
3
Approaches to Evidence Generation
|
Key Points Raised by Speakers
|
PHARMACOGENOMICS CLINICAL TRIALS
A basic question in clinical research is whether an intervention works across populations. In pharmacogenomics research, said Caryn Lerman of the University of Pennsylvania, the question can be reframed as whether the intervention benefits or harms particular patients. Ultimately, the question is whether a genomics-based therapy is worth doing from the perspectives of patients, payers, and other stakeholders. Data to answer these questions can be gathered through observational studies of the association of a genomic marker with an outcome (e.g., a cohort study) or through experi-
mental studies of the efficacy of a pharmacogenomic intervention based on accumulated data (e.g., a randomized controlled trial).
Randomized Controlled Trials
An advantage of randomized controlled trials (RCTs) over cohort studies is that they provide controlled exposure to treatment. In addition, randomization helps avoid the type of confounding that can occur in an observational study, in which treatment may be selected based on patient characteristics.
A retrospective trial of a pharmacogenomic marker is carried out after a RCT of a drug has been completed, with researchers testing patient samples to identify which patients were positive or negative for a particular marker and then comparing that information with the patients’ responses to the drugs being tested (Figure 3-1). Retrospective trials can provide useful data when a marker is unknown at trial initiation. They are also ideal for
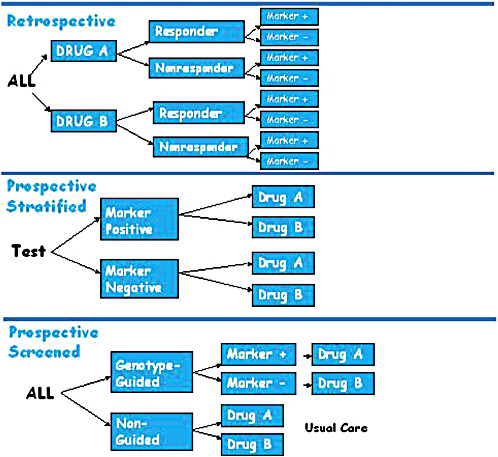
FIGURE 3-1 Pharmacogenomic trial designs, including retrospective, prospective stratified, and prospective screened.
SOURCE: Lerman, IOM workshop presentation on November 17, 2010.
hypothesis generation and can be used for independent validation. There are, however, several important limitations to relying solely on retrospective clinical trials, including unbalanced groups, reduced power based on those unbalanced groups, and missing data (e.g., not all patients may have consented to tissue collection or use of their tissue for further study).
In contrast, prospective stratified trials first test and identify participants as marker positive or marker negative and then randomize each group of participants to therapy arms (i.e., all the marker-positive participants are randomized to either a drug group or a control group, and the same is done for the marker-negative participants) (Figure 3-1). The advantage of this design is that the trial is based on a hypothesis that takes into account prior genomic knowledge about the members of the test population. One important feature of prospective stratification is that it allows for enrichment of more rare genotype groups and balancing of treatment assignment.
A third variant is the prospective screened trial, which, Lerman said, comes closer to a clinical utility model than to a clinical validity model. Some argue, she said, that this is the true test of whether personalized medicine works. In a prospective screened trial, patients are randomized to either a genotype-guided group or a non-guided group. In the genotype-guided group, participants are tested for the presence or absence of the marker under study and then assigned to a treatment group based on the hypothesized association of the marker with the outcome of a particular therapy. The therapy for those who are marker-negative can be an alternate therapy. Results for the genotype-guided groups are compared with those for the non-guided group, which is either randomized to the same two therapies as the guided group or receives the current standard of care (Figure 3-1). The prospective screened design has high ecological (i.e., real life) validity, providing evidence of whether a genome-guided therapy will provide significantly better outcomes than non-guided therapy.
Genome-therapeutic response associations, however, are not necessarily translated into clinical practice. To foster the adoption of genomics-based interventions, it will be important to increase the generalizability of clinical trial designs and results to include clinical practice settings; to demonstrate improvement in health outcomes as well as the cost effectiveness of testing versus not testing; and to establish evidence-based guidelines. Lerman offered several reasons for the reduced generalizability and lack of translation of classic randomized clinical efficacy trials into clinical practice. RCTs have strict eligibility criteria and are conducted in a highly controlled setting, the treatment is protocol-driven, and treatment compliance is very closely monitored. In contrast, in everyday clinical practice the population is very diverse, the practice settings are heterogeneous, treatment is flexible and depends on clinical judgment, and compliance is variable (and likely lower than in the clinical trial setting).
