tests fit into the classification scheme. Manufacturers of Class II devices need to follow FDA guidance documents that detail what manufacturers need to provide in order to receive FDA market clearance of their medical device, quality system regulations, and other special controls. They also must show that their device is substantially equivalent to a device that is on the market, or was on the market before 1976. This process is what the FDA calls “premarket notification (510(k))” (FDA, 2009b). Class III devices are the most complex and pose the highest degree of risk. Manufacturers of Class III devices are required to submit an application for Premarket Approval (PMA) to the FDA that details the safety and effectiveness of their device. The device cannot enter the market until after the FDA reviews and approves this application (FDA, 2009b).
In general, “the nice thing about this regulatory process is that it is quite malleable,” Dr. Gutierrez explained. “We can apply the necessary regulation depending on both the risk of your test and its complexity, so it allows the reviewers the ability to mold their regulatory process to what you have.” The FDA determines a device’s risk classification based on the intended use of the device. If a device has more than one intended use, it will have a separate review process for each use. For example, “you could have a device that is used for monitoring cancer, which will have a lower risk than a device that does screening for cancer, because if you tell somebody they don’t have cancer when, in fact, they do, you can actually put them at very high risk,” Dr. Gutierrez said.
In its review of devices, the FDA considers analytic validity (the accuracy of a test in detecting the specific entity that it was designed to detect) and clinical validity (the accuracy of a test for a specific clinical purpose), but not clinical utility (the clinical and psychological benefits and risks of positive and negative results of a given technique or test). This means that although the FDA evaluates whether a companion diagnostic test can provide accurate information for clinical decision making, it does not thoroughly assess the risks and benefits of using the test on patients. However, Dr. Gutierrez added that sometimes the FDA consults with experts as to whether the risk of a device giving the wrong information outweighs the benefits of allowing the test.
In addition, the FDA regulates companion diagnostic tests by ensuring that all of the claims made on a diagnostic test’s label are accurate and can be supported by evidence. The OIVD review of device performance is transparent with the reviews posted on the FDA website (FDA, 2009c). The FDA also does postmarket surveillance, and takes action to help resolve device failures when they are detected (FDA, 2009d).
In 2005 FDA published a white paper on codevelopment of diagnostics and therapeutics, and established a procedure whereby a codeveloped drug and companion diagnostic could undergo parallel FDA review and approval (FDA, 2005). This process has led to drugs receiving FDA approval based on studies that only tested the drug in marker-positive patients, rather than on unselected populations. However, there are shortcomings to this process, Dr. Gutierrez stressed. This type of testing does not conclusively show that the drug’s effectiveness is linked to the companion diagnostic test result because this conclusion can only be determined by testing the drug in both marker-positive and marker-negative patients. “What we learned from HER2 is that if you do a trial in which you actually have only marker-positive patients, in the end you actually know the positive predictive value of the test, but not much else about the test,” he said. Such a study does not indicate sensitivity, specificity, and the negative predictive value. This poses problems when a competing biomarker is discovered because its comparative value to the older test cannot be fully ascertained given the lack of information on the marker-negative population. However, one participant noted that it would be difficult, if not unethical, to accrue patients who are marker negative to a clinical trial of a targeted agent because they are not likely to receive any benefit.
OVERVIEW OF CMS’S REGULATION OF LABORATORIES PERFORMING PREDICTIVE TESTS
As discussed above, laboratory-developed tests are not currently regulated by the FDA, however the agency has the authority to do so. Instead CMS regulates the laboratories that develop these tests through CLIA. Laboratory-developed tests have historically been conducted in a single laboratory on specimens that come from the nearby patient population. More recently, laboratory-developed tests are being done in a single lab using samples from all over the country. In contrast, FDA-approved companion diagnostic tests are generally developed by industry to be used in laboratories throughout the country, if not the world. FDA companion diagnostic “test kits” need to be more robust because anyone can buy the kit and perform the test, regardless of their expertise, Dr. Leonard said.
The FDA has chosen not to regulate laboratory-developed tests because of a lack of resources, according to Dr. Steven Gutman of the University of Central Florida and former head of OIVD, and not necessarily because they pose less risk than FDA companion diagnostics. When the decision was made not to oversee laboratory-developed tests, most of these tests were for research purposes and were not commercialized. However, today many companies are using laboratory-developed tests because they present an easier method of getting predictive tests on the market.
Dr. Penelope Meyers of CMS explained that there are no CLIA-certified or approved tests because CLIA certifies and regulates the laboratories doing the testing, and not the tests themselves. A laboratory must receive CLIA certification to be reimbursed by Medicare/Medicaid. In addition to CLIA requirements, some laboratories must follow more stringent rules required by certain states for licensure and permits. The purpose of CLIA was to ensure that patients receive the same quality of laboratory testing, regardless of where the test is performed, be it in a hospital laboratory, a large reference laboratory, or a physician’s office, said Dr. Meyers.
CLIAS’s complexity requirements apply to all clinical laboratories and their stringency depends on the complexity of the testing being performed (high, moderate, or waived). FDA risk assessment is based on the impact of the test result on decision making (low, moderate, and high risk based on impact on patient.) Laboratories that perform testing with the lowest degree of complexity (waived testing) are not subject to any routine CLIA oversight, and do not get inspected. These laboratories are only required to follow the manufacturer’s test instructions. Genetic-based predictive tests are all considered to be high complexity. Laboratories that perform these
tests must follow the most stringent CLIA requirements, including the most rigorous personnel requirements. However, there are no specific personnel requirements for genetic-based predictive tests.
