
2
MCM Enterprise and Stakeholder Perspectives
FDA REGULATORY SCIENCE RESEARCH NEEDS
To provide a framework for subsequent discussions, representatives from FDA’s Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research(CBER), and Center for Devices and Radiological Health (CDRH) provided a broad overview of current efforts underway at the agency to support research, development, and evaluation of MCMs, and discussed current challenges and how regulatory science can assist in advancing MCM development and evaluation.
Center for Drug Evaluation and Research (CDER)
The regulatory review process is an ongoing, iterative process, explained Susan McCune, deputy director of the Office of Translational Science at CDER. It is hoped that gaps and questions identified during a regulatory review can be addressed through application of regulatory science and that regulatory science in turn informs the broader regulatory review process.
Within CDER, the Science Prioritization and Review Committee has identified science and research needs across seven key areas. McCune pointed out that although these identified needs are centerwide and not limited to MCMs, all are relevant to the MCM initiative:
- Improve access to postmarket data sources and explore feasibility of their use in different types of analyses.
- Improve risk assessment and management strategies to reinforce the safe use of drugs.
- Evaluate the effectiveness and impact of different types of regulatory communications to the public and other stakeholders.
- Evaluate the links among product quality attributes, manufacturing processes, and product performance.
- Develop and improve predictive models of safety and efficacy in humans.
- Improve clinical trial design, analysis, and conduct.
- Enhance individualization of patient treatment.
McCune briefly reviewed the drug regulatory review life cycle (Figure 2-1) and offered a number of examples of potential areas for Pillar 2 research across the drug life cycle (Table 2-1). CDER scientists are already working in many of these areas. For each of these areas, she said, there are numerous potential scientific studies that could be done. One of the primary challenges is prioritization of studies as they relate to the MCM initiative.
In closing, McCune said, CDER has a robust regulatory science program with significant expertise to support the research agenda of the MCM initiative, and CDER researchers and reviewers are eager to collaborate on efforts to advance the regulatory science needs of the initiative.
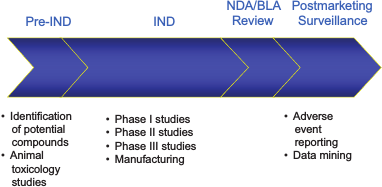
FIGURE 2-1 FDA regulatory review cycle.
NOTE: BLA, biologics license application; IND, investigational new drug application;NDA, new drug application.
SOURCE: Susan McCune. 2011. Presentation at IOM workshop; Advancing Regulatory Science for Medical Countermeasure Development.
Center for Biologics Evaluation and Research (CBER)
Products regulated by CBER include blood, blood components and derivatives, vaccines, allergenic products, cell and gene therapies, xenotransplantation products, human tissues, and various related devices, explained Carolyn Wilson, associate director for research at CBER. MCMs fall into several of these categories. For example, in the area of cell and gene therapies, mesenchymal stem cells are being evaluated for treatment of acute radiation syndrome; pathogen-specific immunoglobulins come under the area of blood components and derivatives; and there are a variety of MCM-related vaccines (e.g., anthrax, botulinum, smallpox, influenza).
Wilson highlighted five areas under Pillar 2 of the MCM initiative in which CBER is conducting research (Table 2-2).1 The anticipated public health outcomes of such research include the development of new scientific tools and biomarkers to facilitate development of safe and effective MCM biologics and improved guidance to sponsors on how to develop and evaluate MCM biologics, including guidance on how to implement the Animal Rule. Research could result in, for example, earlier identification of toxicity, improved means to assess potential for efficacy, and more rapid detection of safety signals, potentially leading to improved decision making regarding benefits and risk.
One example of current CBER research on animal model biomarkers involves luciferase-expressing Bacillus anthracis, which allows for more precise staging of the bacterial infection in mice, and improved design for studies of such things as postexposure prophylaxis and combination therapies. This approach is generalizable, Wilson said, and could be applied to other pathogens and to the development of improved animal models. Other examples of ongoing CBER research mentioned by Wilson include using phage display libraries to evaluate the human immune response, developing faster methods to generate reference reagents for influenza vaccines, and improving detection of adventitious infectious agents in complex samples.
