
Evaluating Biological Mechanisms
of Adverse Events
Charged with reporting on biological mechanisms, the committee reviewed evidence presented in case reports/clinical write-ups, laboratory tests, and animal models. Based on the array of adverse events and types of vaccines being reviewed, the committee compiled a list of mechanisms it deemed most likely to contribute to the development of adverse events after vaccination. The pathophysiologies and, at times, the evidence needed to identify a mechanism as operative were discussed. The mechanisms include immune-mediated reactions, viral activity, and injection-related reactions. The committee also discussed the coagulation cascade and its contribution to disease. In addition, the committee discussed the mechanisms that could lead to the development of adverse events in susceptible individuals, as well as the role vaccination could have in revealing an underlying immunodeficiency. The committee also discussed alterations in brain development that included a discussion of autism. Lastly, the advantages and disadvantages of applying evidence of a mechanism derived from an animal model to a human condition are discussed.
LATENCY BETWEEN ANTIGEN EXPOSURE AND
PEAK ADAPTIVE IMMUNE RESPONSE
Antigen exposure initiates an array of reactions involving the immune system, including the activation of white blood cells called lymphocytes that fight infection. After antigen exposure, two types of lymphocytes, B cells and T cells, differentiate into effector (e.g., antibody-producing B cells and cytotoxic and helper T cells) and memory cells. For both B and T cells in a
typical immune response to an antigen exposure, the latency between the first (primary) exposure and development of the primary response is characterized by a lag phase, logarithmic phase, and plateau phase. The lag phase is characterized by the initial activation of B and T cells upon encounter with the antigen for which they are specific, and this triggers the cells’ differentiation into effector and memory cells. The lag phase between primary exposure to an antigen and the logarithmic phase is classically thought to be 4 to 7 days, but it varies depending on route of exposure and the antigen itself. For B cells, the logarithmic phase is characterized by an increase in serum antibody levels that classically is logarithmic. The plateau phase is characterized by the maintenance of peak antibody levels for a length of time that is followed by a decline in the serum antibody levels. For many antigens the latency (lag phase) between primary exposure and development of the primary antibody response is 7 to 10 days. Due to the development of memory B and T cells during the primary immune response, the latency between subsequent exposure to the antigen and development of the immune response will usually be shorter. The lag phase is generally 1 to 3 days; the logarithmic phase of the secondary antibody response occurs over the next 3 to 5 days. As mentioned for the primary immune response, these time periods will vary depending on the route of exposure, the timing of the subsequent exposure, the antigen itself, and the antigen dose. While this discussion is not specific to a particular antigen, it can be used as a reference point for the latency between antigen exposure and the initiation of some of the immune-mediated mechanisms described below.
Contributing to the activation of B and T cells and the initiation of the adaptive immune response are cells classically associated with the innate immune system (e.g., macrophages and dendritic cells). These cells play roles at each of the stages mentioned above and are usually the first cells of the immune system to be exposed to antigen. Upon antigen encounter, macrophages and dendritic cells engulf the antigen, a process that also activates these innate immune cells to become antigen-presenting cells. Antigen- presenting cells, as their name suggests, present the antigen to T cells (see “Effector Functions of T Cells” below) and release inflammatory mediators (e.g., cytokines and chemokines) that contribute to the recruitment, activation, and proliferation of B and T cells. Activated B and T cells in turn release inflammatory mediators leading to the recruitment and activation of additional immune cells that further amplify the immune response through the release of inflammatory mediators. Regulatory cells and soluble immunoregulatory mediators (not discussed in this report) play roles in suppressing the immune response. Chaplin (2010) provides a review of the immune response including discussion of the interplay between the innate and adaptive arms of the immune system, cells associated with the innate and adaptive immune systems, and inflammatory/immunoregulatory mediators.
Many vaccines, particularly subunit vaccines (e.g., recombinant hepatitis B and tetanus toxoid), contain adjuvants that help to increase the response rates to vaccines and facilitate the use of fewer and smaller doses (Coffman et al., 2010). Currently, two adjuvants (alum as aluminum phosphate or aluminum hydroxide, and ASO4, which is comprised of mono-phosphoryl lipid A and alum) are in vaccines licensed for use in the United States. Although the exact mechanism of action of many adjuvants is not completely understood, it is hypothesized that alum delays systemic absorption of injected antigens, resulting in antigen retention in particulate form and in high concentration at the site of local injection (Tritto et al., 2009). This in turn results in prolonged exposure of the cells of the innate immune system to antigen (Tritto et al., 2009). Furthermore, alum may directly activate cells of the innate immune system through its effect on local inflammasome complexes (Coffman et al., 2010) leading to the release of inflammatory mediators and enhancement of the immune response as described above. The review by Coffman et al. (2010) provides a detailed description of the mechanism(s) of action of clinically approved adjuvants including alum and ASO4.
Several immune-mediated mechanisms have been hypothesized to be involved in the pathogenesis of tissue damage or clinical disease related to natural infection or immunizations. A brief description of some of these mechanisms follows.
Effector Functions of T Cells
T cells are the subset of lymphocytes that develop in the thymus. They are further delineated by the expression of cell surface markers and the production of inflammatory and immunoregulatory mediators. Two T cell subsets, CD8+ and CD4+ T cells, are activated via recognition of peptides derived from antigen. For activation of T cells to occur, the peptides are bound to major histocompatibility complexes (MHCs) expressed on the surface of specialized white blood cells called antigen-presenting cells. T cells have various functions in the immune response.
CD8+ T cells are activated in response to antigens that gain access to the cytosol of cells. These antigens are broken down into peptides. The peptides are presented to CD8+ T cells after being bound to class I MHC molecules. Class I MHC molecules are expressed on nearly all nucleated cells (Harty et al., 2000). CD8+ T cells express a T cell receptor (TCR) that binds peptide-class I MHC complexes. CD8+ T cells that express different TCRs allow for recognition of many different antigens. The binding of
the CD8+ T cell TCR to the peptide-class I MHC complex on professional antigen-presenting cells (e.g., dendritic cells) activates the CD8+ T cells which then respond against cytosolic infections such as viruses, intracytoplasmic bacteria, and protozoa (Harty et al., 2000). Activated CD8+ T cells induce death of infected cells through mechanisms that include (1) release of granules containing the pore-forming molecular perforin or (2) engagement of Fas receptors on target cells (Harty et al., 2000). Both mechanisms induce apoptosis, or programmed cell death, in the target cell. In addition, activated CD8+ T cells secrete cytokines, molecules critical to intercellular communication, that recruit and activate macrophages and neutrophils (Harty et al., 2000).
In contrast to CD8+ T cells, CD4+ T cells are predominantly activated in response to extracellular antigens that are endocytosed or phagocytosed, broken down into peptides, and bound to class II MHC molecules on the surface of professional antigen-presenting cells (Guermonprez et al., 2002). Class II MHC molecules are expressed on dendritic cells, macrophages, B cells, and activated T cells. The CD4+ T cells express TCRs that bind peptide-class II MHC complexes. Recognition of peptide antigen-MHC complexes activates CD4+ T cells against a variety of antigens including, but not limited to, bacteria, parasites, and proteins. Activated CD4+ T cells direct aspects of the immune response via the secretion of immunoregulatory cytokines and other soluble mediators. These inflammatory mediators can induce B cells to undergo immunoglobulin (Ig) class switching (e.g., IgM to IgE); to support the activity of CD8+ T cells; to recruit and activate eosinophils, basophils, neutrophils, mast cells, and macrophages; and to down-regulate immune responses (Koretzky, 2008; Wan and Flavell, 2009). Several lineages of CD4+ T cells, with overlapping and competing effects based on those described above, have been identified (Wan and Flavell, 2009). One CD4+ T cell lineage, referred to as regulatory T cells, functions to maintain self tolerance and immune homeostasis (Wan and Flavell, 2009). In addition, some CD4+ T cells can induce cytolysis via the mechanisms described for CD8+ T cells (Soghoian and Streeck, 2010).
In summary, T cells contribute to the establishment and maintenance of immune responses, the clearance of pathogens, and the maintenance of self-tolerance. T cells play roles in many disease processes including, but not limited to, rheumatoid arthritis, type 1 diabetes, and asthma (Wan and Flavell, 2009).
Effector Functions of Antibodies and Autoantibodies
Antibodies are antigen-binding proteins produced by terminally differentiated effector B cells called plasma cells. Antibodies that bind antigens
derived from the host organism (i.e., self-antigens) are referred to as autoantibodies. Autoantibodies are considered one of the hallmarks of certain autoimmune diseases; however, the presence of autoantibodies does not correlate perfectly with disease. Autoantibodies have been detected in healthy individuals as well as those with autoimmune diseases (Elkon and Casali, 2008; Zelenay et al., 2007). The mechanisms whereby autoantibodies exert their effects in the disease process are the same used by antibodies against foreign antigens (i.e., non-self-antigens). These include, but are not limited to, opsonization, neutralization, complement activation, augmentation, and engagement of constant region (Fc) receptors.
Neutralization of an antigen or pathogen expressing the target antigen is one effector mechanism attributed to antibodies. For example, antibodies against influenza virus hemagglutinin neutralize the virus by blocking the interaction of the virus with the receptor on the target cell, thereby preventing infection (Han and Marasco, 2011). In addition, while not preventing influenza infection, antibodies against influenza neuraminidase restrict replication of the virus by preventing release of virus from infected cells (Han and Marasco, 2011). This is one of the ways vaccines, which induce pathogen-specific antibodies, elicit protection from diseases. However, neutralization of self-antigens by autoantibodies can also contribute to the pathogenesis of some autoimmune diseases. For example, neutralizing auto-antibodies against the cytokine granulocyte/macrophage colony-stimulating factor (GM-CSF) are found in autoimmune pulmonary alveolar proteinosis, which is characterized by dysfunctional alveolar macrophages and functionally impaired neutrophils (Watanabe et al., 2010). Autoantibodies against GM-CSF block interaction of the cytokine with receptors on macrophages, inhibiting their maturation, and on neutrophils, leading to impairment of phagocytosis, adhesion, bacterial killing, and oxidative burst (Watanabe et al., 2010).
Antibodies against surface-bound antigens can lead to the opsonization (coating) of the pathogen or a cell expressing the antigen. For example, antibodies against the capsular polysaccharide of Streptococcus pneumoniae result in the opsonization of the bacteria and clearance of the bacteria by phagocytic cells (Bruyn et al., 1992). In a proinflammatory setting, such as antineutrophil cytoplasmic autoantibody–associated vasculitides, opsonization can lead to the perpetuation of inflammation (van Rossum et al., 2005). For example, opsonization of neutrophils by autoantibodies against proteinase 3 (PR3) and myeloperoxidase (MPO) contributes to the activation of neutrophils resulting in their degranulation, which in turn leads to vessel injury (van Rossum et al., 2005).
Antibody-antigen interactions can lead to complement activation (complement activation is discussed in a subsequent section). Antibodies against
bacteria lead to complement activation resulting in elimination of the bacteria (Bruyn et al., 1992). Similarly, engagement of aquaporin-4, expressed on the surface of astrocytes, by autoantibodies results in complement activation leading to disruption of the integrity of the plasma membrane and astrocyte injury (Cayrol et al., 2009).
Engagement of Fc receptors by antibodies bound to antigen can lead to clearance of the antigen or antigen-expressing pathogen or cell, or to activation of the receptor-expressing cell. The Fc receptors on macrophages, by binding to antibody-coated bacteria, allow the macrophages to engulf and then kill the bacteria. One example, discussed above, is the opsonization of Streptococcus pneumoniae by antibodies against the capsular polysaccharide that leads to the clearance of the bacteria by macrophages (Bruyn et al., 1992). Likewise, the clearance of apoptotic neutrophils opsonized by autoantibodies against PR3 and MPO, as discussed above, is facilitated by engagement of the Fc receptors expressed on the surface of the macrophages (van Rossum et al., 2005). In addition, as described above, opsonization of neutrophils by autoantibodies against PR3 and MPO contributes to the activation of neutrophils. Autoantibodies against PR3 and MPO contribute to neutrophil activation through engagement of Fc receptors by the constant region of the autoantibodies whose variable regions (Fab) are binding either PR3 or MPO on the same cell (van Rossum et al., 2005).
Autoantibodies also have the ability to augment the effects of the target antigen. For example, the autoantibody complex interleukin-8 (IL-8) has been shown to augment IL-8-induced neutrophil migration in acute respiratory distress syndrome (Watanabe et al., 2010). IL-8-induced neutrophil migration is more strongly induced by engagement of Fc receptors by IL-8-autoantibody complexes than by engagement of the IL-8 receptor alone (Watanabe et al., 2010).
As suggested above, autoantibodies use multiple mechanisms during a disease process. Antigen-bound autoantibodies can both (1) engage Fc receptors and (2) induce activation of the complement system. These processes lead to the activation of inflammatory cells such as neutrophils and macrophages, and to generation of proinflammatory mediators that play pathogenic roles in autoimmune diseases.
Complement Activation
The complement system is comprised of more than 30 soluble or membrane-bound proteins. Complement activation, an outcome of a cascade of enzymatic reactions, leads to the generation of inflammatory mediators that play a role in host defense via three physiological processes (Dunkelberger and Song, 2010). First, complement activation leads to the targeted lysis of infectious agents through the generation of the mem-
brane attack complex (MAC), which forms membrane-penetrating pores in pathogens (Dunkelberger and Song, 2010). Second, complement activation leads to the opsonization of infectious agents by complement opsonins and the engagement of complement receptors on phagocytic cells resulting in the clearance of the infectious agent (Dunkelberger and Song, 2010). Lastly, complement activation leads to the generation of proinflammatory anaphylatoxins that act as vasodilators, cytokines, and inducers of smooth muscle contraction; oxidative bursts from neutrophils; and histamine release from mast cells (Sarma and Ward, 2011). In addition to the physiological processes described above, the complement system plays a role in the selection, maintenance, and differentiation of B cells into plasma and memory cells, and in the priming of CD4+ and CD8+ T cells (Dunkelberger and Song, 2010).
Three pathways—classical, lectin, and alternative—lead to complement activation and the generation of inflammatory mediators responsible for the physiological processes discussed above. These pathways converge where C3 convertases cleave the complement component C3 into the anaphylatoxin C3a and the opsonin C3b; from this point, further enzymatic reactions generate additional anaphylatoxins, opsonins, and the MAC (Gros et al., 2008). The pathways are discussed below.
