
2
Food and Drug Administration Postmarket Surveillance Activities and Recall Studies of Medical Devices
This chapter reviews the Food and Drug Administration (FDA) postmarket surveillance activities, including the agency’s current system and future plans for monitoring the safety of marketed devices. FDA’s surveillance activities are focused on identifying potential safety issues with devices currently on the market. If safety concerns are identified through surveillance activities, FDA can take several different types of actions, including recalling devices. The workshop included presentations on two studies that analyzed data on product recalls. The first of these studies was commissioned by the committee (see Appendix C for the commissioned paper describing the study). The second presentation is a summary of a separate study of recall data.
MONITORING DEVICE SAFETY: THE CENTER FOR DEVICES AND RADIOLOGICAL HEALTH’S CURRENT SYSTEM AND VISION FOR THE FUTURE
In addition to getting safe and effective products to market as quickly as possible, FDA must ensure that devices currently on the market remain safe and effective. Susan Gardner, director of the Office of Surveillance and Biometrics of the Center for Devices and Radiological health (CDRH), described the CDRH postmarket program as consisting of postmarket problem identification, postmarket problem assessment, and public-health response. Particularly in the 510(k) program, the diversity of products demands a diverse surveillance strategy, she said. FDA relies heavily on a pas-
sive surveillance system, and, Gardner noted, some initiatives for improving surveillance efforts are under way.
FDA surveillance systems include mandatory reporting though the Medical Device Reporting (MDR) system, voluntary reporting (primarily by health-care professionals or consumers) through the MedWatch system, hospital-based reporting through the Medical Product Safety Network (MedSun), and an international vigilance program in which reports are exchanged with global regulatory authorities.
Voluntary reporting was initiated in 1973 and now accounts for about 3% of the adverse-event reports that FDA receives, Gardner said. Mandatory reporting was initiated in 1984 for manufacturers and importers (accounting for 93% and 1% of reports, respectively) and in 1990, under the Safe Medical Device Act, for user facilities, including hospitals, nursing homes, surgical ambulatory centers, and so on (accounting for 3% of the reports). All together, FDA receives about 200,000 case reports a year and has a database of about 2.5 million reports.
Mandatory Reporting
For all device classes, FDA regulations require manufacturers to report deaths, serious injuries, and malfunctions to FDA within 30 working days of their becoming aware that a device may have caused or contributed to those events. User facilities are required to report deaths to FDA within 10 working days of recognition of an event and deaths and serious injuries to the manufacturer within 10 working days.
In addition to individual reports, FDA initiated in the late 1990s a program called summary reporting, which provides an abbreviated method for reporting device adverse events. The program relies on established codes (rather than text) for device events that are well known and allows the agency to assess the data for trends. Summary-reporting exemptions are granted only for a specific well-known product and a specific well-known adverse event. Whenever there is an incident related to a product that is outside those boundaries, the manufacturer must file a full individual report.
MedSun is a national network of 350 user facilities. Each facility has two liaisons to the program—an engineer and a risk manager—who are trained to recognize and report adverse events. The system uses electronic reporting to reduce the burden on staff. The emphasis of the program is on device use issues. In addition to the mandatory reporting requirements, FDA encourages user facilities to voluntarily report near-misses and close calls, which now account for bulk of the reports. The program has given the agency an additional connection to the clinical community beyond the reporting relationship, Gardner noted.
About 14 people are dedicated to reviewing postmarket surveillance
reports and another 14 or so work for the MedSun program, Gardner said, and 15–18 epidemiologists are closely linked to the MDR staff.
Identifying Signals Among the Array of Device Hazards
When evaluating adverse event reports, FDA looks for a broad array of device hazards (see Box 2-1). The challenge is to identify the issues, or “signals,” that are important among the many reports. A signal is defined as information about a product that FDA regulates that suggests an unexpected risk to patients or users. About 2 years ago, Gardner said, the agency embarked on a “signal escalation” program designed to organize the signals that arise and make them more visible to others in the regulatory centers within FDA. Briefly, as reports come, they are reviewed by analysts who sort them according to an established triage system called Code Blue. Some reports are immediately pulled out and sent to the branch chiefs to review
|
BOX 2-1 Types of Device Hazards
|
(for example, reports of pediatric death, explosions, and burns). Additional information is obtained as needed from the manufacturer or user facility. Reports of interest are further assessed by a specialized group, and a signal is entered into the central tracking system, which is accessible to all staff. A reviewer can, for example, go into the system and see whether signals have been reported on a product being reviewed. Occasionally, reports may also be sent directly to the Office of Compliance if there is a potential compliance issue. Signals come from all offices in CDRH.
