
4
Human Adaptations to Diet, Subsistence, and Ecoregion Are Due to Subtle Shifts in Allele Frequency
ANGELA M. HANCOCK,* DAVID B. WITONSKY,* EDVARD EHLER,*† GORKA ALKORTA-ARANBURU,* CYNTHIA BEALL,‡ AMHA GEBREMEDHIN,§ REM SUKERNIK,|| GERD UTERMANN,# JONATHAN PRITCHARD,*** GRAHAM COOP,*†† AND ANNA DI RIENZO*‡‡
Human populations use a variety of subsistence strategies to exploit an exceptionally broad range of ecoregions and dietary components. These aspects of human environments have changed dramatically during human evolution, giving rise to new selective pressures. To understand the genetic basis of human adaptations, we combine population genetics data with ecological information to detect variants that increased in frequency in response to new selective pressures. Our approach detects SNPs that show concordant differences in allele frequencies across populations with respect to specific aspects of the environment. Genic and especially nonsynonymous SNPs are overrepresented among those most strongly correlated with environmental variables. This provides genome-wide evidence for selection due to changes in ecoregion, diet, and subsistence.
|
* |
Department of Human Genetics, University of Chicago, Chicago, IL 60637; |
|
† |
Department of Anthropology and Human Genetics and Department of Biology and Environmental Studies, Charles University, Prague, 128 00 Czech Republic; |
|
‡ |
Department of Anthropology, Case Western Research University, Cleveland, OH 44106; |
|
§ |
Department of Internal Medicine, Addis Ababa University, Addis Ababa, Ethiopia; |
|
|| |
Laboratory of Human Molecular Genetics, Department of Molecular and Cellular Biology, Institute of Chemical Biology and Fundamental Medicine, Russian Academy of Sciences, Novosibirsk, 630090 Russia; |
|
# |
Institute for Medical Biology and Human Genetics, Medical University of Innsbruck, 6020 Innsbruck, Austria; |
|
** |
Howard Hughes Medical Institute, University of Chicago, Chicago, IL 60637; and |
|
†† |
Section for Evolution and Ecology and Center for Population Biology, University of California, Davis, CA 95616. |
|
‡‡ |
To whom correspondence should be addressed. E-mail: dirienzo@uchicago.edu. |
We find particularly strong signals associated with polar ecoregions, with foraging, and with a diet rich in roots and tubers. Interestingly, several of the strongest signals overlap with those implicated in energy metabolism phenotypes from genome-wide association studies, including SNPs influencing glucose levels and susceptibility to type 2 diabetes. Furthermore, several pathways, including those of starch and sucrose metabolism, are enriched for strong signals of adaptations to a diet rich in roots and tubers, whereas signals associated with polar ecoregions are overrepresented in genes associated with energy metabolism pathways.
Modern humans evolved in Africa approximately 100–200 kya (White et al., 2003), and since then human populations have expanded and diversified to occupy an exceptionally broad range of habitats and to use a variety of subsistence modes. There is wide physiologic and morphologic variation among populations, some of which was undoubtedly shaped by genetic adaptations to local environments. However, identifying the polymorphisms underlying adaptive phenotypes is challenging because current patterns of human genetic variation result not only from selective but also from demographic processes.
Previous studies examined evidence of positive selection by scanning genome-wide SNP data using approaches that are generally agnostic to the underlying selective pressures. These studies detected outliers on the basis of differentiation of allele frequencies between broadly defined populations (Barreiro et al., 2008; Coop et al., 2009), extended regions of haplotype homozygosity (Voight et al., 2006; Wang et al., 2006; Pickrell et al., 2009), frequency spectrum-based statistics (Carlson et al., 2005; Williamson et al., 2007), or some combination of these methods (Sabeti et al., 2007; Jakobsson et al., 2008). These approaches are well suited to detect cases in which selection quickly drove an advantageous allele to high frequency, thereby generating extreme deviations from genome-wide patterns of variation. However, selection acting on polygenic traits may lead to subtle shifts in allele frequency at many loci, with each allele making a small contribution to the phenotype [see Pritchard et al. (2010) for a discussion]. Recent genome-wide association studies (GWAS) support this view in that most traits are associated with many variants with small effects and involve a large number of different loci (Manolio et al., 2009). Given that most phenotypic variation is polygenic, adaptations due to small changes in allele frequencies are likely to be widespread.