The initiation of the classical pathway occurs when the complement component C1q, in complex with the complement components C1r and C1s, bind immune complexes (comprised of antigen bound by IgG or IgM antibodies) (Rus et al., 2005). C1q can also initiate the classical pathway by binding to C-reactive protein, serum amyloid P, gram-negative bacterial walls, and central nervous system myelin (Rus et al., 2005). Autocatalytic activation of C1r and C1s leads to an enzymatic reaction involving the complement components C4 and C2 and the generation of fragments that combine to form C3 convertase (Dunkelberger and Song, 2010).
The lectin pathway is initiated when pattern recognition receptors (PRRs), such as mannose-binding lectin, bind to highly conserved structures in microorganisms termed pathogen-associated molecular patterns (PAMPs) (Dunkelberger and Song, 2010). PAMPs can be found on the surfaces of yeast, bacteria, parasites, and viruses (Sarma and Ward, 2011). Similar to the classical pathway, recognition of PAMPs by PRRs leads to an enzymatic reaction involving the complement components C4 and C2 and the generation of fragments that combine to form C3 convertase (Dunkelberger and Song, 2010).
Initiation of the alternative pathway occurs when C3 undergoes spontaneous hydrolysis on the surface of pathogens or other targets that have neutral or positive charge characteristics and/or that support the binding of activated C3 (Holers, 2008). The altered form of C3, called C3i or C3(H2O), can bind factor B, which in turn is cleaved by factor D, leading
to the generation of C3 convertase (Holers, 2008). In addition to promoting the generation of the inflammatory mediators discussed above, the alternative pathway increases complement activation through an amplification loop (Holers, 2008). The amplification loop is engaged when C3b, generated by C3 convertase from any of the three complement activation pathways, binds factor B, which in turn is cleaved by factor D, leading to further C3 activation (Holers, 2008). Sites of local injury and decreased expression of complement regulatory proteins can promote engagement of the amplification loop (Holers, 2008).
Hypersensitivity Reactions
Hypersensitivity reactions are immune-mediated reactions to substances, termed allergens, which do not generate adverse immune responses in the majority of the population. Individuals who are “atopic” develop immune responses to the allergens that lead to symptoms such as hay fever or wheezing in response to pollens, or vomiting and lip swelling in response to certain foods. These reactions develop after sensitizing exposure(s) and reexposure to an allergen, and are broadly classified as immediate or delayed hypersensitivity reactions. Described below are two mechanisms classified as immediate hypersensitivity reactions involved in allergic reactions, including the severe, potentially fatal, systemic allergic reactions that are rapid in onset and known as anaphylaxis.
Immunoglobulin E–Mediated Hypersensitivity
Definition of immunoglobulin E–mediated hypersensitivity By far the most common mechanism responsible for immediate hypersensitivity reactions involves immunoglobulin E (IgE) and is termed immunoglobulin E– mediated hypersensitivity, in which allergen-specific IgE antibodies undergo synthesis and binding to high-affinity IgE receptors on the surface of mast cells and basophils. Subsequent exposure of allergen to receptor-bound IgE leads to cross-linking of IgE, activation of mast cells and basophils, and release of inflammatory mediators (Simons, 2009).
Evidence needed to conclude that IgE-mediated hypersensitivity is operative in anaphylaxis Positive skin test results and/or the presence of allergen-specific IgE in serum indicate that a patient is sensitized to an allergen but alone are not conclusive of IgE-mediated reactions or anaphylaxis (Simons, 2009); similarly, negative tests do not conclusively exclude clinical reactivity to an allergen. Testing for mediators of allergic reactivity, such as histamine and tryptase, may be useful in confirming an episode of anaphylaxis (Simons, 2009). However, testing for these mediators is frequently not
available, so physicians must rely on the clinical history, and signs and symptoms of a reaction, to make the diagnosis (Sampson et al., 2006).
Examples of allergen exposures thought to cause IgE-mediated anaphylaxis Many allergens have been associated with the development of IgE-mediated anaphylaxis. These include food (e.g., milk, egg, peanuts, tree nuts, shellfish, gelatin), food additives (e.g., some colorants, spices, yeast), venoms (e.g., insect stings), latex, and inhalants (e.g., animal danders and grass pollen) (Simons, 2010).
Adverse events on our list thought to be due to IgE-mediated hypersensitivity reactions Antigens in the vaccines that the committee is charged with reviewing do not typically elicit an immediate hypersensitivity reaction (e.g., hepatitis B surface antigen, toxoids, gelatin, ovalbumin, casamino acids). However, as will be discussed in subsequent chapters, the above-mentioned antigens do occasionally induce IgE-mediated sensitization in some individuals and subsequent hypersensitivity reactions, including anaphylaxis.
Complement-Mediated Hypersensitivity
Definition of complement-mediated hypersensitivity A much less frequent cause of immediate hypersensitivity is due to complement-mediated hypersensitivity, which involves the activation of the complement pathway by dialysis membranes, for example. Complement activation generates the anaphylatoxins C3a and C5a which bind to complement receptors on the surface of mast cells, leading to the release of inflammatory mediators (Noone and Osguthorpe, 2003).
Evidence needed to conclude that complement-mediated hypersensitivity is operative in anaphylaxis Although the clinical history and signs and symptoms of anaphylaxis are typically used to make the diagnosis of anaphylaxis, measurement of inflammatory mediators such as histamine, tryptase, kallikrein, and bradykinin, in addition to others, may be helpful in confirming an episode of anaphylaxis (Sampson et al., 2006; Simons, 2010). During or shortly after an episode of anaphylaxis, the demonstration of an acute elevation of C3a and C5a (both of which can increase vascular permeability and smooth muscle contraction) is useful in implicating complement-mediated hypersensitivity as the operative mechanism in the anaphylactic episode.
Examples of exposures thought to cause complement-mediated anaphylaxis A small number of substances have been associated with the development of complement-mediated anaphylaxis. These include dialysis
membranes, human proteins (e.g., transfusion or other blood product), immune complexes, and oversulfated chondroitin sulfate-contaminated heparin (Noone and Osguthorpe, 2003; Simons, 2010).
Adverse events on our list thought to be due to complement-mediated hypersensitivity reactions The antigens and potential antigens contained in the vaccines that the committee is charged with reviewing are not commonly associated with complement-mediated anaphylaxis.
Immune Complexes
When present in adequate concentrations, antigen and antibody generate large complexes, termed immune complexes, which can lead to initiation of the inflammatory cascade through complement activation and engagement of Fc receptors, and to increased vascular permeability through the release of vasoactive factors upon activation of mast cells and neutrophils (Gao et al., 2006; Malbec and Daeron, 2007; Mayadas et al., 2009; Roubin and Benveniste, 1985; Volanakis, 1990). In addition, at cold temperatures, in vitro, some antibodies can precipitate from serum; they are called cryobglobulins (Tedeschi et al., 2007). The immune complexes may include IgM rheumatoid factor and antibodies against pathogens (Tedeschi et al., 2007). Immune complexes can cause pathologic damage and disease.
Evidence Needed to Conclude That Immune Complexes Are Operative in a Clinical Case or an Animal Model
The first requirement before attributing a symptom complex to the action of immune complexes is to demonstrate their presence. This can be done in plasma, using assays such as the Raji cell assay or the enzyme-linked immunosorbent assay to detect binding to plate-bound C1q, or to look for immune complexes on red cells that transport the complexes to the liver where they are ingested by Kuppfer cells (Bellamy et al., 1997; Crockard et al., 1991; Kohro-Kawata et al., 2002; Zhong et al., 1997). It is also useful to demonstrate immune complexes in the affected tissue when tissue biopsy is available or needed for diagnostic purposes. Im-munohistology showing co-localization of IgG and early components of the complement cascade serves to demonstrate the presence of immune complexes. To conclude that a particular antigen is responsible for immune complex formation, it is necessary to show that the antigen is present at the site of antibody deposition in tissue, or is within the circulating immune complexes in plasma. It is not necessary to show that the entire antigen is present, because serum and tissue proteases may digest much of the antigen that is not protected within the antibody-binding site (Durkin et al., 2009).
Therefore, negative studies for antigen may be considered inconclusive as only a small moiety of antigen may remain and may not be easily detectable (i.e., antibody to the antigen may be targeted to previously digested portions of the antigen).
Examples of Natural Infection, Vaccine, or Drug Exposure Thought to Cause a Clinical Condition or Disease That Is Due to Immune Complexes
There are several conditions in which immune complex–mediated tissue damage occurs.
- Hepatitis B infection is characterized by a number of accompanying comorbidities. Polyarteritis nodosum occurs in individuals with chronic hepatitis, and is thought to be mediated by immune complexes that include viral antigen and specific antibody (Cacoub and Terrier, 2009).
- Some drug allergies can cause serum sickness which is an immune complex disease with deposition of complexes in joints, pleura or pericardium, and glomeruli causing local, generally reversible, inflammation (Naguwa and Nelson, 1985).
- Systemic lupus is characterized by immune complexes in the circulation, skin, pleura, and pericardium. When the immune complexes are present in glomeruli, they cause glomerulonephritis, a serious manifestation of the disease. The target antigens in lupus appear to be apoptotic debris in circulating immune complexes, and both trapped and tissue antigen in the kidney (Munoz et al., 2010). In lupus, antibodies to the complement component C1q can bind to tissue-bound immune complexes, making it difficult to clear the complexes and increasing the consequent inflammation.
- Rheumatoid arthritis is a disease characterized by antibodies to IgG (rheumatoid factor) and cyclic citrullinated peptide. Both antibodies are thought to enhance inflammation in affected tissue, primarily joints (Conrad et al., 2010; Wegner et al., 2010). In mouse models, antibody-mediated enhancement of rheumatoid arthritis has been demonstrated; in the human disease, the model remains speculative.
- Streptococcal infections exhibit many antibody-mediated sequelae. In particular, arthritis and glomerulonephritis are considered to be the consequence of circulating immune complexes that deposit in joints and glomeruli, initiating an inflammatory cascade (Rodriguez-Iturbe and Batsford, 2007). These conditions are self-limited because the immune complexes cease to form once streptococcal antigen is eliminated.
- In many patients, hepatitis C is characterized by the presence of cryoglobulins that are thought to be rheumatoid factors bound to antibodies to the hepatitis C viral antigen (Sansonno et al., 2007). These cryoglobulins are notoriously difficult to treat, and they cause injury in multiple organs.
Adverse Events on Our List Thought to Be Due to Immune Complexes
It is not clear that this mechanism is operative in any adverse event reported secondary to vaccine administration. It is important to note that the immune complexes and ensuing immune complex-mediated symptoms induced by vaccine usually should be short-lived. As vaccine antigen is eradicated by antibody or catabolism, specific antibody is no longer produced and the inflammatory process subsides. Only live vaccines have the potential for continued long-term production of antigen due to viral replication; antigen from nonreplicating vaccines is likely to disappear within a few weeks. The adverse events most suggestive of immune complex-mediated symptomatology are those associated with hepatitis B vaccine, as it is known that the antibodies raised to viral antigen in the course of the natural infection can form damage-inducing complexes. There are no data, however, documenting immune complexes containing hepatitis B surface antigen.
Tissue Responses (Fever and Seizures)
The mechanisms leading to the development of febrile seizures include the induction of fever by inflammatory mediators and the effects of pyrexia, and of the inflammatory response on neuronal excitability. It is now recognized that febrile seizures have significant genetic susceptibility components.
Induction of Fever by Inflammatory Mediators
Fever is a biologic response to a host of extrinsic and intrinsic pyrogenic stimuli (Avner, 2009). Extrinsic pyrogenic stimuli include bacterial toxins and other products of microbial metabolism (e.g., lipopolysaccharide released from the cell wall of gramnegative bacteria). Intrinsic pyrogenic stimuli include antigen-antibody complexes and activated components of the complement system, either of which may result from a microbial infection or immunization with microbial antigens.
These pyrogenic stimuli induce monocytes, macrophages, and other inflammatory cells to release pyrogenic cytokines (e.g., IL-1 alpha, IL-1 beta, TNF, interferon) into the circulation. Acting either directly or indirectly on the specialized neurons of the thermoregulatory center in the preoptic area
of the hypothalamus, these cytokines induce the production of E-series prostaglandins that raise the host’s thermoregulatory set point, resulting in an increase in core body temperature.
Effects of Pyrexia and the Inflammatory Response on Neuronal Excitability
The specific mechanism whereby fever might induce a seizure is not known. It is known that changes in temperature can alter certain ion channels in the brain and potentially cause abnormal or synchronized neuronal discharges and seizures (Shibasaki et al., 2007; Thomas et al., 2009). Moreover, fever-induced hyperventilation and the resulting alkalosis may also play a role in seizure induction (Schuchmann et al., 2006). Furthermore, animal data are emerging on the role of the inflammatory response in astroglial cells after a prolonged febrile seizure. It has been shown that IL-1 beta synthesis is induced after a febrile seizure, and that it has potent proconvulsant effects in both immature and adult rodents (Dube et al., 2010). The role inflammatory mediators play in epileptogenesis is not fully understood and is an area of intense research interest. Recently, Balosso et al. (2008) showed that IL-1 beta is overexpressed in glia and neurons in animal models with seizures. This inflammatory mechanism has a proconvulsant effect, and may influence changes in neuronal excitability (Balosso et al., 2008). Fever-induced factors (e.g., IL-1 beta) may precipitate seizures in the immature brain or in individuals who are genetically susceptible (Heida et al., 2009; Nakayama, 2009).
Genetics and Febrile Seizures
Febrile seizures are the result of a combination of genetic and environmental factors, with polygenic inheritance the most common means of inheritance. Epidemiologic studies have shown that 13–37 percent of children with febrile seizures have a family history of febrile seizures and 4 percent have a family history of epilepsy (Offringa et al., 1994; Sadleir and Scheffer, 2007). In monozygotic twins, the concordance rate is higher (Corey et al., 1991; Tsuboi, 1987).
Although specific susceptibility genes have not been identified in most patients with febrile seizures, several susceptibility loci that are inherited in an autosomal dominant fashion in certain families have been identified (Audenaert et al., 2006; Hedera et al., 2006; Johnson et al., 1998; Kugler et al., 1998; Nabbout et al., 2002; Nakayama et al., 2000, 2002, 2004; Peiffer et al., 1999; Poduri et al., 2009; Wallace et al., 1996). Other genetic factors have been implicated as a link between fever and susceptibility to seizures. Mutations in sodium channels (e.g., splice site variant SCN1A) and gamma aminobutyric acid A receptor genes have been identified in
children with febrile seizures (Petrovski et al., 2009; Sadleir and Scheffer, 2007; Schlachter et al., 2009).
