
10
Footprints of Nonsentient Design Inside the Human Genome
JOHN C. AVISE
Intelligent design (ID)—the latest incarnation of religious creationism—posits that complex biological features did not accrue gradually via natural evolutionary forces but, instead, were crafted ex nihilo by a cognitive agent. Yet, many complex biological traits are gratuitously complicated, function poorly, and debilitate their bearers. Furthermore, such dysfunctional traits abound not only in the phenotypes but inside the genomes of eukaryotic species. Here, I highlight several outlandish features of the human genome that defy notions of ID by a caring cognitive agent. These range from de novo mutational glitches that collectively kill or maim countless individuals (including embryos and fetuses) to pervasive architectural flaws (including pseudogenes, parasitic mobile elements, and needlessly baroque regulatory pathways) that are endogenous in every human genome. Gross imperfection at the molecular level presents a conundrum for the traditional paradigms of natural theology as well as for recent assertions of ID, but it is consistent with the notion of nonsentient contrivance by evolutionary forces. In this important philosophical sense, the science of evolutionary genetics should rightly be viewed as an ally (not an adversary) of mainstream religions because it helps the latter to escape the profound theological enigmas posed by notions of ID.
Department of Ecology and Evolutionary Biology, University of California, Irvine, CA, 92697. E-mail: javise@uci.edu.
The result of these cumulative efforts to investigate the cell—to investigate life at the molecular level—is a loud, clear, piercing cry of “design!” The result is so unambiguous and so significant that it must be ranked as one of the greatest achievements in the history of science… . The observation of the intelligent design of life is as momentous as the observation that the earth goes around the sun or that disease is caused by bacteria …
Michael Behe (1996, pp. 232–233)
Michael Behe’s purported biochemical challenge to evolution rests on the assertion that Darwinian mechanisms are simply not adequate to explain the existence of complex biochemical machines. Not only is he wrong, he’s wrong in a most spectacular way. The biochemical machines whose origins he finds so mysterious actually provide us with powerful and compelling examples of evolution in action.
Kenneth Miller (1999, p. 160)
In Darwin’s Black Box, biochemist Michael Behe (1996) issued a challenge to evolutionary biology by claiming that various molecular apparatuses within cells are “irreducibly complex” and therefore could only have been designed purposefully by a higher intelligence. There is no dispute that molecular systems can be astonishingly complex, but most geneticists attribute such biological complexity to the cumulative effects of evolutionary tinkering by natural forces (including a mindless directive agent, natural selection) rather than to conscious engineering by a supernatural entity. In support of this contention, evolutionary biologists have dissected the genetic mechanisms and the probable step-by-step phylogenetic histories by which complex biological features [including some that Behe deemed to be irreducibly complex, such as the eye (Gehring, 2005; Ayala, 2007), bacterial flagellum (Miller, 2004; Pallen and Matzke, 2006; Liu and Ochman, 2007), and biochemical gadgetry of blood clotting (Doolittle and Feng, 1987; K.R. Miller, 1999)] each could have evolved gradually from simpler precursor systems.
However, every year is witness to renewed pressure on textbook publishers and on state and local education boards to inject intelligent design (ID) into the science curricula of public schools. In recent decades, courts in the United States generally have ruled against such creationist initiatives on grounds that the government should not endorse particular religious beliefs (National Academy of Sciences and Institute of Medicine, 2008) (Table 10.1). The social pressures continue, however, not only from evangelical Christianity but from fundamentalist branches of some other religions, including Islam (Numbers, 2006; Hameed, 2008). This situa-
TABLE 10.1 Decisions or Excerpts from the Four Most Famous Courtroom Cases Involving Attempts to Mandate Creationist Instruction or Exclude Evolutionary Biology from Science Classrooms
|
1 |
Scopes “monkey trial,” Dayton, Tennessee, 1925 |
John Scopes was a high school teacher who admitted violating a state law that forbade the teaching in public schools of “any theory that denies the story of the Divine Creation of man as taught in the Bible.” Three other states (Arkansas, Mississippi, and Oklahoma) likewise had passed laws prohibiting evolutionary instruction in public schools. The Tennessee court found Scopes guilty as charged. |
|
2 |
Epperson vs. Arkansas, Supreme Court of the United States, 1968 |
“Government in our democracy, state and national, must be neutral in matters of religious theory, doctrine, and practice. It may not be hostile to any religion or to the advocacy of non-religion, and it may not aid, foster, or promote one religion or religious theory against another or even against the militant opposite.” |
|
3 |
Edwards vs. Aguillard, Supreme Court of the United States, 1987 |
The “primary purpose [of the Louisiana ‘Creation Act’] was to change the public school science curriculum to provide persuasive advantage to a particular religious doctrine that rejects the factual basis of evolution in its entirety. Thus, the Act is designed either to promote the theory of creation science that embodies a particular religious tenet or to prohibit the teaching of a scientific theory disfavored by certain religious sects. In either case, the Act violates the First Amendment.” |
|
4 |
Kitzmiller vs. Dover Area School District, District Court for Middle Pennsylvania, 2005 |
“ID [Intelligent Design] is not science and cannot be adjudged a valid, accepted scientific theory, as it has failed to publish in peer-reviewed journals, engage in research and testing, and gain acceptance in the scientific community. ID, as noted, is grounded in theology, not science…. Moreover, ID’s backers have sought to avoid the scientific scrutiny which we have now determined that it cannot withstand by advocating that the controversy, but not ID itself, should be taught in science class. This tactic is at best disingenuous, and at worst a canard. The goal of the IDM [Intelligent Design Movement] is not to encourage critical thought, but to foment a revolution which would supplant evolutionary theory with ID.” |
|
SOURCE: National Academy of Sciences and Institute of Medicine (2008). |
||
tion evidences considerable global sympathy for ID (as well as profound misunderstandings about evolutionary biology by the public), no doubt exacerbated by the intuitive appeal of invoking a supreme intelligence to account for superb adaptations, such as the vertebrate eye. At a cursory level, fine-working adaptive traits are easy to rationalize; people of creationist persuasion need only invoke the attentive craftsmanship of a loving God, whereas the science-minded can invoke the unconscious agent, natural selection. Both a Creator God and natural selection are powerful shaping forces that might be expected to have engineered beautiful functionality and efficiency into complex biological features, such as the human genome. The much greater challenge—for proponents of ID and for scientists alike—is to explain complex biological traits that operate inefficiently or even malfunction overtly. On closer inspection, the human genome itself becomes a prime example of a highly complex trait with serious molecular shortcomings.