CLIA oversight focuses on laboratory procedure, on the training of laboratory personnel, and on the credentials needed for test interpretation. CLIA oversees the registration, certification, and accreditation of laboratories, proficiency testing of the lab, regulations governing the physical plan, record retention, and other facility requirements and quality control systems.2 CLIA regulations require 84 listed analytes to have proficiency testing; however, none of the listed analytes is for a genetic test. A current CDC project is attempting to update the analyte list, partly in response to the issues raised by the genetic testing community about proficiency testing, Dr. Meyers said.
CLIA preanalytic requirements include those governing specimen submission and handling. The major postanalytic requirement is that laboratories have systems for collecting, responding to, and acting on communications and complaints about performed tests. All laboratories, except for those doing waived testing, are inspected every 2 years, and CLIA can take enforcement action against labs that do not correct deficiencies detected during inspections.3 It is important to note that CLIA does not have any legal mechanism to enforce CLIA violations in laboratories performing predictive tests that do not have CLIA certificates. However, there have been instances where CMS CLIA offices have sent their state and regional office surveyors into unregulated laboratories that were known to be conducting predictive tests, and have been successful in having them either apply for a CLIA certificate and submit to inspection or cease testing, Dr. Meyers said.
CLIA has a lengthy list of requirements that ensures the analytic validity of the testing a laboratory performs, but CLIA does not regulate the clinical validity of a test, unlike the FDA’s regulation of companion diagnostic tests. Clinical research data are not required to support the claims on the laboratory-developed tests’ label, even if these tests are linked to the same degree of risk and complexity as companion diagnostic tests approved by the FDA. In addition, there is no requirement for reporting adverse events with laboratory-developed tests, nor is there
|
2 |
42 C.F.R. Ch. IV Part 493. http://ecfr.gpoaccess.gov/cgi/t/text/text-idx?c=ecfr&sid=24b0d768489ac16c021cd4c5656568b4&rgn=div5&view=text&node=42:4.0.1.5.29&idno=42. |
|
3 |
42 C.F.R. Ch. IV Part 493, Subparts Q and R. |
public information on their analytic or clinical validity, as there would be for tests that undergo FDA reviews.
“There is regulation that requires a laboratory director to offer testing that is appropriate for the patient population, and this rule is sometimes interpreted loosely to mean that the clinical validity of a test needs to be considered,” said Dr. Meyers. “But CLIA really does not directly regulate clinical validity, and we don’t require specific data on clinical validity for laboratory-developed tests.” If CLIA surveyors notice anything questionable about a laboratory-developed test during an inspection, they can consult with an expert at CMS, CDC, and FDA, Dr. Meyers said.
When a laboratory is going to implement an FDA-approved or -cleared companion diagnostic test, the laboratory must verify that the test’s performance specifications are met. But for laboratory-developed tests, the laboratory establishes its own performance specifications, such as analytical sensitivity, specificity, and other performance characteristics required for test performance, and can begin offering the test once a laboratory director deems these performance specifications suitable.
SHOULD THE FDA DO MORE?
Some companies have tried to use the CLIA regulatory pathway for their predictive tests inappropriately, rather than go through the FDA approval process. For example, LabCorp put its OvaSure test on the market as a laboratory-developed test, even though it was actually an in vitro diagnostic test according to the FDA. Eventually, LabCorp pulled OvaSure from the market due to FDA pressure. Recognizing this confusion in the regulation of laboratory-developed tests, the FDA published the Analyte Specific Reagent rule in 1997 and again in 2007, which specified that the materials used in laboratory-developed tests must follow FDA rules for Class I devices (FDA, 1997, 2007a). More recently, the FDA began working on the In Vitro Diagnostic Multivariate Index Assay (IVDMIA) guidance for high-risk, high complexity tests, such as Oncotype Dx (FDA, 2007b). This guidance defines and specifies the regulatory status of IVDMIAs, and clarifies that even when offered as laboratory-developed tests, IVDMIAs must meet pre- and postmarket device requirements, including premarket review requirements in the case of most Class II and III devices. However, the FDA is not currently enforcing the IVDMIA guidance and it is unclear when or if it will be finalized.
In addition, several speakers and discussants noted that these guidance
documents are insufficient to counter the lack of regulatory parity between FDA-reviewed companion diagnostic test kits and those laboratory-developed genetic tests that fall under CLIA’s purview. “Industry sees a big disparity between those tests that go to market as a laboratory-developed test, and what they have to do to get an FDA-approved or -cleared test,” said Dr. Gutierrez. Genentech recently filed a citizen’s petition with the FDA that asked the agency to review its regulation of laboratory-developed tests, with the aim of having all tests that are used or intended to be used for therapeutic decision making undergo the same scientific and regulatory standards (Genentech, 2008). “There’s been a broad proliferation of assays that are allegedly being used to make decisions about patient care without any type of FDA clearance as it relates to efficacy or safety,” said Dr. Mass. “We think that a lot of these claims are misleading, or certainly unsubstantiated by the kind of data that would be required of a drug manufacturer to get marketing approval for a drug.”
An example of such unsubstantiated claims is a predictive test used to determine the likely responsiveness of lymphoma patients to rituximab therapy. The maker of this test claims it will enable physicians to “confidently predict” whether lymphoma patients will respond to rituximab (PGxHealth, 2009). This claim was supported with data generated by the company that devised the test, which showed that when individuals with follicular lymphoma are homozygous for a specific gene, they will have a 100 percent response rate to the drug, whereas those who are heterozygous or completely lack the gene will only have a 67 percent response rate (Figure 6) (Cartron et al., 2002; PGxHealth, 2009). “The confidence intervals here are quite wide and overlapping so one could really question whether the claims being made by this assay system are relevant,” said Dr. Mass.
