
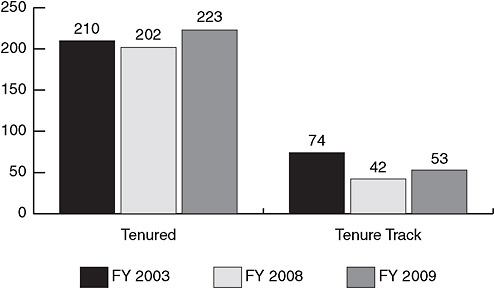
FIGURE 3-1 While the number of senior NIAID/NIH tenured investigators is relatively stable, the number of NIAID/NIH tenure-track principal investigators is decreasing.
SOURCE: Lane, 2009. Courtesy of John Gallin, 2009 (unpublished).
cause publishing results from this work is easier, and the difficulties of getting a clinical trial protocol approved can be avoided. Accordingly, NIAID has created a protocol development program to decrease the burden of regulatory and administrative requirements and optimize the use of existing clinical research tools. The program’s goal is to allow investigators to focus on their work as clinical scientists rather than having to serve as operations managers of a complex regulatory process. When investigators provide a robust scientific idea with a strong hypothesis, appropriate endpoints, and a sound study design, NIAID’s Office of the Clinical Director helps them navigate such regulatory issues as ethics review, technology transfer, safety concerns, and interactions with Institutional Review Boards (IRBs) and the FDA. This support can help investigators implement a trial successfully. In addition to principal investigators, multidisciplinary support staffs are necessary to complete a clinical trial successfully. Biostatisticians, epidemiologists, laboratory technicians, and administrative support personnel are just a few of the types of staff needed.
The importance of involving knowledgeable staff throughout a study was highlighted in a discussion of cardiovascular and depression clinical trials. Califf referred to the crucial role of a well-trained, intelligent data monitoring committee tasked with evaluating interim trial results. Data fluctuations revealed by interim trial monitoring require analysis but do not always
indicate that a trial should be discontinued. For instance, when death is the primary outcome of a trial, data fluctuations may indicate an adverse effect on mortality of the treatment being studied or a regular clinical occurrence unrelated to the study drug. Califf noted that if it were not for a particularly well-informed data monitoring committee, the ISIS-2 (Second International Study of Infarct Survival) trial would have been most recently discontinued at 5,000 patients, with aspirin showing an adverse effect on mortality. William Potter, most recently Vice President of Translational Neuroscience, Merck Research Labs, Merck & Co., Inc., indicated that in the depression studies in which he has been involved, interim data that indicate a possible adverse effect usually result in a trial’s being discontinued.
Because clinical trials are conducted in an ad hoc fashion, and study personnel of varying professional quality are recruited and trained anew at each site, inconsistencies in trial execution across sites are not unusual. Woodcock explained that the failure to execute a clinical trial successfully is often attributable in part to the fact that ensuring proper execution of a single trial is no one’s full-time job. The core activities of a clinical trial are largely supplemental responsibilities assigned to a variety of staff in addition to their full-time work.
Califf noted that clinical investigators often are unsupported by their academic institutions and are left largely to their own devices to design a trial and gather the necessary resources. The major reason for this lack of support, he suggested, is that clinical research is not widely respected among academics as a truly intellectual endeavor. Califf explained that, while investigators who are leading large, multisite trials predicted to have a major impact on clinical practice enjoy such respect, this is not the case for those conducting less visible work or just starting out in their research careers. A number of workshop participants expressed their support for rewarding academic researchers who conduct clinical trials. Early career development at the graduate and postgraduate levels could create incentives for more experts to enter the field of clinical research.
NEED TO NAVIGATE ADMINISTRATIVE AND REGULATORY REQUIREMENTS
The internal requirements of an academic institution, federal agency, or pharmaceutical company for reviewing multiple aspects of a clinical trial can significantly delay its initiation. In the case of an academic institution conducting a clinical trial for a pharmaceutical company, the internal review processes of both organizations are involved. In addition to such internal requirements, myriad federal and state regulatory requirements affect the conduct of clinical trials. Adhering to these many requirements is a significant challenge for investigators. Moreover, the delays incurred
increase the time cost of a trial and decrease its overall efficiency. U.S. academic institutions typically take longer to navigate the approval process (i.e., from budget/contract to IRB approval) compared to private or academic institutions abroad. The protracted timeline to approve a clinical trial through U.S. institutions is one reason industry sponsors look outside the United States to initiate studies.