THUMBNAIL HISTORIES OF THREE ANCIENT WORLD VIEWS
Natural Theology and the Argument from Design
Before Darwin, most scientists, as well as theologians, accepted what seemed obvious: that divine intervention must have underlain nature’s design. The standard “argument from design” traces back at least to the classical Greek philosopher Socrates (Sedley, 2008). Indeed, a common sentiment in recent centuries, and certainly in Western cultures, was that religion and biological inquiry were intellectual allies in a grand mission to explain, and thereby glorify, God and His Creation. Scientists and religious leaders often shared a conviction that the careful study of nature would confirm God’s invention and oversight of life. Many scientists were avowed deists, and many clerics were also science-scholars, all jointly engaged in confirming God through rational inquiry (which frequently was seen as a helpful complement to traditional knowledge of God from gospel truths and religious revelations). When objections to science were raised in theology (and they often were), they usually stemmed from a notion that it was heretical or even dangerous (given a wrathful God) to strive to prove empirically that which required no proof: God’s magnificence as detailed in the Scriptures.
In 1802, Reverend William Paley published an eloquent and thoughtful book (Natural Theology) that formally explained what many scientists of his era sought to accomplish in their studies of nature: proof and glorification of God’s majesty through empirical investigations of His works. These biologists (i.e., natural theologians) typically started with two premises: that life’s beauty and complexity were prima facie evidence
of God’s creative power and that by carefully analyzing living nature, they inevitably would exalt God, and perhaps come better to comprehend His intentions. These themes were developed further in the ensuing Bridgewater Treatises, a set of eight works by different experts in biology, geology, and physics, published between 1833 and 1840. These books were commissioned by Reverend Francis Henry, Earl of Bridgewater, who died in 1829 but whose last will and testament encouraged and funded respected scientists to write treatises “on the power, wisdom, and goodness of God, as manifested in Creation.”
Darwin himself was a natural theologian when he boarded the HMS Beagle in 1831. He later recalls in his autobiography that Paley’s logic “gave me as much delight as did Euclid” and that it was the “part of the Academical Course [at the University of Cambridge] which … was the most use to me in the education of my mind.” The 5-year journey on the HMS Beagle would prove to be a fateful voyage—not just for Darwin but for humanity—into previously uncharted waters in science and philosophy. Later, Darwin’s elucidation of natural selection would launch a revolutionary paradigm in biology, wherein biological outcomes (species and the traits they possess) could be understood as products of natural forces (rather than the supernatural) that are entirely amenable to critical scientific analysis. The Darwinian revolution did for biology what the Copernican revolution three centuries earlier had done for the physical sciences: permit the workings of the universe to be interpreted as reflecting natural laws that could be studied and tested via objective scientific hypotheses, observations, and experiments (Ayala, 2008; Ayala and Avise, 2009).
Creationism and ID
Creationism in its many guises is likewise an ancient human philosophy. Indeed, nearly all human cultures and religions have had their own creation mythologies: about life’s origins in general and/or about human geneses in particular (Eliot, 1990; Avise, 1998). Natural theology (as described previously) and its offshoot, ID, have tended to be movements within Christianity, but many other cultures have held comparable sentiments about how a Creator God or Gods consciously manipulate the biological world. The modern ID version of natural theology can be dated to the publication in 1984 of The Mystery of Life’s Origin (Thaxton et al., 1984). This project was encouraged by the Foundation for Thought and Ethics (FTE), a Dallas-based Christian organization. The book was intended to highlight difficulties with scientific explanations for life’s origins; indeed, it claimed categorically that life could not arise by natural causes. The FTE soon sponsored a high-school textbook, Of Pandas and
People (Davis and Kenyon, 1989), that also took a hostile stance on the scientific evidence for evolution.