TABLE 3-1 Classic Randomized Controlled Trials (RCTs) Versus Practical Clinical Trials (PCTs)
|
|
Classic RCT/Efficacy |
PCT/Effectiveness |
|
Research Question |
Does it work in ideal circumstances? |
Does it work under best practice conditions? |
|
Population |
Selective, homogeneous |
Diverse, heterogeneous |
|
Setting |
Specialized, controlled |
Clinical practice |
|
Intervention |
Fixed, protocol-driven |
Flexible, clinician judgment |
|
Comparator |
Placebo or active |
Usual care, least $ |
|
Compliance |
Closely monitored, high |
Highly variable |
|
Assessments |
Elaborate, complex |
Simple outcomes |
|
Goal |
FDA approval |
Adoption in practice |
|
SOURCE: Lerman, IOM workshop presentation on November 17, 2010. |
||
One approach to addressing these issues, Lerman suggested, is a practical clinical trial model (also called a pragmatic clinical trial; [Brass, 2010]). Classic RCTs focus on establishing the efficacy of the intervention, while practical clinical trials study the effectiveness of the intervention, looking at simple outcomes, such as health outcomes, patient satisfaction, and costs (Table 3-1).
The advantages of the practical approach to clinical trials are that they are more reflective of patients and practice, more efficient and less burdensome, and the results of the trial are more likely to be generalizable. The disadvantages are that practical clinical trials are less experimentally rigorous by design, usual care is not a stable comparator, and increased heterogeneity results in a much lower signal-to-noise ratio, making greater sample sizes necessary.
Case Example
To illustrate these issues, Lerman offered a case example involving the pharmacogenetics of nicotine addiction treatment. The six-month quit rate across a variety of interventions (lozenge, gum, patch, inhaler, nasal spray, bupropion, and varenicline) is very low. Even using best-in-class pharmacotherapy with varenicline (CHANTIX®), only about one-third of smokers will have successfully quit smoking at 6 months (Gonzales et al., 2006).
A marker that could predict which intervention would be optimal for a given patient could have a substantial medical and public health impact, and Lerman and colleagues have validated a novel metabolic biomarker across several clinical trials. The ratio of the nicotine metabolites 3-hydroxycotinine and cotinine is a stable measure of an individual’s nicotine metabolism rate derived from smoking (Ray et al., 2009). This marker reflects a heritable trait and is independent of the time since the last cigarette, and the metabolites can be measured in saliva, plasma, and urine. This metabolic marker is highly correlated with the CYP2A6 genotype (i.e., it is a phenotypic measure of a genomic trait) (Benowitz et al., 2003; Malaiyandi et al., 2006), but the test for the metabolites is less costly and easier to perform than the genomic test. As a phenotypic test, it also reflects environmental influences on nicotine clearance, as well as genetic influences beyond CYP2A6. Lerman and her colleagues obtained evidence of association by a retrospective analysis of four clinical trials, and they then established the clinical validity of the nicotine metabolism ratio test in a prospective stratified RCT.
Next Lerman discussed a hypothetical practical clinical trial of genotype-guided versus non-guided nicotine therapy, comparing a nicotine patch (low cost, low toxicity) to varenicline (higher cost, greater toxicity) (Figure 3-2). Participants in the genotype-guided arm would be tested for their nicotine metabolism ratio, and slow metabolizers would be treated with a nicotine patch, while fast metabolizers would receive varenicline. Participants in the non-guided arm would be randomized to patch or varenicline.
In this hypothetical scenario, about 20 percent of smokers in the population are slow metabolizers. In the genotype-guided arm this would mean that 20 percent of participants would get the patch and 80 percent would receive varenicline. In the non-guided group, however, randomizing between the two medications means that 50 percent of these participants are treated with the patch and 50 percent with varenicline; based on this randomization, half of the slow metabolizers in the non-guided group will receive the same treatment as the slow metabolizers in the guided group, and similarly for the normal metabolizers in the two groups. To have sufficient statistical power to examine the marginal quit rates in the genotype-guided versus non-guided groups, the study would need to enroll thousands of people.