Enhancing Surveillance
In addition to the passive-reporting surveillance systems, FDA is implementing enhanced surveillance capabilities. Using MedSun, for example, the agency is conducting targeted surveillance through surveys; 10 surveys and special studies are going on now. Through the MedSun regional-representative pilot program, FDA representatives are visiting 15 hospitals and working directly with their staff to improve reporting. The agency has also formed networks within MedSun to collect real-time data in targeted areas: LabNet fosters reporting from laboratories, HomeNet from home-care agencies, and KidNet from pediatric and neonatal intensive-care units.
Another example that Gardner cited is the Clinical Assessment Reporting and Tracking program for catheterization laboratories (CART-CL), in collaboration with the Veterans Health Administration.1 With 76 cardiac-catheter laboratories nationwide involved, the program fosters enhanced reporting of unexpected problems with devices by clinicians at the point of care.
FDA is also working on data mining of its vast database of event reports, looking for device–event associations. Gardner noted that FDA’s current Manufacturer and User Facility Device Experience (MAUDE) database, built in the early 1990s, is being revamped.
The agency is building its electronic infrastructure and working toward electronic medical device reporting (eMDR). In 2009, nearly 70,000 reports were submitted electronically via an agency-wide portal. Gardner noted that regulation for eMDR is under review.
A particularly important initiative, Gardner said, is the development of a unique device identification (UDI) system. Most devices are bar-coded, but the bar code is “static,” including only such information as the manufacturer, make, and model. There is no dynamic way to identify individual devices uniquely, for example, by serial number or by lot number and expiration date. Accurate identification is challenging, Gardner noted, given the variety of devices that FDA regulates, including software and implantable devices.
|
1 |
The Department of Veterans Affairs CART system is discussed further in Chapter 3. |
FDA is also conducting discretionary observational studies (for example, in collaboration with such registries as the American College of Cardiology’s [ACC’s] National Cardiovascular Data Registry [NCDR])2 and claims-based studies.
Postapproval Studies
Postapproval studies may be ordered as a condition of approval for the highest-risk premarket approval (PMA) products. The studies are used to address important, but not essential, questions of device safety or effectiveness. All studies that are ordered now for PMAs are hypothesis-driven and have deadlines and deliverables, Gardner said. All postmarket studies are listed on a public database with their status. There are also postapproval studies conducted on devices cleared through the 510(k) clearance process.
Postmarket Surveillance Studies
Section 522 of the Federal Food, Drug, and Cosmetic Act (FFDCA) gives FDA the authority to require a manufacturer to conduct postmarket surveillance studies for class II and class III devices if failure of the device is reasonably likely to have serious adverse health consequences or is expected to have substantial use in pediatric populations, is implanted for longer than 1 year, or has life-supporting or life-sustaining use outside the device user facility.
In accordance with Section 522, FDA asks a company questions about a device, the company returns to FDA with a protocol for answering the questions, and then FDA goes through a process of approving or not approving the protocol. A manufacturer can take a number of surveillance approaches, for instance, if the program protocol does not necessarily require that it conduct a clinical trial. Approaches could include, for example, literature review, nonclinical testing, use of secondary data sources, followup with patients, clinical registries, and observational studies. Gardner noted that FDA can order a postmarket surveillance study for a predicate device if necessary.
There are drawbacks to these “Section 522 studies,” Gardner said. Surveillance can be required for only 3 years. Because discussion, development, and review processes take time, there are no immediate answers to questions about a device. Thus, if there is a safety issue, the answer is not to order a Section 522 study; other FDA tools are more appropriate.
In 2007, as a result of an Institute of Medicine study of postmarket surveillance for pediatrics (IOM, 2005), the FFDCA was amended to allow FDA to order Section 522 studies as a condition of approval or clearance
|
2 |
The NCDR is discussed further in Chapter 3. |
and for longer than 36 months for devices that are expected to have substantial use in pediatric populations. That was a significant change.