Genentech did its own analysis of the test on patients with diffuse lymphoma, and found that when rituximab was added to the cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy protocol sequentially in a few hundred patients, the tumors of 40 percent of those homozygous for the gene progressed on the regimen, compared with 57 percent of those that were heterozygous or completely lacking the gene—a much less striking difference, Dr. Mass noted (Vose et al., 2009). A larger study may have revealed more differences between the homozygous and heterozygous patients. However, Dr. Mass said, unlike the univariate analysis done by the maker of the test, Genentech did a more statistically rigorous multivariate analysis in which it corrected for other prognostic variables not considered in the simpler analysis. “We don’t think physicians or patients
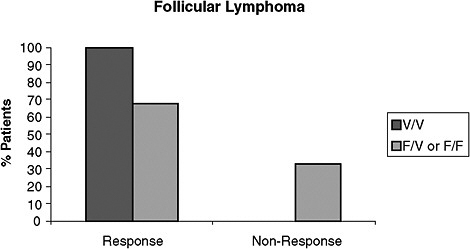
FIGURE 6 Data from PGxHealth on follicular lymphoma patients receiving Rituximab monotherapy. This study shows that when individuals with follicular lymphoma are homozygous for a specific gene, they have a 100 percent response rate to Rituximab, whereas those who are heterozygous or completely lack the gene only have a 67 percent response rate. Response is defined as complete or partial response, and non-response is defined as stable or progressive disease by Cheson criteria.
NOTE: F/F = lack the gene, F/V = heterozygous for the gene, V/V = homozygous for the gene, n = 10 for the V/V group, n = 39 for the F/V or F/F group.
SOURCES: Mass (2009); Cartron et al. (2002); PGxHealth (2009). This research was originally published in Blood. Cartron, G., L. Dacheux, G. Salles, P. Solal-Celigny, P. Bardos, P. Colombat, and H. Watier. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor Fcgamma RIIIa gene. 2002; Vol 99(3):754–758. © the American Society of Hematology.
should be subjected to this test without more rigor around the claims being made about it,” he said. “We think that any test that’s making a claim about clinical effectiveness should be reviewed by the FDA.”
Dr. Mass also questioned the claim that laboratory-developed tests do not have to be reviewed by the FDA because they tend to not pose safety hazards. In the context of predictive tests, safety can be defined as the right patient getting the right drug, and the wrong patient not getting the wrong drug, he noted. However, there are limited examples of these types of safety issues being considered for laboratory-developed tests because CLIA does not require this type of record keeping. As a result, it may take years for safety problems in laboratory-developed tests to become apparent, he pointed out. “Did Mrs. Jones actually get the right therapy based on some assay that was conducted, and was her outcome altered in some way by that
treatment?” To ensure tests are safe, there needs to be some clinical validity or utility measurements, he said.
Dr. Gutierrez concurred that predictive tests that are intimately tied to a therapeutic should be approved by the FDA, whether or not they are laboratory developed, because “for the drug to be safe and effective, the device itself has to be controlled.” Yet several tests that are intimately tied to therapeutics did not undergo FDA review, including Oncotype DX.
FDA regulation of laboratory-developed tests might stifle innovation and prevent the iterative development of these tests that often occurs, Dr. Gutierrez pointed out, but Dr. Mass disagreed that this would be problematic. Dr. Mass recognized the lack of resources that currently prevents FDA from reviewing laboratory-developed tests, but added, “If we believe as a community that this needs to happen, there are certainly ways that the resourcing can be applied to review the tests that we think are important to review. The CLIA process is essential in terms of laboratory quality, but it’s not really closing the loop on proper clinical validation. If we can’t improve this regulation, we can never fully realize the promise that personalized medicine should bring to patients.”
An additional reason for providing more comprehensive regulation of predictive tests is that drug developers want more predictability, Dr. Gutierrez said. Ms. Stack called for increasing clarity on both the regulation and reimbursement of predictive tests so venture capitalists, such as herself, can continue to develop innovative companion diagnostic companies. “I don’t want more regulation. I just want clarity because it’s really hard to develop a business when you’re not clear of how it’s going to be regulated and reimbursed,” she said.
IS THE STATUS QUO APPROPRIATE?
Some speakers and participants questioned whether FDA review of laboratory-developed tests would be sufficient. “Everybody talks about the FDA pathway as the gold standard ideal, but there are lots of problems with the FDA process,” said Dr. Leonard. “Nor do I believe that the CLIA process is perfect,” she added. Dr. Hayes said that when he and others develop ASCO guidelines, “We don’t care if the FDA has or has not approved a test, because FDA approval doesn’t mean the test should be used to take care of patients, and possibly the best test we have in breast cancer now [Oncotype DX] has never been approved by the FDA. The whole system needs to be revamped. We need to review tests the same way we review
drugs.” Ms. Bonoff concurred, saying, “The controlled, randomized clinical trial has been the gold standard for drug treatment. Why shouldn’t that also be the standard for tests that will guide the use of therapy? We really need to know that the tests are dependable because we’re making decisions that affect our lives based on these tests. The current system makes no sense. Predictive tests are now in the clinic without FDA review, and FDA-reviewed companion diagnostic tests may be used for non-approved means. There is an essential need for stronger criteria and oversight to replace the current patchwork system.”