It is much more efficient to assess genomics-guided versus non-guided therapy in a prospective stratified trial, which allows for oversampling of slow metabolizers in order to achieve comparable numbers of slow and fast metabolizers in the various treatment arms (Figure 3-3). Examining efficacy is then a matter of simply comparing patch to varenicline for the slow metabolizers, and patch to varenicline for the fast metabolizers.
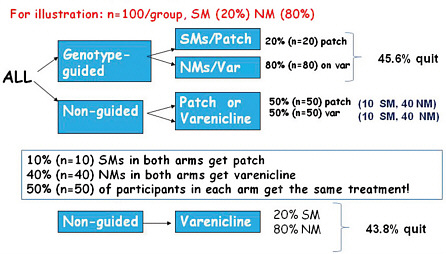
FIGURE 3-2 Hypothetical genotype-guided versus usual care scenario to measure smoking cessation rates using varenicline or patch.
Abbreviations: NM (normal metabolizer); SM (slow metabolizer; var (varenicline).
SOURCE: Lerman, IOM workshop presentation on November 17, 2010.
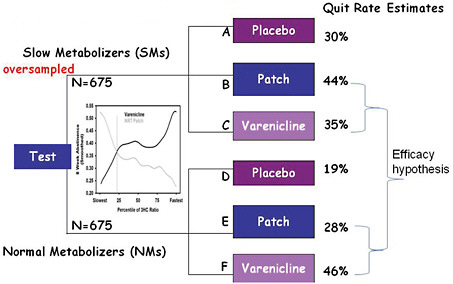
FIGURE 3-3 Prospective stratified RCT scheme.
SOURCE: Lerman, IOM workshop presentation on November 17, 2010.
Principles
In summary, Lerman said that one trial design does not fit all. Retrospective and prospective RCTs are both valid, but prospective trials overcome the limitations of retrospective trials, and population enrichment designs can be used.
Practical clinical trials are an important tool to address the translational gap. However genotype-guided versus non-guided trial designs are inefficient under some scenarios and are not likely to supplant classic RCTs. Once clinical validity has been established in a prospective trial, a practical clinical trial could be part of the validation pathway to help facilitate the transition into practice.
USE OF ARCHIVED SAMPLES TO EVALUATE GENOMIC TESTS
Richard Simon of the National Cancer Institute (NCI) described his work with colleagues on the use of archived specimens to generate new evidence about the clinical utility of prognostic and predictive biomarkers (Simon et al., 2009).
Biomarkers
The term prognostic biomarker is not well defined, Simon noted, and it is used differently in different fields. For the purposes of pharmacogenomics, Simon suggested that prognostic biomarkers are biomarkers that are measured before treatment and are used to predict the long-term outcome for patients receiving standard treatment. The marker may provide information about both the aggressiveness of the disease and the effect of the treatment. A primary intended use of the prognostic marker is to define a subset of patients who have a very good prognosis on the standard treatment and therefore do not require a more aggressive treatment.
An example of the application of a prognostic biomarker is the Oncotype DX gene expression assay initially developed for node-negative, ERpositive women who are receiving tamoxifen. The goal of testing is to identify those patients who are unlikely to benefit from adjuvant chemotherapy in addition to surgery/radiotherapy and hormonal therapy. The Oncotype DX test was initially validated through a retrospective analysis of a previously performed prospective clinical trial. The key to the successful development of the test was that it was done with an indication in mind, Simon said. An important therapeutic decision context was identified, the development and clinical validation separated in a staged manner, and analytical validation emphasized. According to Simon, most prognostic factor studies are not performed with a specific clinical context defined prior to
starting and are consequently very difficult to interpret. A prospective trial, TAILORx, for the validation of Oncotype DX is ongoing (Zujewski and Kamin, 2008).1
Predictive biomarkers are measured before treatment to identify who will or will not benefit from a particular treatment. Many cancer treatments benefit only a minority of the patients to whom they are administered, Simon said, and there is probably no case in which a treatment does not harm at least some of the patients. Being able to predict which patients are likely to benefit (or which are unlikely to benefit) could save patients from unnecessary toxicity, enhance the chances of success, and help control medical costs. Predictive biomarkers are also a critical part of the drug development process for almost all new cancer drugs.
