
C
510(k) Premarket Notification Analysis of FDA Recall Data
William H. Maisel, MD, MPH
EXECUTIVE SUMMARY
The 510(k) process requires a device manufacturer to notify the Food and Drug Administration (FDA) before it intends to market a device and to establish that the device is “substantially equivalent” to a legally marketed “predicate” device that does not require premarket approval (PMA). A recall is an action taken to address a problem with a medical device that violates FDA law. Recalls occur when a medical device is defective and/or when it could be a risk to health.
Analysis of FDA’s 510(k) Database (1996–2009) and Recall Database (2003–2009) revealed the following:
-
48,402 510(k)s were cleared by FDA between January 1, 1996, and December 31, 2009, and were available for analysis.
-
From 2003 to 2009, 3,132 unique 510(k)s were subject to recall. Among 510(k)s affected by recall, 73.9% were recalled a single time and 26.1% were recalled more than once, including nearly 2% that were recalled more than five times.
-
Among 510(k)s cleared in 2003–2009, 98.4% remained free of recall 1 year following the decision. Longer-term follow-up shows that 92.6% and 91.5% of 510(k)s remain free of recall 5 and 6 years, respectively, following regulatory clearance.
-
The annual 510(k) recall rate is highest in the first 3 years following clearance (1.6–1.9%/year). Lower recall rates are observed in years 5 and 6 post clearance (0.9–1.1%/year).
-
More than half the 510(k) recalls are due to manufacturing process
-
errors (28.8%) or device design issues (28.4%). Materials and component issues (16.3%) and change control processes (11.9%) account for the majority of the remaining 510(k) recalls.
-
Compared to 510(k)s unaffected by recall, recalled 510(k)s are more likely to have been reviewed by a third party or submitted as a Special application (rather than Traditional or Abbreviated). Recalls are also more likely to affect 510(k)s involving life sustaining devices and Class III devices.
OVERVIEW OF 510(k) PREMARKET NOTIFICATION PROGRAM AND FDA RECALLS
The 510(k) process requires a device manufacturer to notify FDA before it intends to market a device and to establish that the device is “substantially equivalent” to a legally marketed “predicate” device that does not require a PMA.1
One measure of the success of the 510(k) premarket notification process would be to assess the performance and reliability of the thousands of individual devices that have been cleared via this pathway. This is both impractical and impossible. Recalls and Medical Device Reports represent surrogate markers of device reliability.
A recall is an action taken to address a problem with a medical device that violates FDA law.2 Recalls occur when a medical device is defective and/or when it could be a risk to health. Recalls may be conducted on a firm’s own initiative, by FDA request, or by FDA order under statutory authority. Importantly, recalls may be issued when there is only a small chance (sometimes <1%) of device malfunction or patient injury. Recalls are classified by FDA as follows:
-
Class I recall: a situation in which there is a reasonable probability that the use of or exposure to a violative product will cause serious adverse health consequences or death.
-
Class II recall: a situation in which use of or exposure to a violative product may cause temporary or medically reversible adverse health consequences or where the probability of serious adverse health consequences is remote.
-
Class III recall: a situation in which use of or exposure to a violative product is not likely to cause adverse health consequences.
|
1 |
Government Accountability Office. Medical Devices: FDA Should Take Steps to Ensure That High-Risk Device Types Are Approved Through the Most Stringent Premarket Review Process. January 2009. Accessed June 16, 2009 at http://www.gao.gov/new.items/d09190.pdf. |
|
2 |
U.S. Food and Drug Administration. Recalls, Market Withdrawals, and Safety Alerts. Accessed at http://www.fda.gov/Safety/Recalls/ucm165546.htm. |
Medical Device Reporting (MDR) is a mechanism by which FDA receives information on significant medical device adverse events from manufacturers, importers, and user facilities (hospitals, nursing homes, etc.). Because of incomplete adverse event descriptions, significant underreporting of device-related adverse events, and the absence of denominator data, these data have significant limitations. Nevertheless, MDR analysis can provide some insights into the performance of the 510(k) program.