Center for Devices and Radiological Health (CDRH)
The devices regulated by CDRH include diagnostic and detection devices, personal protective equipment (e.g., N95 respirators), emergency devices (e.g., ventilators, intravenous administration sets, resuscitation equipment, drug or vaccine delivery systems, needles), and combination
_____________
1 Further information on CBER biologics research projects can be found at http://www.fda.gov/BiologicsBloodVaccines/ScienceResearch/default.htm (accessed June 9, 2011).
TABLE 2-1 Examples of Potential Areas for Pillar 2 Research in CDER
| Phase of Development | Potential CDER Research Areas | |
| Identification of Potential Molecules for Study |
Repurposing drugs Molecular modeling Screen approved drugs for other pathogens Effects of combinations of antimicrobials |
|
| Animal Toxicology Studies and Animal Models for MCM |
Animal models: For pregnancy Modeling of disease states in general To study toxin effects on organ systems Qualified through the drug development tools qualification process For combination added benefit studies For placebo studies For postexposure prophylaxis To evaluate potential safety signals For studies of natural history and pathophysiology of disease Conversion of data from animal model studies to standard format Animal model database |
|
| Human Safety Studies for MCMs or Clinical Safety and Efficacy Studies for Influenza |
Evaluation of the effects of genetic variations Studies of dosage forms for special populations Extrapolation models from animal to human, including dose scaling for special populations Pediatric safety studies, including ethical issues Understanding human disease through the world literature Development of clinical endpoints for seriously ill influenza patients Development of threat-based data standards Development of standardized case report forms for data collection Modeling drug interaction studies Modeling of PK/PD to labeled drugs for special populations |
|
| Manufacturing and Product Quality |
Therapeutic protein PK/PD comparability studies Shelf-life extension studies Develop stable product formulations Rapid detection of problems with marketed products |
|
| Incident-Related Studies |
Develop hospital networks for rapid information transfer Communication studies on emergency notification Data mining for adverse events associated with therapy Develop protocols for use during events Real-world use studies on home preparation instructions Real-time epidemiology cluster monitoring |
NOTE: PK/PD, pharmacokinetic/pharmacodynamic.
TABLE 2-2 Examples of Potential Areas for Pillar 2 Research in CBER
| Pillar 2 Program | Research Needs | |
| Animal Model Biomarker Program |
Identify in vitro or in vivo correlates of bioactivity, safety, toxicity Methods development Develop and evaluate animal models |
|
| Clinical Biomarker and Immunology Program |
How to bridge from animal studies to the human immune response to vaccines Insufficient knowledge of human disease Clinical trial design |
|
| Ensuring MCM Product Quality |
Measurable product characteristics that correlate with safety and efficacy Improved methods to assess new cell substrates New methods that incorporate new technology and are faster, use fewer animals, and have improved sensitivity/specificity |
|
| Radiation Injury Protection and Response Program |
Improved tools to assess critical product attributes of cell therapy-based products used to treat radiation injury Animal models to evaluate product safety and potential for efficacy |
|
| Health Informatics/Scientific Computing |
Improved means to detect rare adverse events Improved methods and access to health care data sources to monitor safety of marketed MCM biologics Improved use of high-performance computing Tools/models for risk assessment |
products (a combination of drug or biological and a device), explained Murray Malin, acting director of the Medical Countermeasure Initiative at CDRH. There are MCM devices in each of these categories. The CDRH Office of In Vitro Diagnostics has developed multiple guidance documents that affect MCMs and has improved transparency by posting device clearance reviews on their website.
