
5
The Global Framework for Regulation of Medical Devices
The final session of the workshop focused on how other countries have dealt with some of the medical device regulatory issues that were identified in the United States. Speakers and panelists discussed the global regulatory environment and past and current efforts toward global harmonization.
Comparative Overview of Medical Device Regulatory Systems
David Jefferys, a medical device expert and senior vice president for global regulatory, health-care policy, and corporate affairs at Eisai Europe, Ltd., provided an overview of European device regulations and discussed some of the key procedures in Japan, China, and India and how they differ from current operations in the United States.
European Regulations
In Europe, four directives cover the medical device sector. A directive is an instruction to the member states of the European Union (EU) to implement a law through national regulations. The first directive, 90/35, was concerned with active or powered implants. It was followed by the main general medical device directive, Directive 93/42, and then more recently by Directive 98/79, which covers in vitro diagnostics, and Directive 2000/70, which covers human blood and plasma derivatives. The date of a directive is not the implementation date, Jefferys noted. The general medical device directive of 1993, for example, was not fully implemented until the end of 1998, and the in vitro diagnostic directive of 1998 did not become fully
operational until 2001. More recently, in an effort to consolidate the texts, Directive 2000/747 was issued to bring together the four others and was an “updating directive.” More detailed implementing directives (there are five in Europe) will be enacted by the European Commission, taking into account the views of the member states. There is also Advanced Therapies Regulation 1394/2007, which is automatically binding on the member states and does not have to be transposed into national law.
Those directives are known in Europe as New Approach legislation. That legislation covers all consumer goods except pharmaceuticals. (Legislation concerning pharmaceuticals has been in place since 1965 at the European level since the thalidomide disaster.) The New Approach incorporates self-regulation and imposes the minimum level of regulation that is necessary to protect public health. The legislation reflects the dynamics of the device industry, which are different from those of the pharmaceutical industry.
Key features of the device regulation are that the legislation sets out what are known as the essential requirements, the core elements and procedures that companies need to have in place; sets out and defines the conformity assessment process (how independent bodies will assess whether a device is in conformity with the directives); and lays down precise obligations on the part of manufacturers. The legislation establishes “notified bodies” to evaluate devices and “competent authorities,” which are the agencies that control clinical trials, designate and supervise the notified bodies, and oversee postmonitoring surveillance. The legislation itself is underpinned by “normative standards.” Some are European standards, others are International Organization for Standardization (ISO) standards, and some are parts of a series of European guidelines called MEDDEV.
The European system is similar to the US system in that it is a risk-based device classification system, Jefferys said. In Europe, there are three classes but four categories:
-
Class I—self-regulation (and registration in each member state where they are marketed).
-
Class IIA—selective quality-system review (QSR) (for example, measuring devices and sterile products).
-
Class IIB—full QSR and targeted review of the design dossier (devices are defined in legislation and are not open to interpretation).
-
Class III—full design-dossier review.
Competent Authorities
Each member state in the EU has a competent authority; these are the same agencies that regulate pharmaceuticals (except in the Netherlands).
The first role of the competent authority is to designate and then to supervise the notified bodies (and on occasion to withdraw the approval of a notified body or restrict it). A notified body falls under the supervision of the member state in which its headquarters is; however, at the EU level, the Notified Body Operations Group (NBOG) sets the criteria for inspection and coordinates training and supervision for the shared audits of the notified bodies.
Clinical trials for devices in Europe are controlled by member states under a competent authority. Another role of a competent authority is compliance and enforcement, ensuring that the Medical Devices Act is being complied with and potentially prosecuting anyone who places a device on the market without authorization or a device that is inappropriately labeled. A competent authority also supervises class I devices. Although there is self-regulation, there is a program whereby the agency will visit a manufacturer and review its dossier to make sure that the company’s self-regulation is appropriate. Some of the audits will be unannounced, others will be targeted around complaints or result from vigilance reports or adverse incidents.
