
2
Stakeholder Perspectives on Evidence
|
Key Points Raised by Speakers
|
Personalized medicine has multiple stakeholders, including regulators and policymakers; evidence-based review groups; health-care providers; payers; academic, industry, and government researchers and developers; and patients. Enabling the validation and utilization of genomic-based diagnostic tests involves understanding the views of these interested parties. Stakeholders representing regulators, payers, evidence-based review groups, and providers shared their perspectives on evidence (summarized in Box 2-1), illustrated through case examples.
|
BOX 2-1 Stakeholder Perspectives on Evidence FDA
Payers
Evidence-Based Review Groups
Providers
|
FDA PERSPECTIVE
A Focus on Safety and Effectiveness
The core function of the Center for Devices and Radiological Health (CDRH) at the U.S. Food and Drug Administration (FDA) is the review and clearance or approval of medical devices for marketing, said Robert Becker of the Office of In Vitro Diagnostic Device Evaluation and Safety, and such clearance or approval is based on evaluations of safety and effectiveness. Regulations mandate that for safety there must be a reasonable assurance that the probable benefits outweigh any probable risks [21CFR860.7(d) (1)] and that for effectiveness there is a reasonable assurance that the use of the device will provide clinically significant results [21CFR860.7(e)(1)].
FDA classifies in vitro diagnostic devices on the basis of risk. Class III devices are the most complex and present the highest risk (e.g., cancer diagnostics). Risk in this case, Becker clarified, refers to the potential consequences of an inaccurate test result, not to risks associated with the testing process itself. Makers of Class III devices must submit a premarket approval application demonstrating safety and effectiveness and obtain FDA approval prior to marketing. Moderate-risk Class II devices, such as those used to monitor a patient already diagnosed with cancer, are subject to premarket notification [also known as the “510(k) process”], in which the sponsor must demonstrate substantial equivalence of the device to an already marketed product. Class I devices are common, low-risk devices which are generally exempt from premarket evaluation by the agency. The regulatory aims for in vitro diagnostics, Becker said, are clarity and reliability concerning the test description, its intended use, the instructions for use, performance claims, manufacturing, and detection and resolution of issues that arise after the test is on the market.
Performance Claims
The performance claims and risk-based classification of a diagnostic test are based on how well the test supports the intended use. FDA requires evidence of analytical validity, the accuracy in and reliability of measuring the analyte of interest, and clinical validity, the accuracy and reliability of the test in identifying or predicting the biological and medical significance of the test result. The focus is on safety and effectiveness, and in its general review of devices the agency does not address clinical utility (the impact on patient care and outcomes), costs, or comparative effectiveness. The FDA does keep in mind how the tests are going to be used and whether they will be able to effectively guide medical care, Becker said, although this is not part of the official review process.
|
BOX 2-2 Challenges to Establishing Analytical and Clinical Validity of Diagnostics Analytical Validity
Clinical Validity
|
A participant asked about the way FDA views clinical utility in a regulatory context versus the way other stakeholders view clinical utility. Becker answered that the metrics of clinical utility used by payers or users are not necessarily needed for regulatory review of most medical devices. Clinical validity is a sufficient bar for regulatory clearance, and FDA does not make an explicit decision about clinical utility per se. Rather, it is factored into the discussions concerning safety and effectiveness. However, under the circumstance of considering claims for a device that directs the use of a specific therapy (i.e., the clinical performance of the device is tied to the performance of the drug), that is “tantamount to an aspect of clinical utility” which can be factored into consideration since that kind of trial will have demonstrated the clinical utility of the drug.
Becker highlighted some of the challenges associated with establishing analytical and clinical validity of diagnostic devices (Box 2-2). For many of the types of devices FDA has reviewed over the past 30 years, there are strong reference methods or standards available for analytical validation. This is not the case, however, for many of the emerging technologies that the agency is reviewing now, such as gene expression assays. New multivariate tests involve numerous analytes for which a full-scale analytical validation of each single analyte would be onerous and would not necessarily
be informative regarding how the test will perform as a whole. There are multiple issues regarding the clinical samples needed for validation (e.g., the types and availability of samples and their age and storage), and assessing the full spectrum of a device’s performance is also a challenge. Clinical validity can present similar challenges regarding the availability of samples or patients, with issues potentially arising from sample and verification bias. There may also be limits to the “diagnostic truth” of the test results if information is limited on the patient’s underlying condition, and follow-up and outcome studies may be difficult and costly to perform.
