
2
The Science of Developing Cancer Therapy
A revolution is under way in the development of cancer therapy. Over the past decade, personalized medicine has leveraged scientific advancements in fields such as genomics,1 proteomics,2 molecular biology,3 and metabolomics4 to improve the extent to which medical care is tailored to the individual patient and his or her cancer. This is because most cancer treatments available today are effective in only a minority of patients, in part due to the tremendous variability in the molecular abnormalities that drive tumor formation (IOM, 2007; PCAST, 2008; Spear et al., 2001). As a result, many patients undergo costly treatments and endure the side effects of those treatments without deriving any benefit. Patients also experience an opportunity cost, as an alternative treatment that might be more effective for a patient’s particular disease is delayed or forgone. Better tools are therefore needed to reduce the time and costs wasted by delivering ineffective and toxic treatments. Future treatment decisions will depend on the use of tissue biomarkers5 that can predict outcomes of therapy. By using these
new methods, it will be increasingly possible to group individual cancers into subpopulations with similar characteristics to predict patient outcomes for cancer therapies. That will help to ensure that the treatments prescribed for patients will be more effective.
Rising health care costs, the increasing availability of new therapies, and the promise of delivering more effective care make it more important than ever to advance the science underlying personalized medicine (PCAST, 2008). Identifying those subpopulations that are likely to respond to therapies can improve and hasten the success rate for the development of new treatments (PCAST, 2008). Being able to predict the therapeutic response and, therefore, being able to deliver safer and more effective treatments to patients will reduce the number of adverse drug events and thus provide cost savings to the entire health care system.
Advances in personalized medicine are rooted in the discovery, validation, and qualification of biomarkers that can be measured by in vitro diagnostic tests on samples from patients or through in vivo biomedical imaging. For example, cancer biomarkers can be used to develop and deliver improved patient care by predicting the likelihood of the response to treatment or the likelihood that an adverse reaction to the treatment will develop (IOM, 2007). Examples of biomarkers routinely used in the treatment of cancer are shown in Table 2-1.
Most diagnostics that are in use today assess a single target; however, it is widely believed that as technologies in genomics, proteomics, metabolomics, and molecular profiling mature, diagnostic platforms capable of simultaneously examining a large number of potential markers will improve the predictive powers of these tests (IOM, 2007). For example, the cost of DNA sequencing is continually decreasing with advances in sequencing technologies, making it more feasible to identify the gene defects underlying a particular type of cancer. These technological advancements could dramatically change how cancer is diagnosed and treated (Niederhuber, 2009). For instance, by applying new sequencing technologies to genome analysis,
TABLE 2-1 Examples of Validated Biomarkers Routinely Used to Predict Response to Cancer Therapy
|
Therapeutic Agent |
Biomarker |
Cancer Type |
|
Endocrine therapies (e.g., tamoxifen) |
Estrogen receptor |
Breast |
|
Trastuzumab |
HER-2 |
Breast |
|
Imatinib mesylate |
BCR-ALB |
Leukemia |
|
Cetuximab and panitumumab |
KRAS |
Colorectal |
|
Irinotecan |
UGT1A1 |
Colorectal |
including large-scale genome sequencing, the Cancer Genome Atlas Project6 aims to catalog the pertinent genetic changes that occur in many types of cancer and identify new potential therapeutic targets.
The technologies used to conduct molecular analyses of tissue embedded in paraffin have also improved dramatically, enabling the large-scale genomic profiling of messenger RNA, the DNA copy number, and focused analysis of mutations on small tissue samples. For example, a recent study demonstrated the feasibility of profiling the expression of all the genes in the entire genome using formalin-fixed, paraffin-embedded tissues. Expression of more than 6,000 genes was assessed in tissues from patients with hepatocellular carcinoma, and 90 percent of the samples tested (including some that had been archived for many years) yielded data of high quality (Hoshida et al., 2008).
THE ROLE OF COOPERATIVE GROUPS IN BIOMARKER DEVELOPMENT
The NCI Clinical Trials Cooperative Groups have a long history of collecting highly annotated specimens7 from patients with many different forms of cancer in clinical trials, including all the major pediatric and adult cancers, and have in place effective systems for collecting, storing, and tracking specimens to conduct correlative science.8 Each of the 10 Cooperative Groups has its own repository for biological specimens. These are generally located in group-affiliated academic medical or cancer centers. All the Cooperative Groups’ biorepositories use standard operating procedures, but because these repositories were developed to fit the needs of the individual Cooperative Groups, the structure, methods, governance, and access policies differ among the Groups. The Group Banking Steering Committee, which includes representatives from each of the 10 Cooperative Groups and NCI, aims to address many of these issues by improving and harmonizing operations of the Cooperative Group biobanks and by coordinating banking activities among Groups conducting phase III and large phase II clinical trials. Five subcommittees are set up to focus on specific issues (Best Practices and Operations, Informatics, Access and Marketing, Regulatory, and Technology Development). Most of the biological specimens collected from Cooperative Group trials are formalin-fixed, paraffin-
embedded tumor tissue, but some frozen tissue and body fluid specimens are also collected (Hamilton, 2009).
Because the Cooperative Groups collect specimens under the auspices of a clinical trial, one of their major strengths is that the pathological data and the clinical data for the specimens are linked, making it feasible to conduct retrospective analyses of the clinical and nonclinical factors that influence prognosis and survival. In addition, information on long-term clinical outcomes is often available (Schilsky et al., 2002).
Historically, much of the correlative science performed by the Cooperative Groups focused on the development of prognostic markers (i.e., biomarkers that can predict the progression of disease in the absence of treatment considerations). In recent years, greater emphasis has been placed on identifying predictive markers (i.e., markers that can identify populations that are likely to be sensitive or resistant to specific treatments). Some Cooperative Groups have conducted clinical trials designed to validate the clinical utility of a biomarker, although one (the Marker Validation for Erlotinib in Lung Cancer [MARVEL] trial of lung cancer) was recently discontinued because of a lack of accrual.9 Lastly, some groups have ongoing efforts in pharmacogenetics to correlate variations in germline DNA with treatment-related toxicity.10 From 2000 to 2008, that work has resulted in 1,350 publications and 36 patents (Hamilton, 2009).
The work of the Cooperative Groups has also been instrumental in developing some of the cancer biomarker tests in common use in the clinic (see also Chapter 1). For example, the Cancer and Leukemia Group B (CALGB) Leukemia Correlative Science Committee has a long history (25 years) of conducting key correlative studies of adult leukemia. That group initially focused on the use of immunophenotyping for the diagnosis and prognosis of acute leukemia but has more recently focused on the clinical use of cytogenetic and molecular genetic markers in acute and chronic forms of leukemia. The work of that group has had a major impact on the way clinicians currently diagnose leukemia in adults, predict the outcome, select the appropriate treatment, document complete remission, and monitor residual disease (Bloomfield et al., 2006).
Another example of a widely used biomarker test resulting from the efforts of a Cooperative Group is the Oncotype DX breast cancer assay. About half of newly diagnosed cases of breast cancer are estrogen receptor (ER) positive and lymph node negative, and approximately 75 percent of those cases are adequately treated with surgery and hormonal therapy with
or without radiation. Although additional chemotherapy benefits less than 5 percent of patients with ER positive lymph node negative breast cancers, chemotherapy adds significant toxicity, so a predictive biomarker test to guide the treatment decision has long been sought. The development of the Oncotype DX breast cancer assay, which measures the expression of 21 genes to predict the likelihood of disease recurrence for women with ER-positive and lymph node-negative breast cancer, would not have been possible without the work of the National Surgical Adjuvant Breast and Bowel Project (NSABP) Cooperative Group. NSABP collected and preserved tissue samples collected in the 1990s to test the benefits of treating breast cancer with chemotherapy. Subsequently, those samples were used for the retrospective clinical validation of the Oncotype DX assay (Wickerham et al., 2008). The results of those retrospective studies showed that the Oncotype DX assay could predict the likelihood of breast cancer recurrence (Paik et al., 2004, 2006). As a result, in the United States today, many ER-positive breast cancer tumors are being tested by use of the Oncotype DX assay (or another multi-gene assay known as Mammaprint11), and use of such tests can reduce the rate of chemotherapy use by at least 20 percent (Hayes, 2009). By sparing many women from needless exposure to chemotherapy, the cost savings attained through the use of this test are also substantial (Lyman et al., 2007).
Cooperative Group studies were also instrumental in achieving Food and Drug Administration (FDA) approval for the PathVysion test, which detects amplification of the gene for human epidermal growth factor receptor 2 (HER-2) and is used to select therapies for patients with breast cancer (Mauer et al., 2007).
At present, the Cooperative Groups are actively participating in more than 25 studies of biomarkers (Hamilton, 2009) in five different categories: (1) correlative studies that use clinically annotated biospecimens and research assays; (2) retrospective-prospective studies that use clinically annotated biospecimens, known clinical outcomes, and analytically validated assays; (3) prospective biomarker-drug codevelopment studies; (4) prospective biomarker development studies; and (5) prospective biomarker validation studies (Schilsky, 2009; see also the section on trial design).
CHALLENGES IN BIOMARKER DEVELOPMENT
The process for the discovery and validation of predictive biomarkers is complex. Only about 20 cancer biomarkers have been approved by FDA, and many of these are not routinely used in clinical practice (IOM, 2008). Taking discoveries of potential biomarkers from the laboratory to routine use in clinical care entails multiple steps that require substantial resources and include a multitude of complex scientific and regulatory challenges. Although hypotheses about putative biomarkers are often generated preclinically, true biomarker assessment and validation occur during clinical development and require large numbers of patients who have been treated uniformly in randomized trials. Investigators must show that the biomarker is correlated with a specific biological function, outcome, or characteristic, and they must validate its clinical utility by demonstrating that it provides useful information that can effectively inform clinical decisions by studying large numbers of patients who have been treated uniformly. They must also develop an assay that measures the biomarker and demonstrate that it can reliably be used to guide treatment. An increasing number of candidate markers are being identified and are in various stages of development, yet validation and clinical qualification of these markers is not progressing nearly as rapidly.
Advances in information technology and molecular research have enabled large retrospective correlative studies linking clinical data to molecular data, but a number of obstacles stand in the way of effectively leveraging these advances, including inconsistent access to quality, annotated biospecimens; a lack of standards for assays or analysis of samples in a clinical setting; a lack of standards and templates for the design of correlative and other biomarker studies; a lack of clear and consistent policies that define tissue ownership and access to biospecimens; and a lack of adequate funding or funding that is piecemeal and requires multiple reviews.
Biospecimen Collection, Storage, Annotation, and Access
The quality of biospecimens can significantly influence clinical and research outcomes. Poor-quality biospecimens can generate data that are of poor quality or nonreproducible (Compton, 2009). Selection of a therapy on the basis of the results of a diagnostic test performed with a poor-quality biological specimen could result in patients receiving therapies that are unlikely to be beneficial or not receiving therapies that are likely to be beneficial.
The lack of consistent standards for biospecimen collection and storage is an impediment to improving the quality of those specimens. Maintaining the biological integrity of biospecimens outside of their natural environment is complex, specimens can easily be damaged, and there are many variables
in the collection and storage of specimens (Compton, 2009). In addition, a lack of complete annotation of the biospecimens can create challenges for researchers. The information that a scientist has regarding the characteristics of a biospecimen and the patient from whom it was collected can affect the groupings, the analysis, or the conclusions drawn from the data.
Within a collaborative group, there may be well-established standards for its biorepositories, and some programs funded by NCI, such as the Cancer Genome Atlas, have stringent standards for biorepositories. However, the cancer community and NCI currently lack uniform standards for the collection, processing, and storage of biospecimens; the collection and annotation of the associated data; and consents that govern their use. These obstacles compromise the molecular research that is dependent on biospecimens and impedes the progress of personalized cancer treatment (Compton, 2009).
NCI has launched some initiatives to improve the quality and consistency of biorepositories. In 2007, NCI released the final version of NCI Best Practices for Biospecimen Resources. That document defines state-of-the-science practices, promotes biospecimen and data quality, emphasizes access, recognizes the interest of research participants, and supports adherence to legal and ethical rules and guidelines. However, the current NCI Best Practices do not comprise detailed laboratory procedures; rather, they consist of principles by which such procedures should be developed by biospecimen custodians. The document also does not tackle the cost of recovery or transfer of the samples and does not address custodial rights or sample access policies (NCI, 2007a).
The ability to conduct good correlative science is affected by policies regarding access, governance, and documentation; contractual agreements with commercial partners; extensive review systems for sample use; and complex administrative interactions and oversight (Hamilton, 2009). Hundreds of organizations throughout the United States store tissue samples, and among those organizations, the policies on these issues vary widely (Eiseman and Haga, 1999).
One challenge entails patient consent and authorization for the disclosure of protected health information. The informed-consent documents obtained from patients for their participation in a clinical trial may not adequately specify the use of patient samples for additional, future research studies. Therefore, to test the samples, it may be necessary for the Cooperative Groups to re-contact the patients who provided them to obtain consent and authorization12 (Hamilton, 2009), or to seek permission from
an institutional review board (IRB) to use the samples without consent and authorization.