Notified Bodies
The notified bodies have a variety of backgrounds and competences. As defined by the directive, notified bodies may cover all consumer products or may be selective. As described above, they are supervised by the competent authority of a member state and by the NBOG. Each has a detailed published policy of conflicts of interest regarding internal staff and expert panelists.
In Europe, a manufacturer chooses one notified body, which then undertakes the evaluation of the manufacturer’s product. Evaluation is done once for all Europe. The manufacturer pays the notified body for this service. For example, a German company can go to a Spanish notified body; once the notified body is satisfied, it allows the company to apply the European Conformity (CE) mark, the product is placed on the register (with a note indicating the number of the notified body), and then the product can circulate, with appropriate labeling, anywhere in the EU, the European Economic Area, and some other countries that have mutual agreements with the EU, such as Switzerland and Turkey. Jefferys noted that with good systems of quality assurance there is no concern about a conflict of interest associated with a manufacturer’s paying the notified body for review of its product. The notified body is inspected and supervised by a government agency, so there is a separation, with respect to quality assurance, between those doing the evaluation and those evaluating the evaluation.
Most companies build a relationship with one notified body, and that notified body will inspect its quality systems, risk-management systems, and
other aspects of its operation, Jefferys said. The notified body lives with the product and with any variations or change in the product, and it is involved if there are any vigilance problems.
Many of the notified bodies play an international role, are qualified under the Conformity Assessment Body system to bring a product into the United States, and have a role in the systems in China and Japan.
A notified body assesses only whether the device works according to the manufacturer’s claims. Other bodies will determine whether the device represents a good use of public money and how it fits with other devices, therapies, or interventions already used in the health system.
Postmarket Surveillance in Europe
There are two systems for postmarket surveillance in Europe: the mandatory vigilance procedure and the user reporting system. The vigilance procedure follows the Global Harmonization Task Force (GHTF) Study Group 2 guidance and is compulsory for manufacturers. Evidence suggests that manufacturers in Europe probably report twice as many cases as they need to. Electronic reporting is now used in many member states. The competent authority of the member state in which an adverse event first occurs will become the lead to coordinate European action. The legislation includes a safeguard clause whereby a member state that is particularly concerned can suspend the CE mark with immediate effect, but that action then must be referred to the European Commission within 15 days for a European view.
The user reporting system is built largely around the fact that health care in almost all member states is paid for and generally run by the countries concerned. It is therefore expected that health-care professionals will report adverse events to the competent authorities. Patients are also encouraged to report adverse events directly. In some member states, there is now a system of liaison officers, designated staff members in the health system who are responsible for seeing that health-care device alerts are received and implemented by all health-care professionals. Liaisons are also responsible for quarantine procedures in the event of a device recall.
In many member states, registries allow all patients with a particular device to be followed—for example, joint implants, cardiac pacemakers, heart valves, coronary stents, breast implants, and cephalic shunts. Many member states also have national electronic record databases, which allow consolidated collection of information over a patient’s life span.
Jefferys noted that the European system allows a device’s classification to be upregulated and downregulated as experience accumulates.
Specific Device Issues
In Vitro Diagnostics
The in vitro diagnostics directive has two annexes: annex 1 requires manufacturers to supply the information necessary for the safe and proper use of the device, and annex 2 is a defined list that includes, for example, blood reagents, anything to do with HIV testing, hepatitis testing, and any over-the-counter device that is for self-monitoring, such as blood-glucose monitoring. But many tests that are left out of annex 2 should perhaps be included, Jefferys said, such as biomarkers and genetic tests. There is a move in Europe to make in vitro diagnostics subject to a more risk-based classification system.
In Europe, as in the United States, there are issues related to “home-brew” test kits and to the balancing of compliance with the needs of innovators and health-care professionals, who develop or adapt diagnostic devices as situations require.