Molecular Mimicry
Molecular mimicry is sequence and/or conformational homology between an exogenous agent (foreign antigen) and self-antigen leading to the development of tissue damage and clinical disease from antibodies and T cells directed initially against the exogenous agent that also react against self-antigen. Molecular mimicry as a mechanism that can cause pathologic damage and disease has been demonstrated in several animal models, most notably experimental allergic encephalomyelitis (EAE) in mice and rabbits (Oldstone, 2005).
Evidence Needed to Conclude That Molecular Mimicry Is Operative in a Clinical Case or an Animal Model of Disease
Essential to concluding molecular mimicry contributes to a clinical case or animal model of disease are the following: (1) a susceptible host whose genetic background and adaptive immune responses allows emergence of self-reactive immunity, (2) exposure to an exogenous agent which expresses antigens that are immunologically similar to self-antigen(s), and (3) a host immune response to the exogenous agent that cross-reacts with biologically relevant host tissue structures and causes tissue damage and clinical disease.
Proving that a particular human autoimmune disease is due to molecular mimicry is problematic (Albert and Inman, 1999; Rose and Mackay, 2000). A realistic and consistent temporal relationship between exposure to exogenous antigen and development of disease must be documented. This can be difficult in the case of a natural exposure to pathogen where infection may have been subclinical, making it impossible to define an exact temporal relationship.
Linear amino acid sequence homology or even similar conformational structure between an exogenous agent and a self-antigen alone are not sufficient to prove that molecular mimicry is the pathogenic mechanism for a disease. Many such homologies exist, and the vast majority of these are not associated with biologically relevant autoimmune phenomena or actual human disease (Albert and Inman, 1999).
Finding a tissue-specific antibody response following exposure to an exogenous agent is also, by itself, not proof of molecular mimicry as the pathologic mechanism of disease (Albert and Inman, 1999). Both naturally occurring and postinfectious cross-reactive antibodies and T cells are relatively common and most frequently not pathogenic (Fujinami et al., 2006). Cross-reacting antibodies can also be secondary to nonspecific tissue injury
(and to consequent expression of otherwise occult self-antigens) rather than primary to tissue injury itself. Moreover, in some circumstances, infection with viruses that express antigens having immunologic cross-reactivity with self-proteins can actually protect against autoimmune disease in certain animal models (Barnett et al., 1996; Fujinami et al., 2006).
Neither the in vitro demonstration of cross-reacting antibodies nor T cell activation by antigen-MHC complexes proves pathogenic mimicry. An in vivo pathogenic autoimmune attack would also require the demonstration of local binding of antibody with activation of the complement cascade, activation of the appropriate co-stimulatory T cell signals and cytokines, and/or involvement of other pathogenic effector mechanisms in a biologically relevant tissue site.
Examples of a Natural Infection, Vaccine, or Drug Exposure Thought to Cause a Clinical Condition or Disease That Is Due to Molecular Mimicry
While molecular mimicry is a well-established mechanism in selected animal models, its relevance to human autoimmune disease remains in most cases to be convincingly proven. Nevertheless, there is some experimental evidence (Albert and Inman, 1999; Fujinami et al., 2006; Rose and Mackay, 2000) that suggests or implicates this mechanism in certain human autoimmune diseases including (among others)
- Rheumatic fever associated with group A streptococcal infection;
- HLA B27-associated spondyloarthropathies and several antigens from Shigella, Yersinia, and Klebsiella bacteria;
- Multiple sclerosis and exposure to several different viruses;
- Insulin-dependent diabetes mellitus and Coxsackievirus B4; and
- Demyelinating diseases and hepatitis B:
- Amino acid homology between myelin basic protein (MBP) and hepatitis B virus polymerase (HBVP) has been reported (Fujinami and Oldstone, 1985). In addition, injection of a HBVP immunologic epitope shared with MBP into rabbits resulted in an EAE-like disease, antibodies against MBP, and T cell reactivity (Fujinami and Oldstone, 1985). However, infection with hepatitis B is not associated with the development of demyelinating diseases. Furthermore, the recombinant vaccines contain hepatitis B surface antigen not hepatitis B virus polymerase.
One example of molecular mimicry as the likely mechanism causing clinical autoimmune disease is found in the subtype of Guillain-Barré syndrome (GBS) characterized by acute motor axonal neuropathy (AMAN).
Approximately one fourth of patients with GBS have had Campylobacter jejuni infection in the preceding few weeks, compared to only 1–2 percent of controls (Kuwabara et al., 2004; Rees et al., 1995b). C. jejuni infection is most highly correlated with AMAN, as opposed to the other subtypes of GBS (Griffin et al., 1996; Visser et al., 1995).
In patients who develop AMAN subsequent to C. jejuni enteritis, IgG autoantibodies and complement are found bound specifically to GM1 ganglioside in the axolemma membrane of peripheral nerves (Hafer-Macko et al., 1996; Solomon and Willison, 2003; Willison and Yuki, 2002). These patients often benefit from plasmapheresis, and their anti-GM1 autoantibody titers decrease as the clinical course improves (The Guillain-Barré Syndrome Study Group, 1985). By contrast, patients who develop C. jejuni enteritis not complicated by AMAN do not develop GM1 autoantibodies (Ogawara et al., 2000; Rees et al., 1995a).
GM1 ganglioside antigens in peripheral nerves are structurally identical to the terminal tetrasaccharides of the GM1-like lipo-oligosaccharide (LOS) structures expressed on the surface of certain strains of C. jejuni bacteria isolated from patients with AMAN (Aspinall et al., 1994a,b; Lee et al., 2004; Prendergast et al., 1998; Yuki et al., 1993, 2004). This suggests that autoantibodies bound to neuronal gangliosides in AMAN may result from immunologic cross-reactivity with antigens from C. jejuni lipo-oligosaccharides.
In the 1990s ganglioside preparations extracted from bovine brain tissue or isolated GM1 were occasionally used as therapeutic agents for various neurological disorders and some of these patients developed clinical AMAN with anti-GM1 IgG autoantibodies (Illa et al., 1995).
The association of C. jejuni infection leading to anti-GM1 autoantibody production and the cross-reactivity of those antibodies with nerve roots and clinical disease in vivo is further strengthened by development of a relevant animal model of the disease. Rabbits immunized with C. jejuni expressing GM1-like LOS surface structures develop high titers of anti-GM1 IgG antibody, flaccid limb weakness, and histopathologic features characteristic of AMAN (including IgG deposited on the axons of the ventral roots, internodal axolemmas, and nodes of Ranvier) (Moran et al., 2005; Susuki et al., 2003, 2004; Yuki, 2005; Yuki et al., 2004).
Adverse Events (AEs) on Our List Thought to Be Due to Molecular Mimicry
Some of the vaccine AEs under consideration by our committee share symptoms with human autoimmune diseases for which molecular mimicry has been hypothesized (i.e., arthritis, systemic lupus erythematosus, insulin-dependent diabetes mellitus, central and peripheral nervous system demyelinating diseases). However, the committee found little clinical evidence
(e.g., challenge/rechallenge), diagnostic evidence (e.g., presence of antigen or relevant immune complexes in affected tissue), or experimental evidence (e.g., in vitro evidence of cross-reactive T cells derived from a site of tissue injury) that could be consistent with the hypothesis of molecular mimicry in rare and selected case reports. For example, as will be discussed in subsequent chapters in more detail, Poirriez (2004) reported the absorption of antinuclear antibodies (ANAs), isolated from a single hepatitis B immunized patient who developed lupus, by highly concentrated vaccine antigen suggesting mimicry between vaccine antigen and self-antigen. There were no unimmunized ANA-positive or other controls tested in this study, and others have not reported this finding subsequently. Based on the literature reviewed, molecular mimicry was not confirmed to be a mechanism leading to the development of the adverse events postvaccination.
Antigen Persistence
During a typical immune response, the offending antigen is effectively removed or neutralized, which reduces the immune stimulation and ultimately results in a down-regulation of the immune response. In contrast, ongoing immune responses to persisting antigens can result in continuing inflammation and tissue damage, which may result in the release of self-peptide, and/or activation of previously tolerant autoreactive T cells. The duration of antigen persistence depends on several variables: (1) whether the antigen or antigenic determinants that activate the immune system are derived from a replicating pathogen or are from a transient or intermittently present non-replicating source; (2) the life cycle of the pathogen, assuming it is the source of the antigen or antigenic determinants; and (3) the anatomical and cellular (intracellular or extracellular) location of the antigen source.
Examples of Natural Infection, Vaccine, or Drug Exposure Thought to Cause a Clinical Condition or Disease That Is Due to Antigen Persistence
The best understood reason for antigen persistence is pathogen replication. In many infections, the amount of pathogen-derived antigens often increases initially before decreasing as the pathogen is fully cleared by the immune system. In immunocompromised individuals, whether due to primary (genetic) or secondary (acquired; e.g., via chemotherapy) etiologies, it is possible that pathogens, and therefore pathogen-derived antigens, persist longer or achieve higher levels than they would in immunocompetent individuals. Regardless of the cause, the consequences of a reduced or delayed ability to eliminate a pathogen often, but not always, include more severe pathology at the target tissue or longer duration of illness. Some individuals may fail to eradicate the pathogen from their body.
Two other causes of antigen persistence are pathogen reactivation and persistent infection. Some pathogens persist within host cells without evoking immune responses, reemerging at a later time or creating a depot of antigen that may be released slowly over time. The mechanisms that control persistence, latency, and reactivation are the subject of active research at this time.
Examples of antigen persistence secondary to persistent infection or viral reactivation include hepatitis B and varicella zoster virus, which are discussed later under those topics. A third example of infectious disease associated with antigen persistence is immune reconstitution inflammatory syndrome which is an escalating immune response to chronically persisting antigen in patients with human immunodeficiency virus (HIV) who are co-infected with mycobacteria, cytomegalovirus, Cryptococcus, herpes simplex virus, and so on. Symptoms of inflammatory disease develop after patients begin highly active antiretroviral therapy, which allows reconstitution of the patient’s T cell function and subsequent immune reaction to the co-infecting agent.
Evidence Needed to Conclude That Antigen Persistence Is Operative in a Clinical Case or Animal Model of Viral Infection
Vaccine-derived antigens persist longer when the vaccine is a live, attenuated virus. The vaccine virus, as an intact pathogen, is thought to persist in the host for several weeks, which is in contrast to the more transient presence of split product, recombinant, or killed whole vaccines, which persist for a much shorter period of time. For the discussion of antigen persistence herein, please refer to the persistent viral infection and viral reactivation sections beginning on page 77. In a handful of cases, there was experimental or clinical evidence (e.g., in vitro evidence of cross-reactive T cells derived from a site of tissue injury) that is consistent with the hypothesis of antigen persistence. Based on the literature reviewed, antigen persistence is a possible mechanism leading to the development of a handful of adverse events postvaccination, but only for live virus vaccines.
Epitope Spreading
Epitope spreading is a process in which a T cell response that is initially specific for one epitope spreads to unrelated epitopes. The initial immune response, such as a CD4 T cell response, is directed to one antigen. Chronic tissue destruction from the initial immune response results in production of additional epitopes that become targets for the immune response (Vanderlugt et al., 1998). The new targets are distinct from the original targets (Vanderlugt et al., 1998). Epitope spreading may result from
target antigens that complex with intracellular self-antigen. The result of this could be an autoimmune response that is initially triggered by the exogenous antigen, but then progresses to a sustained autoimmune response against self-antigen (Davies, 2000).
Autoreactivity/Bystander Activation/Hyperresponsiveness
Autoreactivity can result from expression and immune recognition of self-antigens that have been modified by some extrinsic factor (e.g., binding of a reactive chemical or viral element) so that they appear foreign to the immune system. The response to such neoantigens would cease when the transforming agent is removed. Examples include drug modifications of normal proteins, hapten-carrier complexes, and oxidative modification of normal cellular constituents.
In bystander activation, there is a robust or exaggerated immune response to an exogenous agent that induces local tissue inflammation and stimulation of otherwise normal unaffected cells. This inflammation can result in the release of normally sequestered self-antigens. The inflammation can result in nonspecific activation of previously dormant autoreactive Th1 cells that then react against the newly released self-antigens.
Increased Cytokines
Cytokines are a group of molecules involved in intercellular communication. They are classed together as lymphokines, interleukins, and chemokines, based on their function, cell origin, and target of action. When the innate immune system, the adaptive immune system, or both are responding to a pathogen, cytokines activate immune cells to produce even more cytokines and alter function of resident cells in tissues.
The cytokine milieu contributes to the differentiation of T cells to one or another subset. Excessive differentiation of T cells to one or another subset may impair the homeostatic and regulatory mechanisms that limit autoreactivity.
Examples of Increased Cytokines
Normally, the control of cytokine secretion is kept in check by regulatory mechanisms. However, in some instances, the regulatory mechanisms break down and too many immune cells (including those of both the innate immune system and the adaptive immune system) are activated, resulting in local tissue and organ damage, and systemic symptoms. This kind of profound systemic oversecretion of cytokines is called cytokine storm. It may follow infection or other types of massive immune activation includ-
ing bacterial sepsis, avian influenza, acute respiratory distress syndrome, hemophagocytic lymphohistiocytosis, and macrophage activation syndrome (Sriskandan and Altmann, 2008; Szyper-Kravitz, 2009; Wong and Yuen, 2006; Yokoyama et al., 2010). Increased levels of proinflammatory cytokines, or decreased secretion of anti-inflammatory cytokines, are found in the active phase of many of the above-mentioned conditions.
Although the committee is not aware of reports of full-blown cytokine storm following administration of any of the vaccines reviewed, more subtle imbalances of proinflammatory and anti-inflammatory cytokines may occur following immunization against rubella, human papillomavirus, or hepatitis B (Albarran et al., 2005; Garcia-Pineres et al., 2007; Pukhalsky et al., 2003). Moreover, it is possible that the unique immunogenetic makeup of an individual might predispose that individual to an exaggerated cytokine imbalance following immune stimulation such as microbial infection or vaccine administration.
Adverse Events on Our List Thought to Be Due to Increased Cytokines
In review of the relevant literature related to the vaccine and AE combinations considered by the committee, no evidence that directly or indirectly supports the oversecretion of cytokines as an operative mechanism was found.