The ID movement got a media boost from the publication in 1991 of Darwin on Trial (Johnson, 1991), which, although anti-evolutionist in its stance, had a more serious aura of scholarship and did not tout hard-line creationist mantras (e.g., a young Earth, a universal flood) that were blatantly untenable scientifically. In that same year, a conservative think tank known as the Discovery Institute (DI) set up shop in Seattle. Since that time, the DI has largely supplanted the FTE as a primary hub of activity for the ID movement in North America. Another noteworthy publication in the ID movement was William Dembski’s No Free Lunch: Why Specified Complexity Cannot Be Purchased Without Intelligence (Dembski, 2001). It echoes and purports to substantiate standard creationist claims that complex biological traits cannot emerge by natural evolutionary processes. However, the modern ID book with perhaps the biggest public impact is Darwin’s Black Box (Behe, 1996), in which biochemist Michael Behe coined the term irreducible complexity. According to Behe, a cellular structure or other biological feature is irreducibly complex if the removal of any of its parts results in the loss of function. Such a structure, Behe claims, could not have evolved incrementally by natural selection but, instead, must have been engineered—in its entirety and for its current role—by an intelligent agent. That agent is left unspecified; however, the reference is clearly to God. Behe’s arguments in Darwin’s Black Box, although couched in unique language and metaphors and applied at a unique phenotypic level of biochemistry and molecules, essentially reiterate Paley’s (1802) sentiments in Natural Theology.
These and other publications from the ID community have been refuted in the scientific literature, and it is not my intent here to repeat the voluminous evidence for how natural selection in conjunction with other nonsentient evolutionary forces can yield complex adaptations. Instead, my focus in this paper is on a relatively neglected category of argument against ID and in favor of evolution: the argument from imperfection, as applied to the human genome in this case.
Theodicy
Theodicy (from the Greek roots theós for God and dik for justice) is the flip side or the dark side of natural theology. It is the formal term for philosophical attempts to vindicate God’s holiness and justice in establishing a world that is rife with evils and woes. Throughout the ages, theologians have pondered why a loving and all-powerful deity allows human suffering. With respect to human phenotypes, why does God permit diabetes, heart attacks, hemorrhoids, impacted wisdom teeth, difficult childbirths,
or bad backs (not to mention behavioral flaws, ethical shortfalls, and death)? For some Christian denominations, an escape from this conundrum is to claim that such frailties result from humanity’s fall from Grace in the Garden of Eden. Other religions have their own rationalizations. So, too, do the biological sciences. From a scientific perspective, biological imperfections in humans (and in other species) are an understandable byproduct of evolution by unconscious and uncaring natural forces. There are many solid scientific reasons why the biological outcomes of evolution by natural selection are expected to fall routinely short of designer perfection (Table 10.2).
TABLE 10.2 Some Reasons Why Evolution Routinely Yields Suboptimal Biological Outcomes
|
1 |
Natural selection is a nonsentient process of nature, as uncaring and dispassionate as gravity. |
|
2 |
Natural selection is not all-powerful. Instead, it is just one in a nexus of evolutionary forces, others of which can override the adaptation-promoting power of natural selection in particular instances, and thereby yield products that fall far short of designer perfection. |
|
3 |
Random mutations, most of which are either deleterious or fitness-neutral, continually arise. |
|
4 |
Harmful mutations (especially those that are only slightly deleterious individually) often fly below the radar screen of purifying natural selection, especially in small populations. |
|
5 |
Genetic drift can alter the genetic composition of populations in ways that are uncorrelated with adaptive benefits. |
|
6 |
Sexual selection on particular traits often operates in direct opposition to natural selection. |
|
7 |
Genetic correlations and conflicts are common. In such cases, deleterious alleles linked to host-beneficial alleles at other loci can hitchhike with the favorable alleles, and thereby escape eradication by purifying natural selection, at least temporarily. |
|
8 |
Pleiotropy and fitness tradeoffs are common. Thus, a particular genotype often has multiple phenotypic consequences, some of which benefit and others of which may harm the organism. |
|
9 |
In sexual species, natural selection acts not only at the organismal level but at the level of genes. Thus, “selfish DNAs” (e.g., many mobile elements) can persist and proliferate in a genome without enhancing the well-being of a host population. |
|
10 |
Phylogenetic constraints are ubiquitous. At any point in geological time, natural selection going forward can only work with the genetic diversity presented by lineages that have survived from the past. This places severe constraints on what evolution can achieve. |
Theodicy—and the associated “counterargument to design”—also have long histories (probably as ancient as the human species). In 1779, the Scottish philosopher-historian David Hume pithily captured the idea in a verbal exchange between two of his fictional characters in Dialogues Concerning Natural Religion:
[Cleanthes]: The Author of Nature is somewhat similar to the mind of man, though possessed of much larger faculties, proportioned to the grandeur of the work he has executed… . By this argument alone, do we prove at once the existence of a Deity.
[Philo]: What surprise must we entertain, when we find him a stupid mechanic.
Darwin himself was well aware (and at times seemed chagrinned) that biological imperfection was a powerful counterargument to ID. He wrote in chapter 14 of On the Origin of Species by Means of Natural Selection (Darwin, 1859):
on the view of each organic being and each separate organ having been specially created, how utterly inexplicable it is that parts … should so frequently bear the plain stamp of inutility.
Darwin was also aware that biological evolution by mindless natural forces, in effect, could alleviate much of the theodicic paradox.
THE HUMAN GENOME
In 2001, the first draft sequence of a human genome was published (Lander et al., 2001; Venter et al., 2001). It was about 3 billion nucleotide pairs in length, or roughly 50,000 times longer than the article you currently are reading (if each letter character or space can be equated to a nucleotide pair). The human genome that the researchers sequenced in 2001 was actually a composite of DNA sequences assembled from different people; however, collectively, it represented one “genome equivalent” from our species. In 2007, the full genome from a single person was sequenced in its entirety (Levy et al., 2007), and similar reports soon followed of whole-genome sequences from additional individuals (e.g., J. Wang et al., 2008; Wheeler et al., 2008). Recently, a Personal Genome Project was announced (Church, 2005), the goal of which is to use rapid DNA sequencing to gather numerous human genomic sequences. Such investigations are merely the latest generation of scientific inquiries into human genetics and biochemistry, which extend back about a century.