Institutional Review Board Approval
Gaining IRB approval is a requirement of the clinical trial process.3 Lane’s survey of intramural NIH investigators revealed that the top four barriers to clinical research are:
-
Ethical/IRB approval,
-
scientific review/protocol approval,
-
interaction with industry and issues with technology transfer, and
-
adequacy of resources.
Lane noted that there is often a lack of clarity among investigators regarding the roles and responsibilities of different oversight bodies. In focus groups with the investigators polled, it became clear that IRB missions can be difficult to interpret. Institutions have used IRBs for risk management above and beyond what is required for human subjects research, and included in their purview travel policies, conflicts of interest, and other management issues. Investigators often do not know or understand what the IRB expects of them, and the IRB decision-making process can be lacking in timeliness and accountability. Investigators reported that if the IRB process results in a request for changes to a trial, they often lack the resources to fulfill the request.
Paul Hébert, Editor-in-Chief of the Canadian Medical Association Journal (CMAJ) and critical care physician at the Ottawa Hospital, commented on the difficulties associated with IRB ethics review. A key concern is that IRBs are accountable only to their own institution and not to the greater public good. Hébert suggested that, to improve the regulatory system, IRBs should be held accountable to the community for the decisions they make. Moreover, decreasing the regulatory burden surrounding clinical trials does not need to be a zero-sum game. For example, decreasing the number of ethics reviews for a trial from 50 to 10 would be a substantial improvement over the current situation.
Carla Greenbaum, Director of the Benaroya Research Institute Diabetes Program and Clinical Research Center, shared her experiences and insights into the IRB process from the perspective of diabetes research. She noted that regulations vary by geographic location. Depending on location, for example, IRBs have different answers to the question of when research in children is appropriate, and they differ as well in how clinical research terms and phrases such as “minimal risk,” “slightly greater than minimal risk,” and “benefit” are defined. Geographic variation is also seen in IRB definitions of reportable adverse events, definitions of equipoise4 and whether a proposed study satisfies this requirement, and rules regarding whether permission can be granted for clinical trial samples to be retained indefinitely by the pharmaceutical sponsor versus NIDDK. Because multiple IRB approvals are required for most large, multisite clinical trials, these inconsistencies in IRB determinations and standards across the country complicate and delay the process of conducting a clinical trial and can inhibit the ability of investigators to implement the same trial protocol across all study sites—a critical factor for developing valid trial results.
Informed Consent
Informed consent refers to the process and documents associated with educating individuals on the details of a clinical trial and potentially gaining their consent to participate in the study.5 Obtaining informed consent from each subject in a clinical trial requires a significant amount of time. The informed consent process includes developing appropriately worded consent documents, discussing the documents and the clinical trial process with individual patients, obtaining the required patient signatures on the documents, and keeping track of the paperwork generated throughout the enrollment process.
As an example of the time and effort necessary to satisfy informed consent requirements, Greenbaum described a hypothetical scenario from her experience in diabetes research. A family consisting of two parents and four children, one with diabetes, decides to be screened for participation in a diabetes prevention study. The consent process for this family requires a total of 8 separate consent forms, each 6 pages long and requiring 16 signatures, plus 5 Health Information Portability and Accountability Act
(HIPAA) forms. This paperwork is in addition to the extensive monitoring and compliance that accompany the consent process.
Greenbaum stressed the irrationality of the current situation in which an individual’s ability to participate in clinical research is dictated by geographic location. As a result of the level of local control exerted over clinical research, patients who frequent a hospital or medical center in one area of town may have access to certain clinical trials, whereas those at a hospital across town do not. For instance, some clinical trials organized through TrialNet are not approved at one institution in Greenbaum’s area because it is their policy that studies should not be conducted in children until the therapeutic approach has first been demonstrated to work in adults.
Protracted Time from Protocol Approval to Trial Activation
Administrative burdens are not always imposed on investigators by external laws and regulations. As Lane noted, many bottlenecks arise internally and are imposed by institutions that are home to the research workforce. In the government-sponsored Occluded Artery Trial (OAT), for instance, it took 3 years from the first NIH steering committee meeting to the start of the trial. Because clinical research relies on substantial human effort that incurs large labor costs, the timeline for a clinical trial affects overall cost. DiMasi and colleagues estimated that in 2000, the average cost to develop a new drug was $802 million, and time costs associated with the length of research and development accounted for half of this cost (DiMasi et al., 2003).