Superantigens
Superantigens are determinants expressed by a microbe that can bypass T cell receptor signaling pathways and directly activate large numbers of T cells. An example would be TSS-A/B toxins in Staphylococcal and Streptococcal toxic shock syndromes. Superantigens can activate up to 20 percent of circulating T cells (Li et al., 1999). The committee found no evidence that supports superantigen stimulation of immune reactions as an operative mechanism in any of the vaccine adverse events under consideration.
Viral infections cause a host of symptoms in affected individuals. Some of these symptoms are attributable to direct or primary infection, persistent viral infection, and viral reactivation.
Direct or Primary Infection
Primary infection with varicella, for example, results in varicella (chickenpox), manifesting as fever, malaise, listlessness, and a rash consist-
ing of vesicles, scabs, and maculopapules in varying stages of evolution (Whitley, 2010). Complications include secondary skin infections, myocarditis, nephritis, pneumonia, central nervous system involvement (acute cerebellar ataxia, encephalitis), and bleeding diatheses (Whitley, 2010).
Similarly, “[t]ransient polyarthralgia and polyarthritis [from rubella] rarely occur in children but are common in adolescents and adults, especially females. Encephalitis (1:5,000 cases) and thrombocytopenia (1:3,000 cases) are complications” (AAP, 2009c).
The acute complications of measles infection include otitis media, croup, and pneumonia (Gershon, 2010). Approximately 1 of every 1,000 individuals infected with measles virus develops acute encephalitits (AAP, 2009b). Furthermore, “[d]eath, predominantly resulting from respiratory and neurologic complications, occurs in 1 to 3 of every 1,000 cases reported in the United States” (AAP, 2009b).
Persistent Viral Infection
Some viruses are capable of causing permanent, latent infection in nearly all individuals, the herpesviruses and retroviruses being the best-known examples. Reactivation, as discussed below, with production of new virus can occur with such latent viruses.
With other viruses, some infected individuals are unable to clear the viral infection. The classic example of persistent infection is hepatitis B virus (HBV). “More than 90 percent of infants infected perinatally will develop chronic HBV infection” (AAP, 2009a). Between 10 percent and 20 percent of children infected between 1 and 5 years of age become chronically infected, whereas approximately 5 percent of acutely infected adults develop chronic HBV infections (Koziel and Thio, 2010). Immunosuppressed patients or patients with an underlying chronic illness who develop an acute HBV infection are at an increased risk of developing a chronic infection (AAP, 2009a). It is important to note here, however, that the hepatitis B vaccine is not a live virus vaccine and so cannot infect recipients.
Viral Reactivation
Reactivation of infection can occur when the virus, following the acute infection, remains in a dormant or latent state somewhere in the body, where it can subsequently reemerge. Varicella zoster virus (VZV) establishes latency in the dorsal root ganglia, cranial nerve ganglia, and enteric ganglia during primary infection (Gershon et al., 2008). Reactivation results in herpes zoster (shingles), characterized by a unilateral eruption of vesicles with a dermatomal distribution, sometimes accompanied by pain localized to the area (Whitley, 2010).
Viral Activity Attributed to Vaccines Containing Live Attenuated Viruses
Attenuated live viral vaccines such as the ones considered in this report (live attenuated influenza virus, cold-adapted influenza virus, VZV, MMR) can cause some of these same effects through the same mechanisms because the vaccines are live. As detailed further below, these effects occur most frequently in patients with impaired immunity. Varicella vaccine virus, which is distinct from the natural varicella virus, for example, has been recovered from the bronchoalveolar lavage fluid and lung biopsy of immuno-compromised children who developed pneumonia and rash as a primary infection after receiving a varicella vaccine (Galea et al., 2008; Kramer et al., 2001; Levy et al., 2003; Sharrar et al., 2001; Waters et al., 2007). As examples of viral reactivation, children who had previously been vaccinated developed zoster and even encephalitis from which vaccine-strain virus was then recovered (Chan et al., 2007; Chouliaras et al., 2010; Iyer et al., 2009; Levin et al., 2003). Some, but not all, of these children were subsequently shown to be immunocompromised. The salient points in these examples are that the adverse effects observed are complications seen with natural infection and that the causal role of the vaccine virus was demonstrated by its isolation or identification by molecular techniques, typically from sites that are otherwise sterile.
INJECTION-RELATED ADVERSE EVENTS
One or more of the mechanisms described above could play a role in the development of many of the adverse events following vaccination reviewed by the committee. However, mechanisms leading to three adverse events (complex regional pain syndrome, frozen shoulder, and syncope) were considered by the committee to be a potential adverse event of direct trauma from the needle occurring with various injected vaccines and not necessarily attributable to the contents of the vaccine. Mechanisms for these adverse events are described below.
Complex Regional Pain Syndrome
Chronic severe and often burning pain affecting part or all of one or more extremities following an injury defines complex regional pain syndrome (CRPS). The pain is often accompanied by skin discoloration, local edema, fluctuation in skin temperature in the affected limb(s), allodynia (pain from stimuli that would not ordinarily be painful), and abnormal sweating (Bruehl, 2010). Mechanisms involving altered skin innervation, dysfunction of the sympathetic nervous system, local inflammation, and possible psychological factors have been purported to play a role in the development of CRPS.
Altered Skin Innervation
The cascade of events leading to CRPS is widely considered to result from nerve trauma. Needle stick injuries to the distal nerves in rats resulted in reduction in sensory neuron density similar to findings in CRPS patients (Bruehl, 2010). In addition, lower densities of epidermal nerves and abnormal innervation around sweat glands and hair follicles have been reported in CRPS patients (Bruehl, 2010).
Dysfunction of the Sympathetic Nervous System
Skin discoloration and fluctuation in skin temperature in the affected region suggests the involvement of the sympathetic nervous system in CRPS (Bruehl, 2010). In some CRPS patients, increased sympathetic nervous system activity is associated with increases in spontaneous pain and hyperalgesia (Bruehl, 2010). In addition, the expression of adrenergic receptors on pain fibers after trauma (in animal studies) provides a mechanism whereby sympathetic nervous system outflow could trigger a pain signal (Bruehl, 2010).
Inflammation
Improvement of symptoms in CRPS patients treated with corticosteroids suggests inflammation as a contributing factor in the development of the acute phase of CRPS (Bruehl, 2010). Nerve injury could induce the release of proinflammatory cytokines and neuropeptides from nociceptive fibers (Bruehl, 2010). Increased levels of proinflammatory cytokines have been isolated from the serum, cerebrospinal fluid, and blister fluid of CRPS patients (Bruehl, 2010). Proinflammatory cytokines can contribute to the increased plasma loss, leading to localized edema (Bruehl, 2010). In addition, certain major histocompatibility complexes have been reported to be expressed at significantly higher frequencies in CRPS patients, further supporting inflammation as a mechanism (Bruehl, 2010).
Psychological Factors
Initially CRPS, due to its poorly understood pathophysiology and unusual symptomatology, was thought to be purely psychogenic (Bruehl, 2010). While a purely psychogenic model is no longer considered, psychogenic factors could play a role in the development of CRPS. Greater CRPS pain intensity was predicted by increased depression levels in a patient self-study (Bruehl, 2010). In addition, psychological stress in CRPS patients has been associated with altered immune function (Bruehl, 2010). Psychological factors could impact all of the implicated mechanisms.
Comprehensive Mechanism
While the mechanisms purported to contribute to the development of CRPS are studied and discussed in isolation, it has been hypothesized that the mechanisms are interconnected (Bruehl, 2010). Studies testing this hypothesis have yet to be performed.
Deltoid Bursitis
Idiopathic- or injury-induced pain, stiffness, and restricted motion of the shoulder defines deltoid bursitis. The presentation of deltoid bursitis is comprised of shoulder pain and stiffness with restricted motion (Anton, 1993). Pathologic examination has revealed an inflammatory process in the affected shoulder. Increased deposition of growth factors, matrix metalloproteinases, and cytokines has been observed in capsular biopsy specimens from patients with deltoid bursitis (Brue et al., 2007; Dias et al., 2005). In addition, magnetic resonance imaging and arthrography have revealed abnormalities of the shoulder joint and synovial membrane, and thickening of the humeral ligament and joint capsule suggestive of inflammation (Brue et al., 2007). Although likely to be a rare event, direct trauma to the bursa from needle injury from the injected vaccine, independent of the contents of the needle, could lead to the activation and recruitment of inflammatory cells leading to the symptoms of deltoid bursitis.
Syncope
Loss of consciousness resulting from decreased blood flow to the brain is termed syncope. The pathogenesis of syncope varies depending on the precipitating event. Syncope resulting from pain or emotional triggers, for example the sight of blood or administration of a vaccine or treatment via an injection, is termed reflex syncope and more specifically vasovagal syncope (van Dijk et al., 2009). The pathophysiology of vasovagal syncope has not been fully delineated; however, manipulation of the blood flow by the autonomic nervous system is involved. The injection of the vaccine leads to an initial increase in stimulation of the sympathetic nervous system (Arthur and Kaye, 2000). The increase in stimulation of the sympathetic nervous system results in an increased heart rate and arterial pressure (Grubb, 2005). The increased arterial pressure leads to the activation of baroreceptors and transmission of afferent signals from the aortic arch via the vagus nerve resulting in stimulation of the parasympathetic nervous system and the development of nausea, vertigo, facial pallor, dizziness, and epigastric discomfort commonly experienced 30 to 60 seconds prior to the loss of consciousness (Fenton et al., 2000; Wieling et al., 2009). Physiologically, the
stimulation of the parasympathetic nervous system results in a decreased heart rate and arterial pressure leading to decreased blood flow to the brain and the loss of consciousness (Grubb, 2005).
COAGULATION AND HYPERCOAGULABLE STATES
Injury to the vessel wall, regardless of the type of injury, leads to the stimulation or activation of endothelial cells and platelets, and to the generation of a thrombus or blood clot. In response to injury, both cell types increase expression of the adhesion molecule P-selectin on the cell surface (Green, 2006). Through interaction with P-selectin, neutrophils, monocytes, and platelets form a thrombus at the site of injury (Green, 2006). The interaction of additional proteins secreted from the injured endothelial cells and platelets enhance platelet-to-platelet aggregation, leading to the formation of platelet-leukocyte aggregates that are favorable to fibrin formation (Green, 2006).
The generation of fibrin results from a cascade of enzymatic reactions initiated upon injury to the vessel wall. A key component of this cascade is tissue factor (TF), which exists in a soluble form and as a transmembrane protein (Shantsila and Lip, 2009). TF is activated upon vessel wall injury and exposure to the subendothelial tissues to blood (Shantsila and Lip, 2009). Monocytes are a major source of TF and can stimulate TF expression by endothelial cells, thus increasing the supply of tissues expressing the factor (Shantsila and Lip, 2009).
The aforementioned cascade is initiated upon the formation of complexes comprised of circulating factor VII and TF, leading to the activation of factor VII and the generation of factor VIIa (Sidhu and Soff, 2009). TF-factor VIIa complexes continue the cascade, culminating in the generation of the serine protease thrombin (Green, 2006). Thrombin activates integrins (these mediate platelet aggregation and other factors of the coagulation cascade), and it further activates platelets leading to the production of platelet activators (Sidhu and Soff, 2009). In addition, thrombin cleaves fibrinogen to produce fibrin monomers (Sidhu and Soff, 2009).
Monocytes, in addition to producing TF, contribute to prothrombotic effects via other mechanisms. Conjugation of monocytes with platelets induces the expression of integrins on monocytes, amplifying their interactions with platelets (Shantsila and Lip, 2009). During inflammation, stimulation of monocytes by T cells induces the expression of matrix metalloproteinases 1 and 3, which are elements of plaque destabilization (Shantsila and Lip, 2009). Monocytes can activate coagulation factor X, which is responsible for the generation of thrombin (Shantsila and Lip, 2009).
A few proteins facilitate regulation of the coagulation cascade. Protein C, which circulates in the plasma, is activated by the serine protease throm-
bin and its cofactor thrombin-thrombomodulin (Rezaie, 2010). Activated protein C functions as an anticoagulant by proteolytically degrading procoagulant cofactors essential for the generation of thrombin (Rezaie, 2010). The cofactor protein S enchances effects of activated protein C (Anderson and Weitz, 2010). In addition, the serine protease inhibitor antithrombin regulates the coagulation cascade by inactivating thrombin as well as other enzymes in the cascade (Rodgers, 2009).
In individuals with inherited (e.g., antithrombin deficiency, Factor V Leiden) or acquired (e.g., obesity, pregnancy) hypercoagulable states, the function of the enzymes involved in the aforementioned coagulation cascade and its regulation are altered or deficient, leading to excessive coagulability (Anderson and Weitz, 2010). Excessive coagulation can contribute to the development of thrombosis, myocardial infarction, and stroke (Anderson and Weitz, 2010).
Both epidemiologic and mechanistic research suggest that most individuals who experience an adverse reaction to vaccines have a preexisting susceptibility. These predispositions can exist for a number of reasons— genetic variants (in human or microbiome DNA), environmental exposures, behaviors, intervening illness, or developmental stage, to name just a few— all of which can interact as suggested graphically in Figure 3-1.
Some of these adverse reactions are specific to the particular vaccine, while others may not be. Some of these predispositions may be detectable prior to the administration of vaccine; others, at least with current technology and practice, are not. Moreover, the occurrence of the adverse event is often the first sign of the underlying condition that confers susceptibility.
The best-understood vaccine-associated adverse effect is the occurrence of invasive disease (such as meningoencephalitis and arthritis) caused by the vaccine virus itself in individuals with an acquired or genetic immunodeficiency who receive live vaccines such as VZV, MMR, and oral polio vaccine. Although the incidence of such infections may decrease with the introduction of newborn screening for severe combined immunodeficiency, the occurrence of vaccine-related disease can be the trigger that leads to the recognition of immunodeficiency (Galea et al., 2008; Ghaffar et al., 2000; Kramer et al., 2001; Levy et al., 2003). Invasive disease may also occur by viral reactivation in individuals who previously received these vaccines while healthy, but who subsequently become immunocompromised, for example, as a result of chemotherapy should they later develop cancer or leukemia (Chan et al., 2007; Levin et al., 2003). Not all individuals who suffer invasive disease have demonstrated recognized immune deficien-cies, even when vaccine virus is recovered from the patient (Iyer et al.,
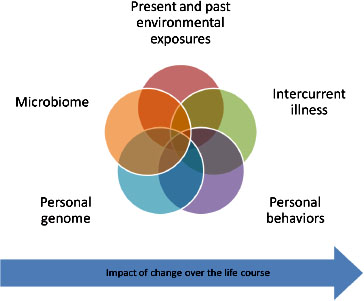
FIGURE 3-1 Present and past environmental exposures.