Combination Products
Combination products are the subjects of active regulation, Jefferys said. Recent research suggests that up to 30 percent of pharmaceutical research and development is now directed toward combination products. There are three basic groups of these products: drug–device combinations, which fall under pharmaceutical law; device–drug combinations, where the lead is device law; and diagnostic–drug combinations or “companion diagnostics,” which Jefferys said may require new legislation to be appropriately handled.
For a device that administers a medicinal product, in Europe as in the United States, the concept of “primary intended purpose” is used. Simply, the medical device directive applies if the device components to deliver the drug could be used separately, such as syringes and infusion pens. A notified body is obliged to get an opinion from a pharmaceutical competent authority. However, if the device and the medicinal product form an integrated element, the product will be covered under the pharmaceutical law, for example, prefilled injectors, such as the EpiPen, in which it is clear that the device is to be used only once for delivering the pharmaceutical product contained.
Some combination products are medical devices that incorporate a pharmaceutical substance with an ancillary action, for example, drug-eluting stents. Those are handled under the device law, but the opinion comes from the drug authority, who looks at the safety, quality, and usefulness of the product. Usefulness, which is determined by a notified body, is
the basic rationale for the product, in light of the contribution that is made by the product, but not the efficacy or the performance itself.
In Europe, Directive 2000/70 deals with combinations of device with stable blood products. That would seem to be a small category and one that might not merit a separate directive, but it was meant to be the legislation that would capture tissue engineering. However, there was not political agreement, and all that was left was a piece of legislation on stable blood products. A combination that involves a blood product is subject to a mandatory consultation with the European Medicines Agency (EMA), not the individual member states.
In summary, there is no European counterpart to the US Food and Drug Administration (FDA) Office of Combination Products. Instead, it is necessary to involve both parts of the system—notified bodies and competent authorities—as appropriate.
Advanced Therapy Products
Europe has legislation regarding advanced therapy products (regulation 1394/2007EC), which covers tissue engineering, cell therapy, and gene-therapy products. The latter two, Jefferys noted, were already controlled under pharmaceutical legislation but have now been brought together with tissue engineering, or human viable cell products, to be included in the regulation of advanced therapies. The legislation covers both allogeneic and autologous products. The Committee for Advanced Therapies has been established and reports to the Committee on Human Medicinal Products in the European Medicines Agency.
Borderline Products
As in the United States, regulations cover medical devices, pharmaceuticals, advanced therapy products, cosmetics, biocides, personal protective equipment, and foods and nutraceuticals. Among each of those, there will be products on the border between classifications (for example, artificial saliva and medicinal wipes). A guideline gives examples of borderline products that are classified as medicines or devices. A product cannot be covered under more than one piece of legislation, so a decision must be made to regulate it under the pharmaceutical or the devices directive.
Japan
Japan has been changing both its pharmaceutical and its device legislation, Jefferys said, working toward following the GHTF classification (additional details about the GHTF system are presented later in this
chapter). How the Pharmaceutical Affairs Law classifies medical device products generally overlaps with the GHTF system. But the Japanese, like the Europeans, have been using third-party certification for class II devices, and Japan has designated 12 certification bodies. Japan’s Ministry of Health, Labour and Welfare (MHLW) receives a dossier at a superficial level and decides whether it is appropriate for third-party assessment. After reviewing a favorable report from a third-party assessor, MHLW issues a certificate. Using the GHTF principles, the third-party assessors use the Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices (STED) for the product application. By the end of 2011, all class II devices will be handled by third-party certification.
Overall, Japan is moving forward in a fashion similar to that in Europe, Jefferys said.
India
In India, medical devices are regulated under the pharmaceutical law by the director general for pharmaceuticals. After extensive consultation, India is introducing comprehensive medical device regulation. Modeled largely on the GHTF, it has the same four classification categories, from low risk to high risk, and will involve a conformity-assessment process, self-regulation, notified bodies, and quality system review, similar to those in Europe. The only difference, Jefferys said, is that India uses type testing, in which is a designated laboratory tests devices. The new legislation is expected to be in place within the next 12 months.