The stewards of biological specimens have competing interests regarding who owns them, how they are used, and who will benefit from them. Policies regarding ownership and access vary by institution, and this impedes progress. As a result, the means of access to biospecimens for research is inconsistent and can entail complex negotiations with the various custodians of the samples. Pharmaceutical companies, in particular, may be reluctant to share patient samples with academic collaborators and may require agreements regarding intellectual property rights that are unacceptable to collaborators. Many hospitals also discard clinical samples after a period of time, so valuable resources are lost to research. According to NCI, a custodianship plan should consider how the integrity of the biospecimens and their associated data are maintained and monitored; how the rules of access and distribution of biospecimen are defined; what values and responsibilities the biospecimen resource has in place; what legacy or contingency plan, if any, the biospecimen resource has in place; and what circumstances, if any, allow the withdrawal or transfer of biospecimens (NCI, 2008).
Because the Cooperative Groups have a long history of responsible stewardship of biorepositories and well-established networks throughout the country with access to large, diverse patient populations, they are a logical choice for playing a central role in the ongoing efforts of NCI to establish consistent policies regarding ownership and access and could be instrumental in conducting future correlative studies. Thus, the committee recommends that NCI mandate the submission of annotated biospecimens to high-quality, standardized central biorepositories when samples are collected from patients in the course of Cooperative Group trials. The accompanying clinical data should be reported on standardized forms, and NCI should establish a national inventory of biological samples held in the central repositories. NCI should also have a defined process for access to biospecimens for research that includes a single scientific peer review linked to funding. All data, including biomarker data from sera, tissues, and biological imaging analyses, should be considered precompetitive and unencumbered by intellectual property restrictions and should be made widely available. This approach would be similar to current practice in the cancer genomics field.13
Lack of Funding for Biomarker Development Within the Cooperative Group Program
There is a growing need for correlative and translational studies that use biospecimen banks, but the funding stream for these studies is inadequate and complex. Funding to support the Cooperative Groups’ biorepositories and correlative science studies is cobbled together from a variety of sources, including the Group’s core U10 grant,14 a U24 grant,15 users’ fees; and other grants, contracts, and institutional commitments. Investigators need a better mechanism to cover the considerable cost of maintaining these important resources.
Furthermore, current NCI policies require that research studies that propose to use specimens collected from intergroup protocols undergo scientific review by a scientific steering committee before the specimens are made available. However, such a review is not linked to funding, and thus, investigators often must seek funding through other mechanisms or from other sources. This process creates many review loops, time delays, and significant double jeopardy, in that each proposal requires at least two scientific reviews (each of which involves many people), one to receive specimens and one to receive funding, that are conducted at different times by different review groups. A consistent and adequate funding source, with appropriate peer review, devoted to biomarker studies that use stored samples is imperative. Broader use of high-quality samples from standardized repositories would speed the pace of scientific and clinical advances, at a much lower expense than would be required if new samples must be collected for the study of each new concept. Thus, the committee recommends that NCI implement new funding mechanisms and policies to support the management and use of Cooperative Group biorepositories for retrospective correlative science. The Cancer Genome Atlas Project provides a model and precedent for the development and adoption of such policies.
The increased cost of including biomarker tests within trials is also substantial and is not well funded. As discussed in Chapter 3, NCI has taken some initial steps to address this need. Nevertheless, the committee recommends that NCI adequately fund highly ranked trials to cover the costs of the trial, including biomedical imaging and other biomarker tests that are integral to the trial design. Although the cost of studying and validating biomarkers is high, the funds are well spent if the effectiveness of therapy is improved and futile therapy can be avoided. Despite the relatively high cost of performing the Mammaprint or Oncotype DX assay (~$4,000),
for example, the cost of chemotherapy is far greater (~$50,000); if only 5 percent of patients assessed by the test would benefit from chemotherapy, then many patients will be spared significant toxicity unaccompanied by any benefit (Hayes, 2009).
Lack of Standards for Biomarker Development and Use
Analytical validation and clinical validation are important for using predictive imaging and other biomarker tests during clinical trials. However, unlike the strategy used for drug development, which has clearly defined preclinical, nonclinical, and clinical milestones accompanied by guidance on how each phase should be conducted, the strategy for biomarker development is significantly less well defined (IOM, 2007; Simon, 2008a). FDA has issued extensive regulatory guidance and procedures to guide drug development, whereas regulatory guidance and procedures for biomarker development are generally lacking. To date, in fact, there are no clear standards for biomarker validation, use, performance, or interpretation (IOM, 2007).
In an effort to improve the efficiency of biomarker development, multiple organizations have developed guidelines and proposed standards for various steps in biomarker development (IOM, 2007). For example, in 2007, NCI released a document titled Performance Standards Reporting Requirements for Essential Assays in Clinical Trials for biomarker assays used in Phase II and III clinical trials (NCI, 2007b). Concept proposals and clinical protocols for Phase II and III trials that use such assays must also include standard operating procedures for the proper collection, preparation, handling, and shipping of clinical biospecimens (NCI, 2007b). In addition, several of the Cooperative Groups, such as CALGB, have taken initiatives to standardize methodologies, interpretation of the results, and reporting of the results to ensure accuracy, uniformity, and the completeness of the data set (Compton, 2006).
This ad hoc and piecemeal approach, however, is not ideal, because the processes and rules used for such things as determining the composition of a standard-setting committee and the voting rules for the members of the committee are being reinvented on a case-by-case basis. This can lead to heterogeneous results and delays. A systematic, multidisciplinary, and dynamic approach fostered by the National Institutes of Health (NIH) and NCI would ensure that unified national standards are rapidly and consistently set as the need arises. Thus, the committee recommends that NCI, in cooperation with other agencies, establish a consistent, dynamic process to oversee the development of national unified standards, as needed, for oncology research. This process should use the input of experts in both subject matter and standards design in the development of standards, and
it should replicate successful aspects of standards development by other standard-setting bodies, both governmental and nongovernmental (e.g., the American Society for Testing and Materials, the National Standards Foundation, National Institute for Standards and Technology, the International Organization for Standardization, and professional societies). NCI could further assist by facilitating the creation of systems and software to aid the process of standards dissemination and implementation.
This proposal builds on a past IOM recommendation that “government agencies (e.g., NIH, FDA, CMS, and the National Institute for Standards and Technology) and nongovernment stakeholders (e.g., academia, the pharmaceutical and diagnostics industry, and health care payors) should work together to develop a transparent process for creating well-defined consensus standards and guidelines for biomarker development, validation, qualification, and use to reduce the uncertainty in the process of development and adoption” (IOM, 2007).
Uncertain Relevance of Primary Tissue for Patients with Metastatic Disease
Tissue samples from tumor biopsies are generally collected and archived at the time of an initial diagnosis and most often consist of primary tumor specimens. Metastatic or recurrent lesions are biopsied much less frequently. With the move toward targeted and personalized therapy, however, questions about whether the primary tumor is representative of the patient’s metastatic disease have arisen, especially in the context of intervening therapy. It is unclear whether primary archived tissue provides biomarker data relevant to predicting the therapeutic response for metastatic cancer or whether another biopsy is required to obtain an accurate and relevant biomarker assessment. It may not be feasible or desirable to ask patients with cancer to undergo multiple invasive procedures to assess recurrent or metastatic disease. However, a second analysis of biological material can also include assessment of samples obtained by noninvasive or less invasive means, such as blood for analysis by blood-based assays (for example, for analysis of serum DNA, the serum protein profile, or circulating tumor cells) or molecular imaging for a relevant target.
Most cancers are not fatal unless they become metastatic, so these questions need to be answered to maximize the effectiveness of cancer therapies. The development of new trial concepts and the provision of funding to compare the molecular pathologies of primary and metastatic tumor tissues and to determine whether it is valid to use archived primary tissue for the selection of therapy for metastatic disease would be helpful. This could be the topic of a Grand Challenge competition, as recommended in Chapter 3.
PROMISES AND CHALLENGES OF COMBINATION PRODUCTS
Developing, testing, and gaining approval for the use of combination products such as multiple therapeutic agents or a therapeutic agent accompanied by a diagnostic test are more difficult than for a single product because of the increased scientific challenges, logistical challenges, unclear regulatory requirements, and high costs. For example, different centers within FDA have jurisdiction over different types of products for use in oncology, and for combination products, more than one center may have jurisdiction (see also Chapter 3 for more details). Furthermore, the traditional and well-established approach to drug development has focused on a single agent (or in some cases, a single agent added to standard chemotherapy), without a paired diagnostic test. In comparison, the stages and important milestones for the development of combination products have not been clearly established by precedent. Although some initial steps have been taken to delineate appropriate development pathways, many challenges remain, as described below.
Diagnostic-Therapeutic Combinations
Cancers have tremendous heterogeneity: even tumors that appear to be the same clinically may have different molecular characteristics and differing mechanisms of development and spread. To deliver effective treatments, it is important to be able to identify the specific molecular characteristics present in a patient’s tumor. To do this, it is necessary to develop biomarker tests that accompany treatments targeting those defects. Alternatively, it might be possible to develop therapeutics for which there are multiple biomarkers to choose from or use in combination. Other diagnostics might indicate that a patient’s cancer could be treated with multiple therapeutic agents. In addition to baseline biomarkers, which are central to predicting the response to treatment, longitudinal biomarkers (e.g., serial measurements) can help to determine whether a particular tumor is responding to a particular therapy.
A good example of the challenges involved in codeveloping an in vitro diagnostic-therapeutic combination is the targeted drug trastuzumab (Herceptin) and the various assays used to measure the overexpression of HER-2. Tumors that overexpress HER-2 are often sensitive to treatment with Herceptin, a monoclonal antibody that blocks the function of this receptor—one of the most well-known applications of personalized medicine. Genentech used a biomarker assay throughout the drug development process to test the efficacy of trastuzumab, but the company was not able to obtain FDA approval for that test at the time that FDA approved the drug. As a result, the sponsor had to rely on another company to develop a diag-
nostic assay that could be paired with its product and that was approved by FDA (IOM, 2008).
Because of the scientific challenges, the increased financial burden, and the increased risk of developing diagnostics in the early phases of clinical trials of a therapeutic agent, such tests are often not incorporated into clinical trials until late in development or even after a therapeutic agent has received FDA approval. Although the development of successful companion diagnostic tests yields substantial benefits for patients, many technical difficulties limit the development of tests that are reliable and clinically meaningful. Complementary work on diagnostic-therapeutic combinations should ideally begin in the early phases of development of a therapeutic agent (Tan et al., 2009).
The ability to streamline the validation process and develop additional tools will depend on better collaboration and coordination among stakeholders (McClellan and Benner, 2009; PCAST, 2008) and adequate funding. The essential tools and resources include access to high-quality biospecimens accompanied by comprehensive disease annotation and study designs, statistical methods, and standards for determining the clinical validity and utility of biomarkers (IOM, 2007; PCAST, 2008).
Some investigators are conducting clinical trials that use novel methods and collaborations. For example, the I-SPY 2 TRIAL,16 which builds on the I-SPY 1 TRIAL,17 is a Phase II adaptive trial that uses Bayesian statistics to predict how the drugs will perform in Phase III studies (see also the section on trial design below). The trial will test various neoadjuvant therapies in patients with primary breast cancer who are at high risk of disease recurrence. The goals of the trial include the identification of biomarkers that predict increased pathologic complete response (pCR) of a therapeutic agent or a combination of agents when added to standard neoadjuvant chemotherapy, modeling of the relationships with imaging to predict pCR, confirmation of the observations to minimize false-positive conclusions, and use of the results to advance biomarker-drug pairs that have a high probability of success to Phase III studies. It is fundamentally a drug screening process and the primary goal is not registration of any particular therapeutic agent or biomarker. A committee determines which therapeutic agents will be tested in the study, and the companies that manufacture the agents then provide them for study. Some of the drug candidates are run through
60 tumor cell lines to see if researchers can predict the response. Three standard biomarkers (estrogen receptor, HER-2, and Mammaprint) are used to categorize patients and determine therapy. Many other biomarkers are assessed in the trial and, depending on the results, may be considered for approval as a predictive marker by the FDA Office of In Vitro Diagnostics within the Center for Devices and Radiologic Health (Barker, 2009; Esserman, 2009).
If the trial proves to be a successful demonstration of the process for testing biomarkers, many scientific challenges will still remain. For example, investigators will need to determine whether this research design is portable to other diseases besides breast cancer. Because breast cancer has more established biomarkers than most other types of cancer, identifying suitable markers for similar studies of other cancers may prove more difficult. It is also not clear whether it is financially feasible to test the current model in other settings (Esserman, 2009).
Combination Therapy
Most cancer drugs have traditionally been broadly cytotoxic and nonselective in their mechanisms of action, resulting in significant toxicity to healthy tissues. In recent years, research has elucidated many of the molecular changes underpinning the initiation and progression of cancer (e.g., changes affecting signaling pathways; cell death mechanisms; cancer spread; and DNA synthesis, repair, and modification). Research also shows that a particular cancer may rely on multiple key pathways or functions to survive, grow, or metastasize. In addition, there is often some redundancy in key pathways, such that if one step in the pathway is inhibited, a compensatory step may be upregulated and overcome the inhibition. Treatments that target multiple pathways or more than one node in a pathway are likely to be more effective than those directed toward a single target.