The age-old theodicy dilemma traditionally was motivated by human frailties at the observable levels of morphology and behavior. Do biological flaws extend to the molecular level also? Especially in the past half century, scientists have answered this question definitively, in the affirmative. Next, I will describe several complex features of the human genome that give compelling evidence for non-ID, and then I will close by highlighting some of the philosophical ramifications of these molecular discoveries. For a more comprehensive treatment of all these topics, see Avise (2010).
FALLIBLE DESIGN: PROTEIN-CODING DNA SEQUENCES
By the early 1900s, doctors had begun to appreciate that biochemical malfunctions inside the human body can produce physical ailments and abnormalities. Archibald Garrod pioneered this revolutionary outlook in two path-breaking books: Inborn Errors of Metabolism (1909) and The Inborn Factors of Inherited Disease (1931). In Inborn Errors of Metabolism, Garrod detailed what then was known about the biochemistry and inheritance of four atypical human conditions: albinism, alkaptonuria, cystinuria, and pentosuria. Today, we know, for example, that alkaptonuria is a rare disorder (1 in 200,000 births) caused by a biochemical defect (in the degradation pathway for phenylalanine and tyrosine) that itself results from various mutational defects in a gene encoding homogentisic acid oxidase, which otherwise catalyzes the breakdown of homogentisic acid. As this acid accumulates, it binds irreversibly to collagen in the patient’s body, eventually producing degenerative arthritis in the large joints and spine usually beginning in midlife. Before Garrod’s time, this and many other disorders often were attributed to ethereal or mystical phenomena, such as bad karma or malevolent demons. After Garrod, the medical profession began to appreciate that careful scientific inquiry into metabolic disorders could reveal their mechanistic (genetic and biochemical) basis, and perhaps someday even lead to treatments or cures.
A modern analogue of Inborn Errors of Metabolism was launched in 1960 with the publication of The Metabolic Basis of Inherited Disease (Stanbury et al., 1960). The book has since seen more than half a dozen updated editions (at roughly 6-year intervals), and the title of the work also expanded to The Metabolic and Molecular Bases of Inherited Disease (MMBID), reflecting the recent explosion of DNA and protein sequence data. A recent edition of MMBID includes more than 6,000 pages in four volumes. In 255 chapters, each on a different heritable disorder or suite of associated genetic disorders, leading biomedical experts encapsulate current knowledge about the molecular mechanisms underlying inborn human diseases. They focus attention on the genetic basis of each disorder and also on the structure and function of each gene’s protein product. More than 500 well-character-
ized genetic disorders are profiled in astonishing detail, and that number will only grow dramatically as the medical profession moves further into the genomics era. A plausible supposition is that at least some harmful mutations exist somewhere in the human species at each of the genome’s ≈ 24,000 protein-coding loci.
The mutational defects profiled in MMBID occur in almost every operational category of protein, including enzyme-mediated energy metabolism, DNA/RNA processing, protein folding and degradation, molecular transport and secretion, signal transduction (mechanisms that link mechanical or chemical stimuli to cellular response), cytoskeletal elements, ribosomal functions, and structures and functions of exported (extracellular) proteins. Various mutations are known to debilitate the nervous system, liver, pancreas, bones, eyes, ears, skin, urinary and reproductive tracts, endocrine system, blood and other features of the circulatory system, muscles, joints, dentition, immune system, digestive tract, limbs, lungs, and almost any other body part you can name. With respect to age of onset, various genetic disorders appear in utero, from birth to the first year (the most commonly diagnosed class), from year 1 to puberty, from puberty to 50 years of age, or in seniors. Approximately two-thirds of the genetic defects described in MMBID shorten human life span, and about three-quarters of these cause death before the age of 30 years.
Another compendium of this sort, launched in 1966, is Mendelian Inheritance in Man (MIM) the current version of which describes thousands of human genes, of which more than 75% are documented to carry mutational defects associated with a disease condition. MIM has appeared in a dozen printed editions and now is also available online (OMIM), where it is updated regularly by computer-based literature searches. Yet another compilation of this sort is the Web-based Human Gene Mutation Database (HGMD) (Stenson et al., 2003), recent versions of which describe more than 75,000 different disease-causing mutations identified to date in Homo sapiens. More than 50% of these molecular damages involve nonsynonymous substitutions in protein-coding segments (exons) of genes, whereas the remaining molecular damages fall into a miscellany of categories, including large and small insertions or deletions of genetic material, DNA rearrangements, regulatory mutations in regions that flank a gene, and alterations in how particular mRNA molecules were spliced together. The HGMD is cross-referenced to OMIM and updated weekly. The HGMD provides a concise summary of all documented mutations that underlie a given metabolic disorder. For example, it describes 86 different disorder-causing mutations known in the glucose-6-phosphatase gene, 63 of which are nonsynonymous substitutions, 18 of which are small additions and/or deletions, 4 involve splicing anomalies, and 1 involves a regulatory region
flanking the coding sequence. Many further molecular details about each mutation are provided, as are electronic links to the original papers.
An apologist for the intelligent designer might be tempted to claim that such deleterious mutations are merely unavoidable glitches or secondary departures from a prototypical human genome that otherwise was designed and engineered to near perfection. As I will briefly describe in the next two sections, however, this excuse would be untenable, because all human genomes are also littered with inherent (endogenous) design flaws.