Predictive biomarkers are usually single gene/single protein markers, such as with HER2 testing to determine the appropriateness of anti-HER2 breast cancer treatments (e.g., Herceptin) (Baselga et al., 1999; Wolff et al., 2007) and KRAS analysis to determine appropriate usage of anti-epidermal growth factor receptor (anti-EGFR) antibodies in treating colorectal cancer (Lee and Chu, 2007).
Validation
Validation is essentially a showing of fitness for intended use. Validation is often broken down into analytical validation, clinical validation, and clinical utility. There is some ambiguity concerning what people mean when they talk about these different terms, especially clinical utility, Simon said. Clinical utility can take into account costs or advantages and disadvantages, but he said that the key factor in utility is whether the result of the test is actionable and informs treatment selection to the benefit of the patient.
The optimal designs for evaluating the clinical utility of a prognostic marker include prospective clinical trials and retrospective analysis of archived specimens from a prospective trial.
In evaluating a predictive biomarker, the optimal design is to measure the marker in all patients to identify them as predicted responsive or predicted non-responsive and then to randomize the patients in each group to treatment and control arms.
Simon also discussed a “marker strategy design,” which was referred to as a “prospective screened trial” by Lerman, and agreed that it is often a very inefficient design and that it requires a very large sample size to have sufficient statistical power.
Prospective–Retrospective Study
Some retrospective analyses of archived samples for biomarker studies can result in highly biased conclusions. To address this, Simon and colleagues have proposed a “prospective–retrospective” trial design which uses archived specimens from a single prospective trial to test a specific intended use of an assay and which meets the following criteria:
(1) adequate amounts of archived tissue must be available from enough patients from an appropriately designed prospective trial (which for predictive factors should generally be a randomized design) for analyses to have adequate statistical power and for the patients included in the evaluation to be clearly representative of the patients in the trial; (2) the test should be analytically and pre-analytically validated for use with archived tissue and the testing should be blinded to the clinical data; (3) the plan for biomarker evaluation should be completely specified in writing before the performance of biomarker assays on archived tissue and should be focused on evaluation of a single completely defined classifier; and (4) the results from archived specimens should be validated using specimens from one or more similar, but separate, studies. (Simon et al., 2009)
Simon also discussed potential revisions to the ASCO LOE scale, which currently classifies retrospective studies as LOE II or lower. He suggested that level 1 evidence could come from either a fully prospective clinical trial or else from two or more prospective–retrospective studies (meeting the proposed criteria above) in which the results were consistent.
In conclusion, analysis of archived tissues for prognostic and predictive biomarkers can provide either a higher or a lower level of evidence in support of clinical utility depending upon several key factors: the analytical validation of the assay; the nature of the study from which the specimens were archived; the number and condition of the specimens; and whether a focused, written plan for analysis of the specified biomarker was developed before assaying any tissue. Studies using archived tissues from prospective clinical trials, when conducted under ideal conditions and independently confirmed, can provide the highest level of evidence (LOE I).
COVERAGE WITH EVIDENCE DEVELOPMENT
The Ontario Model
In 2003 the Ontario Ministry of Health and Long-Term Care implemented a new structure with the goal of implementing an evidence-based approach to policy decision-making regarding medical products and procedures (Figure 3-4) (Goeree and Levin, 2006; Levin et al., 2007). The key component of the new structure is the Ontario Health Technology Advisory
Committee (OHTAC), which receives requests for evidence-based analyses from the Ontario Health System and the Ministry of Health. As Leslie Levin of the Medical Advisory Secretariat (MAS) explained, these requests are passed on to the MAS which coordinates systematic reviews and economic analyses with academic partners. Expert panels are engaged to evaluate the evidence; feedback and input from stakeholders, professionals, the public, and industry is sought; and all evidentiary information is then passed to OHTAC, which develops appraisals based on the evidence and provides recommendations to the Ontario Health System and to the Ministry of Health (Figure 3-4). Adoption of the recommendations can be tracked through a geographic information system.
