
Appendix B
Dissenting Statement and Rebuttal
Dissenting Statement on Mode of Action of Tetrachloroethylene in Mouse Hepatocarcinogenesis
By Rolf Schulte-Hermann
The authors of the Integrated Risk Information System (IRIS) draft conclude in Chapter 4.4.
-
That peroxisome proliferator-activated receptor-alpha (PPARα) activation is not the primary mode of action (MOA) for tetrachloroethylene-induced hepatocarcinogenesis in mice.
-
That the specific mechanisms or MOAs for hepatocarcinogenesis are not known.
-
That it is highly likely that more than one mechanism is operative.
That conclusion is supported in Chapter 6 of the present committee review of the IRIS draft although some deficiencies in the draft are mentioned. They include the lack of coherent flow and an imbalance in critiquing the view that the PPARα MOA is not relevant for human carcinogenesis. This committee member concurs with the criticisms.
However, the member disagrees with the conclusions quoted above. In the members’ opinion, the weight of evidence strongly favors a key role of PPARα activation in tetrachloroethylene-induced hepatocarcinogenesis in mice; furthermore, this MOA lacks relevance for human hepatocarcinogenesis. Because of the deficits in the respective presentation in the IRIS draft, the following paragraphs will briefly compile the essential data supporting the PPARα MOA
for tetrachloroethylene, the role of trichloroacetic acid (TCA) as the major responsible metabolite of tetrachloroethylene, the potential roles of other MOAs, new mechanistic data supporting the lack of relevance of the PPARα MOA for humans. The author hopes that the arguments collected in this dissent will be helpful in revising the IRIS draft.
EVIDENCE THAT TETRACHLOROETHYLENE AND TRICHLOROACETIC ACID ARE PEROXISOME PROLIFERATORS
Relevance of Trichloroacetic Acid vs Dichloroacetic Acid
Both TCA and dichloroaceticacid (DCA) are peroxisome proliferators. TCA is the major metabolite found in the body after exposure to tetrachloroethylene. It is eliminated slowly and therefore accumulates to some extent. In contrast, DCA is present in only tiny amounts after tetrachloroethylene exposure because of low formation and more rapid elimination (IRIS draft, Chapter 3). Thus, after tetrachloroethylene administration in mice, DCA concentrations in blood were below 10 or 25 βg/mL in the initial hours and then undetectable and were undetectable in the liver in the presence of high TCA concentrations (up to 150 βg/mL or 150 βg/g) (Philip et al. 2007; see below for experimental details). TCA and DCA have similar potency as hepatic carcinogens and tumor promoters (Bull 2000; Bull et al. 2004). Overall, therefore, DCA probably contributes little to PPARα-mediated effects of tetrachloroethylene. Other metabolites of tetrachloroethylene are not known to be peroxisome proliferators. The arguments related to the PPARα MOA should therefore focus on TCA.
Peroxisome Proliferator-Activated Receptor-Alpha Transactivation
Tetrachloroethylene (up to 5 mM) did not transactivate mouse and human PPARα in cells transfected with the PPAR genes. Likewise, chloral hydrate and trichloroethanol, minor metabolites of tetrachloroethylene, did not activate PPARα. In contrast, TCA was active at 1 and 5 mM but not at 0.1 mM. Activity was considerable at 1 mM, suggesting that the lowest observed-adverse-effect level (LOAEL) for binding activity is distinctly below 1 mM (Zhou and Waxman 1998; Maloney and Waxman 1999). The maximal activation was only about 50% of that of Wy 14643, a strong activator, but similar to that of mono(2-ethylhexyl) phthalate, the carcinogenic metabolite of di(2-ethylhexyl) phthalate (DEHP). Mouse PPARβ displayed little, and human PPARβ no, responsiveness to TCA. DCA transactivated PPARα with somewhat less potency than TCA, but it showed no effect on mouse or human PPARβ (Zhou and Waxman 1998; Maloney and Waxman 1999). In another study (Walgren et al. 2000), TCA but not DCA was found to activate mouse PPARα at 4 mM.
Tetrachloroethylene as a Peroxisome Proliferator
Tetrachloroethylene induces in mouse liver responses that are known to be mediated by PPARα—such as a 4-fold increase in CN-insensitive palmitoylCoA oxidation (PCO), morphologic evidence of peroxisome proliferation based on morphometric analysis, and hepatomegaly—at doses of 1,000 mg/kg by gavage for 10 days or 200 and 400 ppm by inhalation 6 hours/day for 14, 21, or 28 days (Goldsworthy and Popp 1987; Odum et al. 1988). Those effects also occurred, although much more weakly, in rats (Goldsworthy and Popp 1987; Odum et al. 1988). Dose-dependent increases in hepatomegaly and (not significantly) hepatocyte proliferation after oral treatment of mice were reported by Schumann et al. (1980) and Buben and O’Flaherty (1985). In male mice, tetrachloroethylene at daily oral doses of 150, 500, and 1,000 mg/kg transiently and dose-dependently increased hepatocyte DNA synthesis at 7 and 14 days; at 30 days, the increase was nearly gone (Philip et al. 2007). Tetrachloroethylene itself does not bind to PPARα (see above), so PPARα-mediated responses should be due to active metabolites, predominantly TCA. In the study by Odum (1988), 200 ppm, the higher dose in the NTP (1986) carcinogenicity study, induced pronounced increases in PCO and peroxisome proliferation that suggested that the NOAEL was much lower. Obviously, doses of tetrachloroethylene that are in the range of the carcinogenic doses are transformed to metabolites (mainly TCA) in amounts sufficient to activate PPARα in mouse liver. Evidence supporting the role of TCA is presented later.
Trichloroacetic Acid as a Peroxisome Proliferator and Hepatocarcinogen in Mice
TCA was shown in numerous studies to induce PPARα-mediated responses, such as PCO increases, in the livers of mice of both sexes and to produce liver tumors in mice (Goldsworthy and Popp 1987; Pereira 1996; Bull 2000; Bull et al. 2002; DeAngelo et al. 2008; further references in the IRIS draft). In the first days of administration, TCA induced liver enlargement and an increase in hepatocyte DNA synthesis in male and female mice (Dees and Travis 1994; Pereira 1996; Stauber and Bull 1997). Effects were present when TCA was given at 100 mg/kg orally over 11 days and showed some increase with dose up to 1,000 mg/kg (Dees and Travis 1994). With continued treatment, the enhancement of DNA synthesis disappeared and was reversed to depression (Pereira 1996; Stauber and Bull 1997). TCA induction of the peroxisomal enzymes PCO and acyl-CoA oxidase (by RNA expression) and of CYP 4a depended on the presence of the PPARα gene and were not seen in PPARα-null mice (Laughter et al. 2004). Some studies reported increased lipid peroxidation by TCA (Bull et al. 1990; Larson and Bull 1992; Austin et al. 1996). An increase in 8-OHdG was not found after TCA (Parrish et al. 1996) or after tetrachloroethylene (Toraason et al. 1999).
Hepatic tumorigenesis after TCA administration was studied mostly in male mice but was also demonstrated in female mice (Pereira 1996). TCA was found to promote hepatic-tumor development efficiently in mice after initiation by 1-methyl-1-nitrosourea or vinyl carbamate (Pereira and Phelps 1996; Bull et al. 2004). Foci of altered cells (presumably preneoplastic lesions) and tumors were predominantly basophilic and did not express glutathione S-transferase-pi (GSTP), as found with other peroxisome proliferators (Pereira 1996; Pereira and Phelps 1996; Stauber and Bull 1997). Clonal expansion of anchorage-independent hepatocytes obtained from male B6C3F1mice by administration of TCA in vitro was also reported (Stauber et al. 1998).
In a recent lifetime dose-response study, DeAngelo et al. (2008) found that the TCA-induced increase in PCO correlated with tumor induction, and a linear association occurred between the two effects. A TCA NOAEL of 6 mg/kg per day and a LOAEL of 58-68 mg/kg were reported.
Evaluation of Effects of Trichloroacetic AcidTCA and Tetrachloroethylene for Consistency with Key Events
Klaunig et al. (2003) have defined seven key events in the PPARα MOA of rodent hepatocarcinogenesis. TCA was found to induce most of the key events in mice:
-
Causal relationship to tumor formation:
-
Direct activation of PPARα (resistance to induction of key events in PPARα-null mice).
-
Transient increase in hepatocyte DNA synthesis.
-
Selective clonal expansion of the putative preneoplastic lesions and of tumors.
-
-
Associative relationship to tumor formation:
-
Peroxisome proliferation as indicated by morphologic and biochemical studies (high weight of evidence and specificity for association with tumorigenesis [Klaunig et al. 2003]).