For the pharmaceutical industry, protracted timelines increase cost and reduce revenue as medications typically have a finite life before losing patent protection and creating an opportunity for generic competitors. Moreover, when a trial addresses a question important for medical practice, increasing the time it takes to obtain an answer can reduce the impact of the results. Musa Mayer, breast cancer survivor, advocate, and author (AdvancedBC.org), commented that if clinical trials are subject to significant delays, the standard of care can move on in the absence of phase III data. Thus, obstacles and delays in clinical trials move health care further away from evidence-based practice. Moreover, if the time lag is significant, the results of a lengthy, expensive trial may already have been rendered irrelevant by changes in clinical practice when they finally become available.
The one-off nature of trial organization, mentioned by a number of workshop participants as a major barrier to the efficient conduct of trials, is one factor leading to prolonged trial startup times. Years can elapse from the time researchers begin talking about a study idea to the point at
which they assemble the appropriate investigators, develop collaborations, establish study sites, and initiate the trial.
Renzo Canetta, Vice President of Oncology Global Clinical Research, Bristol-Myers Squibb, provided an example of the internal administrative burdens faced by industry. Historically, Bristol-Myers Squibb has required 8 months, or 34 internal review cycles, to produce and activate a new study protocol. Recent efforts to improve the review cycle have been aimed at reducing this internal process to 150 days (5 months). Some individual institutions have exhibited greater flexibility and have been able to further streamline the protocol approval process. The University of Arkansas has a 70-day timeline for activating a new trial, while M.D. Anderson Cancer Center has a project under way (Project Zero Delay) to turn protocols around in 46 days, according to Canetta.
Case Report Forms
Collecting data for each participant in a clinical trial efficiently and accurately and according to the study objectives is essential for regulatory compliance, as well as the success of the research effort. The case report form (CRF) is the tool used by investigators to collect patient information throughout a clinical trial. Data Safety Monitoring Boards (DSMBs) are tasked with ongoing monitoring of the data collected in CRFs. A portion of the monitoring costs for a trial is directly linked to the complexity of the CRF developed for that trial. Complex CRFs with many data points are more expensive to monitor than simpler CRFs. A number of workshop participants noted that efforts to simplify CRFs so they include only the necessary, biologically relevant details of the trial could decrease trial costs.
Beyond the cost issue, the lack of standardized CRFs and trial procedures can create chaos in some study sites. Woodcock reflected on a recent meeting with the FDA and contract research organizations (CROs) in which the CROs openly discussed the monitoring of study sites. Among the problems they reported, many sites were not conducting critical study procedures correctly or entering all of the data required by the study protocol. According to Woodcock, poor understanding of the study protocol is a common problem in clinical trials and can lead to sloppy data collection and poor data quality. Califf suggested that expending resources and enrolling patients in a clinical trial that does not yield useful information could be considered unethical.
Clinical investigators may be trained to use multiple CRFs depending on the number of trials in which they participate. To reduce costs, Canetta suggested developing a standardized, electronic CRF for use across the research enterprise. Doing so would benefit all stakeholders—government and industry included—because it would help clinical investigators do their
job more efficiently. Cooperative groups supported by the National Cancer Institute (NCI) are currently using standardized CRFs, and the Cancer Biomedical Informatics Grid (caBIG) online network is developing a library of standardized CRFs to be used throughout oncology trials.
RECRUITMENT AND RETENTION OF PATIENTS
A core function of a successful clinical trial is finding patients who fit the predetermined eligibility criteria and getting them to participate. Each disease area addressed during the workshop (cardiovascular disease, depression, cancer, and diabetes) has a unique patient base for clinical trials, and the issues that affect patient enrollment in trials can vary according to features of the disease. In addition, workshop participants identified challenges to patient recruitment that transcend disease status.
Patient Education
Mayer presented the results of a Harris Interactive Survey of 6,000 cancer patients that found that 85 percent were unaware that participation in clinical trials was even an option. Of the patients surveyed, 75 percent said that if participation in a clinical trial had been offered, they would have been receptive to the idea. Of those aware of clinical trials and offered the possibility of participation, 71 percent chose not to participate. However, almost all who participated were satisfied with the experience. Thus, according to these survey results, patients’ preconceived notions about trial participation could pose a barrier to clinical trial enrollment.
Greenbaum noted that the socioeconomic status of patients plays a role in whether they decide to enroll in clinical trials. In addition to income and education, patients’ access to health care services and the network of social support patients have to help them cope with their disease can affect their connection to the medical system and their interest in clinical research. As Mayer noted in her presentation, the online patient network she has developed for metastatic (advanced) breast cancer is composed primarily of younger, better educated, less diverse, and more affluent individuals as compared with the general population. Thus, higher socioeconomic status is associated with having the resources, knowledge, and motivation to seek information about a disease, including access to clinical trials.