Numerous scientific and medical challenges impede the efficient and successful evaluation of combination therapies. One quandary is prioritizing the combinations of agents to be tested. Preclinical studies, primarily studies that use animal models, may be the best means for determining which drugs are the most effective in combination, but there are limitations to this approach, especially since preclinical models do not always predict effects in patients with cancer.
In the past, little was generally known about the target of the therapeutic agent in the early stages of development. The risk of failure was high because the agent might not interact with the intended target or the intended target might not be relevant for tumor growth. Combining products early in their development increased the risk of failure because little is known about each of the agents alone or in combination. As a result,
it has been rare for two or more investigational products to be combined. Rather, typical combination products include at least one product that has already received FDA approval, along with other approved products or experimental products (IOM, 2008). This could change in the future, as targets for a new drug are better understood. For example, AstraZeneca and Merck recently announced plans to collaborate on a novel cancer regimen using two investigational agents early in development, both allosteric kinase inhibitors (Winslow, 2009). According to Merck, this is the first time that two large pharmaceutical companies have established a collaboration to evaluate the potential for combining candidate molecules at such an early stage of development (Merck, 2009). A unique challenge of this collaboration is that to date, co-registration of two unapproved agents for the treatment of cancer is unprecedented (Huang, 2010). GlaxoSmithKline and Novartis have also initiated a similar early-stage collaboration to evaluate the combination of two investigational kinase inhibitors.18 While these types of collaborations are just emerging, it is thought that combining targeted agents early in development may accelerate the delivery of promising cancer therapies and ultimately change the drug development paradigm.
If a single sponsor is developing both therapeutic agents, the developmental and regulatory process is less complicated than it would be if multiple sponsors were involved. Having multiple sponsors creates unique data-sharing, intellectual property, logistical, and marketing challenges (Canetta, 2009). These challenges are further discussed in Chapter 3. Numerous regulatory challenges also exist (IOM, 2008). The conventional approach to regulatory reporting is to attribute adverse events (AEs) and efficacy to particular agents. However, studies indicate that some targeted therapeutics that work effectively in concert with other agents may not evince a response as a single agent, so it can be difficult or impossible to measure and delineate the contribution of each agent to the safety and efficacy of therapy when agents are used in combination, as expected by FDA (IOM, 2008; Lutzker, 2009; Woodcock, 2009).
FDA is developing a guidance document to clarify the codevelopment of agents for combination therapy (Woodcock, 2009). This document is likely to require that combination regimens demonstrate much-improved outcomes compared with the small advances that it currently requires for the approval of new therapies (Woodcock, 2009). The codevelopment of two investigational agents will require an improved understanding of their mechanisms of action and will ultimately involve biomarkers (Woodcock, 2009). The availability of biomarkers that are capable of predicting a patient’s response could be helpful in interpreting the results of combination therapy trials and in determining the safety and efficacy of each compo-
nent. However, as noted, finding such markers will require a great deal of research, focused primarily on studying tumor responses to identify markers that predict the response or resistance to the therapeutic agent (IOM, 2008). Input from experts in clinical pharmacology may also be beneficial in designing trials to test novel combinations.
Furthermore, testing all relevant combinations in traditional two-arm trials will be very time-consuming and expensive. Many researchers have proposed novel clinical trial designs that could allow more efficient development of combination therapies (IOM, 2008; Lutzker, 2009; Parmar et al., 2008). FDA has indicated that innovative trial designs (e.g., adaptive designs; see the section on trial design below) could provide faster answers regarding drug combinations (IOM, 2008). However, stakeholders (including industry, FDA, and NCI) have heretofore generally taken a conservative stance on trials with novel designs. All stakeholders should work together to develop innovative and collaborative approaches to this urgent challenge in cancer clinical trials (IOM, 2007; McClellan and Benner, 2009; PCAST, 2008). The committee recommends that NCI facilitate a process by which stakeholders (NCI, NIH, FDA, industry, investigators, and patients) can define an effective mechanism for the development of targeted cancer therapies, with particular emphasis on combination products.
TRIAL DESIGN
The increasing complexity of cancer clinical trials, along with the great expense and high failure rate of late-stage clinical trials, has spurred innovations in trial design, with the aim of conducting clinical trials more efficiently and with greater likelihood of success. Experts have proposed numerous trial design innovations, including multi-arm trials, adaptive trials, greater reliance on predictive biomarkers, use of the progression-free survival as an endpoint, testing of multiple agents in the same trial, garnering more information from the early stages of clinical trials, and implementing more randomized Phase II trials in which both safety and efficacy are assessed (Adjei et al., 2009; Berry, 2005, 2006; Bria et al., 2009; Rubinstein et al., 2009; Sargent and Taylor, 2009; Schiller, 2004; Simon, 2008b; Tan et al., 2009; Thall et al., 1988, 1989). An explicit goal in designing clinical trials can be shortening the time to develop and reliably assess therapeutic strategies. This increased efficiency might be possible by utilizing emerging information on biomarkers and other intermediate measures in a more deliberate way. Cooperative groups are in a unique position for developing innovative approaches to clinical trial design and for demonstrating the feasibility and utility of innovative, efficient approaches in their clinical trials.
The randomized controlled trial (RCT) was established as the standard
of practice for evaluating new medical treatments in the middle of the 20th century. Randomization was properly hailed as an enormous advance in empirical science and the RCT is widely recognized to be the gold standard of clinical research. Although many enhancements have been made to trial design and analysis over the ensuing decades, the current explosion of biological knowledge demands increased attention to developing trial designs that can take advantage of this knowledge more fully, with a goal of improving the efficiency of trials without reducing the reliability of results. Many clinical trialists are developing approaches to clinical trials that involve multiple stages or otherwise permit increased flexibility by allowing for changes to be made during the trial, based on emerging results. Such design innovations have potential for decreasing the time to study conclusions and improving the likelihood of offering effective treatment to a greater proportion of trial participants.
Although the committee does not endorse any particular type of trial design, one example of design that may be more flexible than the traditional two-armed RCT that has a fixed sample size is sketched in Figure 2-1. The primary endpoint in this example could be the accepted Phase III endpoint, perhaps overall survival. However, early markers of treatment effect (e.g., tumor response, performance status, disease progression, longitudinal biomarkers) might also be used to make decisions about dropping arms.
The use of the multiarm, multistage (MAMS) trial design has been proposed as a way to evaluate treatments faster and more efficiently than is possible by the use of current standard trial designs. By using intermediate outcomes and testing a number of new agents (and combinations of agents) simultaneously against a single control arm, the MAMS design for RCTs requires fewer patients (Parmar et al., 2008). A performance evaluation of the two-stage, multiarm design using four cancer trials conducted at the Clinical Trials Unit (CTU) of the Medical Research Council (MRC) in the United Kingdom found that such a design performs well with regard to the Type I and II error rates19 obtained and is an effective way of speeding up the therapy evaluation process (Barthel et al., 2009). The first MAMS trial to use multiple arms and stages synchronously was recently launched by the MRC CTU. The STAMPEDE trial (Systemic Therapy in Advancing or Metastatic Prostate Cancer: Evaluation of Drug Efficacy) is an open-label, five-stage, six-arm RCT of drug treatments for men with prostate cancer. Although the results of the STAMPEDE trial will not be known for some years, the implementation of this trial may demonstrate the feasibility of
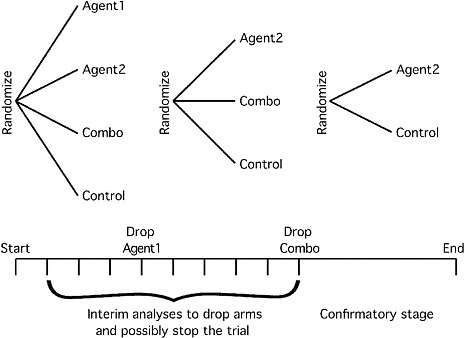
FIGURE 2-1 Example of a Phase II/III trial for investigating several possible treatment strategies. Several treatment arms are considered at the start of the trial (in this case, two individual agents and their combination, but there are many other possibilities). As information accrues, arms that are not performing as well as other arms can be dropped, and if none of the experimental arms are performing well relative to control then the trial can be stopped. A goal may be to reduce to a two-armed trial by particular time points. Accrual continues without stopping for interim analyses, and accrual might be ramped up on the basis of interim analyses as the trial moves into confirmatory mode. The sample size of the confirmatory stage can be determined on the basis of the performance of the experimental arms relative to control in the early stages of the trial.
using the MAMS design to initiate and undertake large-scale trials of cancer therapies (Sydes et al., 2009).
Most clinical trials are designed to employ classical “frequentist” statistical methods.20 Another approach to clinical trial design and analysis is the Bayesian approach, which considers the treatment effect as a random variable with a probability distribution rather than as an unknown constant that the investigator wishes to estimate. FDA has issued draft
guidance on the use of adaptive Bayesian designs for trials of drugs and biologics, and recently issued final guidance on the use of such designs for device trials, with the commissioner of FDA noting, “This final guidance on the use of Bayesian statistics is consistent with the FDA’s commitment to streamline clinical trials, when possible, in order to get safe and effective products to market faster” (FDA, 2010a,b,c). The use of Bayesian methods for clinical trials is increasingly being advocated because many statisticians believe that this approach fosters more flexibility in the conduct of a trial and because adaptation can be incorporated more naturally into trials with Bayesian designs than into those with frequentist designs. For example, the performance of Bayesian analyses midtrial can indicate early in the course of the trial the subpopulations most likely to respond to treatment, enabling enrichment of the study population. They can also suggest which among several treatments is the most likely to be effective or ineffective, enabling researchers to drop those treatment arms likely to be failures and to modify the number of subjects needed to show the efficacy of treatments on the basis of midstream analyses. Frequentist designs can also incorporate such opportunities for adaptation, but Bayesian approaches may be more suitable when substantial adaptation is desired, particularly in the earlier stages of drug development. For example, Phase I studies that use Bayesian adaptive designs may enable more doses to be considered and may be more likely to identify the most effective dose with minimal adverse effects than trials with more traditional designs (Berry, 2006). The use of Bayesian designs has been more controversial in the later, confirmatory stages of drug development. An element of subjectivity is an inherent part of this approach.
Bayesian approaches are increasingly being used in exploratory studies. For example, this approach will be used in the I-SPY 2 TRIAL, described earlier in this chapter, to develop paired neoadjuvant cancer therapies and biomarkers (Barker et al., 2009). Improved computational techniques and the widespread availability of high-speed computers enable researchers to more easily conduct Bayesian clinical trials, and the results from Bayesian trials are increasingly being used in clinical trials and to petition FDA for the approval of drugs and devices (Berry, 2006; Biswas et al., 2009; Gonen, 2009; Katsnelson, 2009).
Another aspect of cancer clinical trials that investigators are reconsidering is the reliance on the tumor response rate as an endpoint in Phase II trials. Phase II cancer trials have historically been single-arm trials aimed at demonstrating tumor responses,21 with the assumption that it will translate into clinical effectiveness in a Phase III trial. However, in many types
of cancer, the tumor response does not reliably predict a survival benefit, and in other types of cancer, the tumor response has proven difficult to measure. In addition, many of the newer molecularly targeted agents are cytostatic rather than cytotoxic and may thus provide a survival benefit in the absence of a significant change in tumor size. For example, some new targeted therapeutic agents, such as sorafenib given to patients with hepatocellular carcinoma, significantly increase patients’ time to disease progression compared with patients receiving standard of care, in the absence of tumor shrinkage, presumably by blocking proliferative signals to the tumor such that much of the tumor mass is comprised of inactive or necrotic tissue (Adjei et al., 2009).
The Clinical Trial Design Task Force of the NCI Investigational Drug Steering Committee recently recommended that alternate Phase II endpoints be studied, including the use of novel imaging modalities. Progression-free survival may be an appropriate measure when newer targeted agents are tested, but researchers need to assess this endpoint at the same, pre-specified intervals in both treated patients and control subjects to avoid biased results (Rubinstein et al., 2009; see also the section on FDA data requirements in Chapter 3). New imaging modalities, such as those that use novel positron emission tomography (PET) imaging agents to highlight the biochemical target of a drug or digital contrast-enhanced magnetic resonance (MR) spectroscopy, which can detect the inhibition of angiogenesis, might also provide more valid endpoints for cancer clinical trials than tumor volume.
The task force also recommended the use of a randomized design rather than historical controls in some circumstances (Adjei et al., 2009). Investigators are increasingly conducting randomized controlled Phase II trials to obtain more reliable preliminary estimates of effectiveness. Such trials reduce the possibility for bias and provide more information than uncontrolled Phase II studies for the purpose of deciding whether to progress to Phase III trials (Rubinstein et al., 2009). Randomized trials are also warranted when appropriate historical control data for a tested targeted cancer treatment are not available, especially for patient subsets identified by specific predictive markers for the treatment. To improve the efficiency of Phase II randomized trials, statisticians have proposed a number of innovative trial designs, including randomization of a small portion of patients to a reference arm, the incorporation of a randomized Phase II trial into the first stage of a Phase III protocol, or the use of a Phase II trial that directly compares two experimental regimens to prioritize candidacy for Phase III studies (Rubinstein et al., 2009).
