
5
Potential Models for Building a Regulatory Science Infrastructure
A theme among the speakers was that collaboration is necessary to building a strong regulatory science infrastructure.
At the heart of the matter, said FitzGerald, is that the current drug development system is no longer sustainable. The traditional, vertically integrated large drug development model represents a siloed approach characterized by limitations of both finance and human capital. Moving a drug to market is already very costly, but the costs increase exponentially when one takes into account failed drugs and the increasing public and political pressure to lower prescription drug prices. The limitations of the siloed approach argue for a paradigm shift toward a modular, disaggregated model that encourages collaboration and distributes risk among the various stakeholders (see Figure 5-1).
Robust regulatory science at FDA will be essential to maintain purpose and focus within the agency and external respect for its scientific mission. FitzGerald suggested that FDA devise new ways to encourage and reward innovation, enhance risk detection to conserve value, and leverage the resources of the academic sector to refine its decision making as drug development grows more disaggregated and globalized. During a panel discussion led by Peter Honig, Head, Global Regulatory Affairs, AstraZeneca, and former Director of the Office of Drug Safety at the Center for Drug Evaluation and Research (CDER), participants considered potential models for building and strengthening regulatory science.
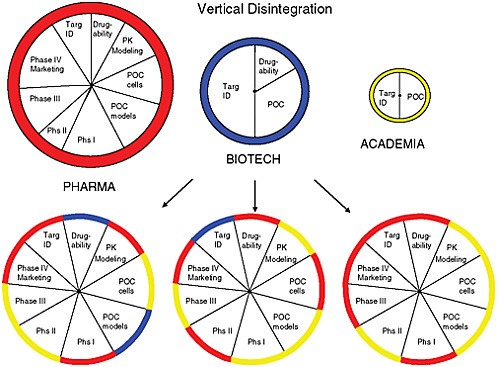
FIGURE 5-1 Visualization of how the current drug development model is growing disaggregated with the involvement of various sectors to reduce risk in innovation.
SOURCE: Skarke and FitzGerald, 2010.
COLLABORATIVE MODELS
According to FDA Commissioner Margaret Hamburg, FDA has been engaged in regulatory science for years, leading to several accomplishments. For example, as noted in Box 2-2, collaboration between FDA and NIH led to a new method of using gene biomarkers to assess the differentiation in stem cell lines; this was an initial step that will ultimately lead to setting standards for use in patients and for ensuring that undifferentiated lines do not contaminate the final product. In 1997, FDA collaborated with academia and industry to create clinical data standards that could be used universally. In the field of drug-induced kidney toxicity, the agency led joint efforts with European health agencies to identify and qualify novel biomarkers for drug assessment. This partnership led to a more sensitive and noninvasive strategy for detecting kidney toxicity in animal models, enabling regulatory bodies to perform assessments early in drug development to help prevent investments in scientific dead-ends.
Judith Kramer, Executive Director, Clinical Trials Transformative
Initiative (CTTI), Duke University, cited the Centers for Education and Research on Therapeutics (CERTs) as a model of collaboration. CERTs came about following the FDA Modernization Act of 19971 and were funded by the Agency for Healthcare Research and Quality (AHRQ). The program formed 60 interdependent centers across the country to provide expertise in therapeutics and to serve as a resource for patients, providers, and agencies, with the goal of optimizing therapeutics in practice. Kramer said that a unique aspect of the CERTs model is the leadership of a steering committee tasked with shaping the initiative and creating synergism across the collaborative efforts of the centers. This differs from the traditional model of funding individual, siloed, centers to conduct independent projects. Kramer indicated that the CERTs model allows the regulatory agency (e.g., AHRQ, FDA), which is required to apply the latest science in its decision-making authority, to collaboratively draw upon the expertise of various centers and meet the needs of both entities through evaluative science. Although the CERTs project was ultimately underfunded, the concept led to the Sentinel Initiative at FDA—a national, active, surveillance system to monitor drug safety.