2009; Levin et al., 2008). This leads to two hypotheses: either immunocompetent individuals can acquire invasive disease from vaccine virus, or further evaluation of these patients would reveal previously unrecognized immunodeficiencies.
Many adverse events appear to be immune-mediated. Anaphylaxis is an obvious example of this. In some patients who experience anaphylaxis, the triggering antigen can be identified with follow-up testing. Known triggering antigens include egg and gelatin. But even when the triggering antigen such as egg or gelatin is known, it is not clear why some people develop anaphylaxis while the vast majority does not. Proposed mechanisms for other immune-mediated adverse responses are many, including molecular mimicry, development of immune complexes, inappropriate cytokine responses, antigen persistence, and epitope spreading, as described above. Here, evidence of predisposing factors to adverse effects from vaccines is beginning to emerge. Some genetic variants that affect immune response have been identified. Reif et al. (2009) demonstrated that genetic variants in ICAM-1, CSF-3, and IL-4 are associated with more severe adverse effects
from the highly reactogenic vaccine for smallpox. Finally, rechallenge cases (those in which a person suffered a particular adverse event after each administration of the same vaccine) also suggest a role for an altered immune response. As noted above, much work remains to be done to elucidate and to develop strategies to document the immunologic mechanisms that lead to adverse effects in individual patients.
Age can also affect susceptibility to adverse responses to vaccines because physiological development, particularly of the immune and nervous systems, continues throughout much or all of life. Some hypothesize so-called critical periods in which adverse reactions to a range of exposures are more likely to occur (IOM, 2006). Young children are more likely than are older children to develop febrile convulsions (Waruiru and Appleton, 2004). This type of rationale led the Japanese three decades ago to delay immunization with whole cell pertussis vaccine until children reached 2 years of age (Gangarosa et al., 1998). Gender can also be a factor. Females, for example, experience less local reactogenicity than males to smallpox vaccine (Talbot et al., 2004) but increased reactogenicity compared to males to anthrax vaccine (Pittman, 2002).
In some metabolically vulnerable children, receiving vaccines may be the largely nonspecific “last straw” that leads these children to reveal their underlying genotype. It was recently discovered that a large majority of children who developed encephalopathy after receiving whole cell pertussis vaccine have mutations in SCN1A, which are associated with Dravet syndrome or severe myoclonic epilepsy of childhood (Berkovic et al., 2006; McIntosh et al., 2010). While it seems likely that the vaccine triggered symptoms in these children by causing high fever, the particular vaccine antigens do not appear to alter the course of the disease. Rather, the ensuing phenotype could and probably would have been precipitated by multiple other fever-inducing triggers (McIntosh et al., 2010; Wiznitzer, 2010). Similarly, Yang et al. (2006) reported a series of seven cases in which children with undiagnosed or inadequately managed metabolic or endocrine disorders suffered acute metabolic crises within hours after administration of a variety of immunizations. Two of these children had adrenal hyperplasia and responded to administration of IV fluid and gluco-and mineralocorticoids.
This list of factors that are known to confer susceptibility is by no means definitive or exhaustive. Rather, we hypothesize that continued study of alleged vaccine-related injuries, the committee informed by epidemiologic studies that identify vulnerable populations and exploration of underlying mechanisms of susceptibility, will provide greater insight into these and other mechanisms and will identify more factors that contribute to vaccine susceptibility.
ALTERATIONS IN BRAIN DEVELOPMENT
The committee was specifically tasked to assess the evidence that vaccines could alter neuronal development, resulting in “secondary autism” or “autistic features” arising from chronic encephalopathy, mitochondrial disorders, or other underlying disorders.1 Some theorize that vaccines can alter the development of the nervous system through inflammatory responses or hyperarousal of the immune system. Most certainly, scientific advances have shown commonalities in the development of and the signaling between the immune and nervous systems.
Development of the human central nervous system is incompletely understood, but certain principles are well established. Development occurs in a predictable sequence, and the earlier in the sequence, the more reliably certain events can be timed. For example, closure of the neural tube is always complete before 28 days gestation. Nutritional factors such as folic acid deficiency or exposure to toxins such as valproic acid during this “critical period” predictably produce neural tube defects.
Nervous system development is under genetic control, and is incompletely understood, but it is clearly a highly complex process in which interactions with the environment beginning in the womb may modify the developmental process. Factors that may modify brain development include maternal, fetal, and infant nutrition; infection; toxins; vascular insults; direct trauma; and aspects of the social environment, in addition to mutations in critical genes regulating development.
Development of the nervous system involves formation of the neural plate and tube, followed by proliferation of neuronal precursors, which must then migrate to their final positions in the nervous system where they establish functional connections with other neurons and glial cells. The neuronal elements ultimately interact to form functional neural circuits. Essentially all of the nerve fiber tracts comprising these circuits are present at birth, but they are not functionally active because the rate of conduction in unmyelinated axons is slow. The process of myelination of fiber tracts occurs in an orderly sequence, most dramatically during the first few years of life, but continues on into the fourth decade. When a nerve fiber (axon) is ensheathed by myelin, the rate of impulse conduction accelerates dramatically, allowing neural circuits to become functionally active. In addition, synapses (the connections that form neural circuits) continue to form at a variable rate that peaks in various parts of the brain at different, but predictable times. These neural circuits exhibit plasticity, and they underlie much of human behavior. The essential stimuli for neural development
___________
1 The list of adverse events the committee was asked to consider by the National Vaccine Injury Compensation Program can be found at: http://www.iom.edu/~/media/Files/Activity%20Files/Research/VaccineAdvEffectReview/Working-List-of-AEs-January-10.pdf.
need not be physical; social and emotional deprivation are wellrecognized causes of impaired development.
It is apparent that interruption of circuits at many different or distinct points may produce similar phenotypes. The mechanisms could be structural, involving improper development or injury to axons, nerve bodies, or dendrites, or they could be functional, implying abnormalities of the neu-rotransmitters or their receptors through which neurons communicate with one another, or implying lack of appropriate stimulation of the otherwise normal circuits. The processes underlying such disruption may be genetic or acquired.
It is important to bear in mind that genetic disorders need not be expressed at birth. Gene expression is regulated throughout life and many genes are expressed selectively only at certain times in specific tissues. Certain developmental sequences appear to be more or less rigidly encoded by the genome, whereas others are more plastic and amenable to environmental influences. These variables are all relevant when considering patterns of both normal and abnormal brain development.
Animal models have been most helpful in understanding disease processes affecting the brain, particularly when these are expressed as structural or motor changes, or as seizures. Advances in molecular genetics have allowed genes to be knocked out completely, temporarily knocked down, or to create milder phenotypes (hypomorphs) by point mutations. Various manipulations of gene function have led to a better understanding of complex gene-gene and genotype-phenotype interactions. Transgenic models, usually generated in mice, permit the study of human gene function, albeit in a different species. However, no animal embodies the repertoire of behaviors seen in the human, and in particular, no animal has language equivalent to that of the human. Although certain behaviors in animals have been compared to human phenotypes, the analogies are always imperfect and may be misleading.
Autism
The terms autism, autism spectrum disorder, and pervasive developmental disorder not otherwise specified embrace a diverse group of children with a common neurobehavioral phenotype, and the first term (autism) will be used to embrace all of these entities in the following discussion. The child psychiatrist Leo Kanner first coined this term in 1943; since that time, varying diagnostic criteria and concepts of autism have been proposed and accepted, and they continue to evolve. Currently, the Diagnostic and Statistical Manual of Psychological Disorders, fourth edition, text revision (DSM-IV-TR) defines the criteria most widely used to diagnose autism and autism spectrum disorders. The criteria require that children
show impairments in three domains: language, social interactions, and restricted interests or repetitive behaviors. Key features include the onset of the phenotype before the third year of life. In about one-third of cases, children who previously appeared to have been developing normally show evidence of regression. However, most of these children likely had not had prior expert evaluation. In the remaining majority of cases, development was never assessed as normal.
Autism is a complex behavioral phenotype, whose neuropathological underpinnings are beginning to be understood. Several lines of evidence, including functional and structural imaging studies (Anagnostou and Taylor, 2011) and neuropathology have pointed to abnormal patterns of neural connectivity as characteristic of autism spectrum disorders (Schipul et al., 2011; Wass, 2011). The autism phenotype can be defined by a trained clinical evaluator using a variety of instruments (Dover and Le Couteur, 2007), particularly the autism diagnostic observational schedule (ADOS) (Lord et al., 1989) and autism diagnostic index–revised, which are widely accepted as the standard for research studies. These instruments have been employed in many, but by no means all, studies of this syndrome. The specialized training required to administer ADOS testing is not universally available. The use of variable diagnostic criteria is a major challenge to interpretation of the burgeoning autism literature. This is particularly pertinent when considering longitudinal trends, since differing criteria have been employed over time. Changes in diagnostic criteria, accompanied by increased social acceptance of this diagnosis, have paralleled marked increases in the number of children receiving this diagnostic label in recent years. It is also important to recognize that autism is frequently accompanied by comorbidities, such as abdominal symptoms, sleep disorders, and seizures, mood disorders, and aggressive disorders.
Genetic variation accounts for many cases of autism; specific genes or genetic loci may be identified in up to 25 percent of patients with autism spectrum disorders (Eapen, 2011; Miles, 2011). Siblings of children with autism have a much higher rate of the disorder, with the highest rate seen in identical twins (Ronald and Hoekstra, 2011). Family members of children with autism have been found to have variants of expressive language suggesting some innate neurologic variant. Several single-gene disorders are associated with autism, including tuberous sclerosis complex, FMR-1 (fragile X), dystrophinopathies, phenylketonuria, Rett disorder (MECP2 mutations), Down syndrome, and oxidative phosphorylation defects (Miles, 2011). The last mentioned, often referred to as mitochondrial diseases, are highly variable multisystem disorders whose complex phenotypes often encompass the autism spectrum (Frye and Rossignol, 2011). In other cases, linkage has been established with genes known to be crucial in modulating neural connectivity, such as neuroligins and neurexins (Sudhof, 2008). It
also appears that the developing brain, particularly early in pregnancy, is subject to environmental insults, including valproic acid and maternal rubella infection, which can result in autism and other developmental disabilities in the offspring (Landrigan, 2010). Such in utero exposures may act by altering the expression of genes regulating development of the nervous system (Dufour-Rainfray et al., 2011). These exposures are less likely to cause autism if experienced later in pregnancy, thus supporting the concept of windows of vulnerability. Maternal antibodies against fetal brain proteins may also be implicated in some cases, raising questions about the possible role of other immune factors such as cytokines (Goines and Van de Water, 2010). There is a growing literature describing inflammatory changes in the autopsied brain in at least a portion of patients with autistic disorders (Pardo et al., 2005), although many of these signs of inflammation are also increased in many other neurodegenerative disorders. The etiology of most cases of autism spectrum disorders is still not understood.
Because the timing of diagnosis or recognition of autism coincides with the administration of many vaccines, questions have been raised regarding potential etiologic relationship(s) between the two. There are several challenges in interpreting existing data. Establishing a temporal relationship between a potential inciting event (such as vaccine administration) and the onset of autism is difficult because dating the onset of the syndrome in most cases is imprecise (although there is a subset of children with acute regression from reportedly normal development). Rechallenge data are not available, since most children do not rapidly (if ever) recover a normal developmental pattern following the onset of their symptoms.
Establishing a mechanistic link is also challenging because it is not understood how known causes of autism lead to this phenotype. Several murine models of genetic disorders have autistic features, and although such models can never reproduce the complete human phenotype, they have added further evidence that disruption of the function of genes participating in brain development may lead to autism spectrum disorders (Ey et al., 2011). Infection of neonatal Lewis rats with Bornavirus (Hornig et al., 2001) has produced a behavioral phenotype with features equated to human autism. One fact of note is that postnatal infections with the vaccine-targeted infectious agents, including measles, mumps, and rubella, are not known to cause autism, although autistic features have been reported in children with congenital rubella syndrome (Chess, 1971); one study reported the use of mathematical modeling and epidemiological data to conclude that MMR immunization had been associated with prevention of substantial numbers of cases of congenital rubella syndrome and associated autism in the period 2001–2010 in the United States (Berger et al., 2011). There are reports of autistic syndromes acquired in children with acute encephalopathic illnesses. DeLong et al. (1981) described three such children, one of
whom had evidence of herpes simplex infection. The two children in whom the etiology of the episode was not discovered made complete recoveries. Additional reports described an autistic syndrome following herpes simplex encephalitis in a 14-year-old girl (Gillberg, 1986) and an 11-year-old boy (Ghaziuddin et al., 2002). The preceding cases were atypical in that the age of onset of autism was between 5 and 14 years; two children with perinatal herpes simplex encephalitis experienced the onset of autism in early childhood (Ghaziuddin et al., 1992). Another series of 14 children with autism included three whose onset of symptoms closely followed episodes of malaria. However, given that malaria is common in Tanzania, where the series originated, this should not necessarily be regarded as evidence of a mechanistic relationship (Mankoski et al., 2006). A single report described a 9-year-old boy who exhibited changes of late-onset autism associated with anti-N-methyl-D-aspartate receptor antibody positive encephalitis; he recovered with monoclonal antibody therapy (Creten et al., 2011).
The foregoing literature suggests that infectious or inflammatory etiologies may underly some cases of autism, although most of the cases described do not meet current diagnostic criteria for autistic disorder, owing to their late onset. Other studies have implicated dysfunction of the innate immune system in the genesis of some cases of autism. Vargas et al. (2005) described a unique pattern of inflammatory changes in brain tissue obtained at autopsy and in cerebrospinal fluid from living patients (Zimmerman et al., 2005) with established diagnoses of autism, using suitable controls and DSM-IV criteria. Herbert (2005) has suggested that the large brains often reported in children with autism in early life could be explained by inflammatory expansion of the white matter that could also contribute to abnormal central nervous system connectivity. The evidence supporting the concept of autism and a neuroimmune disorder has been reviewed recently (Theoharides et al., 2009).
At a minimum, prior to ascribing autism to vaccination, it would be important to rule out known associations with this phenotype. These include both macroscopic and microscopic structural abnormalities of the brain (Casanova, 2007), particularly minocolumnar architecture (Casanova and Trippe, 2009) as well as specific chromosomal and single-gene defects, including a variety of metabolic disorders and inflammatory or infectious antecedants.