The task force recommended more rational incorporation of biomarkers in Phase II clinical trials as well. The development and evaluation of biomarkers for use in patient treatment decisions also warrants innovative trial designs. The current method of biomarker discovery is largely retrospective.
Various hypotheses about potential biomarkers are generally examined in Phase II studies. Some retrospective biomarker validation is generally required because throughout the development of a therapy and even at the time of FDA approval, researchers may not fully understand the biological implications of a therapy and might not be able to identify subgroups of patients who are likely to respond. However, retrospective studies have inherent limitations. The replication of retrospective studies by multiple investigators can provide informative data, but better strategic planning of biomarker evaluation throughout the development phase, especially in Phase II trials, may enable the design and conduct of more efficient Phase III studies, which could lead to the increased success of new therapies in Phase III studies (McShane et al., 2009).
The use of studies with retrospective/prospective designs is one way to improve biomarker development. That type of design requires randomized patient accrual and the use of prespecified hypotheses, techniques for analyses, patient populations, patient subgroups, and large numbers of patients with biospecimens (Sargent, 2009). Figure 2-2 illustrates one approach that is being used for biomarker validation.
On the basis of data from retrospective-prospective studies, FDA recently added information about testing for mutations in the KRAS gene to the labels of two drugs used to treat patients with colorectal cancer (FDA,
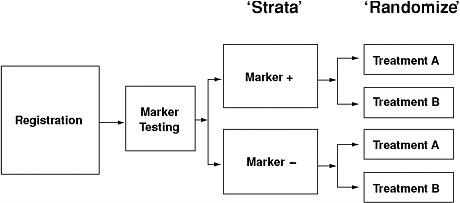
FIGURE 2-2 Example of a retrospective/prospective approach to biomarker validation. The benefit of treatment is tested separately in the two marker-defined patient populations.
SOURCE: Mandrekar, S. J., and D. J. Sargent. 2009. Clinical trial designs for predictive biomarker validation: One size does not fit all. Journal of Biopharmaceutical Statistics 19(3):530–542. Reprinted with permission of Taylor & Francis Group, http://www.informaworld.com.
2009). About 10 percent of patients with metastatic colorectal cancer respond to the monoclonal antibodies panitumumab (Vectibix) and cetux-imab (Erbitux), which target the epidermal growth factor receptor (EGFR). However, the detection of EGFR protein expression by immunostaining does not reliably predict the clinical outcome of EGFR-targeted treatment (reviewed by Modjtahedi and Essapen, 2009). A search for alternative predictive biomarkers led to the discovery that oncogenic activation of signaling pathways downstream of EGFR, such as mutation of the KRAS gene, can render EGFR-inhibiting therapies ineffective (reviewed by Siena et al., 2009). Forty percent of the tumors from patients with colorectal cancer have KRAS gene mutations that result in the continuous activation of the KRAS protein (Allegra, 2009). Retrospective subset analyses of metastatic colorectal cancer trials demonstrated that patients with this mutation do not receive therapeutic benefit from treatment with cetuximab or panitumumab, but those with wild type KRAS do often benefit. In July 2009, FDA announced that the companies that make these two products would be updating the labels so that physicians could use this information to guide treatment (FDA, 2009), although it is not yet clear whether this type of evidentiary data set will lead to similar FDA decisions in the future (Allegra, 2009; Goldberg, 2009).
Prospective randomized clinical trials may be scientifically ideal for the evaluation of predictive biomarkers, but such studies are complex, costly, and challenging to conduct. One reason is that patients may be reluctant to be randomized if they have a biomarker thought to be predictive of benefit from some therapy. Examples of ongoing prospective trials include TAILORx (Trial Assigning IndividuaLized Options for Treatment)22 and MINDACT (Microarray in Node-negative Disease may Avoid Chemo-Therapy),23 which are designed to test the validity of the Oncotype DX test and the Mammaprint tests, respectively. The NCI-sponsored TAILORx trial is a prospective study of the Oncotype DX test that will enroll 4,000 ER-positive, lymph node-negative, and HER-2-negative patients to determine whether women with an intermediate score by the Oncotype DX test will benefit from adjuvant chemotherapy (IOM, 2008). MINDACT is a multicenter, prospective, Phase III trial coordinated by the European Organisation for Research and Treatment of Cancer and run under the Breast International Group (BIG) and TRANSBIG24 networks, which will accrue 6,000 node-negative patients. Patients for whom there is discordance
|
22 |
See http://www.cancer.gov/clinicaltrials/digestpage/TAILORx. |
|
23 |
See http://www.breastinternationalgroup.org/Research/TRANSBIG/MINDACT.aspx. |
|
24 |
BIG is a nonprofit organization for academic breast cancer research groups that facilitates breast cancer research at international level. TRANSBIG is a consortium within BIG that is focused on translational research. |
between the gene signature risk results (high versus low) and the clinical-pathological risk will be randomized to determine which result will be used to make the decision about whether to offer chemotherapy.
Other new strategies being used include “targeted trial designs,” which initially test subjects for the presence of predictive markers and include (and randomize) only those subjects whose biomarker status predicts that they will respond to a treatment (Bria et al., 2009). The number of randomized patients needed for such a targeted design is often much smaller than that needed to conduct a trial with a standard design (Simon, 2008b); however, the target population is also smaller, so these trials will not necessarily be completed more rapidly, and they may be more expensive to conduct since many more potential subjects than will ultimately be enrolled will need to be screened for eligibility. Additionally, one must be very sure that the assays measuring the marker are highly accurate before the decision is made to use such a design to avoid unnecessarily limiting a potentially widely effective treatment to a small population. If a predictive marker is not known or is not validated before the start of a clinical trial, an “adaptive signature” trial design could be used. In a trial with such a design, different subsets of data within the Phase III trial are used to develop a predictive marker and to evaluate the effects of the treatment in populations identified by the marker (Simon, 2008b). Another trial design that enriches the patient population with likely responders is the “randomized discontinuation” design, in which all patients receive a cytostatic drug for one or two cycles, and those with stable disease are then randomized to receive either a placebo or the drug (Schiller, 2004).
As Sargent and Taylor (2009) summarize in a recent paper,
The need to strategically consider drug development as an integrated program as opposed to a collection of isolated studies, the benefits in many cases of randomization earlier in the drug development process, and the need for new endpoints all are challenging the standard paradigms…. Our overall premise is that the potential benefits associated with the oncology clinical trial community moving away from the one-size-fits-all paradigm of trial design are great, and that more flexible and efficient designs tailored to match the goals of each study are currently available and being used successfully.
Thus, the committee recommends that the Cooperative Groups lead the development and assessment of innovative designs for clinical trials that evaluate cancer therapeutics and biomarkers (including combinations of therapies). For example, prospective clinical trial designs that randomize patients based on biomarkers or treatments, or both should be explored and evaluated. For targeted therapies, a predictive hypothesis for a biomarker
should be put forward in the preclinical phase and tested in early-phase clinical trials (Phase I-II trials).
COMPARATIVE EFFECTIVENESS RESEARCH
In addition to developing and testing novel approaches to cancer care, the Cooperative Group Program also plays an important role in further evaluating therapies and preventive strategies that are already in clinical use. For example, FDA registration trials may provide evidence of efficacy in a well-defined test population, but physicians would often like to have additional information for making decisions about patient treatment when more than one therapy is available for similar indications. Comparative effectiveness research (CER) is designed to address this data need by generating evidence about the effectiveness of health care options and clinical outcomes that result from different medical interventions for the same condition. CER can span a spectrum of research methods, from data set mining and observational studies to randomized controlled trials, and can include research on preventive interventions, screening tests, diagnostic tests, treatments, follow-up strategies, and end-of-life care. Cooperative Groups often undertake large prospective randomized trials to rigorously compare outcomes for patients treated with cancer drugs that have similar targets, or are indicated for similar patient populations. Evidence generated from such trials can then be used to make treatment decisions based on relative efficacy or toxicity. Pharmaceutical companies have less incentive to undertake such trials because if a competitor’s drug fares better in the trial, then they are likely to loose market share.
One type of prospective clinical study that can be used to develop high-quality scientific evidence about real-world effectiveness is a “pragmatic” (or “practical”) clinical trial, in which the hypothesis and study design are developed specifically to answer questions faced by decision makers. A pragmatic clinical trial selects clinically relevant alternative interventions to compare; includes a large, diverse population of study participants; recruits participants from heterogeneous practice settings; and collects data on a broad range of health outcomes (although data collection is still greatly minimized compared to standard FDA-style registration trials) (Benson et al., 2009). In some cases, this type of research entails large, simple trials that are constructed to reflect routine clinical practice as closely as possible.
However, the study of biospecimens and biomarkers can greatly increase the value of such trials. If comparisons are studied in a broad, heterogeneous population, it can appear that one treatment is not much better than a comparator, but it may turn out that 10 or 15 percent of the population that has some specific characteristic that results in better outcomes treatment with a particular treatment. Without biomarker studies to identify
the biological subsets of patients, some patients could be at a disadvantage when making treatment decisions.
As noted in Chapter 1, many new cancer therapies have become available in clinical practice in recent years, so the opportunity for developing meaningful CER studies in oncology has never been greater. Congress and the U.S. Department of Health and Human Services have also put a new emphasis on CER as a way to improve the quality of health care.25 In 2009, Congress requested a study to identify priorities for CER across all fields of health care and provided $1.1B in new funding to support such studies (IOM, 2009; Marshall, 2009). Topics identified as high priorities for CER in oncology include comparing the effectiveness of management strategies for ductal carcinoma in situ, comparing the effectiveness of imaging technologies in diagnosing, staging, and monitoring patients with cancer, and comparing the effectiveness of genetic and biomarker testing and usual care in preventing and treating breast, colorectal, prostate, lung, and ovarian cancer (IOM, 2009). The Cooperative Groups have a history of successful trials to address these types of questions.
BIOMEDICAL IMAGING AS A CANCER BIOMARKER
The quest to incorporate innovative science into clinical trial design and to evolve toward personalized medicine demands a reassessment of the role of biomedical imaging. The use of anatomic imaging to assess changes in tumor size has long constituted the main application of imaging in clinical trials. However, because of advances in computational and molecular imaging techniques, imaging now has the potential not only to provide more refined anatomic endpoints of drug efficacy but also to aid decision making in drug development and patient care by providing molecular information about a patient’s cancer.
Imaging studies offer important advantages over in vitro assays for the measurement of biomarkers. Tissue for use in in vitro assays cannot be excised and evaluated for all cancers at all sites at all points along the history of a disease. In contrast, imaging biomarkers can be evaluated for all metastatic sites and have the potential to yield insights into in vivo tumor biology and the heterogeneity of metastatic tumors. Furthermore, imaging can be applied serially, and molecular imaging techniques can provide information about disease activity in real or nearly real time. Thus, expanding the use of imaging biomarkers will play an important role in improving the efficiency of clinical trials and advancing personalized cancer care.
It must be emphasized though, that many of the approaches to molecular imaging discussed in this chapter are being used primarily in the preclinical setting today. Most of these methods must be tested in rigorous clinical trials to prove their utility.
Role of Anatomic Imaging in Clinical Trials
Anatomic imaging is and will remain essential for staging primary cancer, determining the extent of recurrence and metastasis, and assessing the response to treatment in clinical trials. However, enhanced quantification methods could further improve the assessment of treatment response. The standard criteria for assessing treatment response (those proposed by the World Health Organization and the Response Evaluation Criteria in Solid Tumors [RECIST]) rely on one- or two-dimensional tumor size measurements obtained from cross-sectional images (Eisenhauer et al., 2009; Gehan and Tefft, 2000; Miller et al., 1981; WHO, 1979). These criteria can be applied relatively quickly and easily but have substantial limitations. The cutoffs that they specify for identifying response and disease progression were defined when anatomic imaging was considerably less precise than it is now and do not take subtle but potentially important size changes into account. Furthermore, measurement of tumor diameters may be subject to significant interobserver variability (Benjamin et al., 2007; Choi, 2005; Choi et al., 2007; Hohenberger and Wardelmann, 2006; Sevinc and Turhal, 2008; Stacchiotti et al., 2009; Weber, 2009). In addition, one- or two-dimensional tumor size measurements provide reasonable surrogates for the volumes of ellipsoid or spherical tumors but not for the volumes of irregularly shaped tumors, such as those that occur in lung cancer.
Various computerized methods for automated or semiautomated tumor volume measurement have been proposed, and some are now entering clinical use (Galanis et al., 2006). Preliminary studies indicate that volumetric tumor measurement may allow the earlier detection of the response to treatment for lung cancer than conventional tumor measurement techniques (Buckler et al., 2010; Gavrielides et al., 2009; Zhao et al., 2009). Because of its wide availability, CT is likely to remain the standard modality for assessing treatment response in patients with cancer for years to come. Routine use of volumetric CT appears to be feasible in clinical settings, and could allow faster, more accurate assessment of new therapies while sparing patients from the side effects of prolonged ineffective treatments. However, validation, comparison, and standardization of volumetric CT software are needed.