-
Hepatocyte oxidative stress (lipid peroxidation) (low weight of evidence and specificity for association [Klaunig et al. 2003]).
-
Inhibition of gap junctional intercellular communication (GJIC) by TCA in a model with lucifer yellow. The same result was obtained with tetrachloroethylene (Benane et al. 1996)
-
Dependence on Kupffer cells has apparently not been studied directly after TCA. administration. However, that is not a serious deficiency for the purpose of this discussion, because the specificity of Kupffer-cell dependence is low (Klaunig et al. 2003).
-
This set of results was generated in several studies, and dose-response and temporal relationships are consistent with the observation of tumors. In the absence
of evidence on genotoxicity and other plausible MOAs, the induction of 6 of the 7 key events provide strong evidence of a PPARα-dependent MOA of TCAinduced mouse hepatocarcinogenesis. The same conclusion was reached by the National Research Council’s Committee on Human Health Risks of Trichloroethylene (2006).
Data on tetrachloroethylene are less comprehensive. An NOAEL and a LOAEL and studies in PPARα-null mice are not available. Nevertheless, the PPARα MOA is considered probable on the basis of the induction of several key events in mouse liver, including transient increases in DNA synthesis, lipid peroxidation, inhibition of GJIC, and, most important, peroxisome proliferation, an event highly specific for PPARα activation. A major argument supporting the PPARα MOA of tetrachloroethylene is related to the role of TCA as the active metabolite, as will be shown below according to several lines of evidence.
SPECIES DIFFERENCES SUPPORTING THE ROLE OF TRICHLOROACETIC ACID AS THE ACTIVE METABOLITE OF TETRACHLOROETHYLENE
Rats are less sensitive than mice to peroxisome-proliferator effects of the same doses of tetrachloroethylene (Goldsworthy and Popp 1987; Odum et al. 1988) and do not develop hepatic tumors in response to it (NCI 1977; NTP 1986; JISA 1993) or to TCA at doses up to 364 mg/kg per day for 104 weeks (DeAngelo et al. 1989, 1997). Those differences can be explained by the kinetics of tetrachloroethylene in the two species. Mice metabolize the agent and form TCA at concentrations several times higher than do rats (Schumann et al. 1980; Reitz et al. 1996). Thus, the area under the curve (AUC) for blood TCA after exposure to tetrachloroethylene at 400 ppm for 6 hours was 6.7 times higher in mice than in rats (Odum et al. 1988). In addition, mice are more sensitive than rats to induction of peroxisome proliferation by TCA. That may, at least partially, be due to the 10-fold higher binding capacity of rats’ than of mice’s plasma proteins for TCA (maximal binding capacity, 283 μM in rats and 29 μM in mice). As a result, the proportion of TCA available for uptake by the liver will be less in rats than in mice and will produce a weaker response in rats (Lumpkin et al. 2003). The weak peroxisome-proliferator effect seen in rats is obviously insufficient for hepatic-tumor formation. Numerous examples show that low levels of induction of peroxisomes are not necessarily associated with hepatic tumorigenesis (Klaunig et al. 2003). Overall, the striking differences between responses of mice and of rats to tetrachloroethylene can be explained by assuming TCA as the active principle.
CARCINOGENICITY STUDIES WITH TETRACHLOROETHYLENE AND TRICHLOROACETIC ACID
Hepatocarcinogenic doses of tetrachloroethylene in mice are displayed in
Tables 1 and 2. Doses that do not increase rates of hepatocarcinoma were not tested in National Cancer Institute (NCI) and National Toxicology Program (NTP) studies. Rats treated in parallel bioassays did not develop hepatic tumors.
Long-term exposure to TCA was shown to result in hepatic-tumor formation in mice (Table 3A) but not rats (DeAngelo et al. 1997). DeAngelo et al. (2008) exposed male B6C3F1 mice to TCA via drinking water at 0.05, 0.5, 4.5, and 5 g/L for 60 and 104 weeks (Table 3B). Daily doses calculated were 6-8, 58-68, and 572-602 mg/kg. The work consisted of three parts conducted in two Environmental Protection Agency (EPA) laboratories. The authors reported significant increases in the prevalence and multiplicity of hepatic tumors in the two higher dose groups. A TCA NOAEL of 6 mg/kg per day and a LOAEL of 58-68 mg/kg per day were derived for neoplastic and nonproliferative pathology.
Somewhat surprisingly, the IRIS draft does not mention parts 1 and 2 of the study by DeAngelo et al. 2008), which is therefore presented completely in Table 3B. The selection of only one of the two 104-week bioassays has important implications for modeling in Appendix 4A of the IRIS draft because the control group selected shows a dramatically higher hepatic-tumor incidence (64% vs 12%; see parts 3 and 2 in Table 3B). Use of the low-tumor control would increase the fraction of animals affected by TCA (Figure 4A-1 of the IRIS draft) and increase the calculated carcinogenic potency of TCA. To add to the confusion, in the publication of DeAngelo et al. (2008), the allocation of controls and treated groups in parts 2 and 3 of the study is at variance between the methods section and Table 6 of the results section. That discrepancy should be resolved, and all pertinent data should be used in revising the IRIS document. At present, the validity of the modeled TCA potency data as used in Appendix 4A is questionable.
TABLE 1 Carcinogenicity Study in B6C3F1 Mice (Tetrachloroethylene In Corn Oil Was Administered By Gavage 5 Time a Week for 78 Weeks and Followed By an Observation Period of 12 Weeks)
|
Sex |
Bioassay |
Dose, mg/kg (TWA) |
Carcinoma (Incidence) |
Mice at Risk |
|
Male |
NCI |
0 |
2 |
17 |
|
|
|
0 (vehicle) |
2 |
20 |
|
|
|
536 |
32 |
49 |
|
|
|
1,072 |
27 |
48 |
|
Female |
NCI |
0 |
2 |
20 |
|
|
|
0 (vehicle) |
0 |
20 |
|
|
|
386 |
19 |
48 |
|
|
|
772 |
19 |
48 |
|
Source: NCI 1977. |
||||
TABLE 2 Incidence of Hepatocellular Adenomas and Carcinomas in B6C3F1 Mice Exposed to Tetrachloroethylene in Two Inhalation Bioassays
|
Sex |
Bioassay |
Administered Exposures, ppm |
Cumulative LiverTumors at Week 104 |
Total at Riska |
||
|
Adenomas |
Carcinomas |
Adenomas or Carcinomas |
||||
|
Male |
NTP (1986) |
0 |
12 |
7 |
17 |
49 |
|
100 |
8 |
25 |
31 |
47 |
||
|
200 |
19 |
26 |
41 |
50 |
||
|
JISA (1993) |
0 |
7 |
7 |
13 |
46 |
|
|
10 |
13 |
8 |
21 |
49 |
||
|
50 |
8 |
12 |
19 |
48 |
||
|
250 |
26 |
25 |
40 |
49 |
||
|
Female |
NTP (1986) |
0 |
3 |
1 |
4 |
45 |
|
100 |
6 |
13 |
17 |
42 |
||
|
200 |
2 |
36 |
38 |
48 |
||
|
JISA (1993) |
0 |
3 |
0 |
3 |
50 |
|
|
10 |
3 |
0 |
3 |
47 |
||
|
50 |
7 |
0 |
7 |
48 |
||
|
250 |
26 |
14 |
33 |
49 |
||
|
aAnimals that died before the first appearance of a hepatocellular tumor, but no later than week 52, were omitted from the totals because they were presumed not to have adequate time in the study to develop tumors. Source: EPA 2008 (Table 4A-3). |
||||||
TISSUE CONCENTRATIONS OF TRICHLOROACETIC ACID AFTER ADMINISTRATION OF TETRACHLOROETHYLENE OR TRICHLOROACETIC ACID
A key question in identification of the MOA of tetrachloroethyleneinduced hepatic tumors is whether sufficient TCA is formed and available in the target organ for effective induction of peroxisome proliferation and hepatocarcinogenesis. To address that question, a literature search has been conducted for analytic data on TCA concentrations in the liver and, as a surrogate, in the blood. The results are described below and displayed in Tables 4A-D and Figures 1 and 2A-C.
Blood and Liver Concentrations of Trichloroacetic Acid After Administration of Tetrachloroethylene
Blood concentrations of TCA after tetrachloroethylene administration were first analyzed by Odum et al. (1988). After a single exposure at 400 ppm for 6 hours, peak blood concentrations in B6C3F1 mice were 130 μg/mL 3-4 hours after the end of exposure and thereafter declined with a half-life of 7-8 hours. The AUC was calculated (Table 4A).