Diagnostic tests may be submitted to FDA as stand-alone tests or in association with a drug. Becker described a supplemental application for a new use for the already-marketed drug, Herceptin, where the sponsor was seeking a label indication for gastric carcinoma. Included in the application was significant additional analytical information to validate a diagnostic test for stomach cancer. This information was factored into the clinical evaluation of the drug in a collaborative review by CDRH and the FDA Center for Drug Evaluation and Research.1
Fostering Progress in Analytical and Clinical Validation
Analytical validation of new diagnostic devices would be facilitated by standards and a technical assessment of the technology behind the tests. Becker cited the FDA MicroArray Quality Control (MAQC) project as one example of efforts in this area. In the case of MAQC, the National Institute of Standards and Technology, the National Institutes of Health (NIH), industry partners, and others are working with FDA to develop standards and quality measures for microarrays. There is also a need for better coordination of the analytical and clinical specifications during test design, verification, and validation, especially with regard to medical decision points (i.e., the test’s analytical performance needs to align with the clinical performance). Becker also mentioned ongoing initiatives by government, industry, patient groups, and others that are addressing various analytical sample-related issues, such as collection, storage, and annotation.
In the quest for clean and cost-effective clinical validation of diagnostic devices, there is potential for improvement in appropriate clinical sample specification, acquisition, retention, maintenance, and accessibility. For example, clinical validation can be achieved more effectively by having well-matched intended use, claims, and evidence and choosing an intended use that is more amenable to studies that will directly support that intended use. The FDA is also exploring how to promote better coor-
|
1 |
See http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2010/103792s5250ltr.pdf for details on approval. |
dination among the therapeutic product developers, the diagnostic device developers, and reviewers through pre-submission meetings or through collaborative reviews. Finally, the use of more efficient study and trial designs (e.g., adaptive or Bayesian designs) can also help facilitate clinical validation.
PAYER PERSPECTIVE
Margaret Piper of the Blue Cross and Blue Shield Association Technology Evaluation Center (TEC) described the evaluation criteria that TEC uses for its assessment of genetic test evidence. Established in 1985, TEC is housed within the Blue Cross and Blue Shield Association, which is the membership organization for Blue Cross and Blue Shield Plans. The mission of TEC is “to provide health-care decision makers with timely, objective, and scientifically rigorous assessments that synthesize the available evidence on the diagnosis, treatment, management, and prevention of disease.” TEC does not work directly for the Plans, but the Plans help to identify topics of interest for TEC to work on. TEC also makes all of its assessment products publicly available.
Technology Evaluation Criteria
Every TEC assessment involves evaluating the technology according to five general criteria,2 Piper explained:
-
The technology must have the required final approval from the appropriate government regulatory bodies. (This includes FDA clearance as required or CLIA certification as it applies to laboratory-developed tests.)
-
The scientific evidence must permit conclusions concerning the effect of the technology on health outcomes.
-
The technology must improve the net health outcome.
-
The technology must be as beneficial as any established alternatives.
-
The improvement must be attainable outside the investigational setting.
To evaluate genetic testing, TEC uses the ACCE framework developed by the Centers for Disease Control and Prevention (CDC) National Office of Public Health Genomics.3 ACCE refers to the four components of the
|
2 |
See http://www.bcbs.com/blueresources/tec/tec-criteria.html for details on TEC criteria. |
|
3 |
See http://www.cdc.gov/genomics/gtesting/ACCE/index.htm for further details on the ACCE framework. |
framework—analytic validity; clinical validity; clinical utility; and ethical, legal, and social implications—which are addressed with a set of targeted questions. All TEC assessments are also reviewed by an outside medical advisory panel which has final say over the conclusions.
A Focus on Outcomes
Piper clarified the distinction that TEC makes between clinical validity and clinical utility. Clinical validity is the association of a test result with an outcome (e.g., diagnosing disease or predicting drug response) and is described by measures of association, such as sensitivity, specificity, and predictive value as well as odds ratios, risk ratios, and logistic regression analyses, that describe whether a genetic test retains significance when analyzed along with other criteria. Clinical validity is concerned with the significance of the test for populations of patients. Measures of association only quantify how well the test discriminates between populations with and without the selected outcome, and these measures alone are not sufficient to gauge the clinical usefulness of a test. Piper cited a study showing the limitations of the odds ratio in gauging test performance as an example of why single measures of association are not sufficient (Pepe et al., 2004).