Detection of beneficial alleles that evolved under a polygenic selection model may be achieved by an approach that simultaneously considers the spatial distributions of the allele frequencies and the underlying selective pressures. Such an approach was used in the past to identify
several paradigmatic examples of human adaptations. For instance, the similarity between the distributions of endemic malaria and those of the thalassemias and sickle cell anemia led to the hypothesis that disease carriers were at a selective advantage where falciparum malaria was common (Haldane, 1949; Allison, 1954). More recent studies of candidate genes support roles for selection on energy metabolism (Hancock et al., 2008), sodium homeostasis (Thompson et al., 2004; Young et al., 2005), and the ability to digest lactose from milk (Bersaglieri et al., 2004; Tishkoff et al., 2007b) and starch from plants (Perry et al., 2007). Taken together, these examples advance a model whereby exposures to new or intensified selective pressures resulted in physiologic specializations.
Here, we develop and apply an approach that uses information about underlying selective pressures while also controlling for the important effect of population structure in shaping the spatial distribution of beneficial alleles. Our approach allows us to detect subtle but concordant changes in allele frequencies across populations that live in the same geographic region but that differ in terms of ecoregion, main dietary component, or mode of subsistence.
RESULTS
We used genotype data for 61 human populations, including the 52 populations in the Human Genome Diversity Project Panel (Li et al., 2008), 4 HapMap Phase III populations (Luhya, Maasai, Tuscans, and Gujarati) (www.hapmap.org), and 5 additional populations (Vasekela !Kung sampled in South Africa, lowland Amhara from Ethiopia, Naukan Yup’ik and Maritime Chukchee from Siberia, and Australian Aborigines). For each of these populations, we gathered environmental data for four ecoregion variables (Fig. S1, available online at www.pnas.org/cgi/content/full/0914625107/DCSupplemental) and seven subsistence variables (comprising four subsistence strategies and three main dietary component variables; Fig. S2, available online at www.pnas.org/cgi/content/full/0914625107/DCSupplemental).
For each SNP and each environmental variable, we contrasted allele frequencies between the two sets of populations using a Bayesian linear model method that controls for the covariance of allele frequencies between populations due to population history and accounts for differences in sample sizes among populations. The statistic resulting from this method is a Bayes factor (BF), which is a measure of the support for a model in which a SNP allele frequency distribution is dependent on an environmental variable in addition to population structure, relative to a model in which the allele frequency distribution is dependent on population structure alone. For subsequent analyses, we use a transformed rank
statistic based on the location of each SNP in the overall distribution of BFs. Because we rank each SNP relative to SNPs within the same allele frequency range and from the same ascertainment panel, this transformed rank statistic allows us to make comparisons across SNP sets. To conduct analyses for the two types of variables (ecoregion and subsistence) as a whole, we also calculated for each SNP a minimum rank statistic across all of the variables within each category, which results in a summary statistic for ecoregion and subsistence, respectively.
Genic and Nonsynonymous SNPs Are Enriched for Signals of Adaptations to Ecoregion and Subsistence
As with any genome-wide scan for selection, there will be SNPs that fall in the extreme tail of the distribution of the test statistic. Therefore, we asked whether two classes of SNPs that are enriched for functional variation [i.e., genic and nonsynonymous (NS) SNPs] are more common in the lower tail of the minimum rank distribution relative to SNPs that are likely to be evolving neutrally (i.e., nongenic SNPs). As shown in Table 4.1, the ratios of the proportions of both genic and NS SNPs to the proportion of nongenic SNPs are significantly greater than 1 across at least two tail cutoffs of the BF distribution (1% and 0.5%) for both variable categories. Importantly, the enrichment of genic and NS SNPs becomes progressively greater in the more extreme parts of the tail. Furthermore, consistent with the fact that a larger fraction of NS SNPs compared with genic SNPs have functional effects, there is a greater enrichment of NS SNPs compared with genic SNPs in the more extreme tail of the distribution. These patterns suggest that the tail of the BF distribution contains true targets of positive selection.