Malin highlighted the regulatory science priorities in CDRH, noting that they are similar to those of the other centers:
- develop infrastructure to support development of diagnostics and other MCMs;
- characterize the medical device supply chain;
- enable real-time or near-real-time surveillance of supply, utilization, and availability of medical devices to avoid shortages;
- address special needs populations, point of care, and personalized use of MCMs;
- enhance ability to capture, monitor, and analyze large datasets;
- create statistical tools to develop innovative clinical trials, perform comparative effectiveness research, and perform active surveillance of adverse event reports;
- genomic sequencing devices and assays for the detection of pathogens and antimicrobial drug resistance;
- multiplex/microarray diagnostic devices capable of simultaneous detection/identification of multiple organisms;
- develop new tools to evaluate nanotechnology-based devices, including use as diagnostic markers;
- increase scientific capacity and expertise to prepare for and facilitate new technologies;
- develop guidance for development of multiuse products and platforms to expand MCM product pipelines during emergencies;
- infrastructure, comparator sequencing database, data processing, and resources to enhance reviews of MCMs, facilitate innovative statistical techniques and clinical trials, and develop regulatory pathways for MCMs;
- agreement with Centers for Medicare and Medicaid Services (CMS) on data necessary for Clinical Laboratory Improvement Amendments (CLIA) waiver of authorized products during an emergency; and
- develop high-performance/scientific computing to facilitate the following:
- answering questions related to comparative effectiveness associated with patient subsets;
- support for genomic sequencing, multiplex devices;
- identification of improved methods for characterizing failure analysis, validation of factors affecting manufacture of MCMs, and development of forensic evaluation techniques necessary to support multiplatform/product development; and
- development of innovative statistical methods and clinical trial design.
Discussion
Much of the discussion with the FDA panelists focused on communication and collaboration as key components of advancing regulatory science. Wilson pointed out that CBER has “research reviewers” who are actively engaged in both the regulatory science agenda and review activities. Research reviewers can help identify gaps in the science, as well as methods, tools, and reference materials that could help move a technology or a whole product area forward. McCune added that many CDER review team members are actively engaged in research at the agency and bring a significant amount of clinical and scientific expertise to the review process. From the CDRH perspective, Malin noted that formal and informal collaboration is key to understanding needs and opportunities for products. Solving the needs for a certain product may be translated to other types of products as well. The speakers highlighted the numerous interactions among their centers, from sharing supercomputing capabilities to collaborating on the development of modeling approaches, software, and analytic tools.
With regard to dissemination of information, panelists noted that information about a product or process that is generalizable knowledge is released by FDA in the form of guidance documents. The panelists noted that FDA scientists are encouraged to publish their research in the peer-reviewed literature. There is also a wealth of information on the FDA website; for example, the CDER Office of In Vitro Diagnostics has several databases on its website with information about approved and cleared devices.
Professional development for FDA scientists and reviewers is also important to keep agency science on the cutting edge. For example, CDER has weekly “scientific rounds”where novel scientific and regulatory issues are discussed. There are online programs as well as classroom-based programs. Reviewers also need to be able to attend scientific meetings and have access to the latest information.
With regard to potential indicators or metrics of success of the regulatory science initiatives underway, it was noted that it is difficult to measure precisely the public health impact of any particular initiative. While the long-term, big-picture goal is an increased number of approved
MCMs, the metric cannot simply be approvals, because not all products will (or should be) approved. The FDA panelists suggested that metrics should be associated with smaller, incremental steps, such as solving a problem that allows for a potency assay in vitro instead of in hundreds of animals, thereby increasing speed and decreasing the cost of that potency assay.
ENTERPRISE PARTNER AND STAKEHOLDER PERSPECTIVES
Immediately following the panel presentations from FDA representatives, stakeholders representing the other key components of the MCM enterprise provided remarks in which they identified key issues in MCM development and utilization that can be addressed through regulatory science and offered suggestions of regulatory science needs or priorities to advance MCM development.
Department of Defense (DoD)
Gerald Parker of DoD said there is a need for affordable, easy-to-use, rapid, point-of-need diagnostics that can be made available on a global basis and that can be connected to an information backbone so the resulting data can be rapidly shared (within minutes or hours, rather than weeks). This includes diagnostics not only for pathogen identification but also for antibiotic/antiviral resistance patterns, presymptomatic biomarkers, and host response markers.