As one part of the larger structure, a field evaluation program was developed to collect primary data in order to address uncertainties identified in the systematic reviews and to perform post-market assessment of real
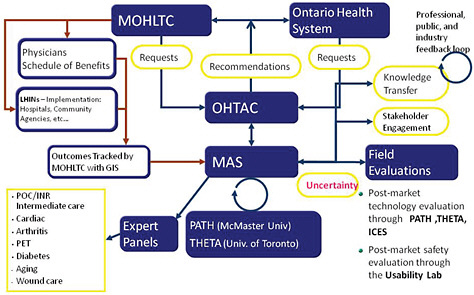
FIGURE 3-4 MAS- and OHTAC-associated structures and linkages. Abbreviations: GIS (geographic information system); ICES (Institute for Clinical Evaluative Sciences); LHIN (local health integration networks); MOHLTC (Ministry of Health and Long-Term Care); PATH (Programs for Assessment of Technology in Health); PET (positron emission tomography); THETA (Toronto Health Economics and Technology Assessment Collaborative).
SOURCE: As modified from Levin et al., 2007 by Levin in IOM workshop presentation on November 17, 2010.
world performance of products. The generation and collection of additional data regarding the utilization and impact of a medical intervention as a requirement of a preliminary coverage decision, or coverage with evidence development, comes under the purview of the Ontario Field Evaluation program. Issues that could trigger a field evaluation include, for example, low quality of evidence, incremental net benefit, generalizability questions, and safety issues. Levin noted that 38 field evaluations have been initiated since 2003, with 19 completed thus far. Of the completed studies, 88 percent affected decision making. Ten of these were coverage with evidence development studies. Most of the studies are published in peer-reviewed journals. Levin said that while saving money was not the original intent of the studies, the resulting coverage decisions have contributed to more than $500 million in cost avoidance.
Examples of Coverage with Evidence Development Field Studies and Recommendations
Levin highlighted several of the ten coverage with evidence development recommendations that have been made thus far (Table 3-2). In response to a published report identifying an increase in restenosis rates for low-risk patients who were treated with bare-metal stents versus those treated with drug-eluting stents, a field evaluation was performed to test the generalizability of this finding. The field study determined that while drug-eluting stents are advantageous for patients at high risk for restenosis, in Ontario there was no significant difference noted for low-risk patients (Tu et al., 2007). As a result, drug-eluting stents are used in only about 30 percent of patients in Ontario, as compared with 90 to 95 percent in the United States. In another case, a systematic review of endovascular abdominal aortic aneurysm repair raised a concern about endoleak (persistent blood flow into the aneurysm sac after the graft procedure). A subsequent prospective field study of 160 patients found that these were primarily Type II endoleaks, which are less serious (Tarride et al., 2008), and an economic analysis showed the procedure is cost effective only for high-risk patients (MAS, 2010). As a result, the decision was made to fund cardiovascular abdominal aortic aneurysm repair only for high-risk patients. MAS has also looked at the clinical utility of positron emission tomography (PET) scanning (Evans et al., 2009). It has insured PET scanning for staging lung cancer, but field studies have not shown clinical utility for head and neck cancer or in staging breast cancer, and these applications of PET are not insured.