Given that we observed evidence of selection for ecoregion and subsistence overall, we next asked which individual variables may be driving
TABLE 4.1 Proportions of Genic and NS SNPs Relative to the Proportion of Nongenic SNPs in the Tail of the Minimum Rank Distribution
|
|
Tail Cutoff |
|||||
|
Genic:Nongenic |
NS:Nongenic |
|||||
|
Variable category |
0.05 |
0.01 |
0.005 |
0.05 |
0.01 |
0.005 |
|
Ecoregion |
1.06a |
1.17a |
1.19a |
1.20a |
1.58a |
1.58b |
|
Subsistence |
1.04c |
1.11a |
1.11b |
1.12 |
1.60a |
1.87a |
|
aSupport from >99% of bootstrap replicate. bSupport from >97.5% of bootstrap replicate. cSupport from >95% of bootstrap replicate. |
||||||
TABLE 4.2 Proportions of Genic and NS SNPs Relative to the Proportion of Nongenic SNPs in the Tails of the Individual Variable Distributions
|
Variable Category |
Variable |
Tail Cutoff |
|||||
|
Genic:Nongenic |
NS:Nongenic |
||||||
|
0.05 |
0.01 |
0.005 |
0.05 |
0.01 |
0.005 |
||
|
Ecoregion |
Dry |
1.06a |
1.12b |
1.14b |
1.18a |
1.02 |
1.33 |
|
|
Polar |
1.05c |
1.10a |
1.19a |
1.19a |
1.54a |
1.78a |
|
|
Humid temperate |
1.06a |
1.11a |
1.11c |
1.15a |
1.14 |
1.17 |
|
|
Humid tropical |
1.01 |
1.05 |
1.08 |
1.06 |
1.28 |
1.25 |
|
Subsistence |
Agriculture |
1.01 |
1.03 |
1.04 |
1.03 |
1.32b |
1.41b |
|
|
Foraging |
1.03 |
1.04 |
1.04 |
1.25a |
1.46a |
1.25 |
|
|
Horticulture |
1.00 |
0.99 |
1.00 |
1.13 |
1.00 |
0.89 |
|
|
Pastoralism |
1.01 |
1.05 |
1.13b |
1.05 |
1.34c |
1.33 |
|
Main dietary component |
Cereals |
1.04 |
1.06 |
1.10 |
1.04 |
1.12 |
1.37b |
|
|
Fats, meat, and milk |
1.03 |
1.09 |
1.07 |
1.13 |
1.14 |
1.29 |
|
|
Roots and tubers |
1.06a |
1.11a |
1.13a |
1.08 |
1.02 |
1.05 |
|
aSupport from >99% of bootstrap replicate. bSupport from >97.5% of bootstrap replicate. cSupport from >95% of bootstrap replicate. |
|||||||
these signals. To this end, we examined the lower tails of the rank statistic distributions for each individual variable to determine which ones showed the strongest enrichment of genic and NS SNPs. Several ecoregion variables exhibited a significant excess of genic and NS SNPs with low rank statistics, with the strongest signals observed for polar domain (Table 4.2). Fewer individual subsistence variables had strong signals, but two variables are worth noting: the foraging subsistence pattern and roots and tubers as the main dietary component. Fig. 4.1 (Figs. S3-S5, available online at www.pnas.org/cgi/content/full/0914625107/DCSupplemental) illustrates the importance of controlling for population structure to expose these signals, many of which are due to subtle, but consistent, allele frequency shifts across geographic regions. These shifts are detectable even in the face of a large effect of population structure in shaping the geographic distributions of allele frequencies.
Two NS SNPs have extremely high BFs (the highest in their respective frequency bins; Materials and Methods) and provide particularly convincing signals of adaptations to dietary specializations. A SNP (rs162036) that is strongly correlated with a diet containing mainly the folate-poor roots
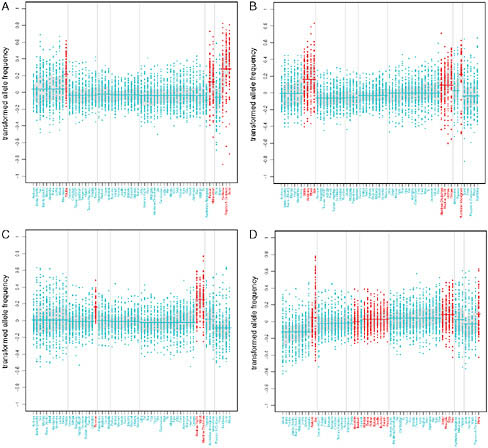
FIGURE 4.1 Transformed allele frequency plotted against population for the variables that showed the strongest enrichment of signal for genic and NS SNPs. Patterns of variation in allele frequencies are shown for (A) the main dietary component roots and tubers, (B) the subsistence strategy foraging, and for (C) polar and (D) dry ecoregions. SNPs were polarized according to the relative difference between the two categories in the first region where both were present; then, transformed allele frequencies were computed by subtracting the mean allele frequency across populations. SNPs with rank <10−4 are included in the plots. Vertical lines separate populations into one of seven major geographic regions (from left to right: sub-Saharan Africa, Europe, Middle East, West Asia, East Asia, Oceania, and the Americas). Dark gray points denote populations that are members of the dichotomous category, and all other populations are denoted by light gray points. Lines are drawn through the mean for the set of populations in a given region that are part of the category of interest, and gray shading denotes the central 50% interval.