Biodefense research at the DoD is focused on both traditional threats and endemic diseases that the nation’s adversaries could choose to use against U.S. forces around the world. The DoD is working in a collaborative manner, seeking to use platform technologies that incorporate rapid pathogen characterization and the ability rapidly to turn that information into a discoverable product. While the DoD program has a sound science and technology base, Parker noted that the department lacks the ability to rapidly develop discoveries and manufacture new candidates against unknown threats.
The DoD MCM Initiative strategy consists of two major elements, each with multiple initiatives: (1) science and technology (novel platform/expression systems for MCMs, regulatory science technologies, manufacturing technologies for biologics that support good laboratory practices [GLP]/good manufacturing practices [GMP]) and (2) advanced development (further maturation of novel platform/expression systems and integration into a production process; innovative, flexible, and agile manufacturing capabilities).
In closing, Parker said that MCM development needs a clearly
defined regulatory pathway for products approved under the Animal Rule, including early and ongoing real-time engagement of all partners. He also noted that a DoD diagnostics leadership meeting held in October 2010 called for more inclusion in discussions and greater collaboration to develop diagnostics and to inform the regulatory roadmap for next-generation diagnostics.
National Institute of Allergy and Infectious Diseases (NIAID)
Michael Kurilla, of NIAID, explained that NIAID is taking a comprehensive approach to MCM development, with certain general criteria for vaccines, therapeutics, and diagnostics. By way of example, he noted that in the case of vaccines, it is important to consider alternatives for immunocompromised persons and special populations such as the elderly and children.
There are several unique aspects of bioterrorism agents that add to the challenge of developing MCMs:
- there are limited facilities to conduct studies under appropriate biological safety containment;
- there is limited prior art on fundamental aspects of specific pathogens (e.g., tularemia);
- there is limited human pathogenesis data available (necessary to inform animal model development); and
- there is increased regulation and oversight of bioterrorism agents (e.g., rules addressing the possession, use, and transfer of select agents).
Kurilla defined MCMs as falling into four broad classes: (1) previously licensed MCMs for which the mechanism of action would support efficacy (e.g., a licensed antibiotic);(2) previously licensed MCMs that are being repurposed for a nonintuitive application(e.g., the oncologic drug, Gleevec, which demonstrates activity against smallpox in vitro); (3) MCMs that are currently in development for other clinical indications(e.g., novel anti-infective agents); and (4) MCMs developed solely for a CBRN application(e.g., an anthrax or Ebola vaccine).
Kurilla posed several questions for consideration regarding the evaluation of new science and technologies:
- What should be done in cases of limited or nonexisting human clinical data with which to define an appropriate animal model?
- How can we study species-specific biological agents, those for which there may be no appropriate animal model?
- What criteria define an animal model correlate?
- Can mechanistic efficacy substitute for disease efficacy? (If the mechanism of an intervention is understood, can that be applied across a wide array of different disease spectrums where that mechanism is identified as crucial for a resolution of that disease?)
In discussion, Kurilla pointed out that much of what was discussed by the panels, and many of the major elements needed, are product-independent regulatory science and product-independent tools. However, the traditional regulatory paradigm is regulation in the context of a product, and there is little interaction with the agency in the absence of a specific product. Product developers approach FDA when they are ready to take a product into pivotal efficacy and safety studies, and it is at that late point that some of the development tools, such as the animal models used thus far, begin to be critically reviewed and questioned (e.g., is the species relevant, is the challenge strain appropriate). Development of acceptable tools has always occurred concomitant with product development, and consequently, Kurilla said, developing these components in a product-independent manner, and in the most expeditious and rational manner, is quite challenging.
Biomedical Advanced Research and Development Authority (BARDA)
The 2009 H1N1 influenza pandemic brought to light some of the challenges of responding to a major public health emergency. Richard Hatchett, chief medical officer and deputy director of BARDA, cited the August 2010 report of the President’s Council of Advisors on Science and Technology (PCAST) on influenza vaccine technology, which identified two response issues directly involving regulatory science: the need to improve sterility testing of influenza vaccine, and the need for new techniques to test potency of vaccine preparations (PCAST, 2010). BARDA is investing in research in these areas, he noted.