China
In China, medical devices are controlled by both the central State Food and Drug Administration (SFDA) and local provincial controls. The system is risk-based and similar to that in India in that class II and class III devices undergo sample testing (type testing) in an approved laboratory. Selected products are required to undergo further clinical evaluation in designated SFDA-approved hospitals.
The SFDA has its own evaluation center and its own expert technical committees. Chinese regulations require a local, Chinese-based distributor. Inspections are handled by provincial authorities. In the case of overseas manufacture, the country of origin must attest its approval. For example, a US-based company cannot bring a new device into China if it is not already registered with the US FDA.
PAST, PRESENT, AND FUTURE GLOBAL HARMONIZATION
Janet Trunzo, the executive vice president for technology and regulatory affairs at AdvaMed and a member of the GHTF steering committee, provided an overview of efforts to harmonize regulatory approaches for medical devices. The call for harmonization came from various stakeholders, including governments, industry, and the public. Harmonization provides for consistent application of regulatory principles and approaches and improves regulatory-system effectiveness and efficiency. There is a reduction in duplication of regulatory activities, which can lead to time and cost savings. New products and technologies enter the marketplace in more streamlined fashion, and there is more transparency in the process.
Many regulatory programs use international standards and guidelines as a basis of their national technical regulations. Trunzo noted that many FDA staff have participated on some of the regulatory-standards committees. It is also important that regulatory systems seek input from stakeholders in the process of harmonization.
The GHTF is a voluntary group that was established in 1992 as a partnership of the regulators and the regulated industry. The founding members were the United States, the EU, Canada, Australia, and Japan. There are liaisons with other bodies throughout the world, including the Asian Harmonization Working Party (AHWP); GHTF has memoranda of understanding with the ISO and the International Electrotechnical Commission, and it works directly with the World Health Organization and the Pan American Health Organization.
The purposes of the GHTF were to encourage convergence in global regulatory practices and to promote technologic innovation and international trade through harmonized regulatory processes. The task force was also designed to serve as an information-exchange forum. (The GHTF does not evaluate the effectiveness of regulatory systems worldwide.)
Structure
The GHTF is governed by a steering committee composed of four regulatory representatives and four industry representatives of each of three geographic areas—North America, Europe, and the Asia Pacific (total, 24 members). Leadership of the steering committee rotates every 3 years. In addition to the steering committee, which directs the work of and defines the strategic plan for the organization, there are five study groups and ad hoc working groups as needed.
Study group 1, the premarket study group, developed many of the documents that were the basis of the harmonized regulatory model. Study group 2, which focused on postmarket issues, had a role in developing the vigilance procedures and adverse-event reporting. Quality systems, the focus
of study group 3, are based on the international standard for quality-management systems, ISO 13485. Basic auditing processes and the standard audit-report format were developed by study group 4. Study group 5 focused on clinical evidence.
A primary subject of activity is principles of classification, especially the establishment of a common vocabulary. Other basic subjects include technical requirements, format and content of marketing applications, assessment and review practices, postmarket activities, and quality-management system requirements and auditing functions.
Ad hoc working groups have been established on medical device software, combination products, training, the global regulatory model, global medical device nomenclature, unique device identifiers, and improvement of GHTF administrative processes.
Accomplishments
Trunzo highlighted several key accomplishments of the GHTF. First is the development of a harmonized regulatory model. Countries that are developing device regulatory systems can use the model as a reference. The model incorporates principles of risk-based classification, harmonized definitions and vocabulary, global medical device nomenclature, the STED format for marketing applications, assessment and review practices, quality-management system requirements, postmarket activities, use of international standards, adverse-event reporting requirements, and the National Competent Authority Report (NCAR) exchange program.