Dr. Leonard agreed that “the systems need to be tweaked to ensure greater safety and demonstration of efficacy where there are holes.” However, she expressed concern that making every laboratory-developed test go through an FDA review process would slow down the development and use of these tests. The pathway for a laboratory-developed test to enter the market is rapid, whereas the FDA-approved test pathway is much slower. “Please don’t eliminate the rapid pathway for test availability. That would be throwing the baby out with the bathwater,” she said. Instead, she suggested that FDA review should be done on those tests with high complexity, and for those tests being performed by institutions on patient specimens collected from outside the local community. An institution does not have the same degree of control over commercialized assays when specimens are coming from all over the country or world.
Ms. Gail Javitt of the Genetics and Public Policy Center, Johns Hopkins University, said, “It’s wrong to think that a one-size-fits-all approach to the regulation of all laboratory-developed tests and genetic tests would work.” She suggested having different regulatory pathways based on risk. In 2008, the SACGHS recommended that HHS convene a multistakeholder, public-and private-sector group to develop criteria for determining the appropriate oversight of laboratory tests, and a process for systematically applying the criteria, said Dr. Ferreira-Gonzalez (SACGHS, 2008a). “Before increasing oversight, the benefits and harms to patient access and cost should be considered,” she said.
However, SACGHS’s predecessor, SACGT, recommended premarket review for all predictive tests, including laboratory-developed tests, said Dr. Wylie Burke of the University of Washington, and former member of SACGT (SACGT, 2000a). It suggested that premarket review should be streamlined, and a template should be developed to standardize the FDA review process for discerning tests that are more complex. The primary goal of this template-driven approach is accurate labeling, according to
Dr. Burke. Labeling of predictive tests should specify the intended use of the test, the specific actions that will follow from use of the test, the clinical condition for which the test is performed, as well as specificity, analytic validity, and any known clinical validity and clinical utility. Transparency about the current state of knowledge for a test might provide protection against unsafe testing, Dr. Burke said.
When it comes to ensuring the quality of the testing itself (analytic validity), Dr. Leonard stressed that the quality of testing done by CLIA-certified labs through laboratory-developed tests is virtually the same as that for FDA-reviewed tests. FDA reviews of tests are done using the same template for providing standard information on a companion diagnostic test as used by laboratories under CLIA, and the proficiency testing is basically the same for both laboratory-developed tests and FDA-regulated tests. One analysis found a 98.1 percent accuracy with samples sent to laboratories for proficiency testing for a broad range of genetic tests, most of which are laboratory developed (SACGHS, 2008b). This is comparable to the 97.6 percent accuracy in proficiency testing done for HIV-1 screening and cardiac markers for heart attacks, Dr. Leonard noted.4
Dr. Leonard added that there is no evidence that laboratory-developed genetic tests are of poorer quality than other laboratory tests. The SACGHS report concluded that genetic testing is not an exceptional type of laboratory test (SACGHS, 2008b). “For the purposes of oversight of genetic testing, that is, analytical validity and clinical validity, we consider that genetic and genomic testing is not different from other testing we do in the laboratory,” said Dr. Ferreira-Gonzalez. “There are quality issues across the board,” Dr. Leonard added. “Stop focusing on laboratory-developed tests and genomic tests as special. There are problems with tests that we currently do that aren’t laboratory-developed tests or aren’t genetic tests. We need to focus on the quality and proper use and interpretation of all tests,” she said.
Dr. Leonard and other participants at the workshop claimed that the main quality issue that needs to be addressed better in regulation is the need to show clinical utility, which neither the FDA nor CLIA requires. “We need to know [whether] these tests [are] usefully affecting the outcome of patients in the clinic. Neither of these processes gets at that,” Dr. Leonard said. However, Dr. Mass pointed out that determining clinical utility is difficult to do because “there’s not consistency of what clinical benefit really means as yet.” Dr. Ralph Coates of the CDC added that a 2007 IOM report
on biomarkers called for defining the translation pathway for biomarkers more clearly (i.e., determining what information on clinical validity and utility is needed, and what kind of research should be done to acquire this information) (IOM, 2007). He noted that in research, “there seems to be more interest in novel findings—what’s new—rather than summarizing what we really do know and don’t know.” He suggested there should be more systematic evidence reviews, and support for research addressing knowledge gaps identified from those systematic evidence reviews.
In addition, personalized medicine and predictive tests are rapidly evolving, and need to be continuously evaluated for testing outcomes, said Dr. Ferreira-Gonzalez. Dr. Herbst added that “we’re dealing with new information that changes maybe not the analytic validity of the test, but the clinical validity and ultimately probably the clinical utility.” Dr. Gutierrez pointed out that the way the FDA deals with new information is to specify what is currently known and unknown on the label of a diagnostic or therapeutic. “The iterative nature of this is very challenging, and we have a mechanism for getting products out in the market quickly, but if it’s investigational, we think patients deserve to know,” he said. “When we know something’s a winner—it’s been hit out of the ballgame—we try to clear or approve it. When we know it’s a loser, we lie down like we are in front of the railroad tracks trying to block it. And when it is in this gray zone, we try to label it.” An example of this regulatory behavior was pointed out by Dr. Amado, who noted that the FDA has agreed to include information about the lack of activity of anti-EGFR antibodies in the setting of KRAS mutations in the labels (i.e., while the indication remains broad, the label states that in a retrospective analysis, patients with KRAS-mutant colorectal tumors did not benefit from panitumumab or cetuximab, drugs that target the EGFR) (Amgen, 2008; ImClone Systems, 2008). This explicit labeling was, in part, a compromise between the test developers, who thought it was not feasible to do a prospective analysis, and the FDA, who wanted prospective data to evaluate the test.