As an example of an ongoing coverage with evidence development study, Levin said that MAS is looking at gene expression profiling with Oncotype DX for guiding adjuvant chemotherapy in early breast cancer. As
TABLE 3-2 Summary of Ontario Field Evaluations
|
Technology (n) |
Field Evaluation Overseen by |
Type of Study |
Reason for Field Evaluation |
Result |
Policy Decision |
|
Drug eluting stents (DES) (21,000) |
PATH, with ICES |
Prospective pragmatic registry |
Generalizability of RCT evidence and cost effective analysis |
Only effective in patients at high risk for restenosis |
Funded; 30% conversion from bare-metal to DES (90% in U.S.A.) |
|
Endovascular abdominal aortic aneurysm repair (160) |
PATH and single AHSC |
Prospective observation |
Safety assessment of endoleak |
No endoleak; CE only for high surgical risk |
Funded for high but not low surgical risk |
|
Multifaceted primary care diabetes program |
PATH, with Oxford University |
Before-after study using micro simulation economic model |
Prioritize investments according to downstream effects and CE following systemic review of diabetes strategy |
Most CE were bariatric surgery, MDT; Least, insulin infusion pumps for type II |
Bariatric program funded and additional funding for MDT; Insulin infusion pumps for type 2 on hold |
|
64-slice CT angiography (CTA) v coronary angiography (CA) (175) |
PATH, with cardiologists, radiologists, selected AHSCs |
Patients for CA also underwent CTA |
Uncertainty regarding indications for use, CE and QA parameters |
Sensitivity lower than reported, reducing CE |
OHTAC recommended slow diffusion until sensitivity issue resolved |
|
PET to stage locally advanced NSCLC (310) |
OCOG |
RCT |
Clinical utility in decisions regarding combined modality therapeutics |
Terminated by efficacy & safety committee |
PET insured for this indication |
|
PET to stage early NSCLC (322) |
OCOG |
RCT |
Resolve inconsistencies to inform decision regarding access |
PET reduces futile thoracotomy rates |
PET insured for this indication |
|
PET to stage breast cancer (320) |
OCOG |
Prospective cohort |
Compare PET to sentinel lymph node biopsy |
No utility in staging |
Not insured |
|
PET for colorectal cancer metastatic to liver (400) |
OCOG |
RCT |
Clinical utility in decision for metastatectomy |
Accrual completed February 2010 |
Awaiting results |
|
PET for head and neck cancer (400) |
OCOG |
Prospective cohort |
Clinical utility presurgery following radiation therapy |
No clinical utility |
Not insured |
|
Extracorporeal photopheresis (120) |
PATH with AHSC |
Prospective observational |
Basis for decision regarding funding for GvH and Sezary |
Effective in GvH; Inconclusive for Sezary |
Insured for GvH; Inconclusive for Sezary -small vol. after backlog dealt with |
|
Abbreviations: AHSC (academic health science center); CE (cost effectiveness); DES (drug eluting stent); GvH (graft vs host); ICES (Institute for Clinical Evaluative Sciences); MDT (multi-disciplinary teams); NSCLC (non-small cell lung cancer); OCOG (Ontario Clinical Oncology Group); OHTAC (Ontario Health Technology Advisory Committee); PATH (Programs for Assessment of Technology in Health); PET (positron emission tomography); QA (quality assurance); RCT (randomized controlled trial). SOURCE: As updated from Levin et al., 2007, by Levin in IOM workshop presentation on November 17, 2010. |
|||||
opposed to the TEC and AHRQ reviews, the MAS evidentiary review found low-quality evidence for its prognostic value and very low-quality evidence for its predictive value in terms of the benefits of a classic chemotherapy regimen. As such, a coverage with evidence proposal has been put forth that would consider three key questions: (1) How does Oncotype DX change treatment? (A prospective cohort study has been proposed.) (2) How does Oncotype DX compare to traditional factors? (Electronically collected data on age, tumor size, grade, ER, PR, and HER-2/neu will make it possible to measure correlations between the Oncotype DX recurrence score and traditional risk classification.) (3) What is the impact of Oncotype DX on breast cancer distant recurrence? (Longitudinal data will be collected.) These studies will be informed by ongoing clinical studies, such as the TAILORx trial being conducted by NCI (Zujewski and Kamin, 2008).
Another example is EGFR mutation testing in non-small cell lung cancer. MAS looked at the predictive value of mutated EGFR based on a retrospective subgroup analysis of archived specimens from a RCT of first-line treatment with gefitinib versus chemotherapy. The results of the analysis suggested a statistically significant improvement in progression-free survival for gefitinib versus chemotherapy in EGFR-mutation-positive patients, but not in EGFR-mutation-negative patients (Zhu et al., 2008). However, results of a similar analysis of second- and third-line chemotherapy (erlotinib versus placebo) were not significant (Shao, 2010), and the studies reviewed were not designed to examine the predictive effects of the mutation. Levin noted that the current pattern of practice is to use erlotinib regardless of EGFR status for second or third-line treatment. As a result, it was recommended that there should be payment for EGFR testing for gefitinib as a first-line treatment and for EGFR testing for erlotinib for second- or third-line treatment, that treatment should be allowed for EGFR-negative patients, but that the response to erlotinib should be monitored by EGFR mutation status and the payment for EGFR testing in this group of patients should be modified based on the findings.