In contrast, clinical utility describes the impact of using the test on patient management and outcomes compared to usual care and therefore describes the significance of the test for individual patient decision-making. From a payer perspective, the focus is on clinical utility. TEC seeks evidence that a genomic test can be used for individual patient management, and it assesses the incremental value of adding the test to usual clinical practice (measured in terms of outcomes). The ideal approach is to obtain direct evidence through randomized controlled trials (outcomes using the test versus not using the test), but such trials are seldom possible. More often, the approach is to establish an indirect evidence chain, such as the assessments conducted by the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) initiative.4
Analytic validity describes the technical performance of the test—the accuracy of the test in measuring the analyte of interest, test repeatability, and reliability in the presence of interfering substances as well as over time. Analytic validity is carefully evaluated by the FDA when tests are submitted for marketing clearance, but such information is not routinely available for laboratory developed tests. “Most laboratory developed tests do not publish their analytic validity, do not make it otherwise publicly available,
|
4 |
EGAPP is discussed further by Calonge below. See also http://www.egappreviews.org/workingrp/reports.htm for more information on EGAPP methodology and reports. Constructing chains of evidence is discussed further by Ransohoff in Chapter 3. |
and therefore we don’t have any evidence of the long-term reliability of the test,” Piper said.
A test should meaningfully improve discriminatory ability when added to existing predictors, or, if it is replacing a currently used test, it should demonstrate superior discrimination. This is easiest to evaluate when test results are classified in a manner that informs decision making (high risk versus low risk). One statistical approach to evaluate the discriminatory ability of a test is concordance, or the “c-statistic,” which is the area under the receiver operating characteristic curve (a measurement that compares the sensitivity of a test to its false positive rate as the discrimination threshold is altered). This is not a very powerful method of analysis, Piper noted, and it can be difficult to determine if improvement in the c-statistic is clinically meaningful with regard to treatment decisions. Another method is the use of a reclassification analysis (Pencina et al., 2008). Risk is first classified by standard methods and then reclassified with the additional information provided by the genetic test results incorporated into the analysis. The net reclassification improvement is then calculated. Analysis must take into account whether the risk is reclassified correctly or incorrectly.
Case Examples of TEC Assessments
Oncotype DX Assay
The Oncotype DX assay is used for predicting response to chemotherapy in women with node-negative, estrogen receptor (ER)–positive breast cancer. TEC first evaluated the relationship between the Oncotype DX Recurrence Score and distant disease recurrence within 10 years. A study published in 2004 first established clinical validity (Paik et al., 2004a), Piper said, but the evidence was deemed insufficient to meet the TEC criteria (TEC, 2005). A subsequent study published in 2006 established the relationship between the Recurrence Score and the likelihood of benefit from chemotherapy (Paik et al., 2006). Again, this evidence was primarily supportive of clinical validity and was not sufficient to meet TEC criteria, Piper said, but the TEC medical advisory panel asked if a better analysis had been done on the existing data.
Reclassification analyses presented in a poster and partially published in a review article found that about half of the patients who were originally classified by the National Comprehensive Cancer Network (NCCN) criteria as being at a high risk of recurrence were subsequently reclassified as being at low risk of recurrence by Oncotype DX testing (Table 2-1) (Paik et al., 2004b).
Before Oncotype DX testing, Piper explained, all of these low-risk patients who were classified as high risk by the original NCCN criteria
TABLE 2-1 Oncotype DX Reclassification of Patients
|
Classification by NCCN |
Reclassification by Oncotype DX |
n |
% Distant Recurrence Free at 10 years (95% CI) |
|
Low (8%) |
Low |
38 |
100 (NR) |
|
|
Intermediate |
12 |
80 (59–100) |
|
|
High |
3 |
56 (13–100) |
|
High (92%) |
Low |
301 |
93 (89–96) |
|
|
Intermediate |
137 |
86 (80–92) |
|
|
High |
178 |
70 (62–77) |
|
SOURCE: Adapted from TEC, 2008a. |
|||
would have received chemotherapy. Since the reclassification analysis was done using retrospective data from completed clinical trials, outcomes were known. The confidence interval for no recurrence in the reclassified population was 89 to 96 percent. The absolute benefit from anthracycline chemotherapy in these low risk patients is 1 to 3 percent at best, she said. In addition, the lower the prior risk of recurrence as indicated by the Oncotype DX result, the less absolute benefit the patient derived from chemotherapy. At such low absolute chemotherapy benefit, the harms may be perceived as greater than the benefit and a woman might reasonably choose to avoid chemotherapy. This analysis, together with the prior information, allowed the test to meet the TEC criteria (TEC, 2008a).