BAROQUE DESIGN: GRATUITOUS GENOME COMPLEXITIES
For natural theologians in centuries past, as well as for adherents to present-day versions of strict religious creationism, biotic complexity is the hallmark—the unquestionable signature—of ID. However, gratuitous or unnecessary, biological complexity—as opposed to an economy of design—would seem to be the antithesis of thoughtful organic engineering. Yet, by objective scientific evidence, gratuitous and often-dysfunctional complexities (both in molecular structure and molecular operations) are so nearly ubiquitous as to warrant the status of hallmarks of the human genome. Here are some representative examples.
Split Genes
The discovery in 1977 that standard protein-coding loci are split into coding regions (exons) interspersed with noncoding regions (introns) came as a complete surprise. So, too, did the discovery of large ribonucleoproteins known as spliceosomes, which biochemically remove the intron-derived segments from each premessenger (prem) RNA and then splice each gene’s exons end-to-end to generate a mature mRNA. Approximately 1% of all known genes in the human genome encode molecular products that our cells employ to build spliceosomes and conduct splicing operations on premRNA. All this rigmarole has some advantages (e.g., opportunities for alternative splicing during ontogeny and exon shuffling during evolution, both of which can generate functional protein diversity), but such benefits do not come without major fitness costs.
There are good reasons to think that cells might be better off without introns, in an ideal world. Introns impose energetic burdens on cells. They are, on average, 30-fold longer than exons and are transcribed into premRNAs before being snipped out; thus, they probably extend the time to produce each mature mRNA by at least 30-fold (compared with the expectation for nonsplit genes). Even if time is not important for somatic cells, the metabolic costs of maintaining and replicating all the extra
nucleotides in introns must be considerable. To these cellular costs must be added the metabolic expense of making spliceosomes and running the extensive premRNA processing machinery. It can also be noted that many organisms (e.g., bacteria) do just fine without split genes and introns, as do the mitochondrial genomes within human cells; thus, there is no universal biological exigency that these features exist. Finally, the human nuclear genome would have ample room to house nonsplit genes for all the proteins it needs (including those that are now alternatively spliced) if an intelligent designer simply would jettison the genome’s junk DNA (see beyond).
Nevertheless, for the sake of argument, let us assume that the metabolic costs imposed by introns are negligible. Do introns otherwise provide evidence of optimal genomic design? No, because premRNA processing also has opened vast opportunities for cellular mishaps in protein production. Such mishaps are not merely hypothetical. An astonishing discovery is that a large fraction (perhaps one-third) of all known human genetic disorders is attributable in at least some clinical cases to mutational blunders in how premRNA molecules are processed (Frischmeyer and Dietz, 1999; Philips and Cooper, 2000). For example, it has long been known that mutations at intron-exon borders often disrupt premRNA splicing in ways that alter gene products and lead to countless genetic disabilities, including various cancers and other metabolic defects (Krawczak et al., 1992). There is also good evidence that the number of introns in human genes is positively correlated with a gene’s probability of being a disease-causing agent (López-Bigas et al., 2005). Avise (2010) summarizes many of the human genetic afflictions that have been documented (in particular clinical instances) to molecular errors in mRNA splicing at specifiable loci. These range from a variety of neurodegenerative diseases to debilitations of the circulatory, excretory, and other body systems. Many of these genetic disorders begin in infancy or early childhood; others are deferred to the elderly. The devastating symptoms of many such disorders, such as Lou Gehrig disease (amyotrophic lateral sclerosis), are simply horrible by any human standard.
Gene Regulation and Nucleic Acid Surveillance
Each protein-coding gene or “structural gene” also has adjoining (cis) regulatory sequences that help to modulate when during development, and where in different tissues or organs, it is expressed. Most notable is a core promoter, usually several dozen base pairs long, to which suites of proteins known as transcription factors bind, to be joined by RNA polymerase molecules that catalyze the fabrication of premRNA from the adjoining structural gene. Other regulatory sequences called enhancers
and silencers, sometimes thousands of nucleotides upstream or downstream from the core promoter, further boost or inhibit transcription. Each gene may have several enhancers and silencers; these can be shared among genes, but different genes have different combinations. The enhancers and silencers influence transcription via their connections to large families of activator and repressor proteins that transpond regulatory signals to RNA polymerase via coactivators and other proteins. Distinct batteries of transcription factors and their molecular associates operate in different cell types, thereby helping to explain how different tissues and organs within an individual can have different patterns of gene expression despite sharing the same underlying genome.
Once a protein-coding gene has been turned on by appropriate regulatory signals, and mRNA has been transcribed, mechanisms of RNA surveillance spring into action. For any of a variety of reasons, some mRNA molecules become mistakenly truncated or otherwise blemished in ways that prevent their effective translation into a useful polypeptide. Somatic cells monitor for such defects and actively degrade many of the damaged mRNA copies. With respect to correcting genetic errors, RNA surveillance is the RNA-level analogue of the cell’s many mechanisms for gene repair that operate directly at the level of DNA. Much of this makes good design sense; if DNA was not repaired routinely, or if faulty mRNAs were not destroyed, dysfunctional rogue proteins might appear in cells far more often than they do. However, if an intelligent designer is responsible for such repair mechanisms, he must also have presaged or understood that his original genomic design would include multitudinous flaws.