In recognition of the potential of quantitative imaging biomarkers to lead to more efficient, effective clinical research and care (Mulshine and Jablons, 2009), the Quantitative Imaging Biomarkers Alliance (QIBA),
sponsored by the Radiological Society of North America, was formed in 2007. QIBA is a consortium of stakeholders from federal, academic, and commercial institutions whose mission is to streamline the development, validation, and standardization of quantitative imaging biomarkers (Mulshine and Jablons, 2009; RSNA, 2010). Validating and standardizing volumetric CT are among the consortium’s highest priorities.
Sometimes, particularly for new therapies that produce cytotoxic or cytostatic effects without substantial tumor shrinkage, tumor size or volume changes alone may not be reliable indicators of treatment response. In such cases, applying conventional imaging techniques in novel, quantitative ways may offer solutions. Thus, to better assess the response of gastrointestinal stromal tumors (GISTs) treated with imatinib mesylate (Gleevec), Choi et al. adjusted the RECIST guidelines and incorporated density changes, as measured by Hounsfield units on contrast-enhanced CT (Benjamin et al., 2007; Choi, 2005; Choi et al., 2007; Hohenberger and Wardelmann, 2006; Sevinc and Turhal, 2008; Stacchiotti et al., 2009). In the long run, however, combining molecular with anatomical imaging is likely to yield even better solutions.
Biological Basis for Improved Cancer Imaging in Oncology
Emerging knowledge about cancer has improved the understanding of the optimal goals for imaging in clinical oncology and clinical trials. First, in vivo, tumors are masses that consist of cancer cells and their supporting nonmalignant cells and blood vessels that are organized into a community of interacting cells. It is this mass that creates the clinical symptoms and that is the target of cancer therapies. This is an important concept and is the biological basis for the continuing value of anatomic imaging, especially volumetric imaging based on ultrasound (US), CT, or MR imaging (MRI).
Second, it is now understood that specific hallmarks of cancer cells cause the clinically evident tumor mass and are fundamental to developing and maintaining the malignant state, including metastases. These include resistance to apoptosis; the ability to replicate indefinitely (immortality); the ability to invade and metastasize; accelerated proliferation, including resistance to growth-stopping signals; and sustained angiogenesis (Hanahan and Weinberg, 2000). The malignant tumor also has an altered metabolism, including accelerated glycolysis, fatty acid synthesis, and lipid synthesis; in short, a metabolic phenotype of malignancy.
Finally, malignancy likely begins as an alteration of key intracellular communications pathways. In some cases there is a transformation of a key gene, the oncogene that encodes a macromolecule that promotes aberrant signaling within the cell. Examples of key modifications include altered EGFR in lung cancer and glioma; HER-2 in breast and gastric cancers;
and c-kit in GIST. In some cases, changes in signal transduction molecules further downstream, such as b-raf mutations, promote proliferation. These alterations are often the basis for targeted tumor therapy and for molecular imaging of normal and aberrant molecules that accompany the malignant state.
Cancer tissues are distinct from the tissues from which they arise, and the differences may be disclosed by both anatomic and molecular imaging methods. Molecular imaging allows in vivo detection, characterization, and quantitative analysis of the key molecules, molecular events, and cellular components that are fundamental to the development and progression of cancer.
Molecular Imaging and Its Potential Applications in Clinical Trials
Recent revolutionary advances in molecular and cell biology, imaging probe development, and imaging technology have rapidly expanded the actual and potential applications of molecular imaging. Today’s sophisticated array of molecular imaging technologies were developed through collaborations across many fields, including radiology, nuclear medicine, chemistry, molecular and cell biology, physics, mathematics, and pharmacology (Bradbury and Hricak, 2005; Grassi et al., 2008; Zakian et al., 2001).
Imaging modalities capable of providing information at the molecular level include PET, single-photon-emission computed tomography (SPECT), MRI, MR spectroscopic imaging, and optical imaging (optical imaging is used extensively in preclinical studies). Apart from diffusion-weighted MRI and MR spectroscopic imaging, which image water molecules and metabolites, respectively, all molecular imaging techniques rely on the use of exogenous probes to provide an imaging signal or contrast. Most probes are composed of an affinity component, which interacts with the target, and a signaling component, which provides image contrast. Although radiolabeled probes are used for PET and SPECT, the signaling component can be a fluorochrome in optical imaging or a chelate containing a paramagnetic atom in MR imaging. Imaging probes can be divided into four main categories, which are described below.
-
Phenotypic imaging probes. Phenotypic imaging probes are used to show general aspects of the physiology of malignant cells and tissues, such as neovascularization (altered blood volume, permeability, perfusion, and vascularity of the tumor tissue) and metabolism. Examples of phenotypic probes include paramagnetic agents such as gadolinium diethylenetriamine-pentaacetic acid, which is used for MR imaging of tumor neovascularity, and metabolic probes, such as the radiolabeled glucose analog fluorine-18 fluorodeoxyglucose (18F-FDG), which is used with PET.
-
Targeted probes. Targeted probes are used to localize specific biomolecules that are characteristic of a tumor or class of tumors, such as signal transduction proteins or tumor-associated antigens. The probes include radiolabeled antibodies, peptides, nanoparticles, and small molecules. An example of a targeted probe is 124-iodine-labeled chimeric monoclonal antibody G250, which is used to image carbonic anhydrase IX in renal cancer and which binds with a high affinity to a cell membrane antigen. Targeted probes include activatable probes, which are made with molecules or nanoparticles that undergo inducible changes. Activatable probes are used to localize enzymes, signal transductors, and downstream effectors.
-
Reporter gene probes. Reporter gene probes allow monitoring of the actions of genes in living biological systems. Gene expression imaging has had a revolutionary impact on the laboratory study of cancer biology and is likely to become important in clinical trials in the future as well. Various bioluminescent-, fluorescent-, and radionuclide-based schemes have been developed, and some of these have already been used to image human cancers.
-
Whole-cell tracking probes. Whole-cell tracking probes are used to localize and follow the movement of cells, such as cancer cells, inflammatory cells, or stromal cells, which may be important for sustaining the tumor in vivo. The cells can be labeled with marker genes or other tags, such as green fluorescent protein, bioluminescent markers, or radiotracers.
Phenotypic probes can be used to image characteristics of the hallmarks of cancer and thus have the potential to provide biomarkers for assessing treatment efficacy and response. Specific biological phenomena that can be visualized and, in some cases, quantified include cell proliferation (Bading and Shields, 2008; Conti et al., 2008), hypoxia (Everitt et al., 2009; Krohn et al., 2008), apoptosis (Blankenberg, 2008a,b,c; Strauss et al., 2008), metabolic changes secondary to oncogenic activation (Plathow and Weber, 2008), angiogenesis (Cai and Chen, 2008; Cai et al., 2008), and the expression of receptors or antigens in tumor cells, such as hormone receptors and peptide receptors (Mankoff et al., 2008; Peterson et al., 2008).
Dynamic contrast-enhanced MRI (DCE-MRI) provides data reflective of tumor vascularity and angiogenesis and is one of the phenotypic imaging techniques most often used in clinical trials and clinical practice. Because changes in tumor vascularity tend to occur earlier than changes in tumor size, DCE-MRI is useful for monitoring the effects of or predicting responses to cancer treatments, including chemotherapy for breast and bladder cancers, radiotherapy for rectal and cervical cancers, and androgen deprivation for prostate cancer (Padhani and Leach, 2005). DCE-MRI is also being used as a biomarker in early clinical trials of antivascular drugs (Zhao et al., 2009). MR spectroscopic imaging, diffusion-weighted MRI,
and blood oxygenation level-dependent functional MRI, which are noninvasive, are also promising sources of biomarkers (Evelhoch et al., 2005; Padhani et al., 2009; Torigian et al., 2007). Multiple MRI techniques can be combined in a single examination.
18F-FDG is widely used in clinical oncology as a marker for the elevated level of glucose metabolism that occurs in most cancers (Gillies et al., 2008). In the case of GIST, 18F-FDG-PET and PET-CT have been used to monitor the response to targeted therapies, which are often cytostatic (Contractor and Aboagye, 2009). 18F-FDG-PET or PET-CT has now been successfully used to assess the response to treatment of malignant lymphomas (Bourre and Vuillez, 2009; Hutchings and Barrington, 2009) as well as lung, breast, cervical, colorectal, and esophageal cancers, among others (Avril et al., 2009; de Geus-Oei et al., 2009; Dose-Schwarz et al., 2009; Everitt et al., 2009; Hicks, 2009; Schwarz et al., 2009; Vriens et al., 2009a,b).
One emerging application of molecular imaging is monitoring the use of pathogens as therapeutic agents against cancer. For example, imaging has been used to assess virus dissemination in preclinical and clinical studies of viral therapies (Brader et al., 2009; Msaouel et al., 2009). Molecular imaging is also suitable for monitoring emerging cellular therapies, which employ stem cells or modified or genetically engineered cells (Arbab et al., 2009). Cells can be labeled, and their movement, growth, and death can then be monitored by optical, radioisotopic, or MR imaging (Akins and Dubey, 2008; Dobrenkov et al., 2008; Kang and Chung, 2008).
Codeveloping Imaging Biomarkers and Cancer Therapeutics: Theranostics and Beyond
There is much interest in the codevelopment of novel therapeutics and related molecular imaging biomarkers for use in diagnosis, treatment monitoring, and therapeutic response assessment. However, unlike in vitro diagnostic tests, most emerging molecular imaging methods rely on probes that must be injected, posing concerns similar to those faced in drug development. In some cases, a single entity serves as both a diagnostic imaging probe and a therapeutic agent, or a theranostic (Kassis et al., 2008).
In one example, receptor-targeting radiopeptides are being developed as single agents for molecular imaging and therapy of tumors that overexpress peptide receptors. To balance the clinical benefits and risks of radionuclide-based therapy, biodistribution, dosimetry, and toxicity must be carefully monitored. Imaging with targeted radiopeptides can help to address these and other issues, including detection of metastases, monitoring of the effects of chemotherapy, and detection of tumor progression or recurrence (de Jong et al., 2009; Mankoff et al., 2008).
Similar approaches are also being developed by the use of nanotechnol-
ogy. The unique properties of nanometer-sized particles can be used to target tumors with high affinity and specificity. When nanoparticles are linked with ligands such as monoclonal antibodies, peptides, or small molecules, they may simultaneously serve as therapeutic and imaging contrast agents, so their distribution can be tracked. Polymerized nanoparticle platform technology, which allows nanoparticles to be loaded with different targeting moieties, contrast agents, and therapeutic agents, could allow the development of highly personalized treatment regimens (Li et al., 2002).
Validation and Standardization
Guidelines and standards are urgently needed to validate new imaging biomarkers and related technologies. In addition, better means of standardizing and harmonizing the clinical use of validated biomarkers and technologies must be developed (Boellaard et al., 2010; Hutchings and Barrington, 2009; Wahl et al., 2009).
Serial measurements used to evaluate treatments need to be reproducible and accurate across institutions and trials to allow meaningful comparisons of different patient populations. During the IOM workshop on Improving the Quality of Cancer Clinical Trials, the participants suggested that imaging and image analysis laboratories for clinical trials be established at NCI-designated Comprehensive Cancer Centers. Such laboratories could ensure the proper execution of experimental imaging protocols and use image response assessment teams to interpret the imaging data from clinical trials and assist with the design of clinical trials (IOM, 2008).
The imaging platforms and techniques used in clinical trials vary widely. The need for standardization of anatomic data collection has been described (Strassburg et al., 2008). Furthermore, a framework for standardizing PET Response Criteria in Solid Tumors (PERCIST) has been drafted (Wahl et al., 2009). Such criteria can provide a template for use in the design of clinical trials and quantitative clinical reporting (Wahl et al., 2009). Although standardizing the hardware used at different institutions may be difficult, harmonization of the methods used by equipment vendors could improve the quality and consistency of the results that are obtained.
For emerging imaging approaches to be useful in clinical trials and clinical practice, acquisition and processing must be straightforward and consistent. However, emerging approaches often demand special expertise. For example, commercial manufacturers often collaborate with investigators in research hospitals to develop new MRI acquisition techniques. To apply these techniques in multicenter clinical trials, technical support must be provided to clinical institutions where it is lacking. In the short term, to facilitate multicenter trials, standards might specify only the minimum requirements for image acquisition (Carson et al., 2003).
Industry-academic partnerships could facilitate broad deployment of new data acquisition and quantitation methods. Innovations that academic researchers make could be shared with industry and incorporated into commercially available platforms. In turn, major industry stakeholders could serve as clearinghouses for data acquisition and analysis software and could refine their selections on the basis of feedback from academia (Carson et al., 2003).