On the Horizon
Gardner highlighted a number of forthcoming initiatives that she said will be important for postmarket surveillance. The Sentinel Initiative is an effort to develop a national, integrated infrastructure of electronic healthcare data systems for medical-product safety surveillance. It will augment, not replace, existing functionality, Gardner noted. The focus is on active surveillance. In the proposed model, the data sources would remain at their remote locations, maintained by their local owners, and FDA would be able to send queries to the data owners about specific safety questions (such as rates of implant revision or reintervention, rates of infection, and selected outcomes, such as myocardial infarction, stroke, and death). The system is not designed, however, to address much of what has been discussed regarding 510(k) products, such as out-of-box failures, software glitches, manufacturing defects, and packaging or labeling errors.
The agency is also forming new critical collaborations and leveraging established partnerships with agencies, academic institutions, and professional societies. Gardner cited one program, MDEpiNet, in which FDA is collaborating with academic centers to advance epidemiologic methods and training related to devices.
FDA is working with multiple stakeholders on the development of registries. As noted earlier, registries are one approach that can be used for Section 522 surveillance. They can be used for active surveillance, for short-term as well as longitudinal data. Other potential capabilities include linkage studies with Medicare claims data and mapping of registry data to electronic health records.
Another effort under way is an evidence synthesis project. The agency acknowledges that there are isolated groups or silos of postmarket data (such as MDRs, observational studies, and published clinical studies), and this project is addressing how to combine the multiple data sources and developing prognostic models of long-term device performance.
CDRH is also focused on better integration of premarket and post-market data on device performance, Gardner said. She described several initiatives, including the Collaborative Review Program, in which center staff spend half their time in premarket review and half their time looking at postmarket adverse-event reports. The program is administratively challenging, Gardner noted, inasmuch as staff are reporting to two different areas, but there has been some success in taking postmarket information back to the premarket function. CDRH also has networks that integrate people across the center in specific fields (such as an orthopedic network and a
cardiovascular network), and it has a embarked on an improved knowledge-management program, using collaborative software and wiki products, to pull together all the information about a given product and make it easily and readily available to all center staff.
Gardner noted that resources, both funding and staffing, continue to be barriers to implementing some of those initiatives.
PREMARKET NOTIFICATION: ANALYSIS OF FOOD AND DRUG ADMINISTRATION RECALL DATA
William H. Maisel, former director of the Medical Device Safety Institute,3 provided an overview of an independent analysis of FDA recall data that he was commissioned to perform for the committee.4 The ideal measure of the success of the device approval or clearance processes, he said, would be the performance and reliability of each individual approved or cleared device. Analysis would need to be done for many thousands of devices, so it would be extremely difficult and impractical. Therefore, surrogates of device performance are used, including recalls and medical-device reports. FDA defines a recall as an action taken to address a problem with a medical device that violates FDA law. Recalls occur when a medical device is defective, when it poses a risk to health, or when it is defective and poses a risk to health.
The purposes of the study by Maisel were to analyze the available FDA recall data affecting 510(k) products, to provide an estimate of 510(k) product recall rates, to describe the causes of recalls affecting the products, and to identify risk factors for product recalls.
Data Analysis
Data for analysis were obtained from the FDA 510(k) database, specifically the 48,402 total 510(k) applications that were cleared by FDA from 1996 through 2009, and from the FDA recall database from 2003 to 2009.
Clearance Data
Maisel used FDA advisory-committee assignments during 1996–2009 to provide some idea of the types of products that were being cleared via the 510(k) process (Figure 2-1). Advisory committees that received the greatest number of applications were those for general and plastic surgery, orthopedic, general hospital, and cardiovascular products.
|
3 |
Dr. Maisel is no longer with the Medical Device Safety Institute as of August 2010. He is currently employed at FDA. |
|
4 |
The complete commissioned paper is available as Appendix C. |
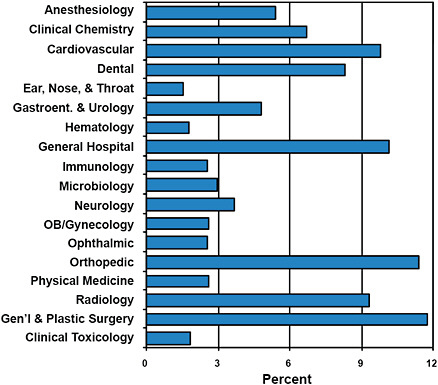
FIGURE 2-1 Breakdown of advisory-committee assignments for 510(k) submissions in 1996–2009.
More than 80% of the devices cleared via the 510(k) program were class II devices, about 10% class I, and just over 1% class III.