The document on principles of classification contains basic principles, but they can be modified by looking at the history of a particular product and considering whether it can be moved into a lower class or a higher class. (It is not based on any kind of predicate system, Trunzo noted.) The principles were defined to allow approval of a product through regulatory systems (for example, notified bodies for the moderate-risk classes); they include basic principles, essential requirements, and conformity-assessment principles for facilitating a determination of whether a product should go onto the market. In its documentation, Trunzo said, the system complements what occurs in the US regulatory system.
With regard to postmarket activities, the GHTF has provided basic guidance in collecting adverse-event reports, the report format, taking field corrective actions (for example, recalls), and vigilance reporting. Part of postmarket activities is the NCAR program whereby regulatory agencies exchange reports of adverse events in their countries. To participate, a country must have an adverse-event reporting system, must be trained by the members of the GHTF who administer the program, and need to understand the various levels of regulatory action.
The GHTF has created over 30 guidance documents that describe all the regulatory processes noted above. There is a consultation system on the GHTF Web site for any guidance document that is proposed and comments are accepted from stakeholders. Every comment is formally addressed by the study group that developed the document. The GHTF is also asked to provide regulatory training on the basic elements of its regulatory model. Trainers are mostly volunteers who work for regulators and the industry.
Challenges
A question often heard at GHTF conferences is why FDA has not fully adopted the GHTF model. The answer, Trunzo said, is that FDA had a regulatory system that was far more mature than any of the regulatory systems of other GHTF founding members. But there is a commitment from the members of the GHTF steering committee toward convergence of their regulatory systems as much as possible with the principles of the GHTF regulatory model.
Another challenge is related to conformity assessment vs type testing. Some countries still want to do type testing, but this is contrary to the quality-management-systems approach. In a quality-management-systems approach, there are procedures that build quality into the system, the product, and the design controls. One cannot test for quality one test at a time, Trunzo said.
In the United States, quality-system regulation is based on the ISO 13485 system adopted by the GHTF, but the QSR is still slightly different. Convergence in this field would be a good step forward, Trunzo said.
Determining when submission of clinical evidence is necessary and the elements that make up clinical evidence is another challenge. The GHTF study group 5 guidance document tries to provide some framework to address this issue.
Finally, adoption of global nomenclature is essential for progress, and continuing funding is needed for GHTF training.
The Importance of the Global Harmonization Task Force’s Work
The guidelines that have been created by the GHTF provide a scientifically sound and internationally harmonized means of establishing quality, safety, and efficacy. The results are improved transparency, predictability, and efficiency of the medical device review process. Harmonization reduces regulatory burden and promotes industry compliance.
The work done by the GHTF promotes trade, innovation, and a more modern risk-based approach to regulation. Harmonization also creates a level playing field for industry in all countries. The GHTF promotes
regulatory communication and cooperation, providing opportunities for regulators to understand what is going on in other countries and for those developing regulatory systems to learn from others’ experiences. Harmonization facilitates earlier availability of new technology and helps to avoid differences in technical requirements. The GHTF fosters productive working relationships among regulators, industry, and other organizations.
Adoption and Expansion of the Global Harmonization Task Force Model
The GHTF founding members are committed to moving their regulatory systems to the GHTF model. The AHWP, which has representatives of 20 countries, has developed its regulatory systems on the basis of the GHTF model, and the Association of Southeast Asian Nations (ASEAN), a group of 10 nations, has agreed to adopt the GHTF model. The Latin American Harmonization Working Party also participates actively in the GHTF.
Expansion is an important factor for the GHTF, and it involves more training and more wider adoption of the guidance documents that have been developed by the GHTF to facilitate broader implementation of the GHTF model. There are also efforts to translate GHTF guidance documents into other languages.
The GHTF has accomplished much in the last 18 years, Trunzo concluded. GHTF discussions today lead to a common regulatory framework of the future. Building on that foundation, we can move forward to the realization of global harmonization.