METHODOLOGY
Methods 2.1
Two primary databases—510(k) database and recall database—were used to conduct the analysis presented in this report. A third database, containing MDR data, was also analyzed. Each database was provided by FDA to the Institute of Medicine. Data analysis was conducted independently of FDA.
Methods 2.1.1 510(k) Database for Years 1996–2009
Consisted of all 510(k) applications that were submitted between January 1, 1996, and December 31, 2009, and were found to be substantially equivalent (SE).
Methods 2.1.2 FDA Recall Database for Years 2003–2009
Consisted of all medical device recalls from January 1, 2003, to December 31, 2009.
Methods 2.1.3 Medical Device Reporting (MDR) Data
Consisted of a subset of all submitted MDRs. A subset of the MDRs occurring between January 1, 2003, and December 31, 2009, were analyzed.
Methods 2.2
All statistical calculations were performed using SAS (Version 9.1, Cary, NC). A two-sided P value of <0.05 was considered to be statistically significant. Chi-square tests, Mantel–Haenszel chi-square tests, and log-rank tests were used where appropriate.
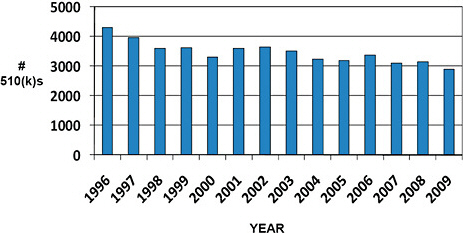
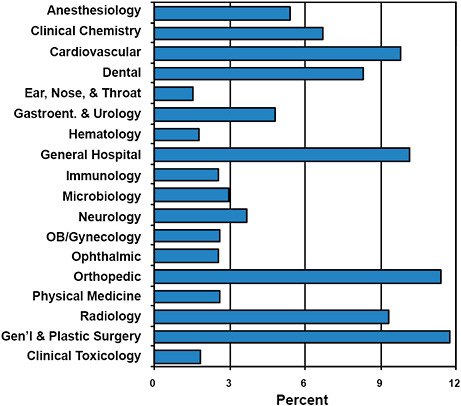
FIGURE C-2 Advisory committee assignments for submitted 510(k)s.
-
All devices submitted for clearance under the 510(k) program are given an Advisory Committee assignment.
-
Devices cleared via the 510(k) program are most often assigned to the General & Plastic Surgery (11.8%), Orthopedic (11.4%), General Hospital (10.2%), and Cardiovascular (9.8%) Advisory Committees.
-
510(k) devices are least often assigned to the Ear, Nose, & Throat (1.5%), Hematology (1.8%), and Clinical Toxicology (1.9%) Advisory Committees.
-
While Advisory Committee assignments inform about the type of devices cleared, the committees are rarely involved in the premarket 510(k) process.
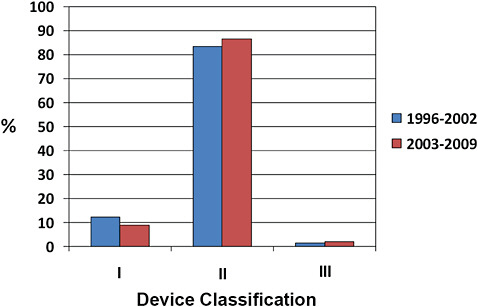
FIGURE C-3 510(k) device classification.
-
More than 80% of 510(k) devices are classified as Class II, ~10% as Class I, and <2% as Class III.
-
Minor changes in the distribution of device classification have occurred from 1996 to 2002 compared to 2003–2009. Specifically, there has been a small reduction in the number of 510(k) devices classified as Class I, and a small increase in the number of Class II and Class III designations.