TABLE 3A Trichloroacetic Acid Drinking- Water Studies in Male Mice: Incidence of Hepatocellular Adenomas and Carcinomas
|
Source |
Weeks of Exposure |
TCA Exposure, g/L |
Equivalent TCA Exposure, mg/kg- day |
N |
Incidence of Adenomas |
Incidence of Carcinomas |
Incidence of Adenomas or Carcinomas |
Proportion Responding with Carcinomas |
|
Bull et al. (1990)a |
37 |
2 |
330 |
11 |
0 |
3 |
3 |
0.27 |
|
52 |
0 |
0 |
35 |
0 |
0 |
0 |
0.0 |
|
|
|
1 |
170 |
11 |
2 |
2 |
NR |
0.18 |
|
|
|
2 |
330 |
24 |
1 |
4 |
NR |
0.17 |
|
|
Bulll et al. (2002) |
52 |
0 |
0 |
20 |
0 |
0 |
0 |
0.0 |
|
|
0.5 |
NR |
20 |
5 |
3 |
6 |
0.15 |
|
|
|
2 |
NR |
20 |
6 |
3 |
8 |
0.15 |
|
|
Herren-Freund et al. (1987) |
61 |
0 |
0 |
22 |
2 |
0 |
2 |
0.0 |
|
|
5 |
NR |
22 |
8 |
7 |
NR |
0.32 |
|
|
Ferreira-Gonzalez et al. (1995) |
104 |
0 |
0 |
16b |
NR |
3b |
NR |
0.19 |
|
|
4.5 |
NR |
11 |
NR |
8 |
NR |
0.73 |
|
|
DeAngelo et al. (2008) |
104 |
0 |
0 |
56 |
10 |
26 |
31 |
0.55 |
|
|
0.05 |
8 |
48 |
10 |
14 |
21 |
0.44 |
|
|
|
0.5 |
68 |
51 |
20 |
32 |
36 |
0.71 |
|
|
aCumulative TCA exposures were provided in grams per kilogram for the mice evaluated at 52 weeks. Those exposures were converted to milligrams per kilogram per day by (1,000 mg/g)/(7 days/[week][52 weeks]). bEstimated from the reported proportion responding by selecting the smallest group size and incidence value consistent with the precision of the reported proportion. NR = not reported. Source: EPA 2008 (Table 4A-1). |
||||||||
TABLE 3B Complete Presentation of Results of the TCA Carcinogenicity Study of DeAngelo et al. (2008)
|
Weeks |
TCA, g/L |
Equivalent TCA, mg/kg |
Na |
Number with Denoma |
Number with Carcinoma |
Number with Adenoma or Carcinoma |
|
60 (part1) |
0 (NaCl) |
0 |
30 |
7 |
7 |
13 |
|
|
0.05 |
8 |
27 |
15 |
4 |
15 |
|
|
0.5 |
68 |
29 |
21 |
21 |
38 |
|
|
5.0 |
602 |
29 |
38 |
38 |
55 |
|
104 (part 2) |
0 (NaCl) |
0 |
25 |
0 |
12 |
12 |
|
|
4.5 |
572 |
36 |
59 |
78 |
89 |
|
104 (part 3) |
0 (1.5 g of acetic acid) |
0 |
42 |
21 |
55 |
64 |
|
|
0.05 |
6 |
35 |
23 |
40 |
57 |
|
|
0.5 |
81 |
37 |
51 |
78 |
87 |
|
aNumber of animals examined. Note: Table 3A from EPA (2008) contains only part 3 and reports higher numbers of animals examined than the publication by De Angelo et al. and somewhat different proportions of carcinomas. |
||||||
TABLE 4A Blood TCA Concentrations After Tetrachloroethylene Treatment
|
1) 1x 400 ppm for 6 hours |
Odum et al. 1988 |
|
|
||
|
Doses (ppm x 6 hours) |
400 |
|
|
|
|
|
Peak concentrations (μg/mL) a |
130 |
|
|
|
|
|
AUC0-24 (μg/mL per hour) b |
1,760 |
|
|
|
|
|
2) 1x i.g. |
Gearhart et al. 1993 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
100 |
536 |
1,072 |
5.36 |
2.0 |
|
Peak concentrations (μg/mL) a |
23 |
80 |
157 |
3.48 |
1.96 |
|
AUC0-24 (μg/mL per hour) b |
368 |
1,317 |
2,840 |
3.58 |
2.16 |
|
3) 1x, i.g.; SW mice |
Philip et al. 2007 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
150 |
500 |
1,000 |
3.33 |
2.0 |
|
Peak concentrations (μg/g) a |
150 |
160 |
170 |
1.07 |
1.06 |
|
AUC0-24 (μg/mL per hour) a |
2,583c |
2,229 |
3,208 |
0.86 |
1.44 |
|
4) 30x, daily i.g.; SW mice |
Philip et al. 2007 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
150 |
500 |
1,000 |
3.33 |
2.0 |
|
Peak concentrations (μg/g) a |
75 |
128c |
130 |
1.71 |
1.02 |
|
AUC0-24 (μg/mL per hour) a |
864 |
2197c |
2,439 |
2.54 |
1.11 |
|
aNumbers read from figure. bCalculated from figure. cData of the first two time points were excluded from the calculation. Note: If not indicated otherwise, male B6C3F1 mice were used. Ratios between doses, peak TCA concentrations, and AUC are indicated. i.g. = intragastric application. Further technical data on the studies is given in the text. |
|||||
TABLE 4B Blood TCA Concentrations after TCA Treatment
|
1) 1x i.g., 4-hour fast |
Larson and Bull 1992 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
|
|
Doses (mg/kg) |
|
20 |
100 |
5 |
|
|
Cmax (μg/mL) |
|
38 ± 1.65 |
130 ± 9.9 |
3.4 |
|
|
AUC0-24 (μg/mL per hour) |
|
333 ± 9.9 |
1185 ± 34.7 |
3.5 |
|
|
2) 1x i.g., 8-hour fast |
Templin et al. 1993 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
5 |
20 |
100 |
4 |
5 |
|
Peak concentrations (μg/mL) a |
10.1 |
40.3 |
80.6 |
4.0 |
2.0 |
|
AUC0-24 (μg/mL per hour) a |
87 |
374 |
934 |
4.3 |
2.5 |
|
3a) 1x i.v., 16-hour fast |
Gonzalez-Leon 1999 |
|
|||
|
Doses (mg/kg) |
|
|
100 |
|
|
|
Cmax (μg/mL) |
|
|
179 ± 30 |
|
|
|
AUC0-24 (μg/mL per hour) |
|
|
2,516 ± 289 |
|
|
|
3b) Pretreatment with TCA at 2 g/L for 14 days, then 1x i.v., 16-hour fast |
|||||
|
Doses (mg/kg) |
|
|
100 |
|
|
|
Cmax (μg/mL) |
|
|
214 ± 17 |
|
|
|
AUC0-24 (μg/mL per hour) |
|
|
2,964 ± 418 |
|
|
|
4) Drinking water, 14 days |
Mahle et al. 2001 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
11.6 |
110 |
268 |
9.5 |
2.4 |
|
Peak concentrations (μg/mL) |
10.3 |
72.9 |
79.9 |
7.1 |
1.1 |
|
5) Drinking water for 5 or 14 days Green 2003 (Data from Sweeney et al. 2009) |
Ratios |
||||
|
|
1 |
2 |
3 |
1 - 2 |
2 – 3 |
|
Doses (mg/kg), 5 days |
|
180 |
443 |
|
2.5 |
|
Peak concentrations (μg/mL) |
|
71.6 |
127 |
|
1.8 |
|
Doses (mg/kg), 14 days |
|
181 |
497 |
|
2.8 |
|
Peak concentrations (μg/mL) |
|
97.5 |
133 |
|
1.4 |
|
aNumbers read from figure. Note: For explanations, see Table 4A. |
|||||
TABLE 4C Liver TCA Concentrations after Treatment with Tetrachloroethylene
|
1) 1x, i.g.; SW mice |
Philip et al. 2007 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
150 |
500 |
1,000 |
3.33 |
2.0 |
|
Peak concentrations (μg/g) a |
53 |
100 |
175 |
1.89 |
1.75 |
|
AUC0-24 (μg/mL per hour) a |
956 |
1,690 |
3,233 |
1.77 |
1.91 |
|
1) 30x, daily i.g.; SW mice |
Philip et al. 2007 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
150 |
500 |
1,000 |
3.33 |
2.0 |
|
Peak concentrations (μg/g) a |
25 |
34 |
42 |
1.36 |
1.24 |
|
AUC0-24 (μg/mL per hour) a |
388 |
563 |
694 |
1.45 |
1.23 |
|
aNumbers read from figure. Note: For explanations, see Table 4A. |
|||||
TABLE 4D Liver TCA Concentrations after Treatment with TCA
|
2) 1x i.g., 8-hour fast |
Templin et al. 1993 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
5 |
20 |
100 |
4 |
5 |
|
Peak concentrations (μg/g) |
6.4 |
21.1 |
28.4 |
3.3 |
1.3 |
|
AUC0-24 (μg/mL per hour) |
55 |
199 |
386 |
3.6 |
1.9 |
|
4) Drinking water, 14 days |
Mahle et al. 2001 |
Ratios |
|||
|
|
1 |
2 |
3 |
1 - 2 |
2 - 3 |
|
Doses (mg/kg) |
11.6 |
110 |
268 |
9.5 |
2.4 |
|
Peak concentrations (μg/mL) |
6.2 |
48.2 |
61.6 |
7.77 |
1.28 |
|
Note: For explanations, see Table 4A. |
|||||
Gearhart et al. (1993) administered a single dose of tetrachloroethylene to male B6C3F1 mice by gavage in corn oil at of 0.1, 0.536, and 1.072 mg/kg. The two higher doses correspond to those used in the NCI oral-carcinogenicity study (Table 1). TCA reached peak blood concentrations of 23, 80, and 157 mg/l; these and the AUC are shown in Table 4A.