and tubers lies within the methionine synthase reductase (MTRR) gene, which activates the folate metabolism enzyme methionine synthase and is implicated in spina bifida (Shaw et al., 2009). Perhaps the most interesting signal comes from a SNP (rs4751995) in pancreatic lipase-related protein 2 (PLRP2) that results in premature truncation of the protein and is strongly correlated with the use of cereals as the main dietary component (Fig. 4.2). Several lines of evidence support an important role for this protein in a plant-based diet. First, unlike other pancreatic lipases, PLRP2 hydrolyzes galactolipids, the main triglyceride component in plants (Andersson et al., 1996; Sias et al., 2004). Second, a comparative analysis found that the PLRP2 protein is found in nonruminant herbivore and omnivore pancreases but not in the pancreases of carnivores or ruminants (De Caro et al., 2008). Our results show that the truncated protein is more common in populations that rely primarily on cereals, consistent with the hypothesis that this variant results in a more active enzyme (Lowe, 2002; Berton et al., 2007) and represents an adaptation to a specialized diet.
Previous analyses have used broad-scale population differentiation, measured by FST, to identify loci that show extreme allele frequency differences between populations and, hence, are candidate targets of natural selection. The approach used here is in some ways similar to an FST-based approach, but it differs in several significant regards (see Discussion). To assess the importance of these differences, we compared our results with those from a simple FST-based analysis. To this end, we calculated global FST for each SNP and compared these values with the minimum transformed rank statistics for ecoregion and subsistence. The correlations were extremely low (−0.024 and −0.034 for ecegion and subsistence,
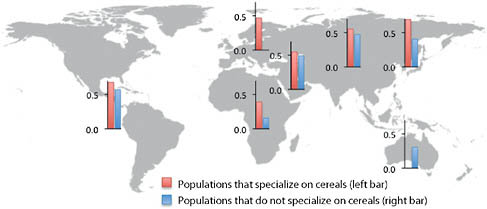
FIGURE 4.2 Average frequencies for PRLP2 W358X (rs4751995) across populations in each major geographic region.
respectively). Further, the amount of overlap in the tails of the distributions (5%, 1%, and 0.5%) was slightly lower than that expected by chance for two independent distributions, suggesting that the environmental contrast approach used here differs from, and is therefore complementary to, a broad-scale FST approach.
Clarifying the Biological Relevance of the Strongest Signals
To identify the pathways that were targeted by selection, we asked whether there is an enrichment of signal for particular canonical pathways. Here, we focused on the individual variables with the strongest enrichment of genic relative to nongenic SNPs: roots and tubers as the main dietary component and polar ecoregion. Because we found that proportionally more genic than nongenic SNPs have strong correlations with environmental variables, an enrichment of signals for SNPs in a particular gene set relative to nongenic SNPs could simply reflect this global genic enrichment. Therefore, in this analysis, we examined the tail of the rank statistic distribution and asked whether the proportion of SNPs from genes implicated in a given canonical pathway was greater than the proportion of genic SNPs from all other genes.