Broad cooperation among all stakeholders, including academia, industry, NCI, FDA, and relevant professional societies, could also play a role in establishing standards for validating and applying imaging technologies. Some efforts to standardize and harmonize molecular imaging biomarkers have been undertaken, for example, by the National Institute of Standards and Technology and by the Oncology Biomarker Qualification Initiative sponsored by FDA, NCI, and CMS; but these efforts have not been coordinated (IOM, 2008). The Radioactive Drug Research Committee, which reviews and approves the use of radioactive drugs for research purposes under a mandate from FDA, could potentially play a larger role in establishing standards for validating and translating novel radioactive imaging agents for clinical use (ORS, 2009). For nanotechnology-based agents, standards for validation and translation might be established through the National Nanotechnology Coordination Office (NNCO) and the Nanoscale Science, Engineering, and Technology Subcommittee (NSET), which falls under the Committee on Technology of the National Science and Technology Council. NNCO and NSET are the central points of contact for federal activities related to nanotechnology research and development and so are in a position to coordinate standards across agencies (NNI, 2009).
This piecemeal approach to standards development is less than ideal, however. The committee therefore recommends that NCI, in cooperation with other agencies, establish a consistent, dynamic process to oversee the development of national unified standards as needed for oncology research. NCI should use this process when standards are required for any important new technology, technique, or breakthrough method, including biomedical imaging and other biomarkers. Standards should be published and updated in a timely manner so that they are useful in clinical trials.
SUMMARY
The recommendations in this chapter support the committee’s goal to incorporate innovative science and trial design into cancer clinical trials. The committee concluded that Cooperative Group clinical trials provide a unique opportunity to enable development of the emerging science of molecular biomarkers through retrospective analyses of archived clini-
cal samples and the prospective evaluation of biomarkers and imaging technologies.
Progress in the treatment of cancer patients depends on the effective incorporation of scientific advances into clinical trials. For example, to achieve the goals of targeted cancer therapy, the use of validated biomarkers will be essential. High-quality annotated biorepositories are needed to gain useful knowledge about the biology of cancer and biomarkers from the analysis of patient samples archived from past trials. The Cooperative Groups have a history of collecting biospecimens from the diverse populations of patients who participate in their clinical trials and maintaining them in repositories with detailed information about patient characteristics, treatment, and outcome. These resources have proven immensely valuable in the development of molecular-based classification schemes and diagnostic tests that now guide decisions on the most appropriate therapy for numerous types of cancer.
However, the maintenance of tissue banks and the analysis of stored samples are costly activities that are not fully covered by the core funding that NCI provides to the Cooperative Groups. Although current NCI policies and funding do support a portion of the costs involved in the collection and storage of samples, the groups must routinely seek supplemental funding to manage and maintain the repositories. Funding mechanisms to conduct retrospective studies of samples that have been collected in previous trials have also been problematic. Current NCI policies require research studies that propose to use specimens collected from intergroup protocols to undergo scientific review by a scientific steering committee before specimens are made available. However, such a review is not linked to funding, so investigators must often seek funding through other mechanisms. This process creates many review loops, time delays, and significant double jeopardy, in that each proposal requires at least two scientific reviews (one to receive specimens and one to receive funding) that are conducted at different times by different review groups.
In addition, access to biospecimens for research is inconsistent and can entail complex negotiations with the various custodians of the samples. Policies regarding ownership and access vary across different institutions, and this impedes progress. Furthermore, many hospitals discard samples after a period of time, so valuable resources are lost to research. Because the Cooperative Groups have a long history of responsible stewardship of repositories, they are a logical choice to play a central role in the ongoing efforts of NCI to establish consistent policies on ownership and access.
The committee recommends that NCI mandate the submission of annotated biospecimens to high-quality, standardized central biorepositories when samples are collected from patients in the course of Cooperative Group trials. NCI should also implement new funding mechanisms and
policies to support the management and use of those resources for retrospective correlative science. For example, all data, including biomarker data from serum, tissue, and imaging analyses should be considered precompetitive, unencumbered by intellectual property restrictions, and be made widely available. NCI should also establish a national inventory of samples held in the central repositories and have a defined process for access by researchers that includes a single scientific peer review linked to funding. In addition, clinical data accompanying biospecimens should be reported using standardized forms.
High levels of evidence are needed to validate and qualify biomarkers for specific uses, and current funding is inadequate to support the research needed to generate that evidence. The availability of a consistent and adequate funding source devoted to correlative studies with stored samples and with appropriate peer review that includes direct input from the group that collected the samples is imperative. The broader use of high-quality, standardized repositories would speed the pace of scientific and clinical advances at a much lower expense than would be required if new clinical samples had to be collected to study each new concept. The creation of a national inventory of samples held by the Cooperative Groups would also greatly facilitate important research in correlative science.
The committee also concluded that the Cooperative Groups are in a unique position to develop innovative designs for clinical trials and to demonstrate the feasibility and utility of using innovative, efficient designs in their clinical trials. The increasing complexity of cancer clinical trials, along with the great expense and high failure rate of late-stage clinical trials, has spurred innovation in trial design, with the aim of conducting clinical trials more efficiently and with a greater likelihood of success. The committee recommends that the Cooperative Groups lead the development and assessment of innovative designs for clinical trials that evaluate cancer therapeutics and biomarkers (including combinations of therapies).
The development and use of innovative trial designs could speed progress in clinical trials in numerous ways. For example, prospective clinical trial designs that randomize patients on the basis of biomarkers or treatments, or both, should be explored and evaluated. For targeted therapies, a predictive hypothesis for a biomarker should be put forward in the pre-clinical phase and tested in early-phase clinical trials (Phase I and II trials). Better Phase II trial designs are needed to more accurately assess which patients benefit from a particular therapy, and thus guide the decisions about whether to move into Phase III trials. Improved designs for Phase III trials, which are the most costly and lengthy trials and entail the majority of Cooperative Group trials, could lead to faster, more accurate conclusions about new therapeutics and in the process reduce costs and conserve resources. For example, recent innovations, such as the use of adaptive
designs for Phase II trials that assess response endpoints during trial accrual in real time, suggest that relevant clinical questions might be addressed more efficiently, with fewer patients required, with less time needed to show differences between groups, and with enhanced confidence in the clinically (and statistically) meaningful differences that are observed between groups. These or related designs may be particularly amenable for the comparison of treatment effects in patients with different biomarker profiles and could hasten the identification of the most promising predictive biomarkers that could be validated in a Phase III trial setting.
As new scientific methods and technologies develop and mature, standards are needed to ensure appropriate and consistent use. However, when new methods or technologies are incorporated into clinical trials, standards to ensure that the results collected at the various trial sites are consistent enough to attain accurate and meaningful conclusions from a study are often lacking. The current approach to standards development is often ad hoc, with the processes and rules for such things as committee composition and voting rules being reinvented on a case-by-case basis. This can lead to heterogeneous and delayed results.
Thus, NCI, in cooperation with other agencies, should establish a consistent, dynamic process to oversee the development of national unified standards as needed for oncology research. This process should be used by NCI when standards are required for any important new technology, tool, or breakthrough method (e.g., biomedical imaging and other biomarkers and biospecimens) and should replicate successful aspects of standards development by other standard-setting bodies, both governmental and nongovernmental (e.g., the American Society for Testing and Materials, the National Standards Foundation, the National Institute for Standards and Technology, the International Organization for Standardization, and professional societies). This process should utilize the input of experts in both subject matter and standards design in developing standards and include consistent operating procedures for developing standards (e.g., representation of stakeholders in committee composition, decision making, and voting rules). The resulting standards should be published and updated in a timely manner so that they are useful for the conduct of clinical trials. A more systematic, multidisciplinary, and dynamic approach to standards development fostered by NIH and NCI would be advantageous for the rapid and consistent setting of unified national standards as the need arises. NCI could further assist by facilitating the creation of systems and software to aid the process of standards implementation.
This need for standards will become increasingly important as the science of cancer research becomes more complex and more dependent on technologies such as imaging and on molecular tools such as biomarkers. In the case of biomedical imaging, many technologies and imaging reagents,
both those in current use and those under development, have the potential to provide information that can aid drug development and clinical decision making by providing improved means of diagnosis and monitoring. However, the lack of standards for image acquisition and quantification of results compromises the validity of the results and the interpretation of those results. In addition, the lack of harmonization of methods among the different vendors of imaging equipment compromises the quality and consistency of results. The consistent development of standard methodologies for established tumor-imaging modalities (e.g., computed tomography, fluorodeoxyglucose positron emission tomography, and conventional magnetic resonance imaging) by expert panels, along with a requirement that manufacturers meet those standards, could significantly improve the accuracy and value of those tests. Validation standards are also needed to continuously evaluate novel imaging methods and modalities to determine their merit and appropriate use.
Similarly, expert panels are needed to establish validation and qualification standards for the development and use of in vitro biomarker tests, to ensure that the results of those tests are consistent and accurate, and for the appropriate interpretation and use of those results. Such standards could also inform FDA guidance for the codevelopment of diagnostic-therapeutic combinations or for the inclusion of a biomarker test on the label for a drug or biologic that is already FDA approved.
Continued progress in the development and incorporation of innovative science into clinical trials will require the efforts of many stakeholders. For example, NCI, NIH, FDA, industry, investigators, and patients all have a role to play in defining an effective mechanism for the development of targeted cancer therapies. Effective collaboration among stakeholders will be particularly important for combination therapies, which may hold the key to successful personalized medicine because most cancers have multiple abnormalities. Companies may be reluctant to work with competitors to test promising combinations at an early and risky stage of development. To date, most combinations tested in Phase III trials have involved at least one agent currently approved by FDA. In addition, the steps needed and the data required to bring target therapies and combinations of products are not well defined. Issues relevant to effective collaboration are addressed in more detail in chapter 3.
REFERENCES
Adjei, A. A., M. Christian, and P. Ivy. 2009. Novel designs and end point for phase II clinical trials. Clinical Cancer Research 15(6):1866–1872.
Akins, E. J., and P. Dubey. 2008. Noninvasive imaging of cell-mediated therapy for treatment of cancer. Journal of Nuclear Medicine 49(Suppl. 2):180S–195S.
Allegra, C. 2009. The Clinical Implications of KRAS Testing in Colorectal Cancer. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, July 22, 2009, Washington, DC.
Arbab, A. S., B. Janic, J. Haller, E. Pawelczyk, W. Liu, and J. A. Frank. 2009. In vivo cellular imaging for translational medical research. Current Medical Imaging Reviews 5(1):19–38.
Avril, N., S. Sassen, and R. Roylance. 2009. Response to therapy in breast cancer. Journal of Nuclear Medicine 50(Suppl. 1):55S–63S.
Bading, J. R., and A. F. Shields. 2008. Imaging of cell proliferation: Status and prospects. Journal of Nuclear Medicine 49(Suppl. 2):64S–80S.
Barker, A. 2009. The FNIH Biomarkers Consortium Cancer Steering Committee: The I-SPY 2 Trial. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, July 22, 2009, Washington, DC.
Barker, A. D., C. C. Sigman, G. J. Kelloff, N. M. Hylton, D. A. Berry, and L. J. Esserman. 2009. I-SPY 2: An adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clinical Pharmacology & Therapeutics in press.
Barthel, F. M., M. K. Parmar, and P. Royston. 2009. How do multi-stage, multi-arm trials compare to the traditional two-arm parallel group design—a reanalysis of 4 trials. Trials 10(21).
Benjamin, R. S., H. Choi, H. A. Macapinlac, M. A. Burgess, S. R. Patel, L. L. Chen, D. A. Podoloff, and C. Charnsangavej. 2007. We should desist using RECIST, at least in GIST. Journal of Clinical Oncology 25(13):1760–1764.
Benson, A., Lyerly K., et al. 2009. Improving Medical Decisions through Comparative Effectiveness Research: Cancer as a Case Study. http://focr.org/files/CER_REPORT_FINAL.pdf (assessed June, 2010).
Berry, D. 2005. Statistical Innovations in Cancer Research. In Cancer Medicine 7th Ed., edited by D. W. Kufe, E. F. III, J. F. Holland, R. R. Weichselbaum, R. E. Pollock, R. C. Bast, W. K. Hong and W. N. Hait. London: BC Decker. Pp. 411–425.
Berry, D. 2006. Bayesian clinical trials. Nature Reviews Drug Discovery 5(1):27–36.
Biswas, S., D. D. Liu, J. J. Lee, and D. A. Berry. 2009. Bayesian clinical trials at the University of Texas M.D. Anderson Cancer Center. Clinical Trials 6(3):205–216.
Blankenberg, F. G. 2008a. In vivo detection of apoptosis. Journal of Nuclear Medicine 49(Suppl. 2):81S–95S.
Blankenberg, F. G. 2008b. In vivo imaging of apoptosis. Cancer Biology and Therapy 7(10):1525–1532.
Blankenberg, F. G. 2008c. Monitoring of treatment-induced apoptosis in oncology with PET and SPECT. Current Pharmaceutical Design 14(28):2974–2982.
Bloomfield, C. D., K. Mrozek, and M. A. Caligiuri. 2006. Cancer and leukemia group B leukemia correlative science committee: Major accomplishments and future directions. Clinical Cancer Research 12(11 Pt 2):3564s–3571s.