POLICY SUGGESTIONS
In addition to suggesting that the FDA review all laboratory-developed tests or all complex tests, speakers and discussants made several suggestions for improving the regulation of predictive tests. These suggestions included
-
strengthening the proficiency requirements for laboratory personnel;
-
increasing the transparency of data collected on laboratory-developed tests;
-
restructuring and coordinating the oversight of companion diagnostic tests;
-
improving FDA and CLIA enforcement of predictive test regulations; and
-
assessing the clinical utility of predictive tests before or after they enter the market.
Improve Laboratory Proficiency
Laboratories performing predictive tests must enroll laboratory personnel in proficiency tests specific to the subspecialty of the tests they will be evaluating. CLIA requires proficiency testing of personnel at least once every 2 years for non-waived tests. However, a major deficiency of CLIA is that it does not require proficiency testing for all tests. Dr. Leonard suggested that one way to improve the regulation of laboratory-developed tests is to require stricter proficiency qualifications and personnel qualifications for predictive tests. She also suggested requiring proficiency testing for any test performed in the laboratory, regardless of whether the tests are FDA approved. Dr. Hayes pointed out before CAP proficiency testing was implemented for HER2 tests, 15 to 20 percent of HER2 analyses were done incorrectly in CLIA-certified labs. “As we began to have CAP proficiency testing, where your feet are put to the fire every 6 months, we’ve seen agreement go from 65 to 70 percent to close to 90 percent,” he said.5
Ms. Javitt described a longstanding concern about the lack of mandatory proficiency testing for genetic tests because there is no specialty for them under CLIA. Dr. Ferreira-Gonzalez added that legally, laboratories are required to perform proficiency testing on only 84 analytes. SACGHS debated whether or not to recommend regulation to require proficiency testing for all analytes for which proficiency testing material is available. However, this requirement would have been problematic because the analytes needed to do proficiency testing for genetic tests are often unavailable, and genetic tests are a rapidly moving target. Consequently, SACGHS decided to recommend that HHS fund studies to assess alternative ways to
conduct proficiency testing for genetic tests, including splitting samples with other laboratories or retesting one’s own samples. SACGHS also recommended that HHS ensure funding for the development of certified or validated reference materials that can be used to validate assays. In addition, it recommended increased funding for the development of assay, analyte, and platform validations that could be used for quality control assessment and standardization of testing among different laboratories (SACGHS, 2008b).
The FDA Center for Devices and Radiological Health (CDRH) has developed many guidance documents on the acceptable levels of performance characteristics for predictive tests. Dr. Ratain suggested that insurers could require that predictive tests be performed in labs that meet the standards specified in these guidance documents, regardless of whether the tests are subject to FDA approval.
Increase Transparency
Ms. Javitt stressed the importance of making proficiency data public. Currently, the CAP collects and compiles these data, but does not release the data to the public. “There should be a way for the public to access proficiency testing data so that they can make decisions about laboratory quality,” she said. Dr. Leonard agreed there should be transparency in how laboratories operate and perform, and added that she understood that under CLIA, CMS is directed to make proficiency testing results public, but it does not currently comply with this requirement.6
The need for transparency of data about predictive tests, including data that have been traditionally considered industrial “trade secrets” not to be divulged to the public, was stressed by Robert Erwin of the Marti Nelson Cancer Foundation. “We are missing huge chunks of information, and the quality of decisions depends a lot on the quality of information going into making those decisions,” he said.
The SACGT report recognized the need for more transparency in data collected on predictive tests, and recommended that genetic test developers be required to provide information on analytic validity, clinical validity, and clinical utility, said Dr. Burke. The committee knew that data on
clinical validity and clinical utility was likely to be very limited (SACGT, 2000a). However, “the point was that there should be transparency. Manufacturers should provide what they know and what they don’t know, including citations to the literature,” she said. SACGHS subsequently also recommended that HHS appoint and fund a lead agency to develop and maintain a mandatory, publicly available, Web-based registry for laboratory tests. It directed that a committee of stakeholders should determine what information should be entered into this registry (SACGHS, 2008b). The Twenty-First Personalized Medicine Coalition and the American Clinical Laboratory Association have also both supported the idea of registering laboratory-developed tests.
A mandatory registry of laboratory-developed tests offers a number of benefits. It would help foster truth in labeling, Dr. Ferreira-Gonzalez said. In addition, Dr. David Parkinson of Nodality, Inc., noted that this sort of registry would be extremely helpful in discerning the underlying biology governing the effectiveness of biomarkers and targeted drugs. “So much of what I hear about in the biomarker world is isolated tests, one point in time. The real information and meaning of these tests come when you are actually following patients longitudinally and when there’s some sort of intelligent life force looking at the result of the biological characterization, the therapeutic action, and the outcome. Then you start to understand what the biology means,” he said. Dr. Herbst added patients and physicians both feel a great deal of confusion or lack of knowledge about recently developed predictive tests. He promoted the development of a registry that would provide real-time information so that patients could be treated in the best possible way.
Restructure and Coordinate Oversight
To address the problem of having two independent regulatory paths for predictive tests under the FDA and CLIA, several presenters and participants suggested ways to restructure and coordinate this oversight. Dr. Darryl Pritchard of the Biotechnology Industry Organization (BIO) suggested reorganizing CLIA as a part of the FDA, and therefore under the same leadership structure to enable a system-wide approach to addressing regulatory gaps. However, Dr. Gutman replied that “it would be both an administrative and statutory challenge to do so because CLIA and the FDA are administratively and legally driven by widely different starting points. Although if you want to think out of the box and push this, it
certainly strikes me that anything is possible if you’re trying to fix a broken system.”