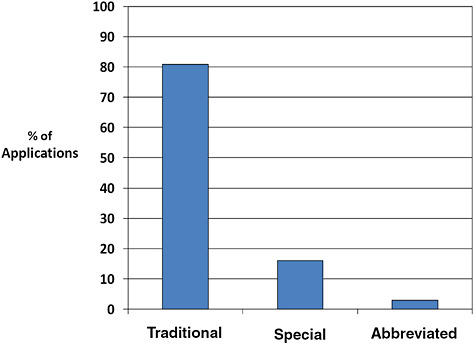
FIGURE C-4 510(k) type, 1996–2009.
-
The majority of 510(k)s are Traditional (80.9%). Fewer are Special (16.0%) or Abbreviated (3.0%).
-
Special 510(k)s are submitted when a manufacturer makes modifications to its own device, design control processes are appropriate, and design validation is performed. Abbreviated 510(k)s are submitted when FDA guidance, a special control, or recognized standard exists and the manufacturer intends to use it. All other 510(k) devices utilize the traditional pathway.
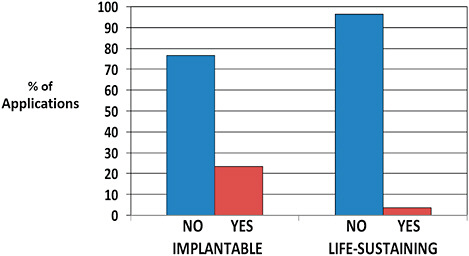
TABLE C-1 Number of Recalls per 510(k)
|
Recalls per 510(k) |
# Occurrences |
Percentage of 510(k) Recalls |
|
1 |
2,298 |
73.9 |
|
2 |
492 |
15.8 |
|
3 |
150 |
4.8 |
|
4 |
74 |
2.4 |
|
5 |
37 |
1.2 |
|
6 |
16 |
0.5 |
|
7 |
15 |
0.5 |
|
8 |
10 |
0.3 |
|
9 |
8 |
0.3 |
|
10 |
2 |
0.1 |
|
>10 |
7 |
0.2 |
-
Among 510(k)s affected by recall, 73.9% were recalled a single time.
-
26.1% of recalled 510(k)s were recalled more than once, including nearly 2% that were recalled more than five times.
-
Importantly, multiple recalls may be due to expansion of an initial recall to additional products with the same potential defect and do not necessarily represent multiple modes of product failure. Additionally, because some 510(k)s contain more than one device, multiple recalls do not necessarily represent product defects repeatedly affecting the same device.
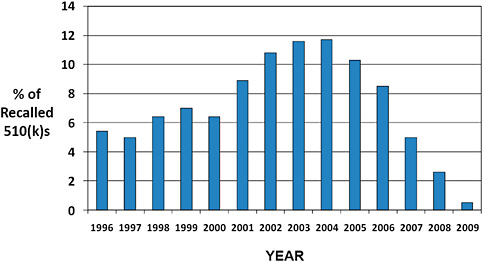
FIGURE C-7 Year of 510(k) decision for recalls occurring in 2003–2009.
-
The original decision year for 510(k)s affected by recall in 2003–2009 is displayed.
-
Nearly half (49.9%) of the 510(k) recalls occurring in 2003–2009 were for products cleared in 1996–2002.
-
40.6% of recalls were for products that were cleared in 2003–2009.
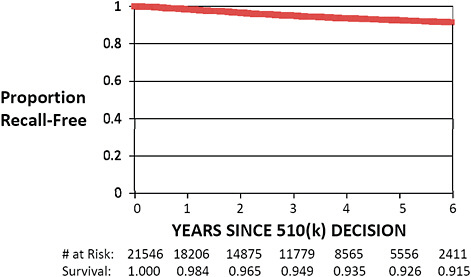
FIGURE C-8 Recall-free 510(k) “survival,” 2003–2009.