THE PRICEWATERHOUSECOOPERS MEDICAL INNOVATION TECHNOLOGY SCORECARD
Trunzo presented an update on the development of a “medical innovation technology scorecard” by PricewaterhouseCoopers (PwC) on behalf of Doug Mowen, managing director of medical device industry practice at PwC, who was unexpectedly unable to attend the workshop.
The goal of developing the scorecard is to inform all medical device industry stakeholders about why the innovation model for medical devices is unique. The project, sponsored by PwC, was announced in spring 2009 at an international conference in Rome, and it is expected to be completed in fall 2010. The final product will be presented at the AdvaMed Technology Conference in October 2010 in Washington, DC.
The framework of the scorecard consists of two basic elements, Trunzo explained. The first is information on the regulatory environment (including policy, compliance, payment, and reimbursement), which is being collected through a survey of medical-technology companies. The second element is a collection of information from publicly available sources (such as the World
Bank) regarding access, demographics, and market factors (Figure 5-1). The markets being studied are the Brazil, China, France, Germany, India, Israel, Japan, the United Kingdom, and United States. All those countries, Trunzo noted, have much medical device technology development.
The questions in the survey are generally focused on infrastructure and investment in medical technology. One question that is being asked with regard to the regulatory environment, for example, is which regulatory and reimbursement environments are the most attractive for the introduction of innovative medical technologies. On access, the survey asks which countries are better equipped with the health-care and technologic infrastructure to deliver innovative medical technologies. On demographics, it asks in which markets the capacity for innovation and the advancement of medical technology is greatest. And on markets, the survey might ask which countries have the most attractive market opportunity for innovative medical technology.
For each of the eight specific focus subjects related to the regulatory environment, access, demographics, and market factors (see Figure 5-1), lists of metrics are being developed. For access to care, for example, metrics could include the number of physicians per capita and the number of clinical trials. For demographics of disease, life expectancy at birth is one metric, and another is access to technology, which refers to the number of Internet users per capita.
The goal is to consolidate all of the information and present it in a usable format. In its analysis, PwC is looking at historical trends and considering the scorecard by dimensions, markets, and future scenarios to develop a technology predictor.
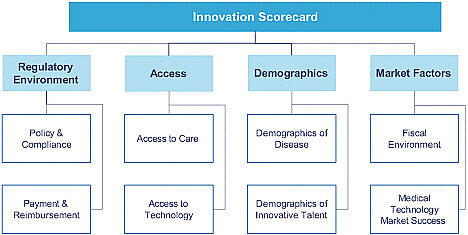
FIGURE 5-1 The PricewaterhouseCoopers innovation scorecard framework.
The findings will be presented in a variety of ways, including “spider charts” or “radar diagrams” for each country. The performance data on the eight subjects will be plotted on a chart, creating a polygon that will allow easy visual comparison of the metrics among countries.
PANEL DISCUSSION: THE GLOBAL REGULATORY ENVIRONMENT
Following the presentations, Jefferys, Trunzo, and Feigal discussed further the favorable outcomes and the challenges of global harmonization, risk-classification issues, other differences between the European and US systems, and concerns during the postmarket period.
Outcomes and Challenges
Feigal noted that when the International Conference on Harmonization (ICH) began to address pharmaceutical harmonization, nearly all countries regulated drugs in some way, but when the GHTF began, 80 countries had no device regulatory scheme whatsoever. The GHTF process was more inclusive than that of the ICH, and its mission included helping countries to develop their medical device regulatory systems. One challenge is to develop systems that are proportional not only to risk but to the resources of the country and of the medical device developers. Another is to build a system that works, in a risk-based way, for thousands of kinds of products. Class II is very broad, ranging from fairly straightforward hospital equipment to complex implants, and the GHTF has taken the stance of trying to separate the higher-risk class II devices from the lower-risk class II devices.