In a broad definitional sense, the genetic regulation of protein-coding genes can also occur at any posttranscriptional stage of protein production, including premRNA editing, the exportation of mature mRNAs from the nucleus, differences in the stability and transport of mRNA molecules after they have reached the cytoplasm, factors impinging on the translation process by which polypeptides are constructed from mRNA on ribosomes, polypeptide assembly into functional proteins, and posttranslational protein modifications or degradations. Many of these regulatory mechanisms involve complex biochemical pathways, and, collectively, they require major expenditures in cellular effort and molecular materials. Probably 50% or more of all coding genes in the human genome could be considered to play some direct or indirect regulatory role in development, for example, in cell signaling and communication, control over gene expression per se, or influences on cell division, structure, or motility.
Protein kinases provide a leading illustration of posttranscriptional regulation in eukaryotic cells. Kinases are enzymes that phosphorylate, and thereby alter the activity of substrate molecules. The human genome contains about 518 functional protein kinase genes that can be arranged
into several dozen functional families and subfamilies of loci, all of which arose, under an evolutionary interpretation, from successive gene duplication events across the long history of vertebrate animals (Manning et al., 2002). By altering the activity profiles of proteins, kinases exert regulatory control over numerous cellular processes, including metabolism, cell-cycle progression, cell movement and differentiation, physiological homeostasis, functioning of the nervous and immune systems, and signal transduction (mechanistic pathways by which chemical or other environmental stimuli evoke cellular responses).
Micro-RNAs (miRNAs) are another important class of loci involved in posttranscriptional genetic regulation (He and Hannon, 2004; Baek et al., 2008; Selbach et al., 2008). Each miRNA is a short (ca. 20-nucleotide) stretch of RNA that can bind to complementary sequences in the messenger RNA molecules of protein-coding genes, and thereby inhibit the translation or induce the degradation of specific genetic messages. Although the exact numbers and precise roles of miRNAs in the human genome remain to be illuminated, more than 500 such loci already are known and findings suggest that miRNAs might be major cellular tuners of protein synthesis. Another interesting class of molecules is long noncoding RNAs, each of which is typically hundreds or thousands of base pairs long (Petherick, 2008). Some geneticists posit that long noncoding RNAs will prove to be important regulators of gene expression; others demur on this possibility for now, pointing to countervailing evidence, such as the fact that cells seem to destroy long RNAs almost as soon as they are produced.
The various mechanisms described here thus help to orchestrate how particular genes and their protein products are expressed within a cell. Many additional routes to gene regulation exist, such as how nucleic acid sequences are spatially organized and packaged into chromatin fibers and chromosomes, how DNA molecules are complexed with histone proteins, and the pattern in which cytosine bases in DNA sometimes are modified via chemical methylation. In short, the sheer complexity of structure and function in the genetic regulatory apparatus of cells is not in dispute.
However, regulatory complexity during development is a double-edged sword for any organism. The molecular machineries of gene regulation are metabolically costly, and they often malfunction with disastrous health consequences. Improprieties in one or another aspect of gene regulation are responsible for many human ailments ranging from particular cases of asthma to various immune disorders, circulatory problems, and heart diseases. Many manifestations of cancer have been traced to aberrant methylation patterns in the promoter regions of particular genes (Jones and Baylin, 2002). Thalassemias—genetic disabilities that arise from inadequate supplies of oxygen-carrying globins in the blood—are another large class of metabolic diseases related to problems in gene regulation
(Weatherall et al., 1984). Protein kinases are also subject to disorder-producing malfunctions, with more than 160 different kinases having been implicated in cancers by their common association with particular tumor types and 80 kinases having been associated at least provisionally with various other disease conditions (Manning et al., 2002). Similarly, research suggests that occasional misregulation of miRNA molecules contributes to the total pool of human metabolic disorders, including perhaps DiGeorge syndrome as well as some cancers (Alvarez-Garcia and Miska, 2005).
Why an intelligent and loving designer would have infused the human genome with so many potential (and often realized) regulatory flaws is open to theological debate. Any such philosophical discussion should probably include the issue of whether the designer was fallible (and if so, why?). It should also address whether the designer might have recognized his own engineering fallibility, as perhaps evidenced, for example, by the DNA and RNA surveillance mechanisms that catch some (but not all) of the numerous molecular mistakes.
From an evolutionary perspective, such genomic flaws are easier to explain. Occasional errors in gene regulation and surveillance are to be expected in any complex contrivance that has been engineered over the eons by the endless tinkering of mindless evolutionary forces: mutation, recombination, genetic drift, and natural selection. Again, the complexity of genomic architecture would seem to be a surer signature of tinkered evolution by natural processes than of direct invention by an omnipotent intelligent agent.
mtDNA
Mitochondria are the only cytoplasmic organelles in humans to house their own DNA (mtDNA). A prototypical molecule of human mtDNA is 16,569 bp long. It is a closed circle of 37 maternally inherited genes, 22 of which encode tRNAs, 13 specify polypeptides, and 2 encode rRNAs.