With regard to regulatory science more broadly, Hatchett said that BARDA is looking to FDA for the following:
- Internal competency—FDA needs to have the expertise to keep up with advances in science, to be able to engage creatively with MCM product developers, and to adapt to new technologies (e.g., nanotechnology, bioinformatics, regenerative medicine, in vivo imaging, new approaches to clinical trial design).
- Capability—FDA needs to have the capability to help BARDA understand the requirements for proving safety, efficacy, sterility,
• and potency of vaccines or other products. This requires FDA to interact with BARDA partners in a collaborative fashion (which the agency is already doing, Hatchett noted).
• Clarity—FDA needs to be confident and assertive in defining the requirements for licensure and approval of new products. BARDA is looking for clear pathways, where those pathways can be defined in advance.
Hatchett also emphasized the need to better understand animal models and apply them in a variety of settings.
Hatchett concluded by drawing attention to a forthcoming BARDA request for proposals(RFP’s) on multiproduct facilities and rapid response manufacturing capabilities. This will require new approaches from FDA, he said, to be able to license products manufactured in facilities where there may be rapid changeover in response to emerging novel threats (e.g., pandemic influenza, sudden acute respiratory syndrome [SARS]).
Centers for Disease Control and Prevention (CDC)
The Laboratory Response Network (LRN) at the Centers for Disease Control and Prevention (CDC), a key national stakeholder in the MCM enterprise, is currently facing a variety of challenges in the area of diagnostics development, said May Chu, director of the Laboratory Science Policy and Practice Program at CDC. Chu described CDC’s most significant current challenge as obtaining FDA clearance of LRN-developed assays (including 11 polymerase chain reaction [PCR]-based assays and seven other assays). Chu noted that an LRN technical review committee oversees assay design, development, validation, and quality assurance prior to deployment, and she anticipated that transformational changes in regulatory science could provide relief while preserving quality and resilience.
Chu also noted that changes to a diagnostic platform require renewed validation. Chu suggested that collaborative discussions are needed to determine what validation is needed when changes occur. A software change, for example, should not necessarily lead to a full reevaluation.
Chu listed the top regulatory science priorities in the diagnostics field as follows:
- Allow for quick and nimble preparedness and response to public health emergencies.
- Prevalidate and preposition diagnostic tests in the field.
- Rapid, real-time, step-in-step MCM development with regulatory oversight. This, Chu explained, would allow data collected during an emergency to be used later to validate test platforms.
-
- Validation methodology for assessing lot-to-lot differences of commercial products.
- Allow for recognition of the diversity of diagnostic test producers.
- Maintain evidence-based quality and postmarket monitoring with stipulated controls and restricted use.
Academia
A perspective from academia was provided by Rick Lyons, director of the Infectious Disease Research Center at Colorado State University. Academicians are now accepting that for maximal benefit, an innovation must be translated to an application, Lyons noted. Regulatory science targets the pathways that are required for this translation (e.g., biomarkers, animal models, correlates of protection).
The most significant challenge, Lyons said, is the extrapolation of animal immunological and pathophysiological data to the human setting. What makes a good animal model is highly dependent on the research question, he said. One must consider, for example, whether there is similar pathophysiology as the human disease or similar mediators of immune protection. For product development, are there well-defined generic animal models or platforms that could be used? Most of the time, Lyons said, researchers are working with a nonvalidated surrogate that is “reasonably likely” to predict clinical efficacy.
Lyons opined that it is unlikely one species model will reflect human disease adequately and suggested a compartmentalization strategy, pooling data from several species models. This systems and pathways approach would require strong comparative immunology and physiology, he noted.
It will be important to educate the public regarding advances in regulatory science, Lyons pointed out, particularly the use of animal models for approval of products. It has been difficult, for example, to convince people to be vaccinated, or to have their children vaccinated, with products that have been FDA approved based on clinical trials in humans. How much more difficult will it be, Lyons asked, to get them to take a vaccine that has not been tested in humans?