The vast majority, about 80%, of the applications in the database are traditional 510(k) applications. Special 510(k) applications, which are for modifications of products that conform to design control standards, make up about 16%, and abbreviated applications, which are for devices that can be cleared on the basis of standards or special controls, make up about 3%.
About 25% of the devices cleared during that period were implantable devices. Over 96% of these 510(k) implantable devices were not life-sustaining.
Recall Rates
Some recalls, Maisel said, can involve more than one 510(k)-cleared product. For the study, recall data were analyzed on the basis of individual 510(k) applications; that is, data were expressed as unique 510(k) applications that were subject to recall.
From 2003 to 2009, 400–500 unique 510(k) applications were affected by recall each year. Maisel reminded participants that several thousand devices are cleared through the 510(k) process each year. About 74% of individual recalled 510(k) devices were recalled a single time, and about 16% were twice. Overall, about 26% were recalled more than once; some were recalled more than 10 times. Analysis of the data is complicated, Maisel noted, in that companies sometimes expand a recall because new information becomes available. Therefore, a large number of recalls of some products should not be interpreted as multiple product failures in the absence of further in-depth analysis. About half the devices recalled in 2003–2009 had been cleared in 1996–2002.
Determining recall rates presents a variety of challenges, Maisel said. For example, how should one account for the fact that some devices have been on the market longer than others? Ultimately, Maisel said, it was decided to apply Kaplan–Meier methods to do a “survival estimate,” essentially thinking of each device as though it were a patient entering a clinical trial.
The analysis of recall-free 510(k) “survival” of devices showed that 98.4% of the devices cleared in 2003–2009 remained on the market, free of recall, 1 year after the 510(k) decision. The proportion that remained recall-free for 2 years was 96.5%; and the proportion for 5 years, 91.5%. That approach, Maisel said, was thought to be the fairest way to conduct an assessment of the rate of the recalls for the 510(k) program.
Another way to look at the data (using Kaplan–Meier analysis) is to ask what percentage of devices are recalled during their first year on the market, their second year on the market, and so on. The data show that there is a higher rate of recall among 510(k) products during their first 2 or 3 years on the market—about 1.6–1.9%. The recall rate drops off to about 0.9–1.1% in years 5 and 6 on the market as device problems are resolved.
Causes of 510(k) Recalls
Analysis of the recall database for FDA’s classification of causes of recalls showed that manufacturing process was the most common cause of a 510(k) recall, accounting for 28.8% of the recalls. Manufacturing-process errors included inadequate control of a process, inadequate environmental controls, and errors in storage, packaging, or labeling. Device design, that is failure of a device to perform as intended despite the product’s meeting all its design specifications, accounted for 28.4% of recalls. Materials and components that were nonconforming, contaminated, degraded, counterfeit, or inadequately tested accounted for about 16%. Change control—changes in specifications, programs, or procedures that adversely affected components or devices—accounted for 11.9%. Employee errors and other miscellaneous concerns accounted for 7.1% and 7.5%, respectively.
Predicates
510(k) devices cleared in 2004–2009 were evaluated for the number of predicates cited by their manufacturers in their applications. More than 80% cited 1–5 predicates, about 10 cited 6–10 predicates, and fewer than 5% cited more than 10 predicate. From Maisel’s analysis, it appears that applications that cited 1–5 predicates were associated with a lower rate of recall, and applications that cited 6–10 predicates were associated with a higher rate.
Maisel noted that some 510(k) submissions include bundled products, and more than one “device” in a 510(k) application might necessitate more than one predicate. Some of the devices with very high numbers of predicates cited are in vitro diagnostics, such as a laboratory analyzer that might perform multiple tests and require multiple predicates.
If there were multiple predicates, the age of the newest predicate cited was less than 5 years in more than 75% of the 510(k) submissions. Maisel said that devices for which the age of the newest predicate was 1–5 years had a slightly higher recall rate than nonrecall rate.
The age of the oldest predicates was less than 5 years in about 50% of cases, 6–10 years in 25%, and more then 10 years in 25%. Some of the predicates cited, Maisel said, were 15-20 years old, but there was no indication that older predicates were associated with an increased recall rate.