The two strongest pathway signals for roots and tubers are with starch and sucrose metabolism and folate biosynthesis (Table 4.3). In light of the fact that roots and tubers are mainly composed of starch and are poor in folates, it is plausible that variation in these pathways is advantageous in populations that rely heavily on these food sources. Among the genes with strong signals in this group, there are several involved in the degradation and synthesis of glycogen (GAA and GBE1). A gene coding for the cytosolic β-glucosidase (GBA3) contains several SNPs strongly correlated with roots and tubers as the main dietary component. This liver enzyme hydrolyzes β-D-glucoside and β-D-galactoside, and it may be involved in the detoxification of plant glycosides, such as those contained in roots and tubers (de Graaf et al., 2001). Several of the pathways with strong signals with polar ecoregion are involved in metabolism (e.g., pyruvate metabolism and glycolysis and gluconeogenesis) (Table 4.3). Among the genes in the pyruvate pathway, we observed particularly strong signals in the gene coding for mitochondrial malic enzyme 3 (ME3), which catalyzes the oxidative decarboxylation of malate to pyruvate. Interestingly, the gene coding for another mitochondrial malic enzyme (ME2) also contains two SNPs strongly correlated with polar ecoregion. These results suggest a link between cold tolerance and energy metabolism and point to specific variants that are likely to influence cold tolerance. Further, our findings are consistent with a previous study that found strong correlations between variants in genes implicated in energy metabolism and winter temperature
TABLE 4.3 Canonical Pathways Enriched in the 1% and 5% Tails of the Minimum Rank Distribution
|
|
|
|
SNPs in Pathway:Other Genic SNPs (tail cutoff) |
||
|
Variable Category |
Variable |
Description |
0.05 |
0.01 |
0.005 |
|
Ecoregion |
Polar domain |
Glycolysis and gluconeogenesis |
5.91a |
4.86a |
2.38a |
|
|
|
Bile acid biosynthesis |
7.04a |
5.53a |
2.61a |
|
|
|
Pyruvate metabolism |
6.92a |
5.10a |
2.72a |
|
|
|
3-Chloroacrylic acid degradation |
17.42a |
12.94a |
4.22a |
|
|
|
Arginine and proline metabolism |
3.42a |
3.39a |
1.86a |
|
Subsistence |
Roots and tubers |
Starch and sucrose metabolism |
2.72a |
2.21a |
1.61a |
|
|
|
Folate biosynthesis |
4.62a |
3.65a |
2.41a |
|
aSupport from >99% of bootstrap replicate. |
|||||
(Hancock et al., 2008) and with studies that show evidence for adaptation in mitochondrial DNA (Ruiz-Pesini et al., 2004; Balloux et al., 2009).
Results of genome-wide association studies with diseases and other complex traits offer an opportunity to connect signals of selection with SNPs influencing specific traits and diseases. To this end, we identified a subset of SNPs with extremely strong correlations with environmental variables that were also strongly associated with traits from 106 GWAS (Table 4.4). We find that several SNPs strongly correlated with subsistence, and main dietary component variables are associated with energy metabolism–related phenotypes [high-density lipoprotein cholesterol, electrocardiographic traits and QT interval (el-Gamal et al., 1995), fasting plasma glucose, and type 2 diabetes]. These signals include a SNP in the type 2 diabetes gene KCNQ1, where we find that the risk allele is at higher frequency in populations where cereals are the main dietary component.
DISCUSSION
This genome-wide scan identified targets of adaptations to diet, mode of subsistence, and ecoregion. The environmental variables in our analysis were chosen to capture the striking diversity among populations in
TABLE 4.4 SNPs with the Strongest Signals of Selection Among Those Associated with Phenotypic Traits in GWAS
ecoregion, diet, and subsistence. Much of this variation is related to major transitions that occurred during human evolutionary history, including the dispersal out of sub-Saharan Africa to regions with different climates and the adoption of more specialized—often less diverse—diets (i.e., farming and animal husbandry vs. foraging). Our results aim to clarify the genetics underlying the adaptive responses to these transitions.
Most human phenotypes, including adaptive traits like height and body proportions, are quantitative and highly polygenic (Manolio et al., 2009), and most human variation is shared across populations. Therefore, the same adaptive allele may often be independently selected in different geographic areas that share the same environment. The environmental aspects considered in this analysis changed dramatically over human evolutionary time. As a result, selection on standing—rather than new—alleles, which afford a faster adaptive response to environmental change (Hermisson and Pennings, 2005), may have played a prominent role in adaptation to new environments. This proposal is supported by expectations of selection models for quantitative traits (Falconer and MacKay, 1996), specifically that selection will generate small allele frequency shifts at many loci until the population reaches a new optimum (Pritchard et al., 2010). Whereas approaches that detect selection under a hard sweep model aim to identify loci that drove a new allele quickly to high frequency in the population (Pritchard et al., 2010), our approach is well suited to detect small shifts in the frequencies of beneficial alleles that have a broad geographic distribution [see Hancock et al. (2010) for a more detailed discussion]. For quantitative traits, the method we use may be particularly appropriate for understanding recent human adaptations. In this sense, our results fill an important gap and are useful for reconstructing the genetic architecture of human adaptations.