Boellaard, R., M. J. O’Doherty, W. A. Weber, F. M. Mottaghy, M. N. Lonsdale, S. G. Stroobants, W. J. Oyen, J. Kotzerke, O. S. Hoekstra, J. Pruim, P. K. Marsden, K. Tatsch, C. J. Hoekstra, E. P. Visser, B. Arends, F. J. Verzijlbergen, J. M. Zijlstra, E. F. Comans, A. A. Lammertsma, A. M. Paans, A. T. Willemsen, T. Beyer, A. Bockisch, C. Schaefer-Prokop, D. Delbeke, R. P. Baum, A. Chiti, and B. J. Krause. 2010. FDG PET and PET/CT: EANM procedure guidelines for tumour PET imaging: version 1.0. European Journal of Nuclear Medicine and Molecular Imaging 37(1):181–200.
Bourre, J. C., and J. P. Vuillez. 2009. Imaging in oncology and international rules for evaluation: The nuclear medicine. Bulletin du Cancer, Nov;96(11):1127–1137. (In French.)
Bradbury, M., and H. Hricak. 2005. Molecular MR imaging in oncology. Magnetic Resonance Imaging Clinics of North America 13(2):225–240.
Brader, P., K. J. Kelly, N. Chen, Y. A. Yu, Q. Zhang, P. Zanzonico, E. M. Burnazi, R. E. Ghani, I. Serganova, H. Hricak, A. A. Szalay, Y. Fong, and R. G. Blasberg. 2009. Imaging a genetically engineered oncolytic vaccinia virus (glv-1h99) using a human norepinephrine transporter reporter gene. Clinical Cancer Research 15(11):3791–3801.
Bria, E., M. Di Maio, P. Carlini, F. Cuppone, D. Giannarelli, F. Cognetti, and M. Milella. 2009. Targeting targeted agents: Open issues for clinical trial design. Journal of Experimental and Clinical Cancer Research 22(28):66.
Buckler, A. J., J. L. Mulshine, R. Gottlieb, B. Zhao, P. D. Mozley, and L. Schwartz. 2010. The use of volumetric CT as an imaging biomarker in lung cancer. Academic Radiology 17(1):100–106.
Cai, W., and X. Chen. 2008. Multimodality molecular imaging of tumor angiogenesis. Journal of Nuclear Medicine 49(Suppl. 2):113S–128S.
Cai, W., G. Niu, and X. Chen. 2008. Imaging of integrins as biomarkers for tumor angiogenesis. Current Pharmaceutical Design 14(28):2943–2973.
Canetta, R. 2009. Testing Therapeutic Combinations. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, July 22, 2009, Washington, DC.
Carson, P. L., M. Giger, M. J. Welch, H. Halpern, K. Kurdziel, M. Vannier, J. L. Evelhoch, G. S. Gazelle, S. E. Seltzer, P. Judy, W. R. Hendee, and J. D. Bourland. 2003. Biomedical imaging research opportunities workshop: Report and recommendations. Radiology 229(2):328–339.
Choi, H. 2005. Critical issues in response evaluation on computed tomography: Lessons from the gastrointestinal stromal tumor model. Current Oncology Reports 7(4):307–311.
Choi, H., C. Charnsangavej, S. C. Faria, H. A. Macapinlac, M. A. Burgess, S. R. Patel, L. L. Chen, D. A. Podoloff, and R. S. Benjamin. 2007. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: Proposal of new computed tomography response criteria. Journal of Clinical Oncology 25(13):1753–1759.
Compton, C. 2006. The cancer and leukemia group B pathology committee at 50. Clinical Cancer Research 12(11 Pt 2):3617s–3621s.
Compton, C. 2009. Biorepositories, Biomarkers and Correlative Science. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, April 23, 2009, Washington, DC.
Conti, P. S., J. R. Bading, P. P. Mouton, J. M. Links, M. M. Alauddin, J. D. Fissekis, H. T. Ravert, J. Hilton, D. F. Wong, and J. H. Anderson. 2008. In vivo measurement of cell proliferation in canine brain tumor using C-11-labeled FMAU and PET. Nuclear Medicine and Biology 35(1):131–141.
Contractor, K. B., and E. O. Aboagye. 2009. Monitoring predominantly cytostatic treatment response with 18F-FDG PET. Journal of Nuclear Medicine 50(Suppl. 1):97S–105S.
de Geus-Oei, L. F., D. Vriens, H. W. van Laarhoven, W. T. van der Graaf, and W. J. Oyen. 2009. Monitoring and predicting response to therapy with 18F-FDG PET in colorectal cancer: A systematic review. Journal of Nuclear Medicine 50(Suppl. 1):43S–54S.
de Jong, M., W. A. Breeman, D. J. Kwekkeboom, R. Valkema, and E. P. Krenning. 2009. Tumor imaging and therapy using radiolabeled somatostatin analogues. Accounts of Chemical Research 42(7):873–880.
Dobrenkov, K., M. Olszewska, Y. Likar, L. Shenker, G. Gunset, S. Cai, N. Pillarsetty, H. Hricak, M. Sadelain, and V. Ponomarev. 2008. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. Journal of Nuclear Medicine 49(7):1162–1170.
Dose-Schwarz, J., R. Tiling, S. Avril-Sassen, S. Mahner, A. Lebeau, C. Weber, M. Schwaiger, F. Janicke, M. Untch, and N. Avril. 2009. Assessment of residual tumour by FDG-PET: Conventional imaging and clinical examination following primary chemotherapy of large and locally advanced breast cancer. British Journal of Cancer 102(1):35–41.
Eiseman, E., and S. B. Haga. 1999. Handbook of Human Tissue Sources: A National Resource of Human Tissue Samples. Santa Monica, CA: Science and Technology Policy Institute, RAND Corporation.
Eisenhauer, E. A., P. Therasse, J. Bogaerts, L. H. Schwartz, D. Sargent, R. Ford, J. Dancey, S. Arbuck, S. Gwyther, M. Mooney, L. Rubinstein, L. Shankar, L. Dodd, R. Kaplan, D. Lacombe, and J. Verweij. 2009. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). European Journal of Cancer 45(2):228–247.
Esserman, L. 2009. Transforming Clinical Trials: The I-SPY TRIAL Process. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, July 22, 2009, Washington, DC.
Evelhoch, J., M. Garwood, D. Vigneron, M. Knopp, D. Sullivan, A. Menkens, L. Clarke, and G. Liu. 2005. Expanding the use of magnetic resonance in the assessment of tumor response to therapy: Workshop report. Cancer Research 65(16):7041–7044.
Everitt, S., R. J. Hicks, D. Ball, T. Kron, M. Schneider-Kolsky, T. Walter, D. Binns, and M. MacManus. 2009. Imaging cellular proliferation during chemo-radiotherapy: A pilot study of serial 18F-FLT positron emission tomography/computed tomography imaging for non-small-cell lung cancer. International Journal of Radiation Oncology and Biological Physics 75(4):1098–1104.
FDA (Food and Drug Administration). 2009. Class Labeling Changes to Anti-EGFR Monoclonal Antibodies, Ccetuximab (Erbitux) and Panitumumab (Vectibix): KRAS Mutations. http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm172905.htm (accessed August 3, 2009).
FDA. 2010a. Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071072.htm (accessed March 3, 2010).
FDA. 2010b. Draft Guidance for Industry: Adaptive Design Clinical Trials for Drugs and Biologics. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201790.pdf (accessed March 3, 2010)
FDA. 2010c. FDA Issues Guidance to Help Streamline Medical Device Clinical Trials. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm199993.htm (accessed March 3, 2010).
Galanis, E., J. C. Buckner, M. J. Maurer, R. Sykora, R. Castillo, K. V. Ballman, and B. J. Erickson. 2006. Validation of neuroradiologic response assessment in gliomas: Measurement by RECIST, two-dimensional, computer-assisted tumor area, and computer-assisted tumor volume methods. Neuro-Oncology 8(2):156–165.
Gavrielides, M. A., L. M. Kinnard, K. J. Myers, and N. Petrick. 2009. Noncalcified lung nodules: Volumetric assessment with thoracic CT. Radiology 251(1):26–37.
Gehan, E. A., and M. C. Tefft. 2000. Will there be resistance to the RECIST (response evaluation criteria in solid tumors)? Journal of the National Cancer Institute 92(3):179–181.
Gillies, R. J., I. Robey, and R. A. Gatenby. 2008. Causes and consequences of increased glucose metabolism of cancers. Journal of Nuclear Medicine 49(Suppl. 2):24S–42S.
Goldberg, P. 2009. KRAS finding changes oncology practice but poses profound regulatory dilemma. The Cancer Letter 35(4):1–8.
Gonen, M. 2009. Bayesian clinical trials: No more excuses. Clinical Trials 6(3):203–204.
Grassi, R., R. Lagalla, and A. Rotondo. 2008. Genomics, proteomics, mems and saif: Which role for diagnostic imaging? La Radiolia Medica 113(6):775–778.
Hamilton, S. 2009. Biorepositories, Biomarkers, and Correlative Science. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, April 23, 2009, Washington, DC.
Hanahan, D., and R.A. Weinberg, 2000. The hallmarks of cancer. Cell 100:57–70.
Hayes, D. F. 2009. Reimbursement Hurdles: Clinician Perspective. Presentation at the National Cancer Policy Forum Workshop on Policy Issues in the Development of Personalized Medicine in Oncology, June 9, 2009, Washington, DC.
Hicks, R. J. 2009. Role of 18F-FDG PET in assessment of response in non-small cell lung cancer. Journal of Nuclear Medicine 50(Suppl. 1):31S–42S.
Hohenberger, P., and E. Wardelmann. 2006. Surgical considerations for gastrointestinal stroma tumor. Der Chirurg 77(1):33–40. (In German.)
Hoshida, Y., A. Villanueva, M. Kobayashi, J. Peix, D. Y. Chiang, A. Camargo, S. Gupta, J. Moore, M. J. Wrobel, J. Lerner, M. Reich, J. A. Chan, J. N. Glickman, K. Ikeda, M. Hashimoto, G. Watanabe, M. G. Daidone, S. Roayaie, M. Schwartz, S. Thung, H. B. Salvesen, S. Gabriel, V. Mazzaferro, J. Bruix, S. L. Friedman, H. Kumada, J. M. Llovet, and T. R. Golub. 2008. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. New England Journal of Medicine 359(19):1995–2004.
Huang, P. 2010. The AZ-Merck Collaboration. Presentation at the National Cancer Policy Forum Workshop on Extending the Spectrum of Precompetitive Oncology Biomedical Research, February 10, 2010, Washington, DC.
Hutchings, M., and S. F. Barrington. 2009. PET/CT for therapy response assessment in lymphoma. Journal of Nuclear Medicine 50(Suppl. 1):21S–30S.
Hylton, N., L. Carey, A. DeMichele, J. Blume, G. Broadwater, S. Madhavan, M. Rosen, S. George, L. Esserman, ISPY Clinical Research, Pathology and Radiology Investigators. 2007. Characterizing the Biology and Response of Locally Advanced Breast Cancer in Women Undergoing Neoadjuvant Therapy: Preliminary Results from the I-SPY Trial, abstr 80. San Antonio Breast Cancer Symposium, December 13–16.
Hylton, N., J. Blume, C. Gatsonis, R. Gomez, W. Bernreuter, E. Pisano, M. Rosen, H. Marques, L. Esserman, M. Schnall, and the ACRIN 6657/I-SPY Trial Team. 2009. MRI tumor volume for predicting response to neoadjuvant chemotherapy in locally advanced breast cancer: Findings from ACRIN 6657/CALGB 150007. Journal of Clinical Oncology 27(Suppl. 15):A-529.
IOM (Institute of Medicine). 2007. Cancer Biomarkers: The Promises and Challenges of Improving Detection and Treatment. Washington, DC: The National Academies Press.
IOM. 2008. Improving the Quality of Cancer Clinical Trials: Workshop Summary. Washington, DC: The National Academies Press.
IOM. 2009. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: The National Academies Press.
IOM. 2010. Policy Issues in the Development of Personalized Medicine in Oncology: Workshop Summary. Washington, DC: The National Academies Press.
Kang, J. H., and J. K. Chung. 2008. Molecular-genetic imaging based on reporter gene expression. Journal of Nuclear Medicine 49(Suppl. 2):164S–179S.
Kassis, A. I., H. Korideck, K. Wang, P. Pospisil, and S. J. Adelstein. 2008. Novel prodrugs for targeting diagnostic and therapeutic radionuclides to solid tumors. Molecules 13(2):391–404.
Katsnelson, A. 2009. Adaptive evolution: A once-rare type of clinical trial that violates one of the sacred tenets of trial design is taking off, but is it worth the risk? The Scientist 23(8):55–57.
Krohn, K. A., J. M. Link, and R. P. Mason. 2008. Molecular imaging of hypoxia. Journal of Nuclear Medicine 49(Suppl. 2):129S–148S.