The Critical Path Initiative (CPI)2 is a similar collaborative effort, aimed at developing strategies to guide innovative medical products through FDA’s regulatory system. CTTI, one of the programs stemming from the CPI, was formed through a memorandum of understanding between FDA and Duke University. The program joins industry, academia, patients, health care providers, investigators, and regulatory law firms as partners in pursuit of the common goal of improving the clinical research enterprise. Involvement of FDA in these collaborations, said Kramer, significantly increases the prospects for producing practicable solutions.
The Biomarkers Consortium (BC) is another collaboration to come out of FDA’s CPI. Mark McClellan, Director of the Engelberg Center for Healthcare Reform at Brookings Institution, cited BC as an example of a successful public-private partnership, which was first undertaken by FDA, NIH, and the industry trade group—Pharmaceutical Research and Manufacturers of America (PhRMA)—in late 2006 to boost the science behind identification of high-impact biological markers for use in drug development, translational research, preventive and predictive medicine, and clinical practice guidelines. BC now consists of 60 partners from the government, industry, and non-profit sectors, and is managed by the
|
1 |
FDA Modernization Act of 1997, Public Law 105-115, 105th Cong. (November 21, 1997). |
|
2 |
More information on FDA’s Critical Path Initiative is available at: http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/default.htm (accessed September 24, 2010). |
Foundation for the National Institutes of Health (FNIH). BC’s research, including FDA’s unique access to and analysis of Phase II data from pharmaceutical companies, has since identified adiponectin, a hormone in fat cells, as a predictive biomarker for type 2 diabetes and superior to the existing standard biomarker, hemoglobin A1C (Wagner et al., 2009).
Two additional collaborative models—in this case, in oncology—were described by Ellen Sigal, Chair and Founder, Friends of Cancer Research. These models are summarized in Box 5-1.
|
BOX 5-1 Collaborative Models in Oncology Two existing partnerships bring government together with other sectors to promote cancer research and explore potential methods and solutions in cancer prevention, treatment, and patient issues:
These cancer-specific collaborative programs can serve as useful case studies for building regulatory science. Evaluation of their successes can be helpful in devising ways to enhance FDA’s regulatory science infrastructure. SOURCE: Sigal, 2010. |
THE CENTERS OF EXCELLENCE MODEL
Establishing a regulatory science infrastructure is a major undertaking that cannot be accomplished by any single group. Workshop participants discussed whether a COE model could be applied to address the barriers to building a regulatory science infrastructure at FDA, as reviewed in Chapter 4. A COE model at its most basic level consists of a network comprising one intramural center, with connections to extramural centers that can then be linked to other networks and/or centers.
Dale Nordenberg, Director, Novasano Health and Science, and former Associate Director, CDC, identified the COE model as a mechanism that could simultaneously support FDA’s regulatory activities, encourage partnerships for research in innovation, and help educate outside groups on regulatory processes. This model could rapidly link the agency with needed expertise—whether internal or external—to enable it to keep pace with emerging science and globalization (see attributes listed in Figure 5-2). The COE model would be funded by FDA and centered in academic institutions. As an FDA-led and community-enhanced initiative, advisory boards from the COE (i.e., academic institutions) and/or the Science Advisory Board within FDA would inform the scientific direction of the
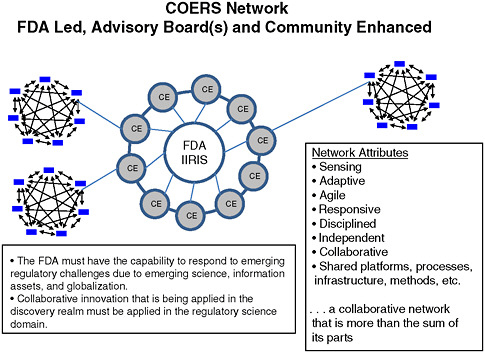
FIGURE 5-2 Centers of Excellence in Regulatory Science Network.
SOURCE: Nordenberg, 2010.
collaboration. Ultimately, the extramural COE and FDA’s intramural center would function as a network with a shared infrastructure.