Dr. Hayes suggested that all oncology regulatory activities within the FDA—both devices and therapies—be consolidated under a single branch or committee. Dr. Mansfield pointed out that the CDRH and the Center for Drug Evaluation and Research do have an intercenter oncology working group that considers both cancer diagnostic and therapeutic issues (FDA, 2009a). However, Dr. Ferreira-Gonzalez noted that the review by SACGHS uncovered a number of duplicate efforts assessing how to improve the oversight of genetic/genomic technologies among government agencies and offices within HHS. “They were not talking to each other. In some instances, they were doing exactly the same fact finding without even sharing some of the information,” Dr. Ferreira-Gonzalez said. This discovery of bureaucratic redundancy led SACGHS to recommend that the Secretary of HHS should coordinate efforts in personalized medicine within the agency, and consider creating a new HHS office for this purpose (SACGHS, 2008a).
“There are several solutions,” Dr. Gutierrez stressed. “But if you don’t have the FDA doing the regulation, you’re going to have to come up with a way to do it that makes sense, that people actually believe in, that is independent of both the laboratories and the manufacturers, and that is credible.” Peter Collins of DxS Ltd. pointed out that globally, FDA regulation is seen as the gold standard to emulate, and any changes to that regulation would have global implications.
Improve Enforcement
Dr. Ferreira-Gonzalez suggested that the current regulations for predictive tests should be more uniformly enforced. She noted that when SACGHS solicited comments from stakeholders, a number suggested the need for better enforcement of current regulations related to laboratory testing. “Sometimes some of the problems we see are due to the regulation not being fully enforced,” she said. Consequently, SACGHS recommended that the gaps in the enforcement of existing regulations for analytic and clinical validity be identified. For example, CLIA surveyors cannot inspect and close down laboratories that are not CLIA certified. They are restricted to providing information about a laboratory to the Government Accountability Office, and must rely on this office to take corrective action. SACGHS also recommended that CMS be empowered to take direct enforcement actions against labs that perform clinical tests without proper CLIA certi-
fication, including those that offer direct-to-consumer testing (SACGHS, 2008b). In addition, increasing the enforcement discretion of the FDA for laboratory-developed tests would not necessarily require new legal authority, Dr. Mansfield noted.
Assess Clinical Utility
Many participants and speakers acknowledged the need to assess the clinical utility of predictive tests. “Clinical utility today is being used by third-party payers to reimburse the testing that we do, but what we’re starting to realize is that we don’t have a lot of clinical utility data because we don’t have the infrastructure to see what information is needed, and fund the collection of that data,” said Dr. Ferreira-Gonzalez. Neither laboratories nor manufacturers have the resources to assess the clinical utility of genetic tests, for example, by building on the CDC’s Evaluation of Genomic Applications in Practice and Prevention (EGAPP) initiative (SACGHS, 2008b). The Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) identified clinical utility as a key issue for evaluation in Medicare coverage decisions, and recommended that Medicare use EGAPP methods in its evaluations of genomic tests (CMS, 2009c). SACGHS recommended that HHS create and fund a public/private entity to assess the clinical utility of predictive tests and develop a research agenda to address gaps in knowledge. SACGHS also recommended that HHS conduct public health surveillance to assess health outcomes or surrogate outcomes, practice measures, and the public health impact of predictive testing (SACGHS, 2008b). In addition, SACGHS recommended that researchers develop more evidence of clinical utility in genetic tests (SACGHS, 2006, 2008b).
Dr. Coates noted that research to assess the population health benefit of genetics or genomics-based tests or treatments comprise less than 3 percent of all published genetics research (Khoury et al., 2007; Woolf, 2008) (Figure 7). “The CDC is currently collaborating with the NCI to assess cancer genomics funding in specific, and how much of it is used for discovery, versus application, versus assessing the population health benefit of new genomics applications.” A separate CDC/NCI effort on the comparative effectiveness of cancer care and prevention included the statement: “To date, there’s been no systematic research conducted to compare the clinical effectiveness and cost effectiveness of cancer care and prevention based on genomic tools and markers compared to existing standards of care and prevention” (NCI, 2009a).
Stakeholders at an NIH/CDC Personal Genomics Workshop in
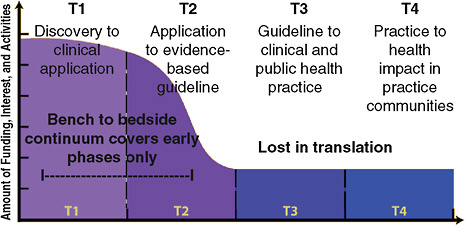
FIGURE 7 The research community’s interest in implementation processes wanes along the continuum of cancer translation research. Ninety-seven percent of genetics research is published in the T0 and T1 phase.
SOURCES: Coates presentation (June 8, 2009); Khoury et al. (2007); Woolf (2008). Adapted from Khoury, M. J., M. Gwinn, P. W. Yoon, N. Dowling, C. A. Moore, and L. Bradley. 2007. The continuum of translation research in genomic medicine: how can we accelerate the appropriate integration of human genome discoveries into health care and disease prevention? Genetics in Medicine 9(10):665–674. Reprinted with permission of Wolters Kluwer Health.
December 2008 agreed that more multidisciplinary research should be done to fill knowledge gaps on the clinical validity and utility of predictive tests, Dr. Coates noted. (Khoury et al., 2009b; NCI, 2008). Participants also recommended that both personal and clinical utility be assessed, and that researchers should link science to evidence-based recommendations (NCI, 2008). The CDC, with the NIH and other organizations, has initiated the Genomic Applications in Practice and Prevention Network (GAPPNet) to increase communications among stakeholders to improve translation of genomic applications (Khoury et al., 2009a). Relevant stakeholders include test developers and others doing translational research; those developing evidence-based recommendations by linking evidence to practice guidance in a transparent and credible way; practitioners in clinics, public health, and community practice; and patient advocates.