Type and Features of 510(k) Submissions
From 2003 through 2009, 75% of the devices that were not recalled were cleared through traditional 510(k) submissions compared with about 62% in the recall group; 22.3% of the devices that were not recalled were cleared through special 510(k) submissions compared with 34.2% of recalled devices. Maisel suggested that that is a signal that may warrant further investigation as to whether there is something about the special 510(k) process that increases risk. (Devices cleared through abbreviated 510(k) applications were infrequent in both devices recalled and not recalled.)
510(k) devices affected that were recalled were less often cleared by the Office of Device Evaluation (67.9%) than devices that were not recalled (77.0%) and were more often cleared by the Office of in Vitro Diagnostic Device Evaluation and Safety (OIVD) (32.1% vs 23.0% of nonrecalled devices). Again, Maisel said, that may be related to the types of products evaluated by each office and does not necessarily reflect the quality of review. There was a slight signal, he said, that applications undergoing third-party review had a higher rate of recall. There was no difference in the recall rate for implantable devices in the percentages that were recalled or not recalled. Life-sustaining devices were more likely to be recalled, but that could be
|
BOX 2-2 Maisel’s Key Findings: Analysis of FDA Recall Data
|
a threshold issue, Maisel suggested; that is, manufacturers are much more likely to issue recalls if they become aware of a potential for serious clinical consequences.
More than 85% of 510(k)-cleared devices were in class II, including devices both affected and unaffected by a recall. Class III devices, which tend to be more often life-sustaining devices, have a higher rate of recall.
Devices assigned to certain advisory committees were more likely to be recalled. Devices assigned to the anesthesia, chemistry, and cardiovascular advisory committees, for example, are generally higher-risk devices and had higher recall rates; but dental, immunology, and microbiology devices were less likely to be recalled. Maisel suggested that this information may be useful in deciding how to allocate resources to the fields in which recalls are more likely (see Box 2-2).
Medical-Device Reporting
As part of his task, Maisel said that he was asked to include MDR data in his analysis but that the available data were not ideally suited to analysis, because of incomplete reporting, insufficient information, and misclassification. He was able to link some of the MDR data with recall data, but Maisel stressed that the data should be interpreted with caution because of the limitations of the sample.
The database included 182,394 MDRs that were associated with 7,823 510(k) devices cleared in 1996–2009. Nearly two-thirds of the reports (66.4%) were associated with a device malfunction; almost one-third (29.5%) were associated with a patient injury, but fewer than 2% involved a patient death.
Of the MDRs in the analysis, 83,000 were associated with products that were ultimately recalled: 750 death adverse-event reports, 19,936 of reported patient injuries, and 60,291 of device-malfunction reports involved 510(k) applications that were associated with recalled devices. There is no information about whether these MDRs came in before or after the recalls, Maisel said. However, the data at least suggest that the recall process is associated with a substantial proportion of the MDRs.
FOOD AND DRUG ADMINISTRATION RECALL-DATA STUDY
The 510(k) clearance process has been subject to substantial criticism, said Ralph Hall, Distinguished Visiting Professor of Law at the University of Minnesota Law School, but there are no systematic means with which to assess whether the system is working. To address that, Hall embarked on a study to determine whether the 510(k) system permits products to enter the market without a “reasonable assurance of safety and effectiveness” and whether specific parts of the 510(k) process lead to greater or lower risk.5
Methods
There are three classes of FDA device recalls, which, Hall noted, with regard to risk are in reverse numerical order from device classification itself. Class I recalls involve the most serious safety issues—situations in which there is a reasonable probability that use of or exposure to a violative product will cause serious adverse health consequences or death. Class II recalls involve temporary or reversible medical issues or remote risks, and class III recalls involve nonsafety issues, such as regulatory violations (for example, marketing without proper clearance). The recall classification is determined by FDA.
Hall said that although it is not perfect, the group of class I recalls offer the best safety-related performance measure of the 510(k) system. MDR data are not a good measure, he said, because the reports include known risks, there is inconsistent reporting, information is incomplete or inaccurate, and there is no quality control or confirmation. Medical-device reports are primarily anecdotal, he added. The number of products involved
in recalls also is not a useful measure, because there is no denominator, it is not possible to differentiate single-use products from multiple-use products or to determine failure rates or rates of harm, and recalls include nondefective products.
For his study, Hall focused on class I recalls in 2005–2009. The study included data derived from FDA databases, including databases of recalls, 510(k) cleared devices, PMA devices, product classification, and the Total Product Life Cycle; the 2009 Government Accountability Office (GAO) report; results of ancillary Internet searches; and direct communication with device companies and FDA.