Lessons Learned
In summary, Levin said, coverage with evidence development works, but more efficient methodologies are needed to expedite conclusions. To this end, Levin suggested that evidence-based analysis should be implemented further upstream in the lifecycle of drugs and technologies. Industry, academia, and health systems should be engaged in the premarket phase so that the important evidentiary questions are addressed ahead of time. In this way, it may be possible to influence the development pipeline toward technologies that are more relevant to health systems and to patient outcomes.
CONSTRUCTING CHAINS OF EVIDENCE
The rules of evidence that apply to genomic information are no different than the rules of evidence for other forms of information about prognosis, prediction, or diagnosis, said David Ransohoff of the University of North Carolina at Chapel Hill. A chain of evidence is a series of questions or evidence that together describe the impact of some activity—in this case, a genomic test. A primary issue is what questions should be in the chain.
Analytic Framework
Ransohoff said that established analytic frameworks should be used to develop chains of evidence for genomic tests, and he referred to a presentation that Steven Woolf had given to the roundtable at its March 2010 workshop. Woolf had discussed standard analytic principles that are applied to the evaluation of screening tests regardless of the type of test and had noted that
groups such as the U.S. Preventive Services Task Force and the World Health Organization generally consider five issues when assessing preventative interventions: (1) the burden of suffering from the target condition; (2) the accuracy and reliability of the test; (3) the effectiveness of early detection of the condition; (4) potential harms; and (5) the balance of benefits and harms. (IOM, 2010)
These questions are simple to ask but difficult to answer, Ransohoff said. As discussed by Piper, Calonge, and others, a RCT addressing questions 2 through 5 would be the ideal source of evidence. If there is no clinical trial that can answer all of these questions at once, then evidence must be pieced together.
Evidence about genomic tests is often limited to the accuracy and reliability of the test. However, it is not sufficient for a test to discriminate. The bottom line is the outcome—the benefits and harms that occur because of an intervention choice based on the discriminatory capability of the test. For efficient discovery and development, researchers must work backward from a specific clinical decision and consider benefits, harms, and the intended use and desired features of the test. “Working backwards from a specific clinical scenario is absolutely critical [but] commonly not done,” Ransohoff said.
Clinical Trials
If no RCT has been performed, the evidence is necessarily limited. It is possible to assess the ability of the test to discriminate between popu-
lations but not to determine whether this discrimination or subsequent action affects outcomes. Much of the current genomic evidence is limited to questions of discrimination. Ransohoff also noted that in reviewing the available evidence he found that many of the studies published in clinical journals do not disclose critical details of the study design and participants and sometimes the discrimination observed is actually due to bias or to error, not to biology.
As an example, Ransohoff cited a proteomics study about differential exoprotease activities which was looking to determine whether peptide patterns are sensitive and specific for prostate cancer (Villanueva et al., 2006). In the study the test arm was 100 percent male prostate cancer patients averaging 67 years of age. However, the control group was 58 percent women with a mean age of 35 years, leading to a potential source of bias in the findings. The publication reported this important detail, though only in supplemental data, but many published “-omics” studies are opaque, making it difficult or impossible to assess the strength of the evidence.
Barriers to Implementing an Analytic Framework
An analytic framework model makes clinical sense as an evidentiary pathway, and there is extensive experience with analytical pathways in other fields. The challenge is gathering the evidence to fill out the framework, Ransohoff said. Investigators may not think of data as a product of a study. If the study design is weak, then the link in the evidence chain is also weak. Studies need to be carefully and prospectively designed. Specimens should also be considered a product of a study, and the source of the specimens should be described in detail in the methods section of the publication.
In many cases the rate-limiting step is funding, infrastructure, or informatics, but in generating evidence for genomic test development, the rate-limiting step is formulating the key clinical questions and designing a study that provides strong evidence or a link in the chain. The question that needs to be addressed, Ransohoff said, is whether existing data can be used in a strong design.