Two examples from EGAPP that highlight the need to do more translational research evaluating the clinical utility of predictive tests are the recent evaluations of (1) breast cancer gene expression profiles, and
(2) the UGT1A1 genotyping. EGAPP used the systematic, evidence-based process it developed for evaluating predictive tests, and other applications of genomic technology in transition from research to practice, to evaluate these two applications of genetic/genomic technology (Teutsch et al., 2009). EGAPP found insufficient evidence to make a recommendation for or against the use of tumor gene expression profiles to improve outcomes in women with Stage I or II node-negative breast cancer (AHRQ, 2008; EGAPP Working Group, 2009a). EGAPP’s evidence review found adequate evidence of the clinical validity of Oncotype DX and MammaPrint, but inadequate evidence of the clinical utility of Oncotype DX and no evidence of the clinical utility of MammaPrint. The analytical validity for both tests was also inadequate. Similarly, EGAPP did not find sufficient evidence to make a recommendation for or against the routine use of UGT1A1 genotyping in metastatic colorectal cancer patients, said Dr. Coates (EGAPP Working Group, 2009b; Palomaki et al., 2009).
However, one participant noted that guidelines and research reviews, such as EGAPP, are often based on a higher degree of efficacy than what doctors use for their clinical decisions. Dr. Hayes suggested that for a test to be clinically useful, its effect must be large enough that clinical decisions based on the test results have acceptable outcomes. These outcomes include cure, improved survival or palliation, or decreased exposure to toxicity due to useless therapy. The most useful tumor markers indicate those patients whose prognosis is so good or so bad that they are not likely to experience improvement with treatment, he said. In these patients the risks of therapy outweigh the benefits.
Ways to Capture Clinical Utility Data
Several suggestions were offered on how to improve the collection of clinical utility data needed to fully evaluate tests. Dr. Leonard suggested creating a registry of test and treatment outcomes, akin to the evidence development that CMS requires for treatments it considers investigational. During the data collection phase, the treatment or test would be reimbursed by Medicare/Medicaid. “This would allow new tests to get out there in medical practice only if you’re collecting data on them,” she said. Such collection and analysis of data would be aided greatly by a national electronic health record system, which would allow real-world data to be collected and used to determine clinical utility. “I would argue that you have to start at the bedside before you go to the bench, if you’re doing healthcare research,
because you have to have research that’s driven by clinically relevant questions,” Dr. Leonard said. If the data do not indicate clinical utility, the predictive test should be taken off the market.
Conceptually, such a registry would be more comprehensive than the registry of predictive tests proposed by SACGHS or SACGT. Dr. Leonard said she envisions a registry that would be disease based, and include all patients receiving the test or treatment, including all age, racial, and ethnic groups. “One of the problems when we do clinical trials is that often they don’t mimic what we do in real clinical practice,” she said. “The registry would create a pathway to acquiring clinical validity and utility data without the need for randomized, controlled clinical trials that are very costly, and, everyone agrees, would be very difficult to do for diagnostics.”
Dr. Hayes pointed out that the proposed registry data would be confounded by the information doctors are given about the tests. Some patients would do well with or without treatment, and a test that predicts they would do well with treatment might lead physicians to assume they subsequently did well because of the treatment. But Dr. Leonard argued that physicians are now using tests for clinical decisions despite the lack of data on how clinically useful the tests are, and the advantage of the registry is that these data would be collected and analyzed. “In our current way of doing it, we don’t get any of that data back,” she stressed. “It’s just not a good data collection process, and we may be able to speed it up if we did have some data collection.” Data analysis for the registry may have to be done differently than for a randomized, controlled clinical trial, she added, because all of the potential confounding factors could not be controlled.
Dr. Austin countered that “health professionals have lots of hypotheses out there, and some prove to be right, and some prove to be wrong. But we don’t know which ones are right and which ones are wrong until we do tests with proper control groups. So why not just do these studies? Why put things out on the market and then try to finagle proper control groups around it, which is very hard to do. What scares me is when doctors come into my lab and say ‘I want this test’ because of some paper they read that was probably of a study performed on just 23 subjects.”
Dr. Leonard disagreed, pointing out the repetitive nature of medical practice. For example, she noted that if the main mutation in cystic fibrosis (CF) had not been discovered and a test for it put into practice, researchers would never have uncovered additional mutations that can cause CF, nor would those with the first mutation have been helped. “Some knowledge does allow us to move forward,” she said. “There are processes that are
alive and well and functioning in medicine, and I don’t know that they’re necessarily bad.”
The FDA could play a serious role in safety determinations of predictive tests, Dr. Friend observed, while still allowing a dynamic, iterative process for determining clinical utility. “Imagine a world where not thousands of patients were enrolled in trials, but millions, and they involved drugs that were already approved,” he said. “Even though a drug was first given for only one indication, 2 years from when it was first approved, the evidence-based data could come back in and change the indication,” without having to undergo the lengthy process that is currently involved in having a drug label changed. For this to occur, patient advocates “would have to step up and say we need care that’s more personal,” and encourage more patients to enroll in trials, he said. Dr. Mass added that the potential evidence that could be developed by a registry similar to what Dr. Leonard proposed is great, but he stressed that the FDA should be involved to ensure postmarketing data are collected. “You’d need to have some leverage, and I don’t think CLIA could do that,” he said.