Some of our most interesting signals seem to be adaptations to dietary specializations. Although cultural adaptations certainly played an important role in our ability to diversify, there is strong evidence that genetic adaptations have been crucial as well. A previous genome-wide analysis of sequence divergence between species found evidence for ancient adaptations along the human lineage in the promoters of nutrition-related genes along the human lineage (Haygood et al., 2007). Examples of more recent genetic adaptations that were integral for dietary specializations include variants near the lactase gene, which confer the ability for adults to digest fresh milk in agropastoral populations, and an increase in the number of amylase gene copies in horticultural and agricultural populations (Bersaglieri et al., 2004; Perry et al., 2007; Tishkoff et al., 2007b; Enattah et al., 2008). Our results indicate that genetic adaptations to dietary specializations in human populations may be widespread. In particular, we find signals of adaptations in populations that heavily depend on roots and
tubers, which are staple foods in places where cereals and other types of crops do not grow well (e.g., in regions with nutrient-poor soils and with frequent droughts). Given that roots and tubers are rich in carbohydrates, it is particularly compelling that the most significant gene set for populations that depend on this food source is the starch and sucrose metabolism pathway. Further, roots and tubers are low in folic acid, a vitamin with an important role in newborn survival and health; accordingly, we find a strong signal for genes implicated in folic acid biosynthesis in populations that specialize on this food source. Additional signals with diet include those observed in populations that specialize on cereals, with SNPs implicated in type 2 diabetes (Table 4.4) and in the hydrolysis of plant lipids.
Foraging, or hunting and gathering, is the mode of subsistence that characterized human populations since their emergence in Africa until the transition to horticulture, animal farming, and intensive agriculture that occurred starting roughly 10,000 years ago (Smith, 1995). With this transition, many aspects of human ecology dramatically changed, from diet and lifestyle to population densities and pathogen loads. Given that our hominin ancestors were foragers, the signal we observe in the contrast between forager and nonforager populations is likely to reflect adaptations to the less diverse, more specialized diets in horticulture, animal farming, and agriculture (Larsen, 2003). Our findings are consistent with the results of an analysis of the NAT2 drug metabolizing enzyme gene, which found a significant difference in the frequency of slow acetylator mutations between forager and nonforager (i.e., pastoral and agricultural) populations. These findings were interpreted as the result of the diminished dietary availability of folates consequent to the subsistence and nutritional shift (Luca et al., 2008).
Ecoregion classifications include information about climatic factors, vegetation, geomorphology, and soil characteristics (Bailey and Hogg, 1986). Therefore, they provide an integrated view of many facets of human environments. Interestingly, the strongest signal was observed for the polar domain classification and, to a lesser extent, for the dry and humid temperate domains. Although polar habitats presented diverse challenges to human survival, including cold temperature, low UV radiation, and limited resources, our gene set enrichment analyses suggest that the signals of selection in the polar domain tend to be due to alleles that conferred adaptations to cold stress. In fact, many of the gene sets significantly enriched for signals with the polar domain are directly relevant to energy metabolism and temperature homeostasis. Adaptations in these genes were probably critical in the establishment of stable human populations in the northernmost latitudes of Europe and Asia. Likewise, signals associated with the dry and humid temperate domains may reflect relatively ancient adaptations that occurred during the dispersal of anatomically
modern human populations. The lack of a significant excess of signals associated with the humid tropical domain may be due to a combination of factors, including the fact that humans reentered the humid tropics outside Africa too recently to generate detectable new adaptations.