Ransohoff also noted that there are opportunities to add well-designed studies onto current practices. As examples, he cited two studies, one a study of prognosis and the other of diagnosis. The prognostic study assessed the five-year risk of developing colon cancer after a negative colonoscopy (Imperiale et al., 2008), while the diagnostic study assessed the ability of colorectal screening to detect advanced proximal neoplasms in asymptomatic adults (Imperiale et al., 2000). Both studies were superimposed on a program that a pharmaceutical company had already implemented as a
clinical benefit for its employees, Ransohoff said, and the prognostic study was done at no additional cost.
Moving Forward
An analytic framework for assessing the impact of a test on outcomes offers an established method for guiding clinical and policy decisions. Conceptualized this way, genetic and genomic information is not exceptional. An RCT to assess the impact of a test on outcome is ideal, but when it is not possible or available, there are other sources of data and evidence that can be used. Banked specimens from clinical trials can be used in prospectively designed studies to address questions about prognosis and prediction, for example, and there are various ways to use other data sources, such as cohort data from a health maintenance organization (HMO). Ransohoff advised participants not to be overly focused on infrastructure, informatics, and data sharing. Rather, the focus should be on answering specific clinical questions and opportunistically designing strong research studies in different settings.
DISCUSSION
Archived Specimens
A participant noted that “you can’t test specimens if you don’t have the specimens to test.” Some fields, such as breast cancer research, collect tissues prospectively when conducting clinical trials, Dan Hayes said. It was noted, however, that the number of patients consenting to this tissue collection has been declining for unknown reasons. Furthermore, collecting other types of samples, such as germ-line DNA, can be very costly. Hayes noted that clinical trials are increasingly run by pharmaceutical companies, which do not necessarily collect and store specimens, and he suggested that the FDA require sponsors of new drug applications to have created specimen banks from their trials, although he acknowledged that intellectual property and other issues would need to be addressed. Simon suggested that, going forward, it will be important to do prospective clinical trials and to store specimens with a prospective–retrospective analysis in mind.
One issue with retrospective sample analysis is that it is not possible to optimize the way in which specimens are acquired for the various purposes that may arise in the wide range of possible future marker investigations. Analyte degradation during storage is another concern. Ransohoff agreed that decay is an issue and cautioned that it is important not to compare newer specimens with older specimens. Researchers need to be aware of the problems that can be caused by such decay, so that bias is not introduced in
the results. “We can be mindful with whatever specimens we have collected in the past and hope to store future ones better,” Ransohoff said. Hayes noted that NCI is developing a prospective systematic funding mechanism to answer some of the more basic questions regarding handling of samples, such as what are the implications if a sample sits for 3 days instead of 3 hours before processing or what happens if the sample is fixed for too long. One approach, Hayes said, is to develop an assay that works in the kind of tissue that is collected and archived currently. Another approach is to develop an assay that is so fundamentally powerful that it will change the way tissue is collected and archived going forward.
Trial Design
Participants discussed what is “clinically relevant.” There are multiple study designs that are valid, and which of them is clinically relevant depends on the particular research question. In the end, the goal is to cross a threshold of evidence based on a combination of observational, retrospective, prospective, and larger, more clinically oriented approaches.
As the panelists noted, many of the studies that are currently being done are not designed to contribute to the evidentiary base in the way that is needed for genomics. A question was raised about how to better train the next generation of clinical investigators to think about biomarker studies. Should the NIH develop some very specific training programs as we move into the genomics era? Ransohoff noted that the system rewards clinicians for getting grants and publishing papers, as opposed to producing products or expanding general knowledge.
Simon said that, in his experience, industry is extremely interested in new clinical trial designs that use predictive biomarkers or candidate predictive biomarkers in new drug development. Industry managers are concerned, however, about what the FDA will require (e.g., prohibitively large clinical trial sizes) and about the potential for more roadblocks in developing new drugs with companion diagnostics. He added that NIH funding is driving much of the basic research on identifying the key targets that could be candidate predictive biomarkers and drug targets.
It was noted that patients are increasingly demanding access to interventions that they regard as essential to their well being and health. Institutions are adopting technologies prematurely, and there is political pressure to approve or cover the latest technologies. This is a knowledge translation problem that needs to be addressed. It is important to consider evidence generation during the premarket phase, as trials are being designed and conducted, before product diffusion into the marketplace.

