In a similar study of male Swiss Webster mice, Philip et al. (2007) applied tetrachloroethylene in aqueous gavage (with Emulphor) daily in three dosages (150, 500, and 1,000 mg/kg) for up to 30 days. Concentrations of tetrachloroethylene, TCA, DCA, and trichloroethanol were analyzed after one and 30 treatments. After the first treatment, peak blood TCA was similar with all three dosages. After 30 doses of tetrachloroethylene at 150 mg/kg, blood TCA ranged from 35 to 75 μg/mL in the 24-hour period, and after 500 and 1,000 mg/kg, from 50 to 135 μg/mL. Peak concentrations and the AUC are displayed in Table 4A. Peak hepatic TCA and AUC tended to be lower than the corresponding blood concentrations, particularly after 30 days of treatment (Table 4C).
Table 4 also shows ratios between different doses compared with ratios between the corresponding peak concentrations and AUC values. Although tissue concentrations in general increased with dose, the relative difference tended to decrease with increasing dose. That reflects the well-known fact that the metabolism of tetrachloroethylene is saturable (Buben and O`Flaherty 1985; Reitz et al. 1996).
Blood and Liver Trichloroacetic Acid Concentrations After Administration of Trichloroacetic Acid
TCA concentrations after administration of TCA in mice and rats have been measured in several studies. After a single oral dose of 20 or 100 mg/kg 14C-TCA in male B6C3F1 mice, TCA Cmax in blood were 38 and 130 μg/mL. Half-lifes (T1/2) were 4.2 and 5.8 hours; for AUC data, see Table 4B (Larson and Bull 1992). In male B6C3F1 mice treated orally with TCA at single doses of 5, 20, and 100 mg/kg, peak blood concentrations were 10.1, 40.3, and 80.6 μg/mL, respectively, and the half-life was 5.4-6.4 hours. Liver concentrations—6.4, 21.1, and 28.4 μg/g—were lower than blood concentrations; it was suggested that this result from plasma-protein binding of TCA. For AUC data, see Tables 4B and D. Liver:blood AUC ratios decreased with increasing dose (Templin et al. 1993).
When given intravenously to male B6C3F1 mice, a “challenge dose” of TCA at 100 mg/kg resulted in a blood Cmax of 179 μg/mL and t½ was 10.0 hours. Other mice received TCA for 14 days at 2 g/L in drinking water. The challenge dose was then administered 16 hours after removal of TCA from drinking water. No significant changes in various kinetic measures occurred: blood Cmax was 214 μg/mL, t½, 9.4 hours; metabolism of TCA in vitro was not altered. The authors concluded that pretreatment with TCA does not affect metabolism and pharmacokinetics of TCA (Gonzalez-Leon et al 1999).
In a similar study, male B6C3F1 mice received TCA at 0.08, 0.8, or 2.0 g/L in drinking water; this resulted in daily dose rates of 11.6, 110, and 268 mg/kg. After 14 days, blood TCA was 10.3, 72.9, and 79.9 μg/mL; they were almost identical after 3 days. Liver TCA at 14 days was 6.2, 48.2, and 61.6 μg/mL (Tables 4B and D) (Mahle et al. 2001).
Available studies of organ TCA concentrations used male mice except that Green et al. (cited from Sweeney et al. 2009) found even lower blood concentrations in female B6C3F1 mice exposed to TCA than in male mice.
In conclusion, blood and liver concentrations after TCA treatment in different studies are fairly consistent at similar doses. Liver concentrations were lower than blood concentrations. The data from Larson and Bull (1992), Templin et al. (1993), Mahle et al. (2001), and Green et al. from CTL (cited by Sweeney et al. 2009) concordantly demonstrate that peak blood and liver TCA concentrations and AUC do not increase linearly with dose. Rather, as shown by the ratios in Tables 4B and D, the increments decreased with increasing dose.
Above 100 mg/kg, little further increase in peak blood concentrations is apparent from the experimental data available. That result is graphically presented in Figure 1. Obviously, bioavailability of TCA administered orally does not increase linearly with dose in mice. Incomplete bioavailability of oral TCA is independently supported by the study of Gonzalez-Leon et al. (1999), in which TCA was administered intravenously at 100 mg/kg. Peak concentrations and AUC were about 2.5 times higher than the mean in studies that used oral administration (Table 4B). Obviously, a large portion of a 100-mg/kg dose of TCA administered orally is not systemically available. Elimination kinetics in blood after various doses of TCA were similar and repeated treatment with TCA (for 14 days) did not significantly modify its metabolism and kinetics (Templin et al. 1993; Mahle et al. 2001; Gonzalez-Leon et al. 1999), so a dose-dependent limit on absorption of TCA seems a likely explanation of the reduced bioavailability of oral TCA.
The fraction of TCA bioavailable after oral exposure was modeled by Sweeney et al. (2009) on the basis of blood-concentration data of Mahle et al. (2001) and Green et al. They concluded that the apparent bioavailability of TCA from drinking water is 25% at low doses (12 mg/kg) and declines to less than 10% at high doses (800 mg/kg).
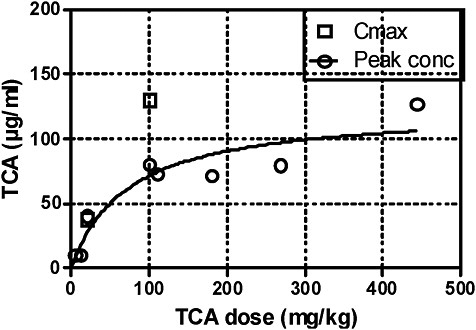
FIGURE 1 Peak TCA concentrations and Cmax in blood after oral administration of TCA. Source: Data from Table 4B.
Conclusions on the Role of Trichloroacetic Acid in Tetrachloroethylene-Induced Hepatocarcinogenesis
Carcinogenic Potency
The validity of the modeled carcinogenic-potency data on TCA in the IRIS draft (Appendix 4A) is questionable, see earlier section on Carcinogenicity studies with tetrachloroethylene and TCA.
Direct Comparison of Trichloroacetic Acid Concentrations in Blood or Target Organ
Tables 4A and B display the available blood TCA concentrations as determined analytically. Peak TCA concentrations and AUC are similar after application of tetrachloroethylene and TCA at carcinogenic doses or perhaps even higher after tetrachloroethylene than after TCA. That point is illustrated by Figures 2A-C. Likewise, the corresponding liver TCA concentrations are similar after both agents or even higher after tetrachloroethylene. That is convincing evidence that TCA can be formed from tetrachloroethylene and be present in blood and target organ in amounts sufficient to induce peroxisome proliferation and hepatocarcinogenesis.
Modeling the Internal Trichloroacetic Acid Dose
In the IRIS draft, a quantitative comparison between hepatic-carcinoma yields after tetrachloroethylene or TCA treatment and the corresponding internal TCA doses is attempted. Internal TCA after tetrachloroethylene was modeled according to Reitz et al. (1996). For modeling internal TCA after TCA treatment, an absorption rate of 95% was estimated (Section 4A1.2, p.4-205). No reference or reason for that estimate is provided, and no support was found in the literature. Clearly the IRIS estimate is not compatible with the available literature reviewed above, which demonstrates that TCA absorption after oral exposure is incomplete and decreases with increasing dose (Tables 4B and D, Figure 1). Moreover, modeling by Sweeney et al. (2009) suggests that in the 10 mg/kgdose range only 25% of oral TCA becomes bioavailable. Apparently, therefore, the internal TCA doses after TCA treatment that are calculated in the IRIS draft are too high. In consequence, tumor yields predicted to result from TCA formed after tetrachloroethylene (Table 4A-4of the IRIS draft) would be too low. Indeed, when they included their bioavailability data in the model, Sweeney et al. (2009) found that TCA in mice exposed to tetrachloroethylene is sufficient to explain the incidence of hepatic tumors. In conclusion, formation of TCA from tetrachloroethylene is probably sufficient to explain tumorigenesis in mouse liver. That adds substantially to the weight of evidence of a key role of
PPARα activation in mouse hepatocarcinogenesis by tetrachloroethylene via a metabolism-mediated pathway.