Laboratory animals have been studied for decades as a means to understand both normal physiology and pathogenesis of diseases. Throughout this time, it has become apparent that animal models can be very useful, or alternatively noninformative, depending on the question being addressed.
Infections
When an infectious organism invades and replicates within a non-human host, there are likely to be many similarities between the human and nonhuman host. In particular, antibody responses appear to be quite similar, often targeting the same antigenic epitopes of the infectious agent. Likewise, tissue and cellular antigenicity is often similar, so the pathogenic or protective potential of human antibodies can be ascertained in animal models. Cellular mechanisms of microbial control and eradication are also often similar, as are mechanisms of microbial evasion of host defenses.
However, multiple differences in response to microbial infection between humans and laboratory animals exist. One major issue in animal models of infection is that not all infectious organisms will infect all hosts; this is particularly an issue for viruses and other intracellular pathogens whenever cellular entry is generally achieved through a particular receptor that may be present in one species and not another. A second issue affecting the response to microbes is the expression of histocompatibility molecules in the infected animal. Histocompatibility molecules vary among species and among individuals of that species; they are expressed on almost all tissues and immune cells. If the histocompatiblity molecules can bind micro-bial antigens, an enhanced immune response to that microbe can develop. Laboratory studies usually focus on a particular strain of mouse or rat and therefore do not mimic the genetic diversity of the human population, either with respect to immune molecules such as histocompatibility molecules or with respect to other aspects of cellular metabolism.
Any observations made in an animal model of infection need to be confirmed in the human host because of the differences between man and animal models discussed above. Nonetheless, animal studies provide the opportunity to sample all body tissue and to ascertain the extent of microbial invasion and the cellular targets with likely correspondence to the human host. Studies of the immune response to microbial agents include vaccine studies that are also subject to the concerns discussed above. One frequent difference between vaccine studies in animals and humans is that vaccine studies in laboratory animals may include adjuvants that are not used in humans.
Immune Response in a Host with a Preexisting Disease
It is well established that individuals with abnormalities in immune function, either genetic or acquired, respond differently to microbial infection and to vaccines compared to the responses of healthy individuals. Several rodent models exist with genetically derived immunodeficiencies or autoimmunity. Some of these models mimic the human condition either
with respect to genetic lesions or with respect to mechanisms and/or phenotypes of disease. It is possible, therefore, to ask whether the genetic lesion or the ensuing disease process renders the host more (or less) susceptible to a particular antigenic challenge. Such studies can provide important information, which must be confirmed in humans. Again, an advantage of an animal model is that one can explore all tissues in the body, including those that are inaccessible to study in living humans.
Relevance to Adverse Events Following Vaccination
There are multiple uses of animal models in vaccine studies. It is possible to study each tissue of the body for microbial invasion and microbe-induced or immune-mediated damage. Fukuda et al. (1994) determined in a hamster model of measles that the measles virus can replicate in the labyrinth, providing a potential explanation of the deafness that occurs with measles infection and providing a biologic mechanism for deafness following vaccination with attenuated measles vaccine. The techniques used to show replication of measles virus in the labyrinth represent an advantage of animal models, as discussed above; techniques to show replication of measles virus in the labyrinth will not be performed on living human patients.
With animal models, it is possible to study whether particular genetic deficiencies or preexisting conditions attenuate, augment, or alter the immune response to infectious agents or microbial antigen, or whether the microbial or antigenic challenge exacerbates the preexisting condition or reveals otherwise unappreciated consequences of the genetic deficiency.
It is possible to look for molecular mimicry between vaccine antigen and self-antigen, although mimicry at the antibody level is more likely to translate to the human situation than molecular mimicry at the T cell level due to the diversity of histocompatibility molecules. Should molecular mimicry be found in an animal model, it still needs confirmation in humans.
AAP (American Academy of Pediatrics). 2009a. Hepatitis B. In Red book: 2009 Report of the Committee on Infectious Diseases, 28th ed., edited by L. K. Pickering, C. J. Baker, D. W. Kimberlin, and S. S. Long. Elk Grove Village, IL: American Academy of Pediatrics. Pp. 337-356.
AAP. 2009b. Measles. In Red book: 2009 Report of the Committee on Infectious Diseases, 28th ed., edited by L. K. Pickering, C. J. Baker, D. W. Kimberlin, and S. S. Long. Elk Grove Village, IL: American Academy of Pediatrics. Pp. 444-455.
AAP. 2009c. Rubella. In Red book: 2009 Report of the Committee on Infectious Diseases, 28th ed., edited by L. K. Pickering, C. J. Baker, D. W. Kimberlin, and S. S. Long. Elk Grove Village, IL: American Academy of Pediatrics. Pp. 579-584.
Albarran, B., L. Goncalves, S. Salmen, L. Borges, H. Fields, A. Soyano, H. Montes, and L. Berrueta. 2005. Profiles of NK, NKT cell activation and cytokine production following vaccination against hepatitis B. APMIS 113(7-8):526-535.
Albert, L. J., and R. D. Inman. 1999. Molecular mimicry and autoimmunity. New England Journal of Medicine 341(27):2068-2074.
Anagnostou, E., and M. J. Taylor. 2011. Review of neuroimaging in autism spectrum disorders: What have we learned and where we go from here. Molecular Autism 2(1):4.
Anderson, J. A., and J. I. Weitz. 2010. Hypercoagulable states. Clinics in Chest Medicine 31(4):659-673.
Anton, H. A. 1993. Frozen shoulder. Canadian Family Physician 39:1773-1778.
Arthur, W., and G. C. Kaye. 2000. The pathophysiology of common causes of syncope. Postgraduate Medical Journal 76(902):750-753.
Aspinall, G. O., S. Fujimoto, A. G. McDonald, H. Pang, L. A. Kurjanczyk, and J. L. Penner. 1994a. Lipopolysaccharides from Campylobacter jejuni associated with Guillain-Barré syndrome patients mimic human gangliosides in structure. Infection and Immunity 62(5):2122-2125.
Aspinall, G. O., A. G. McDonald, H. Pang, L. A. Kurjanczyk, and J. L. Penner. 1994b. Lipo polysaccharides of Campylobacter jejuni serotype O:19: Structures of core oligosaccharide regions from the serostrain and two bacterial isolates from patients with the Guillain-Barré syndrome. Biochemistry 33(1):241-249.
Audenaert, D., C. Van Broeckhoven, and P. De Jonghe. 2006. Genes and loci involved in febrile seizures and related epilepsy syndromes. Human Mutation 27(5):391-401.
Avner, J. R. 2009. Acute fever. Pediatrics in Review 30(1):5-13.
Balosso, S., M. Maroso, M. Sanchez-Alavez, T. Ravizza, A. Frasca, T. Bartfai, and A. Vezzani. 2008. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain 131(Pt 12):3256-3265.
Barnett, L. A., J. L. Whitton, L. Y. Wang, and R. S. Fujinami. 1996. Virus encoding an encephalitogenic peptide protects mice from experimental allergic encephalomyelitis. Journal of Neuroimmunology 64(2):163-173.
Bellamy, J. D., D. J. Booker, N. T. James, R. Stamps, and R. J. Sokol. 1997. Measurement of red blood cell-bound C3b and C3d using an enzyme-linked direct antiglobulin test. Immunohematology 13(4):123-131.
Berger, B. E., A. M. Navar-Boggan, and S. B. Omer. 2011. Congenital rubella syndrome and autism spectrum disorder prevented by rubella vaccination—United States, 2001-2010. BMC Public Health 11:340.
Berkovic, S. F., L. Harkin, J. M. McMahon, J. T. Pelekanos, S. M. Zuberi, E. C. Wirrell, D. S. Gill, X. Iona, J. C. Mulley, and I. E. Scheffer. 2006. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: A retrospective study. Lancet Neurology 5(6):488-492.
Brue, S., A. Valentin, M. Forssblad, S. Werner, C. Mikkelsen, and G. Cerulli. 2007. Idiopathic adhesive capsulitis of the shoulder: A review. Knee Surgery, Sports Traumatology, Arthroscopy 15(8):1048-1054.
Bruehl, S. 2010. An update on the pathophysiology of complex regional pain syndrome. Anesthesiology 113(3):713-725.
Bruyn, G. A., B. J. Zegers, and R. van Furth. 1992. Mechanisms of host defense against infection with streptococcus pneumoniae. Clinical Infectious Diseases 14(1):251-262.
Cacoub, P., and B. Terrier. 2009. Hepatitis B-related autoimmune manifestations. Rheumatic Diseases Clinics of North America 35(1):125-137.
Casanova, M., and J. Trippe. 2009. Radial cytoarchitecture and patterns of cortical connectivity in autism. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 364(1522):1433-1436.
Casanova, M. F. 2007. The neuropathology of autism. Brain Pathology 17(4):422-433.
Cayrol, R., P. Saikali, and T. Vincent. 2009. Effector functions of antiaquaporin-4 autoantibodies in neuromyelitis optica. Annals of the New York Academy of Sciences 1173:478-486.
Chan, Y., D. Smith, T. Sadlon, J. X. Scott, and P. N. Goldwater. 2007. Herpes zoster due to Oka vaccine strain of varicella zoster virus in an immunosuppressed child post cord blood transplant. Journal of Paediatrics & Child Health 43(10):713-715.
Chaplin, D. D. 2010. Overview of the immune response. Journal of Allergy and Clinical Immunology 125(2 Suppl. 2):S3-S23.
Chess, S. 1971. Autism in children with congenital rubella. Journal of Autism and Childhood Schizophrenia 1(1):33-47.
Chouliaras, G., V. Spoulou, M. Quinlivan, J. Breuer, and M. Theodoridou. 2010. Vaccine-associated herpes zoster ophthalmicus [correction of opthalmicus] and encephalitis in an immunocompetent child. Pediatrics 125(4):e969-e972.
Coffman, R. L., A. Sher, and R. A. Seder. 2010. Vaccine adjuvants: Putting innate immunity to work. Immunity 33(4):492-503.
Conrad, K., D. Roggenbuck, D. Reinhold, and T. Dorner. 2010. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmunity Reviews 9(6):431-435.
Corey, L. A., K. Berg, J. M. Pellock, M. H. Solaas, W. E. Nance, and R. J. DeLorenzo. 1991. The occurrence of epilepsy and febrile seizures in Virginian and Norwegian twins. Neurology 41(9):1433-1436.
Creten, C., S. van der Zwaan, R. J. Blankespoor, A. Maatkamp, J. Nicolai, J. van Os, and J. N. Schieveld. 2011. Late onset autism and anti-NMDA-receptor encephalitis. 378(9785):98.
Crockard, A. D., J. M. Thompson, M. B. Finch, T. A. McNeill, A. L. Bell, and S. D. Roberts. 1991. Immunoglobulin isotype composition of circulating and intra-articular immune complexes in patients with inflammatory joint disease. Rheumatology International 11(4-5):169-174.
Davies, J. M. 2000. Introduction: Epitope mimicry as a component cause of autoimmune disease. Cellular and Molecular Life Sciences 57(4):523-526.
DeLong, G. R., S. C. Bean, and F. R. Brown, 3rd. 1981. Acquired reversible autistic syndrome in acute encephalopathic illness in children. Archives of Neurology 38(3):191-194.
Dias, R., S. Cutts, and S. Massoud. 2005. Frozen shoulder. British Medical Journal 331(7530): 1453-1456.
Dover, C. J., and A. Le Couteur. 2007. How to diagnose autism. Archives of Disease in Childhood 92(6):540-545.
Dube, C. M., T. Ravizza, M. Hamamura, Q. Zha, A. Keebaugh, K. Fok, A. L. Andres, O. Nalcioglu, A. Obenaus, A. Vezzani, and T. Z. Baram. 2010. Epileptogenesis provoked by prolonged experimental febrile seizures: Mechanisms and biomarkers. Journal of Neuroscience 30(22):7484-7494.
Dufour-Rainfray, D., P. Vourc’h, S. Tourlet, D. Guilloteau, S. Chalon, and C. R. Andres. 2011. Fetal exposure to teratogens: Evidence of genes involved in autism. Neuroscience and Biobehavioral Reviews 35(5):1254-1265.
Dunkelberger, J. R., and W. C. Song. 2010. Complement and its role in innate and adaptive immune responses. Cell Research 20(1):34-50.
Durkin, M., L. Estok, D. Hospenthal, N. Crum-Cianflone, S. Swartzentruber, E. Hackett, and L. J. Wheat. 2009. Detection of coccidioides antigenemia following dissociation of immune complexes. Clinical and Vaccine Immunology 16(10):1453-1456.
Eapen, V. 2011. Genetic basis of autism: Is there a way forward? Current Opinion in Psychiatry 24(3):226-236.
Elkon, K., and P. Casali. 2008. Nature and functions of autoantibodies. Nature Clinical Practice Rheumatology 4(9):491-498.
Ey, E., C. S. Leblond, and T. Bourgeron. 2011. Behavioral profiles of mouse models for autism spectrum disorders. Autism Research 4(1):5-16.
Fenton, A. M., S. C. Hammill, R. F. Rea, P. A. Low, and W. K. Shen. 2000. Vasovagal syncope. Annals of Internal Medicine 133(9):714-725.
Frye, R. E., and D. A. Rossignol. 2011. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatric Research 69(5 Pt 2):41R-47R.
Fujinami, R. S., and M. B. Oldstone. 1985. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 230(4729):1043-1045.
Fujinami, R. S., M. G. von Herrath, U. Christen, and J. L. Whitton. 2006. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clinical Microbiology Reviews 19(1):80-94.
Fukuda, S., K. Ishikawa, and Y. Inuyama. 1994. Acute measles infection in the hamster cochlea. Acta Oto-Laryngologica 514:111-116.
Galea, S. A., A. Sweet, P. Beninger, S. P. Steinberg, P. S. LaRussa, A. A. Gershon, and R. G. Sharrar. 2008. The safety profile of varicella vaccine: A 10-year review. Journal of Infectious Diseases 197(Suppl. 2):S165-S169.
Gangarosa, E. J., A. M. Galazka, C. R. Wolfe, L. M. Phillips, R. E. Gangarosa, E. Miller, and R. T. Chen. 1998. Impact of anti-vaccine movements on pertussis control: The untold story. Lancet 351(9099):356-361.
Gao, H., T. Neff, and P. A. Ward. 2006. Regulation of lung inflammation in the model of IgG immune-complex injury. Annual Review of Pathology 1:215-242.