Mitochondria are the primary seat of energy production in cells. The principal biochemical pathway in mitochondria by which this is carried out is oxidative phosphorylation, of which the respiratory chain is a key component. The respiratory chain consists of five enzyme complexes (I-V) plus coenzyme Q and cytochrome c. Complexes I and II oxidize NADH and succinate, respectively; complexes I, III, and IV pump protons to effect an electrochemical gradient; and complex V uses energy from that gradient to synthesize adenosine triphosphate from adenosine diphosphate. A remarkable fact is that four of these five enzyme complexes are composed of combinations of polypeptides from the mitochondrial and nuclear genomes (Graff et al., 1999). In complex IV, for example, 3 of the 13 polypeptides are encoded by mitochondrial loci (COI, COII,
and COIII), whereas the remaining polypeptides are encoded by nuclear genes. Only in complex II are all the necessary enzymatic subunits (four in this case) encoded by just one genome (the nuclear). Nuclear genes are also intimately involved in other basic mitochondrial functions. Indeed, mtDNA does not encode any of the proteins that are directly involved in its own replication, transcription, translation, surveillance, or repair. In short, mtDNA is just a tiny snippet of DNA that by itself would be absolutely helpless to itself and to the organism in which it is housed. None of this makes any biological sense, except in the light of evolutionary science (which has discovered that modern mitochondria are remnants of a microbe that invaded or was engulfed by a protoeukaryotic cell in an endosymbiotic merger that took place billions of years ago).
Like the other genetic systems we have considered thus far, the mitochondrial genome is plagued by mutations that often compromise molecular operations. Indeed, on a per-nucleotide basis, mtDNA experiences about 5–10 more mutations per unit time than do typical protein-coding nuclear genes (Brown et al., 1979). Many mtDNA mutations are of little or no consequence to a person’s health, but many others have negative effects ranging from mildly debilitating to deadly. Clinical disabilities from mtDNA mutations disproportionately involve high-energy tissues and organs (Wallace, 2005; McFarland et al., 2007): brain, eye and other components of the peripheral nervous system, heart, skeletal muscle, kidney, and the endocrine system. Mutations in mtDNA have also been implicated in a spectrum of cancers (Copeland et al., 2002). In short, an emerging paradigm is that many of the degenerative diseases of aging have their etiologies in mitochondria, either as deleterious mutations in the mtDNA molecules themselves or as operational flaws in nuclear-mitochondrial interactions.
The serious health problems that arise from mtDNA mutations immediately challenge any claim for omnipotent perfection in mitochondrial design. Perhaps these mutational aberrations can be viewed as unfortunate but inevitable byproducts of molecular complexity. However, the intellectual challenges for ID go much deeper. Considering the critical role of cellular energy production in human health and metabolic operations, why would an intelligent designer entrust so much of the production process to a mitochondrion, given the outrageous molecular features this organelle possesses? Why would a wise designer have imbued mtDNA with some but not all of the genes necessary to carry out its metabolic role (and then put the remaining genes in the nucleus instead)? Why would a wise engineer have put any crucial genes in a caustic cytoplasmic environment in which they are exposed routinely to high concentrations of mutagenic oxygen radicals? Why would he have dictated that the mitochondrial genetic code must differ from the nuclear genetic code,
thereby precluding cross-translation between two genomes for which effective communication would seem to be highly desirable? Why would an intelligent designer have engineered mtDNA structures (e.g., closed-circular genome, no introns, no junk DNA, lack of binding histones) and mtDNA operations (e.g., little or no genetic recombination, production of a polygenic transcript, limited ability to mend itself, no self-sufficiency in transcription or translation) to differ so fundamentally from their counterpart features in the nuclear genome? In a nutshell, the underlying design of the whole mitochondrial operation seems to make no (theo)logical sense. Not only is the overall design of mtDNA suboptimal, but it appears downright ludicrous!
WASTEFUL DESIGN: REPETITIVE DNA ELEMENTS
Before scientists gained direct access to DNA sequences from the modern tools of molecular biology, it was widely assumed that nuclear genomes were composed of sleek and efficient protein-coding genes strung together along chromosomes like tight beads on strings. In truth, however, structural genes have complex internal structures in which the exons typically are like small islands in much larger hereditary rivers of noncoding introns and regulatory regions. An even bigger surprise came with the discovery that the vast majority of human DNA exists not as functional gene regions of any sort but, instead, consists of various classes of repetitive DNA sequences, including the decomposing corpses of deceased structural genes and legions of active and retired transposable elements.
Duplicons and Pseudogenes
At least 4,000 protein-coding genes and other lengthy stretches of DNA (up to 200,000 bp in length) are present not just once but in small to moderate numbers of copies per genome. At least 5% of the human nuclear genome consists of such gene families in which the redundant elements (termed duplicons, which arose through gene duplication processes) are typically more than 90% identical to one another in nucleotide sequence (Eichler, 2001; Bailey et al., 2002). Duplicate genes often perform useful functions, but we are concerned here with the evidence for genomic faults rather than benefits.
Because duplicate genes show close sequence similarity, they predispose chromosomes to pair abnormally during meiosis. Such homologous recombination can generate deletions, additions, inversions, or translocations of genetic material in the resulting gametes, which, in turn, can generate health problems in the resulting offspring. Metabolic disturbances
that result from duplicon-mediated genomic rearrangements typically stem from dosage imbalances attributable to the presence of too many or too few copies of a gene or to an altered orientation of particular genes relative to their regulatory regions.
Numerous metabolic disorders have been traced to duplicon-mediated recombination. No organ system seems immune to damage; various disorders are known to affect elements of the circulatory, respiratory, hormonal, skeletal, muscular, reproductive, excretory, or nervous systems. Some genetic conditions, such as red-green color blindness, are rather benign, whereas others, such as Prader-Willi syndrome, are severely debilitating. Many severe disorders tend not to be transmitted through families (because the afflicted seldom are able to reproduce) but, instead, recur in the human population from de novo mutations in paternal or maternal germlines. Approximately 0.1% of humans who survive to birth carry a duplicon-related disability, meaning that millions of people worldwide are afflicted by this category of metabolic errors. Many more afflicted individuals probably die in utero. Clearly, humanity bears a substantial health burden from duplicon-mediated genomic malfunctions.