Genetic Test for Long QT Syndrome
Another example of a TEC assessment concerns congenital long QT syndrome, which can lead to major cardiac events and sudden death. Although beta-blockers are effective as a low-risk preventative treatment, the clinical diagnostic criteria are not well established, and the syndrome is difficult to detect. There is, however, a genetic test that can detect 60 to 70 percent of people with long QT syndrome. Individuals who are defined as having long QT syndrome by genetic marker testing only (i.e., no clinical signs) also have a high risk of catastrophic cardiac events (Moss et al., 2007). If an individual has been diagnosed with congenital long QT syndrome, either following an event or by clinical means, or if there is a known mutation in the family, then relatives with possible long QT syndrome can be identified through genetic testing for the mutation and treated with betablockers to reduce the risk of adverse cardiovascular outcomes (Roden, 2008). The disease can be ruled out with confidence for those who test negative for a known mutation. Based on these findings, the TEC assessment was affirmative for use of the genetic test (TEC, 2008b).
Epidermal Growth Factor Receptor Mutation Testing
As a final example, Piper described TEC’s assessment of whether epidermal growth factor receptor (EGFR) mutations can prospectively predict response to tyrosine kinase inhibitor therapy for patients with non-small-cell lung cancer. The majority of the early data relate to the drugs gefitinib (which is no longer available in the United States) and erlotinib, both of which inhibit EGFR activation. The first assessment of genetic testing to predict response to EGFR inhibitors did not meet TEC criteria because there were not enough data to separate the responder and nonresponder populations through mutation testing (TEC, 2007). It also appeared that the test did not reliably identify nonresponders, as some patients with wildtype EGFR genes who were not supposed to respond to therapy actually did respond (TEC, 2007). There was also a concern that the inhibition mechanisms of gefitinib and erlotinib might be slightly different. As only erlotinib is available in the United States, the TEC medical advisory panel requested independent assessment of erlotinib, and the subsequent assessment concluded that EGFR mutation testing to predict response to erlotinib treatment does meet TEC criteria. Outcomes for progression-free survival and overall survival showed a much better separation between responders and nonresponders, and showed that patients with the wild-type EGFR gene were not likely to respond to erlotinib, thus, indicating it is best for them to move to an alternative treatment (TEC, 2011).
EVIDENCE-BASED REVIEW GROUP PERSPECTIVE
The EGAPP initiative is an independent, non-regulatory CDC-funded project to develop evidence-based recommendations on the appropriate use of genetic tests, said Ned Calonge, chair of the EGAPP Working Group. The EGAPP process is transparent and publicly accountable, and its methods integrate knowledge from existing processes of evaluation and appraisal, such as those of the ACCE systematic review process, the Evidence-Based Practice Centers program of the Agency for Healthcare Research and Quality (AHRQ), and the U.S. Preventive Services Task Force, to assess the quality of individual studies, the adequacy of overall evidence, and the magnitude of net benefit. EGAPP also takes contextual issues into account. In addition to providing recommendations, EGAPP identifies gaps in the evidence in order to inform the research agenda.
The EGAPP Working Group Process
The basic steps in the EGAPP working group process are:
-
Select the topic or genomic application for evaluation.
-
Define the clinical scenario (diagnosis, disease screening, risk assessment, prognosis, or pharmacogenetics).
-
Create an analytic framework of key questions to guide the evidence review.
-
Find, synthesize, and evaluate the quality and adequacy of existing literature.
-
Determine net benefit (benefit minus harms) of test application.
-
Create a recommendation based on the certainty of net benefit.
Analytic Framework
Often, Calonge noted, there is no overarching or direct evidence of health outcomes associated with the use of a genomic test (such as the evidence from a randomized controlled trial). In these cases, EGAPP uses the ACCE criteria to develop an indirect chain of evidence to address key questions of analytic validity, clinical validity, and clinical utility. As an example, Calonge presented an analytic framework and a set of key questions for the assessment of pharmacogenomic testing for selective serotonin reuptake inhibitor (SSRI) therapy (Figure 2-1) (Teutsch et al., 2009).
Quality of Evidence
After developing the framework and the key questions, the working group grades the quality of the evidence. The quality assessment takes into account the hierarchical level of the study design as well as study flaws and threats to internal validity, Calonge said. Evidence is classified as either convincing (the observed effect is likely to be real), adequate (a higher risk exists that the effect may be influenced by study flaws), or inadequate (too many flaws exist to confidently assign the results to the factors under study).