As an open innovation model, centers would not function in silos but rather as a network to draw out the broad expertise necessary to conduct regulatory science. With the understanding that a single system cannot fully meet FDA’s broad needs, the COE model creates a network of networks to safeguard FDA’s ability to access scientific expertise. Ultimately, this model would:
-
Enhance FDA’s regulatory activities—Nordenberg characterized FDA as an applied organization, meaning that the agency delivers services to broad groups of stakeholders. Should FDA not have access to the requisite expertise or be unable to provide the necessary number of experts, the COE structure would enable the agency to reach out to a support network on an ad hoc basis to fill the gaps. FDA would benefit from such consultations in both regulatory review and policy determination (e.g., development of guidance documents).
-
Advance research in innovation—A COE model would make available to FDA experts from all scientific sectors, permitting strategic development of research and innovation plans. The model would establish a natural leadership role for FDA with its regulatory authority and scientific base. Figure 5-2 portrays such a model—the Centers of Excellence in Regulatory Science Network (COERS)—whose hub is FDA IIRIS, as discussed in Chapter 4.
-
Help FDA educate about regulatory processes—A useful result of relationships formed in the COE model is mutual education and understanding. By better understanding FDA’s regulatory processes, sponsors could more efficiently meet the safety requirements imposed by the agency. The agency could also utilize these connections to develop fellowship programs and construct curricula collaboratively.
Nordenberg also stressed that clear outcome measures will be necessary for evaluating, assessing, and ultimately documenting the impact of this collaborative model. A COE model for FDA’s regulatory science framework, said Nordenberg, could be used for clinical trials, safety surveillance, and comparative effectiveness research. “The structure and operational model for the COE is an important enabler of innovation, outcomes, and ultimately public health impact,” concluded Nordenberg.
Moving Toward a COE Structure
It was suggested during the workshop that some centers already in place today can serve as models of the open collaborations being sought in COE. For instance, CERTs (as described earlier in Chapter 5) were highlighted as a unique model for leveraging the resources of a network of centers to assist regulatory agencies. As opposed to funding individual centers to conduct independent research, the CERTs model presents a unique structure in which a steering committee provides leadership and guides the efforts of the centers, creating synergies through the network that are ultimately to the benefit of regulatory science and decision making.
Moving toward a successful COE model will largely rely on the human capital available and able to be activated in the name of regulatory science. Margaret Anderson, Executive Director of FasterCures, discussed strengthening human capital through the engagement of a broad population with different perspectives. For example, the unique communication of online communities such as Facebook and PatientsLikeMe provide an opportunity to harness the energy, engagement, and informatics expertise of a new generation.
Examples of successful implementation of the COE model also exist. The Observational Medical Outcomes Partnership (OMOP) is a public–private partnership designed to help improve monitoring of the safety of drugs (OMOP, 2010). Its membership consists of all stakeholders in drug safety, including FDA, NIH, and industry. The collaboration is focusing on detection of safety signals. OMOP is also building artificial and synthetic data sets and running simulations to test new methods. Likewise, disease-based organizations, such as the Cystic Fibrosis Foundation, are driving collaborative science and conducting their own research within their networks, aided by readily available patient population data. According to Nordenberg, FDA and its collaborative partners could study these and other examples to anticipate and develop solutions to potential barriers to the successful establishment and operation of a collaborative COE model.
The COE model is not without obstacles, said Ellenberg. FDA review staff are subject to stringent conflict-of-interest rules that could discourage collaboration. The current environment includes particular scrutiny of relationships with industry and suspicion of potential bias arising from collaboration. To implement a collaboration model, it will be necessary to merge differing approaches to confidentiality of information, as academic culture tends toward open sharing of scientific discoveries, while the economics of successful drug development mandate that scientific discoveries be treated as proprietary and confidential, said Ellenberg. A delicate balance should be struck between collaboration and confidentiality, with confidentiality being maintained in all linked COE.