-
Only 510(k)s cleared from January 1, 2003, to December 31, 2009, are included in this graph. Kaplan–Meier “survival” estimates were calculated using standard methodology.
-
Recall-free survival estimates demonstrate that 98.4% of 510(k)s cleared in 2003–2009 remained free of recall 1 year following the decision.
-
Longer term follow-up shows that 92.6% and 91.5% of 510(k)s remain free of recall 5 and 6 years, respectively, following regulatory clearance.
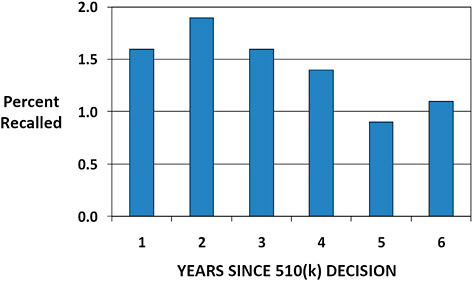
FIGURE C-9 Annual 510(k) recall rate based on years since decision.
-
The annual rate of recall for 510(k)s cleared January 1, 2003, to December 31, 2009, is displayed.
-
The annual recall rate is highest in the first 3 years following clearance (1.6–1.9%).
-
The lowest recall rates are observed in years 5 and 6 post clearance (0.9–1.1%).
TABLE C-2 Causes of 510(k) Recalls
|
Cause |
Description |
Recalls (%) |
|
Manufacturing Process |
Manufacturing process inadequately controlled, inadequate environmental controls, storage, packaging, labeling, equipment maintenance, material removal, etc. |
28.8 |
|
Device Design |
Failure of device to perform as intended despite meeting design specifications. |
28.4 |
|
Materials/Components |
Materials/components that are non-conforming, contaminated, degraded, counterfeit, or inadequately tested |
16.3 |
|
Change Control |
A change made to a specification, program, procedure, vendor, etc. that adversely affects a component, finished device, packaging, leveling, etc. |
11.9 |
|
Employee Error |
Employee error (not a systematic problem). Usually corrected by retraining. |
7.1 |
|
Miscellaneous |
|
7.5 |
|
TOTAL |
|
100 |
-
FDA classifies the cause of each recall based on the available information.
-
More than half the 510(k) recalls are due to manufacturing process errors (28.8%) or device design issues (28.4%).
-
Materials and component issues (16.3%) and change control processes (11.9%) account for the majority of the remaining recalls.
-
Employee error is a less common cause of 510(k) recalls (7.1%).
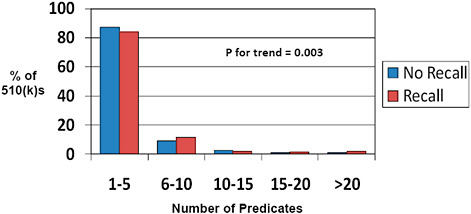
FIGURE C-10 Impact of predicate number on 510(k) recalls, 2004–2009.
-
The number of predicates cited by each individual 510(k) is displayed.
-
Most 510(k)s utilize 1-5 predicates (>80%). Fewer than 5% utilize more than 10 predicates.
-
Fewer predicates (1-5) are associated with a lower rate of recall while a higher number of predicates (6-10, for example) are associated with an increased 510(k) recall rate.
-
Many of the submissions that cite multiple predicates contain multiple products (for example, submissions to the Office of In Vitro Diagnostic Device Evaluation and Safety [OIVD] for diagnostic tests). Therefore, one cannot conclude from these data that multiple predicates are unsafe without indexing the recall rate to the number of unique products at risk. This latter analysis could not be performed based on the available data.
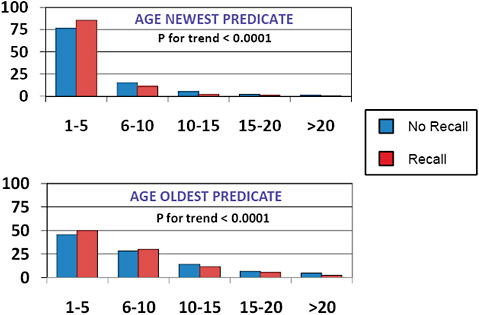
FIGURE C-11 Impact of predicate age on 510(k) recalls, 2004–2009.