Net Benefit
Determining the net benefit involves balancing the benefits or potential benefits with the harms or potential harms. This often requires comparing harms and benefits that are very different in terms of health, value, or metrics, Calonge said. Net benefit is classified as small, moderate, or substantial. Based on the overall assessment of evidence, EGAPP also determines the certainty of net benefit. Calonge described certainty as the opposite of the risk of being wrong. The higher the level of certainty, the less likely that the recommendation is incorrect or will be changed because of future research. An example of high certainty is the value of blood pressure screen-
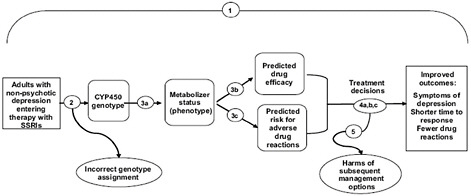
FIGURE 2-1 Sample analytic framework.
NOTE: Key questions (shown as numbered circles) addressed at each stage are: (1) overarching, direct evidence; (2) analytic validity [technical performance, including analytic sensitivity/specificity, reliability, and assay robustness]; (3) clinical validity [ability to identify or predict the disorder of interest: clinical sensitivity, specificity, and predictive value relating to expression/phenotype]; and (4 and 5) clinical utility [balance of benefits and harms with clinical use: efficacy/effectiveness and net benefit].
SOURCE: Teutsch et al., 2009.
ing. It is highly unlikely that at some point in the future it will be decided that people should not be screened and treated for high blood pressure. Moderate certainty means that there are some questions about the evidence and some risk that future research could lead to a change in the recommendation, but in the judgment of the EGAPP working group the evaluation of net benefit has met the criteria for making the recommendation. Finally, low certainty is when there is inadequate evidence of a net benefit to make a recommendation.
A Focus on Net Benefit
If there is high or moderate certainty concerning a small to substantial net benefit, EGAPP will recommend use of the genomic application, Calonge said. If there is high or moderate certainty of a zero benefit, or of a net harm, EGAPP recommends against the use of the application. Low certainty results in a conclusion of insufficient evidence and no recommendation for or against is made.
EGAPP further classifies insufficient evidence conclusions according to contextual factors. Such applications are classified as neutral, indicating that it is not possible to predict what future research will find; discouraging,
in cases where either the risk of harm is so high that EGAPP discourages use of the application until specific knowledge gaps are filled or the topic is not likely to ever meet evidentiary standards; or encouraging, in cases where the working group believes that the test is likely to meet evidentiary standards with further study and reasonable use in limited situations is appropriate while awaiting further evidence.
Case Examples of EGAPP Assessments
Lynch Syndrome
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, is an inherited condition that increases an individual’s risk of colon and other cancers, including those of the stomach, small intestine, liver, gallbladder ducts, upper urinary tract, brain, skin, prostate, endometrium, and ovaries (IOM, 2010). The EGAPP working group “found sufficient evidence to recommend offering genetic testing for Lynch syndrome to individuals with newly diagnosed colorectal cancer in order to reduce mortality and morbidity in relatives.” This is a very specific use, Calonge noted.
Three genetic testing strategies showed evidence of analytic validity: microsatellite instability (MSI) testing, immunohistochemistry (IHC) testing, and testing for mutations in the BRAF gene. The analytic performance of MSI testing is high, according to College of American Pathologists (CAP) external proficiency testing. IHC testing for Lynch syndrome mismatch repair gene proteins is not currently subject to CAP testing, but IHC testing for other proteins is, and therefore an assumption was made that IHC testing for Lynch syndrome has adequate analytic validity. Finally, BRAF mutation testing is based on single-gene mutation sequencing, and analytic validity in that setting is high. Based on existing clinical studies, EGAPP also found adequate evidence of clinical validity for all three tests.
Clinical utility was first assessed for the probands themselves, but EGAPP found insufficient evidence to support differential treatment options based on a proband being identified as having Lynch syndrome. The working group did note, however, that a small body of evidence suggests that MSI-high tumors might be resistant to treatment with 5-fluorouracil and more sensitive to irinotecan (Palomaki et al., 2009), and it highlighted this as a research gap worthy of further study.
Lynch syndrome testing had the greatest clinical utility, Calonge said, with regard to first- and second-degree relatives of patients testing positive for Lynch syndrome. Seven studies found an increase in colonoscopy testing of between 53 and 100 percent in test-positive relatives (Palomaki et al., 2009). The benefit outweighed the harms, which were judged as no more harmful than colonoscopy testing in general. Evidence for utility was pro-
vided by a randomized controlled trial that showed a 62 percent reduction in colorectal cancer and related mortality in relatives with Lynch syndrome mutations who then chose colonic surveillance (Jarvinen et al., 2000) and from an observational study showing a 73 percent mortality reduction in a study involving nearly 3,000 persons across 146 Lynch syndrome families (de Jong et al., 2006). There was similar indirect evidence for increased ovarian and endometrial cancer screening in women (Schmeler et al., 2006). Together, these pieces supported the EGAPP recommendation for Lynch syndrome testing (Palomaki et al., 2009).