Another issue is off-label use of drugs or tests. Dr. Hayes noted that such use makes it difficult to accrue patients to trials assessing new indications. For example, it took 13 years to accrue enough subjects to conduct a randomized, controlled trial on the use of the prostate-specific antigen (PSA) test for prostate cancer screening because the PSA test was already approved for monitoring the progress of prostate cancer (Andriole et al., 2009; Schroder et al., 2009). “Because the assay was out there and being used, it took much longer to get the data we wanted,” he said. The off-label use of a questionably useful test also made the predictive test regulation appear inconsistent, Dr. Quinn said.
Off-label use of tests can also be risky for patients, stressed Mr. Collins of DxS Ltd. The use of a test without sufficient evidence can lead to patients being denied treatment based on an unproven test that indicated the individual was unlikely to respond to the drug. “There has to be a better mechanism for dealing with this,” he said. Dr. Leonard pointed out that European countries with universal health care coverage control test use based on evidence. “It’s not that you don’t get the test paid for. You don’t get the test period” if there’s no evidence, she said. “Part of healthcare reform has to look at the decision-making process of who gets what, when, and not just whether it gets paid for or not.”
One participant noted that creating a prospective registry of test consumers—primarily practitioners—could indicate which predictive
tests are being used by doctors for which purposes, and what clinical decisions are being influenced by the test results. Although a registry would not be as robust as a clinical trial, it could still provide some useful information. Another participant pointed out that the registry of treatment outcomes created by the Cystic Fibrosis Foundation, which includes data from 150 cystic fibrosis centers throughout the country, has led to dramatic improvements in treatments and outcomes for cystic fibrosis (CFF, 2009). “It led to a tremendous dialogue between patients and centers as to what they are actually doing, in terms of quality care, which has now led to many centers improving their performance,” he said. Similar non-regulatory strategies for collecting and improving the clinical utility of predictive tests could be implemented. Ms. Stack noted that many predictive tests, such as Oncotype DX, provide risk information, but ultimately, doctors make the final call on what that risk means—they determine their own cutoff points for risk that warrants treatment or not. “Ultimately, the doctor’s going to make that treatment call and have a lot of data about it,” she said, so a registry that collects that data would be useful in ascertaining the clinical utility of a test.
Dr. Ferreira-Gonzales noted the catch-22-like nature of clinical utility determinations. “Some third-party payers are making decisions on the lack of information that we have in these areas. But if we don’t offer the testing and know how it was actually being used, we will never know this information. So we need to be able to gather this information on what the test does for the patients, and SACGHS discussed the possibility of making a decision to allow a test to enter the market dependent on evidence development via a registry, as has been done with CT scans and in other areas of medicine.” Dr. Burke added that “there is a need to know, not only what we know, but what we don’t know, because it’s what we don’t know that points to the critical research that needs to be done. Clinicians on the front lines are a very good source for that kind of information.”
A participant stressed the need for a test registry to report not only positive results of studies, but also negative results. “Another registry concept would be the registration of validation studies prior to their initiation so that we could follow up and make sure that the results of those studies are subsequently presented and published.” Dr. Ferreira-Gonzalez responded by pointing out that one of SACGHS’s recommendations was that both positive and negative results should be shared in a Web-based system (SACGHS, 2008b).
Dr. Friend agreed that the experimental evidence should be posted in registries and made available to the public. However, he cautioned against
having the FDA be responsible for the registry. “I’m not sure the FDA needs to do that,” he said. The FDA should be responsible for regulating analytical and clinical validity, but not clinical utility. Mr. Erwin added, “When I hear about proposals that the FDA should regulate everything, on the one hand you don’t want to see innovation delayed or stifled. On the other hand, if the standards are not high, the promise of personalized medicine will never be realized, because there will be no real incentive to put the money and time that’s necessary to do it right in order to get rewarded financially or professionally. We need rigor without rigidity. As technology evolves and as unexpected things come along, the regulatory framework has to be adaptable enough to deal with that without extremely long delays. If new technology can’t fit into a box that’s so rigid that we can’t derive the benefit from it, then there’s something wrong with regulation.”
Dr. Parkinson concurred, calling for more flexible regulation of predictive tests. “These tests are going to have to continue to evolve as the therapeutics are evolving, and it’s almost like an iterative process. There needs to be informed regulation. This requires a tighter link between biological characterization, and predictive test development and therapeutic applications—a strategic approach to the regulation recognizing that these diseases are being redefined by the tests, and by the effect of therapeutics on patients characterized by these tests.” Dr. Parkinson called for having some ongoing review of new information, akin to what EGAPP does.
Dr. Mansfield cautioned against just putting tests on the market before adequately assessing their safety and effectiveness, and relying on a registry to determine clinical utility. “There is already a mechanism for tests to go to market before we know all their performance mechanisms, and it’s called an investigational device exemption. It seems to work very well,” she noted.
Regardless of how clinical utility is assessed, it is a costly endeavor that needs more federal financial support. Dr. Burke noted that SACGT asked all federal agencies to indicate how much work they did in research relevant to evaluating tests, and found “there was tremendous room for growth in federal funding of research around the assessment of clinical utility. We noted that healthcare funding decisions often function as an oversight mechanism.” SACGT also recommended more federal government support for evidence-based guideline development related to predictive tests. This recommendation has been realized to some degree through the U.S. Preventive Services Task Force (USPSTF) and EGAPP reviews of predictive tests, Dr. Burke noted.
