-
The age of the newest predicate cited for 510(k) clearance was <5 years in most (>75%) cases and <10 years in >90%.
-
The age of the oldest predicate was <5 years in ~ 50%, 6-10 years in ~ 25%, and >10 years ~25% of the time.
-
The newest predicate and the oldest predicate tended to be younger among recalled 510(k)s than among those 510(k)s that had been unaffected by recall.
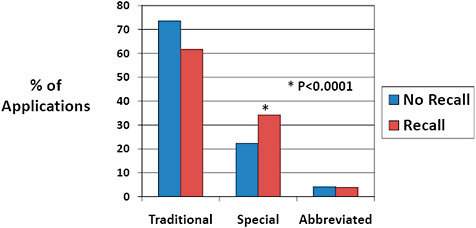
FIGURE C-12 Impact of 510(k) type on recall rate, 2003–2009.
-
Traditional 510(k) were most common for both 510(k)s affected and unaffected by recalls.
-
Special 510(k)s represented a higher percentage of 510(k) type among recalled 510(k)s than among 510(k)s unaffected by recall (34.2% vs 22.3%).
-
Abbreviated 510(k)s were infrequent for both 510(k)s affected and unaffected by recall.
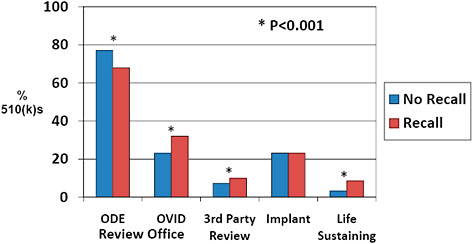
FIGURE C-13 Association of 510(k) features with recall rate, 2003–2009.
-
510(k)s affected by recall were less often cleared by the Office of Device Evaluation (67.9% vs 77.0%) and more often cleared by the OIVD (32.1% vs 23.0%) than devices unaffected by recall.
-
510(k)s were more often cleared via third party review among recalled 510(k)s than among those unaffected by recall (9.9% vs 7.3%).
-
510(k)s affected by recall were more often life-sustaining devices than 510(k)s unaffected by recall (8.5% vs 3.2%).
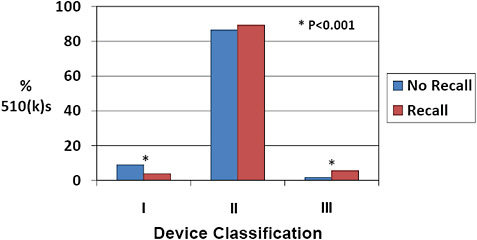
FIGURE C-14 Device classification and 510(k) recall rate, 2003–2009.
-
More than 85% of 510(k)s were Class II devices for devices both affected and unaffected by recall.
-
There was a more than 3-fold increase in the percentage of Class III devices among 510(k)s affected by recall compared to those unaffected (5.6% vs 1.7%). In contrast, there was a reduction in the percentage of Class I devices among recalled 510(k)s compared to 510(k)s unaffected by recall (5.6% vs 1.7%).
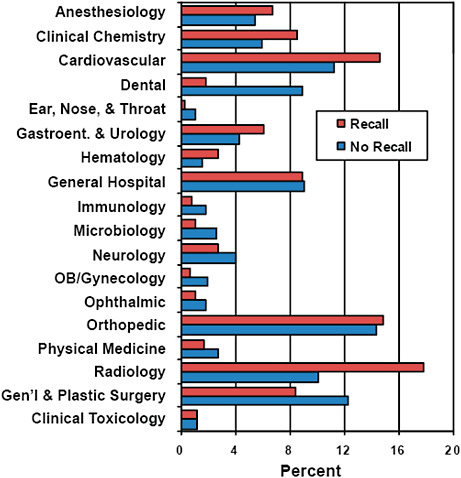
FIGURE C-15 Association of advisory committee assignments with 510(k) recall rate, 2003–2009.