OTHER MODES OF ACTION
The operation of additional, non-PPARα-mediated mechanisms does not seem necessary to explain hepatocarcinogenesis by tetrachloroethylene but from a scientific point of view cannot be excluded. The question is whether evidence exists which supports a significant contribution of other MOAs to hepatocarcinogenesis.
Cytotoxicity
Tetrachloroethylene causes some hepatotoxicity in mice. It may be due to formation of reactive metabolites, including trichloroacetyl chloride, which have shown protein binding in rodents (Pähler et al. 1999; Green et al. 2001). However, hepatotoxicity has been found to disappear almost completely within 30 days (Philip et al. 2007), and the available long-term carcinogenicity studies revealed little evidence of hepatic damage or inflammation (NTP 1986; JISA 1993). Nevertheless, because the relation between cytotoxicity, inflammation, and cancer is not sufficiently understood, this point should receive attention in future studies. TCA also exerts little hepatotoxicity (Bull et al. 1990; DeAngelo et al. 1989). Overall, current evidence does not indicate that hepatotoxicity of tetrachloroethylene or TCA contributes to hepatocarcinogenesis to a substantial extent. Protein binding in humans was below the level of detection (Pähler et al. 1999).
Genotoxicity
Hypothetically, genotoxic activity could produce initiated hepatocytes, whose development to tumors might be promoted by TCA. Genotoxic activity could thereby enhance the carcinogenic potential of TCA. However, although some metabolites of tetrachloroethylene are genotoxic, there is no convincing evidence of genotoxic or mutagenic effects of tetrachloroethylene in vivo, and no initiating potential has been detected in appropriate assays (committee report, Chapter 5). Thus, a contribution of genotoxicity to hepatic-tumor formation by tetrachloroethylene is not supported by current evidence.
DCA as the Active Metabolite
As described in section on Relevance of TCA vs DCA, substantial contribution to PPARα-mediated tumor formation is unlikely. The potential MOAs of DCA include genotoxicity, but this activity is weak and probably not relevant at the low levels formed (IARC 2004).
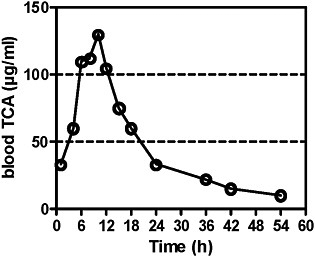
FIGURE 2A TCA concentrations in blood after single exposure of mice to tetrachloroethylene at 400 ppm for 6 hours. Source: Odum et al. 1988. Reprinted with permission; copyright 1988, Toxicology and Applied Pharmacology.
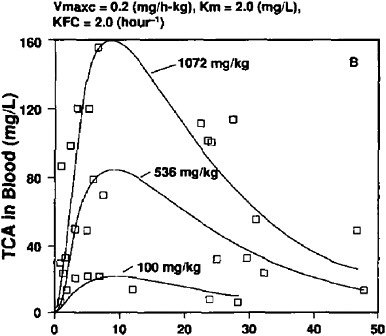
FIGURE 2B TCA concentrations in blood of male mice after single dose of tetrachloroethylene at 0.1, 0.536, and 1.072 mg/kg in corn oil by gavage. Experimental data shown as symbols; computer simulations shown as solid lines. Source: Gearhart et al. 1993. Reprinted with permission; copyright 1993, Toxicology Letters.
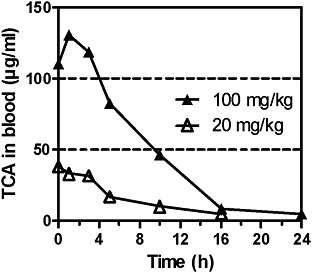
FIGURE 2C TCA concentrations in blood of male mice after single doses of TCA at 20 or 100 mg/kg by gavage. Data read from figure. Source: Larson and Bull 1992. Reprinted with permission; copyright 1992, Toxicology and Applied Pharmacology.
Other Mechanisms
Several other effects of tetrachloroethylene or TCA have been discussed as potential MOAs. Among these, changes in DNA methylation occur during PPARα activation. Therefore, that effect, although not specific for the PPARα MOA, does not necessarily support a contribution of other MOAs to hepatocarcinogenicity of tetrachloroethylene. TCA slightly transactivated mPPARβ, but this effect was much weaker than seen with PPARα and therefore is considered to have little or no relevance in mouse hepatocarcinogenesis. Importantly,TCA had no effect on human PPARβ (see section on PPARα Transactivation). In conclusion, there is no evidence available tosuggest that MOAs other than PPARα activation have asignificant impact on mouse hepatocarcinoma formation by tetrachloroethylene. Therefore, the weight of evidence supports the PPARα MOA.
SOME RECENT FINDINGS CONCERNING THE ROLE OF PPARα ACTIVATION IN MOUSE AND HUMAN HEPATOCARCINOGENESIS
The evidence suggesting that PPARα activation plays a causal role in rodent hepatic-tumor formation by many peroxisome proliferators but is not relevant for human hepatocarcinogenesis has been compiled in recent reviews (Klaunig et al. 2003; Meek et al. 2003; Peters et al. 2005; EU 2008; Corton 2008).
The authors of the IRIS draft present two recent publications that in their opinion raise questions about the causal relationship between activation of PPARα and rodent hepatic-tumor formation (p. 4-31). First, Yang et al. (2007) used transgenic mice (LAP-VP16PPARα) that target constitutively activated PPARα specifically at hepatocytes. The transgenic mice exhibited various PPARα-mediated effects—changes in fatty acid metabolism peroxisome proliferationand hepatocyte proliferation—but, surprisingly, not hepatic tumors after 1 year. Transgenic mice showed no hepatocyte hypertrophy and eosinophilia and no induction of proliferation of nonparenchymal liver cells. Those results indicate that PPARα-dependent induction of hepatocyte proliferation alone is not sufficient for hepatocarcinogenesis and that additional effects, such as activation of nonparenchymal cells, are required. Activation of Kupffer and other nonparenchymal cells had been found necessary for optimal induction of proliferation of normal and preneoplastic hepatocytes (Rose et al. 1997; Parzefall et al. 2001; Hasmall et al. 2001; Drucker et al. 2006). Thus, the study of Yang et al. does not refute the PPARα MOA but confirms and extends current knowledge.
Second, Ito et al. (2007) found that a low dose of DEHP (0.05% in diet) known to be noncarcinogenic in wild-type mice produced a low rate (26%) of hepatic adenomas in PPARα-null mice after 22 months. The tumors apparently were induced by oxidative stress and inflammation, as indicated by histopathologic changes and increases in 8-OHdG, NF-ĸB, and c-jun RNA, all of which were particularly high in the null mice. Activation of PPARα can have anti-inflammatory effects, resulting in higher vulnerability to tumorigenesis in PPARα-null mice (Ito et al. 2007). 8-OHdG was not increased after tetrachloroethylene or TCA (see earlier section on TCA as a Peroxisome Proliferator and Hepatocarcinogen in Mice). Thus, the results of Ito et al. suggest that DEHP, an agent unrelated to tetrachloroethylene, can induce (benign) hepatic tumors through a second, previously unsuspected PPARα-independent pathway. They do not contradict the causal role of PPARα activation in many instances of rodent hepatocarcinogenesis induced by peroxisome proliferators, which is supported by overwhelming evidence.
Some important new findings are missing in the IRIS draft. Thus, the generation of transgenic mice in which the mouse PPARα is replaced by the human counterpart provided substantial progress. The hPPARα mice were essentially resistant to hepatocarcinogenesis when fed a potent peroxisome proliferator (WY-14643) for 44 weeks, whereas corresponding wild-type mice developed tumors in 38 weeks. Gene-expression analysis for peroxisomal fatty-acidmetabolizing enzymes revealed that both receptors were functional. The findings suggest that structural differences between human and mouse PPARα are responsible for the different susceptibility of mice and humans to hepatocarcinogenesis by peroxisome proliferators (Morimura et al. 2006).