In some ways our approach is similar to previous analyses based on FST, but there are two important differences. First, we compare populations on the basis of environmental variables rather than their geographic origin, thus providing greater power to detect allele frequency differences that track the underlying selective pressure. Second, unlike other analyses of spatial patterns of variation, we use a test statistic (the BF) that detects a signal relative to a null model that captures aspects of human population structure. Taken together, these two features of our approach allow us to detect novel loci where SNPs show subtle, but consistent, patterns across populations. As a result, our findings differ substantially from the results of previous analyses based on broad-scale population differentiation. The overlap in the tails from global FST and the minimum ranks for subsistence and ecoregion, respectively, are slightly less than expected by chance.
A possible caveat to the results presented here is that they are due solely to background selection, whereby the elimination of strong deleterious alleles continually arising in genic regions effectively reduces the effective population size of these regions compared to the less constrained nongenic regions. As a result, genic regions may be expected to experience higher rates of genetic drift and to exhibit greater differentiation between subdivided populations compared with neutrally evolving loci (Charlesworth et al., 1997; Hu and He, 2005). Therefore, purifying rather than positive selection could potentially account for the excess of genic SNPs strongly correlated with environmental variables. Although we cannot formally rule out this possibility, we note that two features of our data suggest that background selection does not entirely account for the observed enrichment. One is that the enrichment of genic and NS SNPs becomes more pronounced in the more extreme lower tails of the BF distribution, as expected if at least some of the SNPs were indeed targets of positive selection. The other feature is that the enrichment of NS SNP is quantitatively greater than the enrichment of genic SNPs; because a larger fraction of NS SNPs affect gene function compared with genic SNPs, this is the pattern expected if at least some of the NS SNPs increased in frequency because of a selective advantage.
Our results extend upon and are complementary to results of previous scans for natural selection in humans. By conducting multiple contrasts between populations that differ with respect to ecoregion or subsistence to identify genetic variants that show concordant changes in allele fre-
quencies across populations, we find a set of adaptive SNPs that differs compared with previous analyses that were agnostic to the underlying selective pressure. Further, because the SNPs we identify tend to have a global distribution and to show subtle, but consistent, differences in allele frequencies across populations, loci we identify are likely to represent cases of selection on standing variation. As a result, the findings presented here represent an important step toward clarifying the genetic basis of human adaptations.
MATERIALS AND METHODS
Environmental Variables
Ecoregion data were obtained for each population on the basis of coordinates where samples were collected, except for the Vasakela !Kung and the Gujarati, who had recently relocated. For these populations, we used coordinates of their most recent homeland. The individuals who were sampled from the !Kung population were known to have recently relocated to Schmidtsdrift, South Africa, from the Angola/Namibia border, so we used coordinates that reflected their location before this migration. Each population was classified into one of four ecoregion domains, which are defined according to a combination of ecologically important aspects of climate. Therefore, the ecoregion variables are closely related to climate, but they may be a more informative representation of climatic variation. The ecoregion domains comprise polar, humid temperate, humid tropical, and dry. We classified each population on the basis of the coordinates of the population using Bailey’s Ecoregion Map (Bailey and Hogg, 1986).
When available, data from Murdock (1967) were used to classify populations according to their main mode of subsistence and dietary specialization. In cases in which Murdock did not have information about a population, we obtained information from the Encyclopedia of World Cultures (Levinson, 1991–1996). We classified each population into one of four subsistence categories (foraging, horticultural, agricultural, or pastoral) and into one of three categories based on the main dietary component (cereals; roots and tubers; or fat, meat, or milk). Each population was classified into subsistence and main dietary component categories by two independent researchers, and the small number of discrepancies that were found were resolved by further research. For the five populations that were genotyped by our group, individuals who oversaw collection gave input for classification.
Detecting Signals Between SNPs and Dichotomous Environmental Variables
To assess evidence for selection related to each dichotomous environmental variable, we contrasted the allele frequencies for each SNP across populations that differ with respect to the environmental variable. More specifically, we used a Bayesian linear model method that controls for population history by incorporating a covariance matrix of populations and accounts for differences in sample size among populations. This method yields a BF that is a measure of the weight of the evidence for a model in which an environmental variable has an effect on the distribution of the variant relative to a model in which the environmental variable has no effect on the distribution of the variant. On the basis of these BFs, for each SNP and each environmental variable, we calculated a transformed rank statistic that was scaled to be between 0 and 1 (with 0 and 1 corresponding to the highest and lowest BF, respectively); this transformed rank statistic is sometimes referred to as an empirical P value. Calculating this transformed rank statistic allowed us to control for some aspects of SNP ascertainment and differences in allele frequencies across SNPs. The Illumina 650Y platform used for genotyping is made up of three panels of tagging SNPs that were ascertained in different ways (Eberle et al., 2007). To calculate the transformed rank statistic for each SNP for a given variable, we found the rank of the SNP relative to all other SNPs in the same ascertainment panel and within the same allele frequency bin, where there were 10 allele frequency bins, based on the global derived allele frequency.