Patient Recruitment
According to Woodcock, sites for clinical trials are frequently selected on the basis of where the investigators are located, as opposed to where the patients are, creating difficulties in patient recruitment. When patient
recruitment is impeded, the trial is delayed, sometimes by years, until the number of patients required by the study protocol can be enrolled. Once a trial protocol has been activated, the recruitment of patients requires a significant amount of time and money. Canetta reported that the ability to recruit patients into a trial successfully is similar for the pharmaceutical industry and NCI. Regardless of the trial sponsor, recruitment of patients who meet the requirements of the protocol is difficult: in one study of 14 cancer centers approximately 50 percent of study sites failed to recruit a single patient (Durivage et al., 2009). Thus, patient enrollment can directly affect the number of trials that are completed.
4
Clinical Trials in Cardiovascular Disease
To inform the workshop discussion of ways to improve the overall clinical research enterprise in the Unites States, speakers offered insight into their research efforts in the four disease-specific areas noted in Chapter 1. Gaining an appreciation of the differences in clinical trials by disease helped participants identify aspects of the clinical research enterprise that are working well and those that are not. According to clinicaltrials.gov, cardiovascular trials currently account for 10 percent of all clinical trial participants. The acute nature of many of the health-related events associated with cardiovascular disease and the large number of individuals with the disease make this area of medical research unique in important ways. Presentations summarized in this chapter described a number of different approaches to conducting clinical research in the area of cardiovascular disease and illuminated the overall evolution of clinical trials in this area as compared with other disease areas. This first of four chapters on clinical trials in disease-specific areas begins with a discussion of clinical research models for coronary syndromes. Next, the Thrombolysis in Myocardial Infarction (TIMI) Study Group is discussed as an academic research organization model—a type of clinical research network. The National Institutes of Health (NIH)—sponsored Occluded Artery Trial (OAT) is then presented as an example of the unique strengths and weaknesses of government-sponsored trials. Finally, the chapter summarizes a discussion of the usefulness of large, simple clinical trials in cardiovascular disease.
CLINICAL RESEARCH MODELS FOR CORONARY SYNDROMES
Acute coronary syndromes (ACS) is a broad term referring to a group of conditions ranging from unstable angina, to myocardial infarction (heart
attack), to sudden cardiac death. The condition depends on the degree to which the coronary artery has been obstructed and the health effects the obstruction has caused. A diagnosis of ACS is made by evaluating the results of an electrocardiogram (ECG) and the presence or absence of certain enzymes in the body.
Clinical research efforts in ACS provide a useful model for examining large, multicenter effectiveness trials in an acute, life-threatening disease. Robert Califf, Vice Chancellor for Clinical Research and Director of the Duke Translational Medicine Institute, reflected on the notable successes of the ACS field in translating basic science into early clinical trials, and then into definitive trials that evaluate outcomes related to key clinical questions. Once effective treatments have been identified and disseminated, the final step is measuring their uptake in hospitals and making the results publicly available, which improves adherence to the treatments. A 2004 study examining hospital compliance with quality guidelines (those of the American College of Cardiology/American Heart Association) and in-hospital mortality rates revealed that a 10 percent increase in guideline adherence corresponded to an 11 percent reduction in mortality rates (Peterson et al., 2004). Califf also cited papers based on data from a national registry of myocardial infarction showing that U.S. hospitals show close to 100 percent uptake of evidence-based therapies for ST elevation myocardial infarction (STEMI) and non-ST elevation myocardial infarction (non-STEMI) at both hospital admission and discharge. The result has been an approximately 30 percent reduction in the risk of death if a patient presents at a hospital with chest pain. Califf stressed that, to establish evidence-based therapies that individuals and institutions can be held accountable for using, clinical trials should be focused on answering the critical questions in that disease area; conversely, trials that are poorly designed and seek to answer peripheral or irrelevant questions should be avoided.
Califf reflected on the evolution of clinical trials in ACS. Califf was part of a small group of people who formed the TAMI Group to address the area of STEMI trials. The group received a small amount of money ($100,000) from Genentech to conduct a randomized controlled trial (RCT) with 340 participants. The trial protocol, or study plan, included three cardiac catheterizations per subject, and the trial results were published in the New England Journal of Medicine. Califf highlighted this example of an early ACS trial because he believes the same trial could not be conducted today given the extremely high cost of conducting trials and the large number of patients now required for cardiovascular outcome trials.
The International Study of Infarct Survival (ISIS) group at Oxford had a similar early trial experience that Califf likewise claimed could not be replicated in today’s clinical trial environment. The minimum sample size