A total of 474 class I recalls during the period were identified. There were often multiple records on a single recall event (involving, for example, different sizes, model numbers, or trade names); when these had been consolidated, it was determined that there were 118 unique class I recalls. Those recalls were then coded by Hall for a variety of factors, including approval or clearance pathway, whether a device was implantable, reason for the recall, device class, third-party review, and medical specialty. Hall indicated that in some instances the coding of the reasons for recall in the system was unclear. Therefore, he developed criteria to determine which category to place devices for which the reason for recall was unclear.
Recalls, Hall said, have three broad root causes: premarket issues, issues that the PMA and 510(k) processes are intended to check and prevent, such as design issues and clinical data gaps; postmarket issues, such as manufacturing issues, labeling mistakes, and sterilization issues; and miscellaneous actions, often taken by unrelated third parties, such as counterfeit products. Hall stressed that the 510(k) system can be expected to prevent only premarket issues, and any assessment of the correctness of clearance decisions or of the robustness of the 510(k) process should look only at premarket issues.
Hall acknowledged several challenges to the method. First, the study relied on public data and the accuracy of the databases. Second, there may be “missing” recalls that are not reported; this is a violation of FDA regulation, and Hall opined that there were probably few cases and that they were probably not major. Third, the study focused on class I recalls because they are the potentially high-end or high-impact safety issues, but there are questions about the consistency of the FDA recall class determinations.
Data Analysis
Of the 118 unique class I recalls in 2005–2009, six involved counterfeit devices, leaving 112 core recalls, for an average of 22.4 class I recalls per year. According to GAO, there are more than 50,000 listed devices (GAO, 2009), so there was a 0.2% recall rate over the 5 years, Hall said.
Hall organized the 118 recalls by primary reason for recall (see
Table 2-1). Of the 13 categories used, four are premarket issues: device design, software design, failure to identify a clinical risk, and failure to warn or inadequate instructions. The other categories are postmarket issues or other concerns, including counterfeit devices. Within categories, recalls were divided according to approval pathway (PMA, 510(k), or class I device).
When all class I recalls are considered, including those for counterfeit devices, premarket issues accounted for about 45% of the recalls involving 510(k) devices. If counterfeits are excluded and only the 112 valid devices are considered, about 48% of the class I recalls of 510(k) devices and about 43% of the PMA device recalls were due to premarket issues. Hall noted that his results indicated that the numbers for 510(k) and PMA devices are not statistically different. Correspondingly, about 55% of the recalls involve postmarket issues.
Most of the premarket issues leading to recall were design issues (including software design). On the basis of his results, Hall said, it is clear that there is a critical role for quality-system regulation (QSR), including bench testing and design controls to identify design issues without endangering patients.
TABLE 2-1 Primary Reason for 118 Class I Recalls
|
Primary Reason for Recall |
PMA |
510K |
Class I |
Other or Unknown |
TOTAL |
|
Manufacturing |
6 |
31 |
2 |
1 |
40 |
|
Labeling error |
0 |
4 |
0 |
0 |
4 |
|
Design issue |
6 |
25 |
1 |
0 |
32 |
|
Software design |
1 |
9 |
0 |
0 |
10 |
|
Software manufacturing |
0 |
2 |
0 |
0 |
2 |
|
failure Supplier issue |
2 |
5 |
0 |
0 |
7 |
|
Failure to identify clinical risk |
0 |
0 |
0 |
0 |
0 |
|
Failure to warn or inadequate |
0 |
8 |
0 |
0 |
8 |
|
instructions Missing parts |
0 |
0 |
0 |
0 |
0 |
|
Sterilization |
1 |
4 |
2 |
0 |
7 |
|
Regulatory violation |
0 |
1 |
1 |
0 |
2 |
|
Packaging or handling |
0 |
0 |
0 |
0 |
0 |
|
Other (such as counterfeit or sham) |
0 |
6 |
0 |
0 |
6 |
Hall pointed out that recalls related to newly discovered clinical risks were identified. Although “inadequate labeling or instructions” could possibly be used to describe unidentified clinical risks, Hall was not able to assess that further with the available data. Overall, about 7% of the recalls were attributed to unidentified clinical issues. Those data suggest, Hall said, that conducting additional human clinical trials would have very little effect on the number of class I safety recalls.