Mobile Elements
Perhaps the most surprising genomic finding of recent decades concerns the abundance of mobile elements. These stretches of DNA have—or previously had in many cases—the ability to colonize unique chromosomal locations by moving, replicatively, from one genomic position to another in a cell lineage. Incredibly, mobile elements constitute at least 45% of the human genome, and the true fraction is probably 75% or more if the tally were to include (i) processed pseudogenes that originated as a byproduct of mobile element activity (Esnault et al., 2000) and (ii) other intergenic DNA regions that probably originated long ago as mobile elements but are no longer identifiable as such because of postformational mutations.
Mobile elements have the potential to cause human diseases by several mechanisms. When a mobile element inserts into a host genome, it normally does so at random with respect to whether or not its impact at the landing site will harm the host. If it happens to land in an exon, it can disrupt the reading frame of a functional gene with disastrous consequences. If it jumps into an intron or an intron-exon boundary, it may cause problems by altering how a gene product is spliced during RNA processing. If it inserts into a gene’s regulatory region, it can also cause serious mischief. The potential for harm by such insertional mutagenesis is great. It has been estimated, for example, that an L1 or Alu mobile element newly inserts somewhere in the genome in about 1–2% and 5%,
respectively, of human births (Brouha et al., 2003; Cordaux et al., 2006). Another problem is that when a mobile element lands in a functional gene, genetic instabilities are sometimes observed that result in deleted portions of the recipient locus. Several genetic disorders have been traced to genomic deletions associated with de novo insertions of mobile elements (Chen et al., 2005). Finally, mobile elements (or their immobile descendents that previously accumulated in the human genome) can also cause genomic disruptions via nonallelic homologous recombination (Burwinkel and Kilimann, 1998). Serious metabolic disorders can result (Hedges and Deininger, 2007).
Despite the relatively recent discovery of mobile elements, the list of genetic disorders associated wholly or in part with their activities already is long. Still, any such list provides only a minimum estimate of these elements’ collective toll on human health. This is because some of the most serious medical difficulties probably arise so early in ontogeny as to cause miscarriages that normally will remain of unknown etiology. Indeed, most mobile elements are especially active in the germline; thus, many of their deleterious effects probably register in gametic deaths and lowered fertility.
A RECONCILIATION: EVOLUTION AS A SALVATION FOR THEOLOGY
From scientific evidence gathered during the past century, and especially within recent decades, we now understand that the human genome and the metabolic processes it underwrites are riddled with structural and operational deficiencies ranging from the subtle to the egregious. These genetic defects register not only as deleterious mutational departures from some hypothetical genomic ideal but as universal architectural flaws in the standard genomes themselves. The findings of molecular biology thus offer a gargantuan challenge to notions of ID. They extend the age-old theodicy challenge, traditionally motivated by obvious imperfections at the levels of human morphology and behavior, into the innermost molecular sanctum of our physical being.
Exactly how a fall from Grace in the Garden of Eden might have become translated into these molecular defects is mechanistically unclear (to say the least). How such genomic flaws arise and persist poses no insuperable mystery from the scientific perspectives of genetics and evolution, however. Herein, I suggest, lies a wonderful opportunity for nonfundamentalist religions.
Evolution by natural causes in effect emancipates religion from the shackles of theodicy. No longer need we agonize about why a Creator God is the world’s leading abortionist and mass murderer. No longer need we
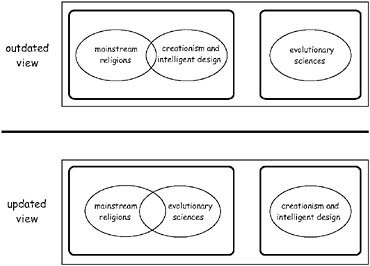
FIGURE 10.1 (Upper) Traditional placement of evolutionary biology as the oddman-out to the spheres of mainstream religion and ID in many philosophical discourses about the human condition. (Lower) Unique and perhaps enlightened perspective in which ID is the odd-man-out to mainstream religions and the evolutionary sciences (whose spheres or magisteria may overlap to arguable degrees).
query a Creator God’s motives for debilitating countless innocents with horrific genetic conditions. No longer must we anguish about the interventionist motives of a supreme intelligence that permits gross evil and suffering in the world. No longer need we be tempted to blaspheme an omnipotent Deity by charging Him directly responsible for human frailties and physical shortcomings (including those that we now understand to be commonplace at molecular and biochemical levels). No longer need we blame a Creator God’s direct hand for any of these disturbing empirical facts. Instead, we can put the blame squarely on the agency of insentient natural evolutionary causation. From this perspective, the evolutionary sciences can become a welcome partner (rather than the conventionally perceived adversary) of mainstream religion (Fig. 10.1).
The evolutionary-genetic sciences thus can help religions to escape from the profound conundrums of ID, and thereby return religion to its rightful realm—not as the secular interpreter of the biological minutiae of our physical existence but, rather, as a respectable philosophical counselor on grander matters, including ethics and morality, the soul, spiritualness, sacredness, and other such matters that have always been of ultimate concern to humanity.




