Furthermore, it was shown that induction of hepatocellular proliferation by peroxisome proliferators involves downregulation of the microRNA let-7c gene by mPPARα. That in turn allows increased expression of c-myc protein, which is essential for hepatocyte proliferation and tumor formation. Human PPARα
apparently cannot suppress let 7c expression, and c-myc was not increased in hPPARalpha mice after WY-14643 treatment (Shah et al. 2007; Gonzalez and Shah 2008). Overall, the findings provide mechanism-based support for the concept that the PPARα MOA of rodent-hepatocarcinoma induction is not relevant to human hepatocarcinogenesis.
SUMMARY
This dissent has critically reviewed evidence related to MOAs of mouse hepatocarcinogenesis after exposure to tetrachloroethylene. The following conclusions can be drawn from findings in the literature:
-
TCA is the major metabolite in the body after exposure to tetrachloroethylene. DCA concentrations in blood and liver were lower than those of TCA by an order of magnitude, or DCA was completely undetectable.
-
TCA transactivates PPARα, while tetrachloroethylene does not. DCA also activates PPARα, but, because of its low occurrence, arguments related to the PPARα MOA should focus on TCA as the dominant active metabolite.
-
Effects of tetrachloroethylene and TCA associated with peroxisome proliferation were compiled and evaluated for consistency with the PPARα MOA as suggested by Klaunig et al. (2003). TCA induces the three key causal events, as well as peroxisome proliferation, and other associatedkey events. Data were generated in several studies, and dose-response and temporal relationships are consistent with the observation of tumors. The weight of evidence of this MOA was considered strong for TCA (in agreement with the National Research Council trichloroethylene committee) and probable for tetrachloroethylene although studies of PPARα-null mice are not available. Major support of the PPARα MOA of tetrachloroethylene rests on the role of TCA as the active metabolite.
-
Rats are less sensitive than mice to PPARα-mediated effects of tetrachloroethylene and do not develop hepatocarcinoma in response to tetrachloroethylene or TCA. That species difference can be explained by kinetic differences in TCA formation and availability in the target organ. In mice, formation of TCA is much higher and binding to plasma proteins much lower than in rats. Therefore, the mouse-rat difference can be explained by assuming that TCA is the active metabolite of tetrachloroethylene.
-
A key question is whether sufficient TCA is produced from tetrachloroethylene to induce peroxisome proliferation and tumor formation in the liver. To address that question, analytic data on blood and liver concentrations of TCA were collected from the literature. The data revealed that peak and AUC levels of TCA in mouse blood after tetrachloroethylene were similar to or even higher than those after TCA when carcinogenic doses of the two agents were compared. That constitutes direct evidence that TCA can be generated from tetra-
-
chloroethylene and be present in blood and target organ in amounts sufficient to induce peroxisome proliferation and hepatocarcinogenesis.
-
Analytic data from all of five available studies consistently demonstrate that absorption of TCA after oral application is incomplete and decreases with increasing dose. Moreover, published modeling work based on some of those studies suggests that only 25-10% of oral TCA bioavailable. The analytic and modeling data are not compatible with the estimate in the IRIS draft that 95% of oral TCA is absorbed—an estimate apparently not founded on experimental data. Apparently, the internal TCA doses derived from that estimate are too high. Consequently, the tumor yields predicted for tetrachloroethylene-derived TCA would be too low. Indeed, modeling studies taking into account the limited bioavailability of TCA suggest that TCA generated from tetrachloroethylene is sufficient to explain the incidence of hepatic tumors.
In conclusion, the weight of evidence clearly favors a key role of PPARα activation by TCA in tetrachloroethylene-induced mouse hepatocarcinogenesis.
-
The available evidence does not support a substantial contribution of other MOAs to hepatocarcinogenesis by tetrachloroethylene.
-
Transgenic mice carrying the human PPARα gene were found to be essentially resistant to hepatocarcinogenesis by a model peroxisome proliferator. This and other recent molecular data provide mechanism-based support for the concept that the PPARα MOA lacks relevance to human hepatocarcinogenesis.
COMMITTEE REBUTTAL
The committee greatly appreciates the dissenting member’s thoughtful and careful review of the scientific literature and presentation of the arguments with respect to the MOA of tetrachloroethylene in mouse hepatic tumors and its relevance to humans. As noted by the dissenter and in Chapter 6 of the committee’s report, the committee agrees that the EPA MOA characterization for hepatic cancer is inadequate and should be revised to provide a more focused and integrated analysis of the available evidence on tetrachloroethylene and its metabolites. The dissenter’s statement is an attempt to provide an example of how such an analysis might be performed. The committee supports much of the dissenter’s approach, but the dissenting member’s conclusions go beyond those drawn by the full committee.
The dissenting member holds the opinion that PPARα mediation of tetrachloroethylene-induced hepatocarcinogenesis in mice is the plausible predominant MOA and that this MOA lacks relevance to human hepatocarcinogenesis. The committee believes that the arguments presented are reasonable and advises EPA to review the considerations presented by the member and the recent literature cited carefully. However, the committee does not support the apparent conclusions regarding mouse hepatic cancer that TCA is the sole carcinogenic metabolite of tetrachloroethylene, that the only MOA of TCA is peroxisome proliferation, and that there is unmistakable concordance in the carcinogenic
potency of tetrachloroethylene in the National Toxicology Program and Japan Industrial Safety Association bioassays and the corresponding studies of TCA. Overall, the committee judges that many gaps in knowledge remain with regard to the MOA of tetrachloroethylene and that the relevance of the peroxisomeproliferator MOA to tetrachloroethylene-induced mouse hepatic cancer and to tetrachloroethylene-induced human hepatic cancer remains hypothetical and requires further rigorous testing.
The committee generally supports the comprehensive literature review and analyses conducted by the dissenting member and recommends that EPA use them when reassessing its own evaluation. However, there are aspects of the dissenter’s analysis that the committee believes require more rigorous assessments before definitive conclusions can be drawn. They include the following:
-
The committee does not agree that a role of DCA in tetrachloroethylene-induced hepatic carcinogenesis in mice can be ruled out solely on the grounds that it is detected at much lower concentrations than TCA in the blood and liver. First, there are few data on DCA formation from tetrachloroethylene. Second, there is some evidence that DCA is formed via a metabolic pathway that does not involve the liver. Third, there is some debate on whether DCA is formed from TCA. In Chapter 6, the committee stated that the conclusions regarding potential relevance or lack of relevance of DCA to hepatic carcinogenesis by tetrachloroethylene would be strengthened by the comparison of tetrachloroethylene hepatocellular-tumor data with predictions based on DCA carcinogenesis studies (in a way similar to that presented in Appendix 4A of the draft IRIS assessment). Such an analysis would provide a strong quantitative rationale for DCA’s potential involvement, or lack thereof, in hepatic cancer.
-
A more critical look at the quantitative differences in metabolic activation of tetrachloroethylene to TCA between mouse and rat, species that are generally believed to be almost equally sensitive to peroxisome proliferation, and in induction of hepatic cancer by other compounds in this class should be conducted by EPA. Chapter 6 recommends that EPA consider performing additional analyses with the rat data similar to those done with the mouse in Appendix 4A of the draft and including a table that shows the quantitative differences in affinity to mouse, rat, and human PPARα of both tetrachloroethylene and its key metabolites in comparison with the known peroxisome proliferators. Such analyses and data would greatly facilitate the discussion of quantitative differences between compounds and species.
-
The committee supports the use of the weight-of-evidence analysis and the need for evaluation of the key events in hepatic carcinogenesis by tetrachloroethylene and its key metabolites. However, important knowledge gaps remain to be addressed with regard to key events in the PPARα MOA, especially those with causal and associative relationship to tumor formation and tetrachloroethylene or its key metabolites (see dissenter’s statement and Chapter 6). Indeed, the committee is not yet convinced of the proof of the hypothesis that
-
the PPARα MOA is the sole MOA of tetrachloroethylene in inducing mouse hepatic cancer. Hence, it is premature to draw conclusions on the relevance of the PPARα MOA to tetrachloroethylene-induced human hepatic carcinogenesis.
-
The committee agrees that the issues of TCA bioavailability, absorption, and blood and liver concentrations in various exposure scenarios are critical for the consideration of MOA of tetrachloroethylene. The current analysis by EPA is important but is inadequate in its current form. The committee recommends that EPA reconsider the analyses performed and consider using the data suggested by the dissenting member.
-
The committee disagrees with the dissenter that the available evidence is sufficient to conclude that other MOAs are unlikely to contribute substantially to hepatocarcinogenesis by tetrachloroethylene. As noted in Chapter 6, the committee recommends that EPA strengthen and clarify the description of the degree, rather than the “significance,” of the contribution of other plausible molecular events, in addition to activation of PPARα, to mouse hepatic tumors produced by tetrachloroethylene.