Breast Cancer Tumor Gene Expression Profiles
Calonge also described the EGAPP assessment of breast cancer tumor gene expression profiles. In this case, after a review of the Oncotype DX, MammaPrint, and H:I ratio tests, EGAPP “found insufficient evidence to make a recommendation for or against the use of tumor gene expression profiles to improve outcomes in defined populations of women with breast cancer.” This insufficient evidence conclusion was classified as encouraging, however, as EGAPP “found preliminary evidence of a potential benefit of testing results to some women who face the decisions about treatment options (reduced adverse events due to low risk women avoiding chemotherapy) but could not rule out the potential for harm for others (breast cancer recurrence that could have been prevented).”
The evidence regarding analytic validity was deemed inadequate. There were some data on technical performance, but it was not possible to make estimates of analytic sensitivity or specificity, and testing failed on 14.5 to 19 percent of fresh samples. Despite the inability to establish analytic validity, there was enough evidence for the clinical validity of Oncotype DX based on three studies and for the clinical validity of MammaPrint based on two studies (Marchionni et al., 2008).
There were no studies of clinical utility for MammaPrint, which was a critical evidence gap, Calonge said. In contrast to the conclusion by TEC that Oncotype Dx met their evaluation criteria, the EGAPP assessment of a retrospective analysis of one arm of a prospective clinical trial found the evidence promising but ultimately unconvincing. And although women are likely to benefit by avoiding unnecessary chemotherapy, the potential for harm (recurrence and perhaps death) is significant for a small number of low- and intermediate-risk women who might benefit from chemotherapy but forego it based on test results. There were no data available on use in women with high risk on conventional assessment but with low risk on Oncotype DX (the type of reclassification analysis evaluated by TEC).
The conclusions were that there was encouraging indirect evidence for Oncotype DX (Marchionni et al., 2008) and a plausible potential use for
MammaPrint (Marchionni et al., 2008). Calonge noted that two randomized controlled trials are under way, the results of which may allow for updating the recommendation.
Common Research Gaps
In closing, Calonge listed several hurdles to evaluation. One challenge to establishing analytic validity is that necessary information is often missing or unavailable due to proprietary issues or because these may be laboratory-developed tests. Clinical validity is often based on testing subjects with the potential for sources of bias (e.g., selection process, study design) to influence the results. The clinical utility evaluation is a major source of insufficient-evidence conclusions. Furthermore, there are few randomized controlled trials, observational studies present bias issues, and recommendations based on observational study results run a higher risk of being wrong than those based on randomized controlled trial findings.
HEALTH-CARE PROVIDER PERSPECTIVE
A Focus on Value
As an oncologist, Daniel Hayes of the University of Michigan Comprehensive Cancer Center looks at genomic diagnostic tests from the perspective of value. The goal is to improve cancer outcomes by focusing the “right therapy on the right patient,” thereby increasing the chances of cure, survival, or palliation and decreasing exposure to toxicity from unnecessary or inappropriate therapy.
Hayes began with data showing that the odds of dying of breast cancer in the developed world gradually increased from 1950 until about 1980, reached a plateau for about a decade, and then began a steady decline around 1990 (Peto et al., 2000) (see Figure 2-2). Hayes said that the plateau and early decline was due not to screening, which did not begin until the late 1980s, but rather to the widespread application of adjuvant chemotherapy and hormone therapy which began in the late 1970s and early 1980s. Subsequent analysis has shown that the overall decline can be attributed equally to the use of screening and the use of adjuvant therapy (Berry et al., 2005) (see Figure 2-2).
The decision to treat differs from patient to patient and is affected by patient, provider, and societal perspectives regarding risks, benefits, and costs. There is no clearly defined level of benefit below which treatment becomes “not worth it.” Two patients presented with the same Oncotype DX Recurrence Score, for example, may come to different treatment decisions.
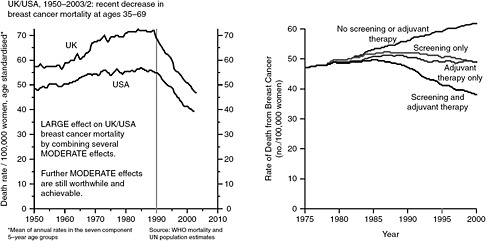
FIGURE 2-2 Breast cancer mortality rates from 1950-2003 and the effect of screening and adjuvant therapy.