To assess the robustness of FDA device review broadly, one must look at the rate of recalls compared with submissions, Hall said. The number of submissions, not approvals or clearances, is a better measure of robustness of the process because it includes situations in which products were not cleared (thus eliminating safety risks).
A denominator is needed to be able to determine how many class I recalls are related to each type of approval pathway. However, finding an exact denominator is impossible because there is no precise time relationship between submission, clearance, and initiation of a recall.
Thus, Hall looked at the total 510(k) submissions for the 10-year period 2000–2009, computed the per-year average, and then multiplied that average by 5 to estimate the average submissions over a 5-year period. Using the calculated denominator of 19,873 submissions over the 5-year study period 2005–2009 and the total of 89 class I recalls of 510(k) devices during that time, Hall concluded that 99.55% of 510(k) submissions did not result in a class I recall (recall rate, 0.45%). The total of 43 class I recalls due to premarket issues represented 0.22% of total submissions, so 99.78% of 510(k) submissions did not result in class I recalls due to premarket issues.
For comparison, Hall noted that 2.3% of Medicare hospitalizations result in a patient-safety event, there is a 2–4% risk of hospital-acquired infection, and more than 15% of patients over 65 years old receive a potentially unsafe prescription.
A similar analysis of PMA products resulted in similar findings. Class I recalls relative to submissions during the study period were comparable: 99.71% of devices were not subject to recall, and 0.12% were recalled for premarket issues.
Roughly 1% of all medical devices are PMA products. Not surprisingly, given the higher complexity and higher risk of PMA products than of other products, PMA products accounted for 14% of the class I recalls during the study period. About 67% of all devices are exempt or class I device products; these lower-technology, lower-risk products accounted for only 6% of recalls.
Hall further subdivided PMA devices by type of product according to the applicable section of the Code of Federal Regulations (CFR). The bulk of recalls involved cardiovascular devices (21 CFR 870) and hospital and personal use devices (21 CFR 880), and these had higher than average
|
BOX 2-3 Hall’s Key Findings: Using Recall Data to Assess the 510(k) Process
|
rates of premarket issues. There were very few recalls of orthopedic devices although these are long-term implantable products, no recalls of obstetrical and gynecologic devices, and only one recall of a radiology device. Hall posed the question of whether the data support the need for a fourth device classification. He opined that, on the basis of safety data, there is no clear, discrete delineation of products that would logically fit into a new “class IIB,” that is, an enhanced 510(k) or limited PMA process.
The same data were analyzed by medical specialty, and the same general pattern was observed: recalls occurred predominantly in cardiovascular and general hospital specialties.
If the three-letter product codes are looked at, 54.2% of recalls were concentrated in five categories: automatic external defibrillators (AEDs), anesthesia products, cardiovascular devices (a broad catchall category), catheters, and infusion pumps. Two PMA product types, AEDs and infusion pumps, accounted for 28% of all class I recalls during the study period.
Hall raised the question of whether product-specific guidance and special controls would be an appropriate response, and he noted that a detailed root-cause investigation of the products may be warranted.
In summary, Hall concluded that FDA has an excellent safety record on
the basis of his analysis of class I recalls. About 99.8% of device submissions did not experience a class I recall during the 5-year period studied (see Box 2-3).
He stressed that QSR is extremely important given the prevalence of design issues and is probably more important than additional human clinical studies.
Hall highlighted several questions for consideration, including
-
What aspects of postmarket surveillance have the greatest effect on identifying recall needs?
-
What are the true root causes of safety recalls (such as common factors, human factors, and complexity)?
-
What are the potential effects of changes in the 510(k) system (such as effects on FDA resources and time, on whether the added burden of changes would be proportional to safety benefits, and on effects on patient access)?
-
Which parts of clearance and approval submissions make a difference relative to improving safety decisions?
-
What is the role of multiple predicates in recall situations?
REFERENCES
FDA (Food and Drug Administration). 2000. Device Advice: Premarket Notification. Washington, DC, Food and Drug Administration. http://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketnotification510k/default.htm (accessed August 31, 2010).
GAO (General Accountability Office). 2009. FDA Should Take Steps to Ensure That High-Risk Device Types Are Approved Through the Most Stringent Premarket Review Process. http://www.gao.gov/new.items/d09190.pdf (accessed August 16, 2020).
IOM (Institute of Medicine). 2005. Safe Medical Devices for Children. Washington, DC: The National Academies Press.


