Garcia-Pineres, A., A. Hildesheim, L. Dodd, T. J. Kemp, M. Williams, C. Harro, D. R. Lowy, J. T. Schiller, and L. A. Pinto. 2007. Cytokine and chemokine profiles following vaccination with human papillomavirus type 16 L1 virus-like particles. Clinical and Vaccine Immunology 14(8):984-989.
Gershon, A. A. 2010. Measles virus (rubeola). In Mandell, Douglas, and Bennett’s principles and practice of infectious diseases. 7th ed. 2 vols. Vol. 2, edited by G. L. Mandell, J. E. Bennett, and R. Dolin. Philadelphia, PA: Churchill Livingstone Elsevier. Pp. 2229-2236.
Gershon, A. A., J. Chen, and M. D. Gershon. 2008. A model of lytic, latent, and reactivating varicella-zoster virus infections in isolated enteric neurons. Journal of Infectious Diseases 197(Suppl. 2):S61-S65.
Ghaffar, F., K. Carrick, B. B. Rogers, L. R. Margraf, K. Krisher, and O. Ramilo. 2000. Disseminated infection with varicella-zoster virus vaccine strain presenting as hepatitis in a child with adenosine deaminase deficiency. Pediatric Infectious Disease Journal 19(8):764-766.
Ghaziuddin, M., I. Al-Khouri, and N. Ghaziuddin. 2002. Autistic symptoms following herpes encephalitis. European Child and Adolescent Psychiatry 11(3):142-146.
Ghaziuddin, M., L. Y. Tsai, L. Eilers, and N. Ghaziuddin. 1992. Brief report: Autism and herpes simplex encephalitis. Journal of Autism and Developmental Disorders 22(1):107-113.
Gillberg, C. 1986. Onset at age 14 of a typical autistic syndrome. A case report of a girl with herpes simplex encephalitis. Journal of Autism and Developmental Disorders 16(3): 369-375.
Goines, P., and J. Van de Water. 2010. The immune system’s role in the biology of autism. Current Opinion in Neurology 23(2):111-117.
Green, D. 2006. Coagulation cascade. Hemodialysis International 10(Suppl. 2):S2-S4.
Griffin, J. W., C. Y. Li, T. W. Ho, M. Tian, C. Y. Gao, P. Xue, B. Mishu, D. R. Cornblath, C. Macko, G. M. McKhann, and A. K. Asbury. 1996. Pathology of the motor-sensory axonal Guillain-Barré syndrome. Annals of Neurology 39(1):17-28.
Gros, P., F. J. Milder, and B. J. Janssen. 2008. Complement driven by conformational changes. Nature Reviews Immunology 8(1):48-58.
Grubb, B. P. 2005. Neurocardiogenic syncope. In Syncope: Mechanisms and management. 2nd ed., edited by B. P. Grubb and B. Olshansky. Malden, MA: Blackwell Publishing. Pp. 47-71.
Guermonprez, P., J. Valladeau, L. Zitvogel, C. Thery, and S. Amigorena. 2002. Antigen presentation and T cell stimulation by dendritic cells. Annual Review of Immunology 20:621-667.
The Guillain-Barré Syndrome Study Group. 1985. Plasmapheresis and acute Guillain-Barré syndrome. Neurology 35(8):1096-1104.
Hafer-Macko, C., S. T. Hsieh, C. Y. Li, T. W. Ho, K. Sheikh, D. R. Cornblath, G. M. McKhann, A. K. Asbury, and J. W. Griffin. 1996. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Annals of Neurology 40(4):635-644.
Han, T., and W. A. Marasco. 2011. Structural basis of influenza virus neutralization. Annals of the New York Academy of Sciences 1217:178-190.
Harty, J. T., A. R. Tvinnereim, and D. W. White. 2000. CD8+ T cell effector mechanisms in resistance to infection. Annual Review of Immunology 18:275-308.
Hedera, P., S. Ma, M. A. Blair, K. A. Taylor, A. Hamati, Y. Bradford, B. Abou-Khalil, and J. L. Haines. 2006. Identification of a novel locus for febrile seizures and epilepsy on chromosome 21q22. Epilepsia 47(10):1622-1628.
Heida, J. G., S. L. Moshe, and Q. J. Pittman. 2009. The role of interleukin-1 beta in febrile seizures. Brain and Development 31(5):388-393.
Herbert, M. R. 2005. Large brains in autism: The challenge of pervasive abnormality. The Neuroscientist 11(5):417-440.
Holers, V. M. 2008. The spectrum of complement alternative pathway-mediated diseases. Immunological Reviews 223:300-316.
Hornig, M., M. Solbrig, N. Horscroft, H. Weissenbock, and W. I. Lipkin. 2001. Borna disease virus infection of adult and neonatal rats: Models for neuropsychiatric disease. Current Topics in Microbiology and Immunology 253:157-177.
Illa, I., N. Ortiz, E. Gallard, C. Juarez, J. M. Grau, and M. C. Dalakas. 1995. Acute axonal Guillain-Barré syndrome with IgG antibodies against motor axons following parenteral gangliosides. Annals of Neurology 38(2):218-224.
IOM (Institute of Medicine). 2006. Genes, behavior, and the social environment: Moving beyond the nature/nurture debate. Washington, DC: The National Academies Press.
Iyer, S., M. K. Mittal, and R. L. Hodinka. 2009. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Annals of Emergency Medicine 53(6):792-795.
Johnson, E. W., J. Dubovsky, S. S. Rich, C. A. O’Donovan, H. T. Orr, V. E. Anderson, A. Gil-Nagel, P. Ahmann, C. G. Dokken, D. T. Schneider, and J. L. Weber. 1998. Evidence for a novel gene for familial febrile convulsions, FEB2, linked to chromosome 19p in an extended family from the midwest. Human Molecular Genetics 7(1):63-67.
Kohro-Kawata, J., M. H. Wener, and M. Mannik. 2002. The effect of high salt concentration on detection of serum immune complexes and autoantibodies to C1q in patients with systemic lupus erythematosus. Journal of Rheumatology 29(1):84-89.
Koretzky, G. A. 2008. T lymphocyte signaling mechanisms and activation. In Fundamental immunology. 6th ed., edited by W. E. Paul. Philadelphia, PA: Lippincott Williams & Wilkins. Pp. 347-375.
Koziel, M. J., and C. L. Thio. 2010. Hepatitis B virus and hepatitis delta virus. In Mandell, Douglas, and Bennett’s principles and practice of infectious diseases. 7th ed. 2 vols. Vol. 2, edited by G. L. Mandell, J. E. Bennett, and R. Dolin. Philadelphia, PA: Churchill Livingstone Elsevier. Pp. 2059-2086.
Kramer, J. M., P. LaRussa, W. C. Tsai, P. Carney, S. M. Leber, S. Gahagan, S. Steinberg, and R. A. Blackwood. 2001. Disseminated vaccine strain varicella as the acquired immunodeficiency syndrome-defining illness in a previously undiagnosed child. Pediatrics 108(2):e39.
Kugler, S. L., E. S. Stenroos, D. E. Mandelbaum, T. Lehner, V. V. McKoy, T. Prossick, J. Sasvari, K. Swannick, J. Katz, and W. G. Johnson. 1998. Hereditary febrile seizures: Phenotype and evidence for a chromosome 19p locus. American Journal of Medical Genetics 79(5):354-361.
Kuwabara, S., K. Ogawara, S. Misawa, M. Koga, M. Mori, A. Hiraga, T. Kanesaka, T. Hattori, and N. Yuki. 2004. Does Campylobacter jejuni infection elicit “demyelinating” Guillain-Barré syndrome? Neurology 63(3):529-533.
Landrigan, P. J. 2010. What causes autism? Exploring the environmental contribution. Current Opinion in Pediatrics 22(2):219-225.
Lee, G., Y. Jeong, I. Wirguin, A. P. Hays, H. J. Willison, and N. Latov. 2004. Induction of human IgM and IgG anti-GM1 antibodies in transgenic mice in response to lipopolysaccharides from Campylobacter jejuni. Journal of Neuroimmunology 146(1-2):63-75.
Levin, M. J., K. M. Dahl, A. Weinberg, R. Giller, A. Patel, and P. R. Krause. 2003. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus, in an immunosuppressed child. Journal of Infectious Diseases 188(7):954-959.
Levin, M. J., R. L. DeBiasi, V. Bostik, and D. S. Schmid. 2008. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella-zoster virus vaccine. Journal of Infectious Diseases 198(10):1444-1447.
Levy, O., J. S. Orange, P. Hibberd, S. Steinberg, P. LaRussa, A. Weinberg, S. B. Wilson, A. Shaulov, G. Fleisher, R. S. Geha, F. A. Bonilla, and M. Exley. 2003. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. Journal of Infectious Diseases 188(7):948-953.
Li, H., A. Llera, E. L. Malchiodi, and R. A. Mariuzza. 1999. The structural basis of T cell activation by superantigens. Annual Review of Immunology 17:435-466.
Lord, C., M. Rutter, S. Goode, J. Heemsbergen, H. Jordan, L. Mawhood, and E. Schopler. 1989. Autism diagnostic observation schedule: A standardized observation of communicative and social behavior. Journal of Autism and Developmental Disorders 19(2): 185-212.
Malbec, O., and M. Daeron. 2007. The mast cell IgG receptors and their roles in tissue inflammation. Immunological Reviews 217:206-221.
Mankoski, R. E., M. Collins, N. K. Ndosi, E. H. Mgalla, V. V. Sarwatt, and S. E. Folstein. 2006. Etiologies of autism in a case-series from Tanzania. Journal of Autism and Developmental Disorders 36(8):1039-1051.
Mayadas, T. N., G. C. Tsokos, and N. Tsuboi. 2009. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation 120(20):2012-2024.
McIntosh, A. M., J. McMahon, L. M. Dibbens, X. Iona, J. C. Mulley, I. E. Scheffer, and S. F. Berkovic. 2010. Effects of vaccination on onset and outcome of Dravet syndrome: A retrospective study. Lancet Neurology 9(6):592-598.
Miles, J. H. 2011. Autism spectrum disorders—a genetics review. Genetics in Medicine 13(4):278-294.
Moran, A. P., H. Annuk, and M. M. Prendergast. 2005. Antibodies induced by ganglioside-mimicking Campylobacter jejuni lipooligosaccharides recognise epitopes at the nodes of Ranvier. Journal of Neuroimmunology 165(1-2):179-185.
Munoz, L. E., C. Janko, C. Schulze, C. Schorn, K. Sarter, G. Schett, and M. Herrmann. 2010. Autoimmunity and chronic inflammation—two clearance-related steps in the etiopathogenesis of SLE. Autoimmunity Reviews 10(1):38-42.
Nabbout, R., J. F. Prud’homme, A. Herman, J. Feingold, A. Brice, O. Dulac, and E. LeGuern. 2002. A locus for simple pure febrile seizures maps to chromosome 6q22-q24. Brain 125(Pt 12):2668-2680.
Naguwa, S. M., and B. L. Nelson. 1985. Human serum sickness. Clinical Reviews in Allergy 3(1):117-126.
Nakayama, J. 2009. Progress in searching for the febrile seizure susceptibility genes. Brain and Development 31(5):359-365.
Nakayama, J., Y. H. Fu, A. M. Clark, S. Nakahara, K. Hamano, N. Iwasaki, A. Matsui, T. Arinami, and L. J. Ptacek. 2002. A nonsense mutation of the mass1 gene in a family with febrile and afebrile seizures. Annals of Neurology 52(5):654-657.
Nakayama, J., K. Hamano, N. Iwasaki, S. Nakahara, Y. Horigome, H. Saitoh, T. Aoki, T. Maki, M. Kikuchi, T. Migita, T. Ohto, Y. Yokouchi, R. Tanaka, M. Hasegawa, A. Matsui, H. Hamaguchi, and T. Arinami. 2000. Significant evidence for linkage of febrile seizures to chromosome 5q14-q15. Human Molecular Genetics 9(1):87-91.
Nakayama, J., N. Yamamoto, K. Hamano, N. Iwasaki, M. Ohta, S. Nakahara, A. Matsui, E. Noguchi, and T. Arinami. 2004. Linkage and association of febrile seizures to the IMPA2 gene on human chromosome 18. Neurology 63(10):1803-1807.
Noone, M. C., and J. D. Osguthorpe. 2003. Anaphylaxis. Otolaryngologic Clinics of North America 36(5):1009-1020.
Offringa, M., P. M. Bossuyt, J. Lubsen, J. H. Ellenberg, K. B. Nelson, F. U. Knudsen, J. F. Annegers, A. S. el-Radhi, J. D. Habbema, G. Derksen-Lubsen, et al. 1994. Risk factors for seizure recurrence in children with febrile seizures: A pooled analysis of individual patient data from five studies. Journal of Pediatrics 124(4):574-584.
Ogawara, K., S. Kuwabara, M. Mori, T. Hattori, M. Koga, and N. Yuki. 2000. Axonal Guillain-Barré syndrome: Relation to anti-ganglioside antibodies and Campylobacter jejuni infection in Japan. Annals of Neurology 48(4):624-631.
Oldstone, M. B. 2005. Molecular mimicry, microbial infection, and autoimmune disease: Evolution of the concept. Current Topics in Microbiology and Immunology 296:1-17.
Pardo, C. A., D. L. Vargas, and A. W. Zimmerman. 2005. Immunity, neuroglia and neuroinflammation in autism. International Reviews of Psychiatry 17(6):485-495.
Peiffer, A., J. Thompson, C. Charlier, B. Otterud, T. Varvil, C. Pappas, C. Barnitz, K. Gruenthal, R. Kuhn, and M. Leppert. 1999. A locus for febrile seizures (FEB3) maps to chromosome 2q23-24. Annals of Neurology 46(4):671-678.
Petrovski, S., I. E. Scheffer, S. M. Sisodiya, T. J. O’Brien, and S. F. Berkovic. 2009. Lack of replication of association between SCN1A SNP and febrile seizures. Neurology 73(22): 1928-1930.
Pittman, P. R. 2002. Aluminum-containing vaccine associated adverse events: Role of route of administration and gender. Vaccine 20:S48-S50.
Poduri, A., Y. Wang, D. Gordon, S. Barral-Rodriguez, C. Barker-Cummings, A. Ulgen, V. Chitsazzadeh, R. S. Hill, N. Risch, W. A. Hauser, T. A. Pedley, C. A. Walsh, and R. Ottman. 2009. Novel susceptibility locus at chromosome 6q16.3-22.31 in a family with GEFS+. Neurology 73(16):1264-1272.