SOURCE: Berry et al., 2005; Peto et al., 2000.
To help guide decisions regarding the value of genomic diagnostic tests, the American Society of Clinical Oncology (ASCO) developed tumor marker guidelines (Harris et al., 2007). The guidelines are rather conservative, Hayes said, and the ASCO panel recommended only those markers for which results would change clinical decisions. To facilitate evidence analysis, the panel developed a tumor marker utility grading scale which classifies studies into one of five levels of evidence (LOEs). LOE I studies are prospective, highly powered studies designed specifically to test a tumor marker or a meta-analysis of lower-level studies. LOE II studies are prospective studies of a therapeutic product where the study of the marker is a secondary objective. LOE III through V are retrospective analyses. Only level I or level II studies should be used for evidence evaluation. Unfortunately, Hayes said, most of the tumor marker studies that are available are LOE III.
For those already diagnosed with cancer, a tumor marker is clinically useful when it is prognostic or predictive of cancer outcomes or predicts toxicity; when the magnitude of the effect is sufficiently large that clinical decisions based on the test data result in acceptable outcomes; and when the estimate of that magnitude of effect is reliable. Cancer diagnostics allow clinicians to identify those patients for whom the benefits of a treatment do not outweigh risks, in which case they can safely recommend withholding that treatment. “We are trying to identify patients who would forgo or discontinue therapy to avoid toxicities,” Hayes said. As an example, he noted that ER testing of tumors is regularly done in the clinic because tamoxifen is effective only in ER-positive patients (Berry et al., 2006), which allows
ER-negative patients to avoid the toxicity associated with treatment. However, only half of ER-positive patients are responsive to the therapy, and the test does not indicate which particular ER-positive patients will benefit from treatment.
Evidence Generation
In oncology, evidence is generated in two ways, Hayes said. The first is by developing prospective clinical trials to “test the test,” where the marker is the primary objective. There are very few such trials to test cancer diagnostics, however, as they are large, costly, and lengthy. Another, more innovative way to generate evidence is by using archived specimens.5 Hayes said that ideally these specimens would be from a previously conducted prospective trial and when testing predictive factors, the specimens would have originated from a trial that specifically addressed the effectiveness of the therapy in question in a randomized fashion. Such studies, when designed and conducted with as much rigor as one would put into a prospective trial and when appropriately confirmed, can generate LOE I data. Hayes and colleagues have authored a proposal to revise the LOE scale to provide a more detailed account of the use of archived specimens to generate a sufficiently high level of evidence to achieve clinical utility (Simon et al., 2009).
One of the barriers to generating the necessary evidence is that tumor marker research, especially the clinical component, is not perceived to be as exciting or as important as research on new therapeutics. Marker studies garner less academic credit and less funding and are often conducted with less rigor. There is also less evidence required for clinical use (e.g., by FDA or guideline panels), less quality control/quality assurance and proficiency testing, and much less reimbursement.
This has led to a vicious cycle, Hayes said. Tumor marker utility is poorly valued, which leads to the low level of reimbursement. This means lower funding for tumor marker research and little incentive to do properly designed and controlled clinical studies. The lack of trials leads to lower levels of evidence and less certainty concerning the data, resulting in few recommendations for use, and the cycle repeats.
Breaking the cycle of undervalue, Hayes said, requires increasing the level of reimbursement for marker testing, increasing funding for tumor marker research, and creating incentives to conduct well-designed trials. This will result in the generation of level I evidence that can support guidelines and recommendations for use, which in turn will lead to marker utility that is highly valued. Hayes called for tumor marker publications to have increased rigor, comparable to that of therapeutic trials. He also suggested
|
5 |
Discussed further by Simon in Chapter 3. |
reforms to the regulatory review of tumor markers, including requiring analytical validity and clinical utility, eliminating laboratory-developed test discretion, and requiring that new drug registration trials include a biospecimen bank. Overall, he concluded, it is necessary to think about tumor markers in the same way that therapeutics are considered.