Trunzo concurred, noting that not every country can set up a regulatory system comparable with one used by FDA. Many organizations, such as ASEAN and AHWP, are looking at ways to develop systems that are more streamlined. On the premarket side in many cases, one of the factors that enters into a country’s decision to approve a product is whether it has already been approved in a major market, for example, if it has a CE mark or FDA approval. ASEAN is considering a similar approach to that in Europe, using third-party certifications and having a CE-like mark that will allow marketing in all 10 ASEAN countries after one approval.
Those approaches not only move toward harmonization but allow a country to be confident that a product on its market has gone through some kind of regulatory review in a manner that is based on the country’s available resources.
Jefferys noted that roughly 750,000 types of devices are on the European market compared with no more than 10,000 active pharmaceuticals. The evidence base is different for devices, and a risk-based approach is ap-
propriate. But one has to remember that although a device may be regarded as being in a lower-risk category, for a variety of reasons, including user issues, the risks may still be great. Therefore, the European legislation makes it clear that the same degree of testing and the same requirements for clinical data are present, although for some the emphasis shifts from premarket regulation to postmarket surveillance.
With regard to combination products, Trunzo noted that the GHTF has established an ad hoc working group to look specifically at combination products in which the device constitutes the primary mode of action because that is in the purview of the GHTF. It was recognized, however, that there needs to be outreach to the ICH and others and that common terminology would be helpful. Jefferys noted that there is a coming together between and within the agencies, for example, in the advanced tissue regulation in Europe. Feigal added that the two therapeutic manufacturing cultures are learning from each other.
Feigal supported the notified-body process and said there are consequences of the US government’s tendency to want to do everything itself. There is an opportunity, Feigal said, to re-examine available approaches and to take the best from each.
One factor that has to be taken into account more in the case of devices than pharmaceuticals is user error. The important issues are not usually about design but rather about education of users. It is a bigger challenge for regulators than are standards or designs.
Risk Classification
Number of Categories
The present system of three or four risk categories is about right, Jefferys said. Most would agree that there is a class of low-risk (not no-risk, he emphasized, but lower-risk) devices for which registration and self-regulation are appropriate and that there is a class of potentially higher-risk devices. It is the middle that is up for discussion, and Jefferys supported dividing class II devices into two groups, as is done in Europe.
Trunzo said that whether it is a three-class or a four-class system matters less than how the classification system is implemented and how regulator assign a level of regulatory oversight to the risk associated with a particular device. In any class of devices—whether class A, B, C, or D or class IIA or IIB—there will always be variation. The three-class system in the United States works well, she said, and FDA has applied it appropriately.
Another way to think of the question, Feigal said, is that the United States has 1,800 classes because there are 1,800 device types. Risk assessment is performed product by product. Once a product is on the market,
there are no annual reports, medical device reporting (MDR) is variable, and manufacturers can make changes without notifying FDA.
Differences Between US and EU Risk-Classification Systems
Class III devices are required to have a full design dossier, which will be fully evaluated; whether it is by a notified body or by FDA, the process is the same, and the postmarket requirements are the same.
For class IIA and IIB in Europe, or class II in the United States, a manufacturer has to have a full design dossier and full quality-review systems. In Europe, there is a targeted quality-review system for class IIA devices; a class IIB device will have a full quality review by a notified body, at whose discretion there is a partial or full evaluation of the design dossier.
Feigel said that the classification processes are more similar than different. The major differences between the United States and Europe pertain to a manufacturer’s responsibility to obtain periodic third-party regular certification of manufacturing quality and to meet other kinds of standards.
Trunzo added that the GHTF outlines principles for classifying devices with respect to risk, intended use, and a number of other factors.
Other Differences Between the European and US Systems
In addition to the differences in risk-classification systems, several differences between the US and EU systems were mentioned.
During implementation of the new EU device directives, Jefferys said, there was no grandfather clause. Rather, a manufacturer had up to 5 years to obtain a CE mark for an existing product. That admittedly placed a burden on industry, but a similar approach was taken after implementation of pharmaceuticals legislation, and companies complied in both cases.