Poirriez, J. 2004. A preliminary experiment of absorption of antinuclear antibodies by the hepatitis B vaccine components, in a case of neurolupus. Vaccine 22(23-24):3166-3168.
Prendergast, M. M., A. J. Lastovica, and A. P. Moran. 1998. Lipopolysaccharides from Campylobacter jejuni O:41 strains associated with Guillain-Barré syndrome exhibit mimicry of GM1 ganglioside. Infection and Immunity 66(8):3649-3655.
Pukhalsky, A. L., G. V. Shmarina, M. S. Bliacher, I. M. Fedorova, A. P. Toptygina, J. J. Fisenko, and V. A. Alioshkin. 2003. Cytokine profile after rubella vaccine inoculation: Evidence of the immunosuppressive effect of vaccination. Mediators of Inflammation 12(4):203-207.
Rees, J. H., N. A. Gregson, and R. A. Hughes. 1995a. Anti-ganglioside GM1 antibodies in Guillain-Barré syndrome and their relationship to Campylobacter jejuni infection. Annals of Neurology 38(5):809-816.
Rees, J. H., S. E. Soudain, N. A. Gregson, and R. A. Hughes. 1995b. Campylobacter jejuni infection and Guillain-Barré syndrome. New England Journal of Medicine 333(21): 1374-1379.
Reif, D. M., A. A. Motsinger-Reif, B. A. McKinney, M. T. Rock, J. E. Crowe, Jr., and J. H. Moore. 2009. Integrated analysis of genetic and proteomic data identifies biomarkers associated with adverse events following smallpox vaccination. Genes and Immunity 10(2):112-119.
Rezaie, A. R. 2010. Regulation of the protein C anticoagulant and antiinflammatory pathways. Current Medicinal Chemistry 17(19):2059-2069.
Rodgers, G. M. 2009. Role of antithrombin concentrate in treatment of hereditary antithrom-bin deficiency. An update. Thrombosis and Haemostasis 101(5):806-812.
Rodriguez-Iturbe, B., and S. Batsford. 2007. Pathogenesis of poststreptococcal glomerulonephritis a century after Clemens von Pirquet. Kidney International 71(11):1094-1104.
Ronald, A., and R. A. Hoekstra. 2011. Autism spectrum disorders and autistic traits: A decade of new twin studies. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics 156B(3):255-274.
Rose, N. R., and I. R. Mackay. 2000. Molecular mimicry: A critical look at exemplary instances in human diseases. Cellular and Molecular Life Sciences 57(4):542-551.
Roubin, R., and J. Benveniste. 1985. Formation of prostaglandins, leukotrienes and pafacether by macrophages. Comparative Immunology, Microbiology and Infectious Diseases 8(2):109-118.
Rus, H., C. Cudrici, and F. Niculescu. 2005. The role of the complement system in innate immunity. Immunologic Research 33(2):103-112.
Sadleir, L. G., and I. E. Scheffer. 2007. Febrile seizures. British Medical Journal 334(7588): 307-311.
Sampson, H. A., A. Munoz-Furlong, R. L. Campbell, N. F. Adkinson, Jr., S. A. Bock, A. Branum, S. G. Brown, C. A. Camargo, Jr., R. Cydulka, S. J. Galli, J. Gidudu, R. S. Gruchalla, A. D. Harlor, Jr., D. L. Hepner, L. M. Lewis, P. L. Lieberman, D. D. Metcalfe, R. O’Connor, A. Muraro, A. Rudman, C. Schmitt, D. Scherrer, F. E. Simons, S. Thomas, J. P. Wood, and W. W. Decker. 2006. Second symposium on the definition and management of anaphylaxis: Summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Journal of Allergy and Clinical Immunology 117(2):391-397.
Sansonno, D., A. Carbone, V. De Re, and F. Dammacco. 2007. Hepatitis C virus infection, cryoglobulinaemia, and beyond. Rheumatology 46(4):572-578.
Sarma, J. V., and P. A. Ward. 2011. The complement system. Cell and Tissue Research 343(1):227-235.
Schipul, S. E., T. A. Keller, and M. A. Just. 2011. Inter-regional brain communication and its disturbance in autism. Frontiers in Systems Neuroscience 5:10.
Schlachter, K., U. Gruber-Sedlmayr, E. Stogmann, M. Lausecker, C. Hotzy, J. Balzar, E. Schuh, C. Baumgartner, J. C. Mueller, T. Illig, H. E. Wichmann, P. Lichtner, T. Meitinger, T. M. Strom, A. Zimprich, and F. Zimprich. 2009. A splice site variant in the sodium channel gene SCN1A confers risk of febrile seizures. Neurology 72(11):974-978.
Schuchmann, S., D. Schmitz, C. Rivera, S. Vanhatalo, B. Salmen, K. Mackie, S. T. Sipila, J. Voipio, and K. Kaila. 2006. Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nature Medicine 12(7):817-823.
Shantsila, E., and G. Y. Lip. 2009. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thrombosis and Haemostasis 102(5):916-924.
Sharrar, R. G., P. LaRussa, S. A. Galea, S. P. Steinberg, A. R. Sweet, R. M. Keatley, M. E. Wells, W. P. Stephenson, and A. A. Gershon. 2001. The postmarketing safety profile of varicella vaccine. Vaccine 19(7-8):916-923.
Shibasaki, K., M. Suzuki, A. Mizuno, and M. Tominaga. 2007. Effects of body temperature on neural activity in the hippocampus: Regulation of resting membrane potentials by transient receptor potential vanilloid 4. Journal of Neuroscience 27(7):1566-1575.
Sidhu, G., and G. A. Soff. 2009. The coagulation system and angiogenesis. Cancer Treatment and Research 148:67-80.
Simons, F. E. 2009. Anaphylaxis: Recent advances in assessment and treatment. Journal of Allergy and Clinical Immunology 124(4):625-636; quiz 637-628.
Simons, F. E. 2010. Anaphylaxis. Journal of Allergy and Clinical Immunology 125(2 Suppl. 2):S161-S181.
Soghoian, D. Z., and H. Streeck. 2010. Cytolytic CD4(+) T cells in viral immunity. Expert Reviews of Vaccines 9(12):1453-1463.
Solomon, T., and H. Willison. 2003. Infectious causes of acute flaccid paralysis. Current Opinion in Infectious Diseases 16(5):375-381.
Sriskandan, S., and D. M. Altmann. 2008. The immunology of sepsis. Journal of Pathology 214(2):211-223.
Sudhof, T. C. 2008. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455(7215):903-911.
Susuki, K., Y. Nishimoto, M. Koga, T. Nagashima, I. Mori, K. Hirata, and N. Yuki. 2004. Various immunization protocols for an acute motor axonal neuropathy rabbit model compared. Neuroscience Letters 368(1):63-67.
Susuki, K., Y. Nishimoto, M. Yamada, M. Baba, S. Ueda, K. Hirata, and N. Yuki. 2003. Acute motor axonal neuropathy rabbit model: Immune attack on nerve root axons. Annals of Neurology 54(3):383-388.
Szyper-Kravitz, M. 2009. The hemophagocytic syndrome/macrophage activation syndrome: A final common pathway of a cytokine storm. Israel Medical Association Journal 11(10): 633-634.
Talbot, T. R., J. T. Stapleton, R. C. Brady, P. L. Winokur, D. I. Bernstein, T. Germanson, S. M. Yoder, M. T. Rock, J. E. Crowe, Jr., and K. M. Edwards. 2004. Vaccination success rate and reaction profile with diluted and undiluted smallpox vaccine a randomized controlled trial. Journal of the American Medical Association 292(10):1205-1212.
Tedeschi, A., C. Barate, E. Minola, and E. Morra. 2007. Cryoglobulinemia. Blood Reviews 21(4):183-200.
Theoharides, T. C., D. Kempuraj, and L. Redwood. 2009. Autism: An emerging “neuroimmune disorder” in search of therapy. Expert Opinion on Pharmacotherapy 10(13):2127-2143.
Thomas, E. A., R. J. Hawkins, K. L. Richards, R. Xu, E. V. Gazina, and S. Petrou. 2009. Heat opens axon initial segment sodium channels: A febrile seizure mechanism? Annals of Neurology 66(2):219-226.
Tritto, E., F. Mosca, and E. De Gregorio. 2009. Mechanism of action of licensed vaccine adjuvants. Vaccine 27(25-26):3331-3334.
Tsuboi, T. 1987. Genetic analysis of febrile convulsions: Twin and family studies. Human Genetics 75(1):7-14.
van Dijk, J. G., R. D. Thijs, D. G. Benditt, and W. Wieling. 2009. A guide to disorders causing transient loss of consciousness: Focus on syncope. Nature Reviews. Neurology. 5(8):438-448.
van Rossum, A. P., P. C. Limburg, and C. G. Kallenberg. 2005. Activation, apoptosis, and clearance of neutrophils in Wegener’s granulomatosis. Annals of the New York Academy of Sciences 1051:1-11.
Vanderlugt, C. L., W. S. Begolka, K. L. Neville, Y. Katz-Levy, L. M. Howard, T. N. Eagar, J. A. Bluestone, and S. D. Miller. 1998. The functional significance of epitope spreading and its regulation by co-stimulatory molecules. Immunological Reviews 164:63-72.
Vargas, D. L., C. Nascimbene, C. Krishnan, A. W. Zimmerman, and C. A. Pardo. 2005. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology 57(1):67-81.
Visser, L. H., F. G. Van der Meche, P. A. Van Doorn, J. Meulstee, B. C. Jacobs, P. G. Oomes, and R. P. Kleyweg. 1995. Guillain-Barré syndrome without sensory loss (acute motor neuropathy). A subgroup with specific clinical, electrodiagnostic and laboratory features. Dutch Guillain-Barré study group. Brain 118 (Pt 4):841-847.
Volanakis, J. E. 1990. Participation of C3 and its ligands in complement activation. Current Topics in Microbiology and Immunology 153:1-21.
Wallace, R. H., S. F. Berkovic, R. A. Howell, G. R. Sutherland, and J. C. Mulley. 1996. Suggestion of a major gene for familial febrile convulsions mapping to 8q13-21. Journal of Medical Genetics 33(4):308-312.
Wan, Y. Y., and R. A. Flavell. 2009. How diverse—CD4 effector T cells and their functions. Journal of Molecular Cell Biology 1(1):20-36.
Waruiru, C., and R. Appleton. 2004. Febrile seizures: An update. Archives of Disease in Childhood 89(8):751-756.
Wass, S. 2011. Distortions and disconnections: Disrupted brain connectivity in autism. Brain and Cognition 75(1):18-28.
Watanabe, M., K. Uchida, K. Nakagaki, B. C. Trapnell, and K. Nakata. 2010. High avidity cytokine autoantibodies in health and disease: Pathogenesis and mechanisms. Cytokine and Growth Factor Reviews 21(4):263-273.
Waters, V., K. S. Peterson, and P. LaRussa. 2007. Live viral vaccines in a DiGeorge syndrome patient. Archives of Disease in Childhood 92(6):519-520.
Wegner, N., K. Lundberg, A. Kinloch, B. Fisher, V. Malmstrom, M. Feldmann, and P. J. Venables. 2010. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunological Reviews 233(1):34-54.
Whitley, R. J. 2010. Varicella-zoster virus. In Mandell, Douglas, and Bennett’s principles and practice of infectious diseases. 7th ed. 2 vols. Vol. 2, edited by G. L. Mandell, J. E. Bennett, and R. Dolin. Philadelphia, PA: Churchill Livingstone Elsevier. Pp. 1963-1969.
Wieling, W., R. D. Thijs, N. van Dijk, A. A. Wilde, D. G. Benditt, and J. G. van Dijk. 2009. Symptoms and signs of syncope: A review of the link between physiology and clinical clues. Brain 132(Pt 10):2630-2642.
Willison, H. J., and N. Yuki. 2002. Peripheral neuropathies and anti-glycolipid antibodies. Brain 125(Pt 12):2591-2625.
Wiznitzer, M. 2010. Dravet syndrome and vaccination: When science prevails over speculation. Lancet Neurology 9(6):559-561.
Wong, S. S., and K. Y. Yuen. 2006. Avian influenza virus infections in humans. Chest 129(1): 156-168.
Yang, Y., S. Sujan, F. Sun, Y. Zhang, Y. Jiang, J. Song, J. Qin, and X. Wu. 2006. Acute metabolic crisis induced by vaccination in seven Chinese patients. Pediatric Neurology 35(2):114-118.
Yokoyama, T., K. Tateishi, K. Tsushima, T. Agatsuma, H. Yamamoto, T. Koizumi, and K. Kubo. 2010. A case of severe ARDS caused by novel swine-origin influenza (A/H1N1pdm) virus: A successful treatment with direct hemoperfusion with polymyxin B-immobilized fiber. Journal of Clinical Apheresis 25(6):350-353.
Yuki, N. 2005. Carbohydrate mimicry: A new paradigm of autoimmune diseases. Current Opinion in Immunology 17(6):577-582.
Yuki, N., K. Susuki, M. Koga, Y. Nishimoto, M. Odaka, K. Hirata, K. Taguchi, T. Miyatake, K. Furukawa, T. Kobata, and M. Yamada. 2004. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barré syndrome. Proceedings of the National Academy of Sciences of the United States of America 101(31):11404-11409.
Yuki, N., T. Taki, F. Inagaki, T. Kasama, M. Takahashi, K. Saito, S. Handa, and T. Miyatake. 1993. A bacterium lipopolysaccharide that elicits Guillain-Barré syndrome has a GM1 ganglioside-like structure. Journal of Experimental Medicine 178(5):1771-1775.
Zelenay, S., M. F. Moraes Fontes, C. Fesel, J. Demengeot, and A. Coutinho. 2007. Physiopathology of natural auto-antibodies: The case for regulation. Journal of Autoimmunity 29(4):229-235.
Zhong, W., P. Oschmann, and H. J. Wellensiek. 1997. Detection and preliminary characterization of circulating immune complexes in patients with Lyme disease. Medical Microbiology and Immunology 186(2-3):153-158.
Zimmerman, A. W., H. Jyonouchi, A. M. Comi, S. L. Connors, S. Milstien, A. Varsou, and M. P. Heyes. 2005. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatric Neurology 33(3):195-201.














