PANEL DISCUSSION
Applying Population-Based Data to Individuals
Participants discussed the challenges of applying results from clinical studies and population-based data to individual patients. When applying new risk stratification methodology, there will inevitably be some patients who were correctly classified with the old methodology but who are incorrectly classified with the new methodology. “How do you deal with patients who actually are disadvantaged as we move to what is, from a population standpoint, a better test?” asked a participant. Hayes said that the focus should be to improve the odds of being cured. He referred participants to Adjuvant! Online (http://www.adjuvantonline.com) which allows providers to enter patient data and make estimates of various risks (e.g., odds of recurrence, benefit from systemic adjuvant therapies, and adverse events). Quantifying those risks allows the patient to better understand the treatment options being offered, Hayes said. Calonge added that while the goal is to balance the potential benefits and harms, people tend to be benefit oriented and do not examine harms as often or in as much depth as they examine benefits. The potential harms include providing therapy to someone who does not need it. In effect, “the cost of the benefits for a person who would have benefited from therapy in the old scheme is the harms to all those who did not benefit.” Becker noted that survival curves are not step functions and that some patients in the apparently responsive group do die. It is always difficult to project what a population study means at the individual patient level. Another challenge to consider is how to move forward when tests results suggest actions in different directions (e.g., test A predicts a high risk, while test B indicates a low risk).
Clinical Utility
Another topic of discussion was clinical utility versus personal utility, which refers to the value of information to the individual for use in decision making. Calonge noted that the potential value of information is included in the EGAPP outcomes set, but understanding the actual value is difficult. If ending the diagnostic odyssey provides a health benefit to an individual,
what is the value of that benefit, and how much should we pay for that additional information?
Hayes added that many patients have tests done primarily because the tests are available to them, under the presumption that they will predict something and that there will be an action taken on the results. In some cases, there is a marker and an implied outcome, but there is not a high level of evidence that the outcome is associated with the marker or that some action should be taken based on the marker. Hayes said that patients are often mistreated based on presumed information, and many people make decisions that end up harming them instead of helping them. The role of a physician is to help patients avoid making decisions that will harm them. From a societal perspective, it is not prudent to spend a lot of money on tests that, while new and exciting, are still unproven. Tests should be done because they have clinical utility. One could argue that if the patient is willing to pay for the test, then it is the patient’s right to have it done. But issues appear downstream with the potential for mistreatment and with the added costs to the system of such treatment (e.g., to payers and to the insured in the form of increased premiums). A participant noted that genetic counseling is based on the concept of personal utility. Genetic counselors spend a lot of time talking about the patient’s motivation for wanting the test. Performing the procedure may not change the result of a particular situation, but people may “perceive what they want to perceive” even if there is not a direct health outcome.
Piper added that insurance companies look at health outcomes and draw fine lines for what qualifies for reimbursement. Information for information’s sake is not health outcome oriented. Similarly, information that is of value for life planning (e.g., making wills, buying insurance, or making job decisions) is not health outcome oriented. A health outcome would instead be a case when the information reduces the diagnostic odyssey or is useful for reproductive decision making. However, if the information predicts the risk of a disease for which there is no preventive or ameliorative treatment, then there is no health outcome. “Where personal preferences come in, is in the final decision-making after you have all the data,” said Piper.
Evidence
Participants also discussed whether there is enough commonality across stakeholders to set a common evidentiary bar. In general, panelists felt that the different stakeholders who were represented were not that far apart. Calonge noted that EGAPP has started looking at the reclassification approach described by Piper, but it has been very careful not to drop the evidentiary standard too low and thus increase the risk of being wrong and
harming individuals. Bringing critical appraisal questions concerning coherence, consistency, strength of association, and precision of the estimate into observational or post hoc analysis studies will allow evidence to be moved closer toward adequate and convincing levels.
Piper said that there is no one blueprint to follow in all situations. The analysis for long QT syndrome was different from the analysis for Oncotype DX, for example. The analysis must be adapted to the clinical setting and the evidence needed for that particular application. The bottom line is to establish that there is evidence of improved patient outcomes. One does not stop at clinical validity and associational evidence, Piper said.
Becker agreed that there is a fair amount of commonality about the kind of evidence that helps in reaching a decision. One of the things that sets the FDA approach apart from the approaches that can be employed in other settings, he said, is that the agency needs to look at tests on a deviceby-device, test-by-test basis and make regulatory decisions about the individual test that the device sponsor has brought to the agency. Stakeholders outside of FDA are more able to synthesize information across the literature and across tests. However, there are some circumstances in which FDA can handle class-specific issues across all of the devices of a particular type, rather than individually for each specific test.
Participants encouraged including subject matter experts in the process of evidence analysis. While such experts may not be part of the final evidence decision, they should be part of the gathering of the evidence.




