Clearance or approval in one market does not necessarily translate to others. It was noted that products that have been cleared by FDA in the United States have been turned down or not taken forward by notified bodies in the EU and vice versa, Jefferys said.
Innovation is taken up much more rapidly in United States than in Europe, partly because of how doctors are trained. The differences in insurance systems also come into play with regard to the uptake of new technology.
Postmarket Reporting
The European system includes timelines for manufacturers to report adverse events for medical devices and penalties that can be leveled if they do not report in a timely manner. Health-care professionals are obliged to
report adverse events and are expected to report immediately. Europe has a no-blame culture, Jefferys said, and people are encouraged to report.
Trunzo added that in the United States, manufacturers are required to report adverse events and malfunctions to FDA and to analyze complaints from the field as part of the quality-management system. In addition, FDA has put into place a sentinel initiative and a signal escalation program whereby the agency analyzes the events that are in the reports database.
A committee member noted that in the United States, health professionals do a small amount of the actual reporting, deferring the task to a ward clerk or other staff who have little information. One of the more successful programs in FDA has been the Medical Product Safety Network (MedSun), in part because risk managers are trained by FDA. Feigal noted that MedSun complements the MDR system. The system recruits risk managers from hospitals and extended-care facilities, such as nursing homes. That gives the agency the ability to query a group of health professionals about an issue. In the United Kingdom, 80–90 percent of adverse-event reports go directly from health providers to the device authority. In the United States, manufacturers collect the information from providers. The MDR system is best at identifying signals that need to be followed up more systematically (these systems do not attempt to determine numerators and denominators). There is no system that will address all the issues, Feigal noted.
With regard to notifiable changes in a device, Jefferys said that in both the United States and Europe, the definition of a reportable change is difficult to determine. In Europe, a company is required to document every small change; a significant change must be reviewed by a notified body, and the design dossier must be updated. But it can be hard to tell which is a minor change and which could result in substantial adverse events.
Trunzo noted that the GHTF documents do not directly address making changes. They focus on the quality-management systems approach to documenting change and control of change.
In many cases, the issue is not postmarket lack of information but what to do with the information, Feigal said. For example, when drug-eluting stents were introduced into the market, cases of thrombosis that resulted in death were reported to the agency within a matter of months. FDA issued a statement that said essentially that it was unclear whether a problem was related to the stents but that deaths had been linked to the products, so adverse events should be reported. Even the agency was not sure what the signals meant. Using the national Medicare database (because the drug-eluting stents have a unique billing code), researchers were able to compare the entire stented population before and after the introduction of the drug-eluting products and to quantify the magnitude of the problem. The problem, it turned out, was discontinuation of platelet drugs after a year, not in-stent thrombosis at the time of insertion.
Whereas pharmacy systems track drugs and drug exposures that can be linked to medical outcomes in pharmacoepidemiology, it is extremely difficult to track devices. They generally are not tracked at the model level or in some cases even identified. From procedure codes in computerized medical-records systems, it will be apparent that a patient has received a hip implant, but tracing it to a specific model or specific manufacturer change is difficult with the current system. Tracking systems are needed not just for the assurance of safety but to find the rare signals that do not appear even in large clinical trials of devices.
Electronic record capture is coming in many countries and will be extremely helpful in this regard, Jefferys said. In some areas, registries are also important in that they they provide both numerators and denominators for analysis.
Feigal noted that FDA has the authority to require studies during the postmarket period, but it is not done very often. That, he said, could be looked at more systematically, specifically to determine types of products in class II that are more likely to need postmarket surveillance.
Ultimately, Feigal said, not all the problems can be solved by tweaking the 510(k) clearance process. There needs to be a systems approach to ensuring safety in a system that includes billing, reporting, and postmarket research.


