-
The committee agrees with the dissenter that recent findings reported with PPARα-null mice (Ito et al. 2007; Takashima et al. 2008; Eveillard et al. 2009), PPARα humanized transgenic mice (Morimura et al. 2006), and hepatocyte-specific constitutively activated PPARα transgenic mice (Yang et al. 2007) are valuable contributions to the discussion of the relevance of the PPARα MOA in human hepatic carcinogenesis. The dissenter cites those studies to draw a conclusion that the PPARα MOA lacks relevance to human hepatocarcinogenesis. However, alternative conclusions that can be drawn from the studies mentioned above are that the short-term carcinogenesis studies in the PPARα-null mouse model have important limitations, that activation of PPARα is necessary but not sufficient for the development of mouse hepatic tumors, and that additional molecular events may be important parts of the peroxisome-proliferator MOA. Thus, the committee believes that it is premature to draw definitive conclusions regarding the relevance of the PPARα MOA to human hepatocarcinogenesis. In Chapter 6, the committee has encouraged EPA to strengthen the discussion of this matter in the draft IRIS assessment.
REFERENCES
Austin, E.W., J.M. Perrish, D.H. Kinder, and R.J. Bull. 1996. Lipid peroxidation and formation of 8-hydroxydeoxyguanosine from acute doses of halogenated acetic acids. Fundam. Appl. Toxicol. 31(1):77-82.
Benane, S.G., C.F. Blackman, and D.E. House. 1996. Effect of perchloroethylene and its metabolites on intercellular communication in clone 9 rat liver cells. J. Toxicol. Environ. Health 48(5):427-437.
Buben, J.A., and E.J. O’Flaherty. 1985. Delineation of the role of metabolism in the hepatotoxicity of trichloroethylene and perchloroethylene: A dose-effect study. Toxicol. Appl. Pharmacol. 78(1):105-122.
Bull, R.J. 2000. Mode of action of liver tumor induction by trichloroethylene and its me-
tabolites, trichloroacetate and dichloroacetate. Environ. Health Perspect. 108(Suppl 2): 241-259.
Bull, R.J., I.M. Sanchez, M.A. Nelson, J.L. Larson, and A.J. Lansing. 1990. Liver tumor induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology 63(3):341-359.
Bull, R.J., G.A. Orner, R.S. Cheng, L. Stillwell, A.J. Stauber, L.B. Sasser, M.K. Lingohr, and B.D. Thrall. 2002. Contribution of dichloroacetate and trichloroacetate to liver tumor induction in mice by trichloroethylene. Toxicol. Appl. Pharmacol. 182(1):55-65.
Bull, R.J., L.B. Sasser, and X.C. Lei. 2004. Interactions in the tumor-promoting activity of carbon tetrachloride, trichloroacetate, and dichloroacetate in the liver of male B6C3F1 mice. Toxicology 199(2-3):169-183.
Corton, J.C. 2008. Evaluation of the role of peroxisome proliferator-activated receptor α (PPARα) in mouse liver tumor induction by tetrachloroethylene. Crit. Rev. Toxicol. 38(10):857-875.
DeAngelo, A.B., F.B. Daniel, L. McMillan, P. Wernsing, and R.E. Savage, Jr. 1989. Species and strain sensitivity to the induction of peroxisome proliferation by chloroacetic acids. Toxicol. Appl. Pharmacol. 101(2):285-298.
DeAngelo, A.B., F.B. Daniel, B.M. Most, and G.R. Olson. 1997. Failure of monochloroacetic acid and trichloroacetic acid administered in the drinking water to produce liver cancer in male F344/N rats. J. Toxicol. Environ. Health 52(5):425445.
DeAngelo, A.B., F.B. Daniel, D.M. Wong, and M.H. George. 2008. The induction of hepatocellular neoplasia by trichloroacetic acid administered in the drinking water of the male B6C3F1 mouse. J. Toxicol. Environ. Health A 71(16):1056-1068.
Dees, C., and C. Travis. 1994. Trichloroacetate stimulation of liver DNA synthesis in male and female mice. Toxicol. Lett. 70(3):343-355.
Drucker, C., W. Parzefall, O. Teufelhofer, M. Grusch, A. Ellinger, R. Schulte-Hermann, and B. Grasl-Kraupp. 2006. Non-parenchymal liver cells support the growth advantage in the first stages of hepatocarcinogenesis. Carcinogenesis 27(1):152-161.
EPA (U.S. Environmental Protection Agency). 2008. Toxicological Review of Tetrachloroethylene (Perchloroethylene) (CAS No. 127-18-4) in Support of Summary Information on the Integrated Risk Information System (IRIS). External Review Draft. U.S. Environmental Protection Agency, Washington, DC.
EU (European Union). 2008. Risk Assessment Report on Tetrachloroethylene (PERC). CAS No. 127-18-4. EINECS No. 204-825-9. Draft Human Health Report. R021_0712_hh_SCHER. January 2008 [online]. Available: http://ecb.jrc.ec.europa.eu/documents/Existing-Chemicals/RISK_ASSESSMENT/DRAFT/R021_0712_env_hh.pdf [accessed Oct. 21, 2009].
Eveillard, A., L. Mselli-Lakhal, A. MOgha, F. Lasserre, A. Polizzi, J.M. Pascussi, H. Guillou, P.G. Martin, and T. Pineau. 2009. Di-(2-ethylhexyl)-phthalate (DEHP) activates the constitutive androstane receptor (CAR): a novel signalling pathway sensitive to phthalates. Biochem. Pharmacol. 77(11):1735-1746.
Ferreira-Gonzalez, A., A.B. DeAngelo, S. Nasim, and C.T. Garrett. 1995. Ras oncogene activation during hepatocarcinogenesis in B6C3F1 male mice by dichloroacetic and trichloroacetic acids. Carcinogenesis 16(3):495-500.
Gearhart, J.M., D.A. Mahle, R.J. Greene, C.S. Seckel, C.D. Flemming, J.W. Fisher, and H.J. Clewell, III. 1993. Variability of physiologically based pharmacokinetic (PBPK) model parameters and their effects on PBPK model predictions in a risk assessment for perchloroethylene (PCE). Toxicol. Lett. 68(1-2):131-144.
Goldsworthy, T.L., and J.A. Popp. 1987. Chlorinated hydrocarbon-induced peroxisomal enzyme activity in relation to species and organ carcinogenicity. Toxicol. Appl. Pharmacol. 88(2):225-233.
Gonzalez, F.J., and Y.M. Shah. 2008. PPARalpha: Mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 246(1):2-8.
Gonzalez-Leon, A., J.L. Merdink, R.J. Bull, and I.R. Schultz. 1999. Effect of pretreatment with dichloroacetic or trichloroacetic acid in drinking water on the pharmacokinetics of a subsequent challenge dose in B6C3F1 mice. Chem. Biol. Interact. 123(3):239-253.
Green, S.M., M.F. Khan, B.S. Kaphalia, and G.A. Ansari. 2001. Immunohistochemical localization of trichloroacylated protein adducts in tetrachloroethene-treated mice. J. Toxicol. Environ. Health A 63(2):145-157.
Green, T. 2003. The Concentrations of Trichloroacetic Acid in Blood Following Administration of Trichloroacetic Acid in Drinking Water. Central Toxicology Laboratory, Alderley Park Macclesfield, Cheshire, UK (as cited in Sweeney et al. 2009).
Hasmall, S., N. James, K. Hedley, K. Olsen, and S. Roberts. 2001. Mouse hepatocyte response to peroxisome proliferators: Dependency on hepatic nonparenchymal cells and peroxisome proliferator activated receptor alpha (PPARalpha). Arch. Toxicol. 75(6):357-361.
Herren-Freund, S.L., M.A. Pereira, M.D. Khoury, and G. Olson. 1987. The carcinogenicity of trichloroethylene and its metabolites, trichloroacetic acid and dichloroacetic acid, in mouse liver. Toxicol. Appl. Pharmacol. 90(2):183-189.
IARC (International Agency for Research on Cancer). 2004. Some Drinking-Water Disinfectants and Contaminants, Including Arsenic. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 84. Lyon: IARC.
Ito, Y., O. Yamanoshita, N. Asaeda, Y. Tagawa, C.H. Lee, T. Aoyama, G. Ichihara, K. Furuhashi, M. Kamijima, F.J. Gonzalez, and T. Nakajima. 2007. Di(2-ethylhexyl) phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor alpha-independent pathway. J. Occup. Health 49(3):172-182.
JISA (Japanese Industrial Safety Association). 1993. Carcinogenicity Study of Tetrachloroethylene by Inhalation in Rats and Mice. Data No. 3-1. Kanagawa, Japan: Japanese Industrial Safety Association.