-
Devices associated with certain advisory committee assignments were more likely to be affected by recall.
-
For example, Radiology (17.8% vs 10.1%), Cardiovascular (14.6% vs 11.2%), Anesthesiology (6.8% vs 5.4%), and Clinical Chemistry (8.6% vs 5.9%) represented a higher percentage of devices among recalled than unaffected 510(k)s.
-
In contrast, Dental (1.8% vs 8.9%), Ear, Nose & Throat (0.3% vs 1.1%), and General & Plastic Surgery (8.4% vs 12.3%) represented a lower percentage among recalled devices compared with devices unaffected by recall.
TABLE C-3 Medical Device Reporting and 510(k)s
|
Description |
Number |
|
510(k)s with MDR |
7823 |
|
Total MDRs |
182,394 |
|
MDRs with Death |
2361 |
|
MDRs with Injury |
53,879 |
|
MDRs with Malfunction |
12,110 |
|
MDRs Other |
5051 |
-
182,394 MDRs associated with 7,823 510(k)s from 1996 to 2009 have been filed.
-
Nearly two-thirds of the reports (66.4%) are associated with a device malfunction.
-
29.5% of the reports are associated with a patient injury, although <2% involve a patient death.
-
Data on MDRs are included in this report because they were specifically requested by the Institute of Medicine. These data are subject to incomplete reporting, insufficient information, and misclassifications and should be interpreted with caution.
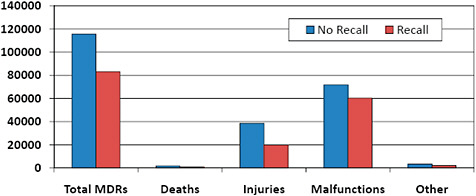
FIGURE C-16 Medical device reporting and recalls.
-
41.8% of 510(k) MDRs were associated with 510(k)s that were subject to recall.
-
30.0% of death adverse event reports, 34.0% of reported patient injuries, and 45.6% of device malfunction reports involving 510(k)s were associated with recalled 510(k)s.
-
Data on MDRs are included in this report because they were specifically requested by the Institute of Medicine. These data are subject to incomplete reporting, insufficient information, and misclassifications and should be interpreted with caution.
SUMMARY OF KEY FINDINGS
-
48,402 510(k)s were cleared by FDA between January 1, 1996, and December 31, 2009, and were available for analysis.
-
From 2003 to 2009, 3,132 unique 510(k)s were subject to recall. Among 510(k)s affected by recall, 73.9% were recalled a single time and 26.1% were recalled more than once, including nearly 2% that were recalled more than 5 times.
-
Among 510(k)s cleared in 2003–2009, 98.4% remained free of recall 1 year following the decision. Longer term follow-up shows that 92.6% and 91.5% of 510(k)s remain free of recall 5 and 6 years, respectively, following regulatory clearance.
-
The annual 510(k) recall rate is highest in the first 3 years following clearance (1.6–1.9%/year). Lower recall rates are observed in years 5 and 6 post clearance (0.9–1.1%/year).
-
More than half the 510(k) recalls are due to manufacturing process errors (28.8%) or device design issues (28.4%). Materials and com-
-
ponent issues (16.3%) and change control processes (11.9%) account for the majority of the remaining 510(k) recalls.
-
Compared to 510(k)s unaffected by recall, recalled 510(k)s are more likely to have been reviewed by a third party or submitted as a Special application (rather than Traditional or Abbreviated). Recalls are also more likely to affect 510(k)s involving life sustaining devices and Class III devices.
