Li, K. C., S. Guccione, and M. D. Bednarski. 2002. Combined vascular targeted imaging and therapy: A paradigm for personalized treatment. Journal of Cellular Biochemistry Supplement 39:65–71.
Lutzker, S. 2009. Clinical Evaluation of 2NME Combinations for Oncology Indications. Presentation at the Conference on Clinical Cancer Research, Engelberg Center for Health Care Reform of the Brookings Institution and Friends of Cancer Research, September 14, 2009.
Lyman, G. H, L. E. Cosler, N. M. Kuderer, J. Hornberger. 2007. Impact of a 21-gene RT-PCR assay on treatment decisions in early-stage breast cancer: An economic analysis based on prognostic and predictive validation studies. Cancer 109(6):1011–1018.
Mankoff, D. A., J. M. Link, H. M. Linden, L. Sundararajan, and K. A. Krohn. 2008. Tumor receptor imaging. Journal of Nuclear Medicine 49(Suppl. 2):149S–163S.
Marshall, E. 2009. Medicine under the microscope. Science 326:1183–1185.
Mauer, A. M., E. S. Rich, and R. L. Schilsky. 2007. The role of cooperative groups in cancer clinical trials. Cancer Treatment and Research 132:111–129.
McClellan, M., and J. S. Benner. 2009. Four important steps toward 21st century care for patients with cancer. The Oncologist 14(4):313–316.
McShane, L. M., S. Hunsberger, and A. A. Adjei. 2009. Effective incorporation of biomarkers into Phase II trials. Clinical Cancer Research 15(6):1898–1905.
Merck. 2009. AstraZeneca and Merck & Co., Inc. Form Pioneering Collaboration to Investigate Novel Combination Anticancer Regimen. http://www.merck.com/newsroom/news-release-archive/research-and-development/2009_0601.html (accessed March 11, 2010).
Miller, A. B., B. Hogestraeten, M. Staquet, and A. Winkler. 1981. Reporting results of cancer treatment. Cancer 47(1):207–214.
Modjtahedi, H., and S. Essapen. 2009. Epidermal growth factor receptor inhibitors in cancer treatment: Advances, challenges and opportunities. Anticancer Drugs 20(10):851-855.
Msaouel, P., A. Dispenzieri, and E. Galanis. 2009. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: An overview. Current Opinion in Molecular Therapy 11(1):43–53.
Mulshine, J. L., and D. M. Jablons. 2009. Volume CT for diagnosis of nodules found in lung-cancer screening. New England Journal of Medicine 361(23):2281–2282.
NCI (National Cancer Institute). 2007a. Best Practices for Biospecimen Resources. Bethesda, MD: National Cancer Institute. http://biospecimens.cancer.gov/global/pdfs/NCI_Best_Practices_060507.pdf (accessed September 18, 2009).
NCI. 2007b. Performance Standards Reporting Requirements for Essential Assays in Clinical Trials. Bethesda, MD: National Cancer Institute. http://www.cancerdiagnosis.nci.nih.gov/pdf/PACCT_Assay_Standards_Document.pdf (accessed September 11, 2009).
NCI. 2008. Custodianship and Ownership Issues in Biospecimen Research: Symposium-Workshop Summary. Rockville, MD: National Cancer Institute.
Niederhuber, J. 2009. Presentation at the Conference on Clinical Cancer Research, Engelberg Center for Health Care Reform of the Brookings Institution and Friends of Cancer Research, September 14, 2009.
NNI (National Nanotechnology Initiative). 2009. National Nanotechnology Initiative. http://www.nano.gov (accessed November 17, 2009).
ORS (Office of Research Services, National Institutes of Health). 2009. Radioactive Drug Research Center. http://drs.ors.od.nih.gov/services/rsc/rdrc (accessed November 17, 2009).
Padhani, A. R., and M. O. Leach. 2005. Antivascular cancer treatments: Functional assessments by dynamic contrast-enhanced magnetic resonance imaging. Abdominal Imaging 30(3):324–341.
Padhani, A. R., G. Liu, D. M. Koh, T. L. Chenevert, H. C. Thoeny, T. Takahara, A. Dzik-Jurasz, B. D. Ross, M. Van Cauteren, D. Collins, D. A. Hammoud, G. J. Rustin, B. Taouli, and P. L. Choyke. 2009. Diffusion-weighted magnetic resonance imaging as a cancer biomarker: Consensus and recommendations. Neoplasia 11(2):102–125.
Paik, S., S. Shak, G. Tang, C. Kim, J. Baker, M. Cronin, F. L. Baehner, M. G. Walker, D. Watson, T. Park, W. Hiller, E. R. Fisher, D. L. Wickerham, J. Bryant, and N. Wolmark. 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New England Journal of Medicine 351(27):2817–2826.
Paik, S., G. Tang, S. Shak, C. Kim, J. Baker, W. Kim, M. Cronin, F. L. Baehner, D. Watson, J. Bryant, J. P. Costantino, C. E. Geyer, Jr., D. L. Wickerham, and N. Wolmark. 2006. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. Journal of Clinical Oncology 24(23):3726–3734.
Parmar, M. K., F. M. Barthel, M. Sydes, R. Langley, R. Kaplan, E. Eisenhauer, M. Brady, N. James, M. A. Bookman, A. M. Swart, W. Qian, and P. Royston. 2008. Speeding up the evaluation of new agents in cancer. Journal of the National Cancer Institute 100(17):1204–1214.
PCAST (President’s Council of Advisors on Science and Technology). 2008. Priorities for Personalized Medicine. Washington, DC: Office of Science and Technology Policy.
Peterson, L. M., D. A. Mankoff, T. Lawton, K. Yagle, E. K. Schubert, S. Stekhova, A. Gown, J. M. Link, T. Tewson, and K. A. Krohn. 2008. Quantitative imaging of estrogen receptor expression in breast cancer with PET and 18F-fluoroestradiol. Journal of Nuclear Medicine 49(3):367–374.
Plathow, C., and W. A. Weber. 2008. Tumor cell metabolism imaging. Journal of Nuclear Medicine 49(Suppl. 2):43S–63S.
RSNA (Radiological Society of North America). 2010. Quantitative Imaging Biomarkers Alliance. http://www.rsna.org/Research/qiba_intro.cfm (accessed January 4, 2010).
Rubinstein, L. V., J. Crowley, P. Ivy, M. LeBlanc, and D. Sargent. 2009. Randomized phase II designs. Clinical Cancer Research 15(6):1883–1890.
Sargent, D. 2009. Challenges in Validating Biomarkers for Approved Agents. Presentation to the Institute of Medicine Committee on Cancer Clinical Trials and the NCI Cooperative Group Program, July 22, 2009, Washington, DC.
Sargent, D. J., and J. M. Taylor. 2009. Current issues in oncology drug development, with a focus on phase II trials. Journal of Biopharma Statistics 19(3):556–562.
Schiller, J. 2004. Clinical trial design issues in the era of targeted therapies. Clinical Cancer Research 10(12 Pt 2):4281s–4282s.
Schilsky, R. 2009. Technological Hurdles. Presentation to the National Cancer Policy Forum, Workshop on Policy Issues in Development of Personalized Medicine in Oncology, June 8, 2009, Washington, DC.
Schilsky, R. L., L. M. Dressler, D. Bucci, L. Monovich, S. Jewell, S. Suster, M. A. Caligiuri, P. W. Kantoff, and C. Compton. 2002. Cooperative group tissue banks as research resources: The cancer and leukemia group B tissue repositories. Clinical Cancer Research 8(5):943–948.
Schwarz, J. K., P. W. Grigsby, F. Dehdashti, and D. Delbeke. 2009. The role of 18F-FDG PET in assessing therapy response in cancer of the cervix and ovaries. Journal of Nuclear Medicine 50(Suppl. 1):64S–73S.
Sevinc, A., and N. S. Turhal. 2008. Please, desist RECIST criteria in GIST, at least in me. Onkologie 31(10):556.
Siena, S., A. Sartore-Bianchi, F. Di Nicolantonio, J. Balfour, and A. Bardelli. 2009. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. Journal of the Cancer Institute 101(19):1308–1324.
Simon, R. 2008a. Lost in translation: Problems and pitfalls in translating laboratory observations to clinical utility. European Journal of Cancer 44(18):2707–2713.
Simon, R. 2008b. The use of genomics in clinical trial design. Clinical Cancer Research 14(19):5984–5993.
Spear, B. B., M. Heath-Chiozzi, and J. Huff. 2001. Clinical application of pharmacogenetics. Trends in Molecular Medicine 7:201–204.
Stacchiotti, S., P. Collini, A. Messina, C. Morosi, M. Barisella, R. Bertulli, C. Piovesan, P. Dileo, V. Torri, A. Gronchi, and P. G. Casali. 2009. High-grade soft-tissue sarcomas: Tumor response assessment—Pilot study to assess the correlation between radiologic and pathologic response by using RECIST and Choi criteria. Radiology 251(2):447–456.
Strassburg, J., T. Junginger, T. Trinh, O. Püttcher, K. Oberholzer, R. J. Heald, and P. Hermanek. 2008. Magnetic resonance imaging (MRI)-based indication for neoadjuvant treatment of rectal carcinoma and the surrogate endpoint CRM status. International Journal of Colorectal Disease 23(11):1099–1107.
Strauss, H. W., F. Blankenberg, J. L. Vanderheyden, and J. Tait. 2008. Translational imaging: Imaging of apoptosis. Handbook of Experimental Pharmacology 185(Pt 2):259–275.
Sydes, M. R., M. K. Parmar, N. D. James, N. W. Clarke, D. P. Dearnaley, M. D. Mason, R. C. Morgan, K. Sanders, and P. Royston. 2009. Issues in applying multi-arm multi-stage methodology to a clinical trial in prostate cancer: The MRC STAMPEDE trial. Trials 10(39).
Tan, D. S., G. V. Thomas, M. D. Garrett, U. Banerji, J. S. de Bono, S. B. Kaye, and P. Workman. 2009. Biomarker-driven early clinical trials in oncology: A paradigm shift in drug development. Cancer Journal 15(5):406–420.
Thall, P. F., R. Simon, and S. S. Ellenberg. 1988. Two-stage selection and testing designs for clinical trials. Biometrika 75:303–310.
Thall, P. F., R. Simon, S. S. Ellenberg. 1989. A two-stage design for choosing among several experimental treatments and a control in clinical trials. Biometrics 45(2):537–547.
Torigian, D. A., S. S. Huang, M. Houseni, and A. Alavi. 2007. Functional imaging of cancer with emphasis on molecular techniques. CA: A Cancer Journal for Clinicians 57(4):206–224.
Vriens, D., L. F. de Geus-Oei, W. T. van der Graaf, and W. J. Oyen. 2009a. Tailoring therapy in colorectal cancer by PET-CT. Quarterly Journal of Nuclear Medicine and Molecular Imaging 53(2):224–244.
Vriens, D., H. W. van Laarhoven, J. J. van Asten, P. F. Krabbe, E. P. Visser, A. Heerschap, C. J. Punt, L. F. de Geus-Oei, and W. J. Oyen. 2009b. Chemotherapy response monitoring of colorectal liver metastases by dynamic Gd-DTPA-enhanced MRI perfusion parameters and 18F-FDG PET metabolic rate. Journal of Nuclear Medicine 50(11):1777–1784.
Wahl, R. L., H. Jacene, Y. Kasamon, and M. A. Lodge. 2009. From RECIST to PERCIST: Evolving considerations for PET response criteria in solid tumors. Journal of Nuclear Medicine 50(Suppl. 1):122S–150S.
Weber, W. A. 2009. Assessing tumor response to therapy. Journal of Nuclear Medicine 50(Suppl. 1):1S–10S.
WHO (World Health Organization). 1979. WHO Handbook for Reporting Results of Cancer Treatment. Offset Publication No. 48. Geneva, Switzerland: World Health Organization.
Wickerham, D. L., M. J. O’Connell, J. P. Costantino, W. M. Cronin, S. Paik, C. E. Geyer, P. A. Ganz, N. Petrelli, E. P. Mamounas, T. B. Julian, and N. Wolmark. 2008. The half century of clinical trials of the National Surgical Adjuvant Breast and Bowel Project. Seminars in Oncology 35(5):522–529.
Winslow, R. 2009. AstraZeneca, Merck to test cancer drugs in “cocktail.” Wall Street Journal June 1, 2009: B1.
Woodcock, J. 2009. Combining Investigational Agents in Drug Development. Presentation at the Conference on Clinical Cancer Research, Engelberg Center for Health Care Reform of the Brookings Institution and Friends of Cancer Research, September 14, 2009.
Zakian, K. L., J. A. Koutcher, D. Ballon, H. Hricak, and C. C. Ling. 2001. Developments in nuclear magnetic resonance imaging and spectroscopy: Application to radiation oncology. Seminars in Radiation Oncology 11(1):3–15.
Zhao, B., L. H. Schwartz, and S. M. Larson. 2009. Imaging surrogates of tumor response to therapy: Anatomic and functional biomarkers. Journal of Nuclear Medicine 50(2):239–249.











