Klaunig, J.E., M.A. Babich, K.P. Baetcke, J.C. Cook, J.C. Corton, R.M. David, J.G. DeLuca, D.Y. Lai, R.H. McKee, J.M. Peters, R.A. Roberts, and P.A. Fenner-Crisp. 2003. PPARalpha agonist-induced rodent tumors: Modes of action and human relevance. Crit. Rev. Toxicol. 33(6):655-780.
Larson, J.L., and R.J. Bull. 1992. Metabolism and lipoperoxidative activity of trichloroacetate and dichloroacetate in rats and mice. Toxicol. Appl. Pharmacol. 115(2):268-277.
Laughter, A.R., C.S. Dunn, C.L. Swanson, P. Howroyd, R.C. Cattley, and J.C. Corton. 2004. Role of the peroxisome proliferator-activated receptor alpha (PPARalpha) in responses to trichloroethylene and metabolites, trichloroacetate and dichloroacetate in mouse liver. Toxicology 203(1-3):83-98.
Lumpkin, M.H., J.V. Bruckner, J.L. Campbell, C.E. Dallas, C.A. White, and J.W. Fisher. 2003. Plasma binding of trichloroacetic acid in mice, rats, and humans under cancer bioassay and environmental exposure conditions. Drug Metab. Dispos. 31(10): 1203-1207.
Mahle, D.A., R.J. Godfrey, G.W. Buttler, L. Narayanan, J.W. Fischer, and A. Taylor. 2001. Pharmacokinetics and Metabolism of Dichloroacetic Acid and Trichloroacetic Acid Administered in Drinking Water in Rats and Mice. AFRL-HE-WP-TR-
2001-0059. United States Air Force Research Laboratory, Wright Patterson Air Force Base, OH.
Maloney, E.K., and D.J. Waxman. 1999. trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol. Appl. Pharmacol. 161(2):209-218.
Maronpot, R.R., T. Fox, D.E. Malarkey, and T.L. Goldsworthy. 1995. Mutations in the ras proto-oncogene: Clues to etiology and molecular pathogenesis of mouse liver tumors. Toxicology 101(3):125-156.
Meek, M.E., J.R. Bucher, S.M. Cohen, V. Dellarco, R.N. Hill, L.D. Lehman-McKeeman, D.G. Longfellow, T. Pastoor, J. Seed, and D.E. Patton. 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Crit. Rev. Toxicol. 33(6):591-653.
Morimura, K., C. Cheung, J.M. Ward, J.K. Reddy, and F.J. Gonzalez. 2006. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor alpha to Wy-14,643-induced liver tumorigenesis. Carcinogenesis 27(5):1074-1080.
NCI (National Cancer Institute). 1977. Bioassay of Tetrachloroethylene for Possible Carcinogenicity. NCI-CG-TR-13. DHEW(NIH)77-813. U.S. Department of Health, Education, and Welfare, Public Health Service, National Institute of Health, Bethesda, MD [online]. Available: http://ntp.niehs.nih.gov/ntp/htdocs/LT_rpts/tr013.pdf [accessed Oct. 14, 2009].
NRC (National Research Council). 2006. Assessing the Human Health Risks of Trichloroethylene. Washington, DC: The National Academies Press.
NTP (National Toxicology Program). 1986. Toxicology and Carcinogenesis Studies of Tetrachloroethylene (Perchloroethylene) CAS No. 127-18-4) in F344/N Rats and B6C3F1 Mice (Inhalation Studies). NTP TR 311. NIH Publication No. 86-2567. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, Research Triangle Park, NC.
Odum, J., T., Green, J.R. Foster, and P.M. Hext. 1988. The role of trichloroacetic acid and peroxisome proliferation in the differences in carcinogenicity of perchloroethylene in the mouse and rat. Toxicol. Appl. Pharmacol. 92(1):103-112.
Pahler, A., J. Parker, and W. Dekant. 1999. Dose-dependent protein adduct formation in kidney, liver, and blood of rats and in human blood after perchloroethene inhalation. Toxicol. Sci. 48(1):5-13.
Parrish, J.M., E.W. Austin, D.K. Stevens, D.H. Kinder, and R.J. Bull. 1996. Haloacetateinduced oxidative damage to DNA in the liver of male B6C3F1 mice. Toxicology 100(1-3):103-111.
Parzefall, W., W. Berger, E. Kainzbauer, O. Teufelhofer, R. Schulte-Hermann, and R.G. Thurman. 2001. Peroxisome proliferators do not increase DNA synthesis in purified rat hepatocytes. Carcinogenesis 22(3):519-523.
Pereira, M.A. 1996. Carcinogenic activity of dichloroacetic acid and trichloroacetic acid in the liver of female B6C3F1 mice. Fundam. Appl. Toxicol. 31(2):192-199.
Pereira, M.A., and J.B. Phelps. 1996. Promotion by dichloroacetic acid and trichloroacetic acid of N-methyl-N-nitrosourea-initiated cancer in the liver of female B6C3F1 mice. Cancer Lett. 102(1-2):133-141.
Peters, J.M., C. Cheung, and F.J. Gonzalez. 2005. Peroxisome proliferator-activated receptor-alpha and liver cancer: Where do we stand? J. Mol. Med. 83(10):774-785.
Philip, B.K., M.M. Mumtaz, J.R. Latendresse, and H.M. Mehendale. 2007. Impact of repeated exposure on toxicity of perchloroethylene in Swiss Webster mice. Toxicology 232(1-2):1-14.
Reitz, R.H., M.L. Gargas, A.L. Mendrala, and A.M. Schumann. 1996. In vivo and in vitro studies of perchloroethylene metabolism for physiologically based pharmacokinetic modeling in rats, mice, and humans. Toxicol. Appl. Pharmacol. 136(2):289306.
Rose, M.L., D.R. Germolec, R. Schoonhoven, and R.G. Thurman. 1997. Kupffer cells are causally responsible for the mitogenic effect of peroxisome proliferators. Carcinogenesis 18(8):1453-1456.
Schumann, A.M., J.F. Quast, and P.G. Watanabe. 1980. The pharmacokinetics and macromolecular interactions of perchloroethylene in mice and rats as related to oncogenicity. Toxicol. Appl. Pharmacol. 55(2):207-219.
Shah, Y.M., K. Morimura, Q. Yang, T. Tanabe, M. Takagi, and F.J. Gonzalez. 2007. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol. Cell Biol. 27(12):4238-4247.
Stauber, A.J., and R.J. Bull. 1997. Differences in phenotype and cell replicative behavior of hepatic tumors induced by dichloroacetate (DCA) and trichloroacetate (TCA). Toxicol. Appl. Pharmacol. 144(2):235-246.
Stauber, A.J., R.J. Bull, and B.D. Thrall. 1998. Dichloroacetate and trichloroacetate promote clonal expansion of anchorage-independent hepatocytes in vivo and in vitro. Toxicol. Appl. Pharmacol. 150(2):287-294.
Sweeney, L.M., C.R. Kirman, M.L. Gargas, and P.H. Dugard. 2009. Contribution of trichloroacetic acid to liver tumors observed in perchloroethylene (perc)-exposed mice. Toxicology 260(1-3):77-83.
Takashima, K., Y. Ito, F.J. Gonzalez, and T. Nakajima. 2008. Different mechanisms of DEHP-induced hepatocellular adenoma tumorigenesis in wild-type and Ppar alpha-null mice. J. Occup. Health 50(2):169-180.
Templin, M.V., J.C. Parker, and R.J. Bull. 1993. Relative formation of dichloroacetate and trichloroacetate from trichloroethylene in male B6C3F1 mice. Toxicol. Appl. Pharmacol. 123(1):1-8.
Toraason, M., J. Clark, D. Dankovic, P. Mathias, S. Skaggs, C. Walker, and D. Werren. 1999. Oxidative stress and DNA damage in Fischer rats following acute exposure to trichloroethylene or perchloroethylene. Toxicology 138(1):43-53.
Walgren, J.E., D.T. Kurtz, and J.M. McMillan. 2000. Expression of PPAR(alpha) in human hepatocytes and activation by trichloroacetate and dichloroacetate. Res. Commun. Mol. Pathol. Pharmacol. 108(1-2):116-132.
Yang, Q., S. Ito, and F.J. Gonzalez. 2007. Hepatocyte-restricted constitutive activation of PPAR alpha induces hepatoproliferation but not hepatocarcinogenesis. Carcinogenesis 28(6):1171-1177.
Zhou, Y.C., and D.J. Waxman. 1998. Activation of peroxisome proliferator-activated receptors by chlorinated hydrocarbons and endogenous steroids. Environ. Health Perspect. 106 (Suppl. 4):983-988.



