To summarize the evidence for selection for each SNP for the two categories of variables (subsistence and ecoregion), we calculated a minimum rank statistic by finding the minimum of the transformed rank statistics across all subsistence and ecoregion variables, respectively. Using these minimum rank statistics, we could ask questions about the evidence of selection for subsistence and for ecoregion overall.
Assessing the Evidence for an Excess of Functional SNPs in the Tail of the Distribution
To determine whether the lower tail of the rank statistic distribution contains an excess of SNPs enriched for function, compared with that expected by chance, we calculated the proportions of genic and NS SNPs relative to the proportion of nongenic SNPs in the tail. Rather than arbitrarily choosing a single tail cutoff, we examined the enrichment at three tail cutoffs (5%, 1%, and 0.5%). To assess significance for an observed excess, we used a bootstrap resampling technique to obtain confidence intervals on the estimated excess. Because positive selection can result
in increased linkage disequilibrium near a selected variant, we bootstrap resampled across 500-kb segments of the genome. For each of 1,000 bootstrap replicates, we calculated the proportion of genic and NS SNPs relative to the proportion of nongenic SNPs in the tail of the distribution. We consider an excess significant for a given tail cutoff if at least 95% of the bootstrap replicates support an excess of SNPs enriched for function.
Comparison of Results from Environmental Contrasts and FST
We calculated global FST values (Weir and Cockerham, 1984) for the complete set of 61 populations. Then, for each SNP, we calculated a transformed rank statistic as we had done for the environmental variable contrasts. Next, we calculated Spearman correlation coefficients between FST values and the minimum transformed rank statistic from the environmental contrast analyses. In addition, we assessed the amount of overlap in the tails of the distributions for FST and environmental contrasts relative to chance.
Canonical Pathway Analysis
To determine whether there was an enrichment of signal for a particular canonical pathway, we used a method similar to that used to test for an excess of genic and NS SNPs relative to nongenic SNPs in the tails of the test statistic distribution. Here, we compared the proportion of SNPs from a given pathway with the proportion of all other genic SNPs in the tail of the minimum rank distribution and of the transformed rank distributions for the individual variables with the strongest genic enrichment. To assess significance for the findings and to ensure that the results are not driven by one or a few genomic regions, we applied the same bootstrap approach described above. The lists of genes included in each of the 438 canonical pathways were obtained from the Molecular Signatures Database (Subramanian et al., 2005).
Comparison with GWAS Results
We downloaded the Catalog of Published Genome-Wide Association Studies (Hindorff et al., 2009) on July 14, 2009, which includes information about SNPs with reported associations with P < 1 × 10−5. We filtered this database for SNPs found on the Illumina HumanHap650Y platform; there were entries for 800 unique autosomal SNPs implicated in 61 traits. From among these SNPs, we identified a set of SNPs with extremely low rank statistics (<5 × 10–4) for each of the subsistence and ecoregion variables. Given that most GWAS are performed in populations of European ances-
try, we binned the SNPs in the Illumina panel on the basis of the allele frequency in Europeans rather than the global allele frequency to calculate the transformed rank statistics.
ACKNOWLEDGMENTS
We thank members of the Di Rienzo laboratory, John Novembre, and Molly Przeworski for helpful discussions during the course of this project; and Molly Przeworski for thoughtful comments on the manuscript. This work was supported by National Institutes of Health (NIH) Grants DK56670 and GM79558 and an International Collaborative Grant from the Wenner-Gren Foundation (to A.D.R.). A.M.H. was supported in part by American Heart Association Graduate Fellowship 0710189Z and by NIH Genetics and Regulation Training Grant GM07197. G.C. was supported in part by a Sloan Research Fellowship. J.K.P. acknowledges support from the Howard Hughes Medical Institute.


