Industry
A biotechnology industry perspective on regulatory science was provided by Eric Rose of SIGA, Inc. The biotechnology industry has developed and manufactures essentially all of the new CBRN countermeasures that have been procured into the SNS, Rose said. Companies are BARDA partners for most of the advanced development contracts. Most compa-
nies, however, are small and not profitable, sustained by private capital and government grants and contracts. Biotechnology companies are, Rose stated, “an essential effector limb of the PHEMCE implementation plan.”
The goals of regulatory science, Rose said, should be development of a broad array of safe and effective MCMs; alignment of stakeholders, products, and product uses;appropriate transparency throughout the process; and speed (i.e., there should be a sense of urgency as these agents are not just causes of illness, they are potential weapons of mass destruction).
In industry, there is a science to process improvement that Rose suggested can be applied to the process of MCM development. The first step is to design a process (hypothesize). That process is tested by use (experiment), assessed (analyzing performance metrics), redesigned (refine hypothesis), and the cycle continues. The ultimate validation of animal efficacy models requires clinical trials during an outbreak situation. This is something that should be planned for, he said. Rose also noted that, with regard to development of animal models, criteria for euthanizing animals when they have reached a certain degree of illness needs to be transparent and prespecified.
Rose concluded by noting that while robust evidence of meaningful outcomes from controlled clinical trials remains the scientific gold standard for efficacy, the challenges facing MCM development are not necessarily new, and FDA has helped foster other industry segments in the absence of feasible trials, including, for example, orphan drugs, complex medical devices (e.g., artificial hearts), and diagnostics.
Discussion
There was some discussion around reverse engineering to evaluate what processes might work best. One suggestion was to select several products that are currently in the SNS and retrospectively simulate the process as if they had been new molecular entities. It was also noted that there is a lot of animal data available that is associated with approved products. Although these products are not MCMs, it might be helpful to look back at the animal data submitted for product approval and evaluate which models were predictive and which were less so.
Participants also discussed benefit-risk assessments for MCMs and whether the criteria should be the same or different from that for routine products. George Korch of HHS noted that a challenge is conducting a benefit-risk calculus that captures rare and yet highly consequential events. Hatchett added that it is very hard to define, in advance of a real event, criteria for a benefit-risk analysis that are able to take into account the operating environment that exists once the event has occurred. Cal-
culating the risk and benefit of an anthrax antitoxin today, for example, is very different from calculating the risk and benefit of the anthrax antitoxin once there has been a widespread anthrax release. Jesse Goodman of FDA said that the language around FDA’s EUA authority regarding the known and potential benefits outweighing the known and potential risks leaves a lot of room for different interpretations. Goodman, Parker, and others all noted that defining the emergency scenario up front would allow for different benefit-risk decisions than those that would be reached for use of common products by generally healthy people. Hatchett cautioned that the calculus done in anticipation of an emergency tends to be much more stringent than that which would actually be made during a real event. To aid benefit-risk decisions, Rose suggested, reviewers of MCMs ought to have the requisite security clearances to be allowed to read the associated confidential population threat assessments. In later discussion about safety assessment, Richard Forshee of the Office of Biostatistics and Epidemiology at CBER, mentioned current agency efforts to develop risk assessment models to support regulatory decision making. These probabilistic quantitative computer simulation models can help explore how different regulatory science options could ultimately have an impact on public health, and thereby improve decision making. FDA is also engaged in a number of computational toxicology computer simulations to help assess, for example, potential risk from vaccine adjuvants.
Key Messages: Enterprise Stakeholder Perspectives
- The regulatory paradigm for MCM development needs to be supported by new regulatory science and evaluative tools that are product-independent. There is a need for a format to permit engagement between product developers and FDA outside the context of a specific product approval.
- MCM development needs more clearly defined regulatory pathways. Priorities include:
- Products approved under the Animal Rule, and
- Diagnostics—prevalidated and pre-positioned in the field
- The benefit-risk calculus may be different for MCMs to be used in low-probability/high-consequence events than for traditional products.
- Repurposing of previously licensed products needs to be studied in a systematic and comprehensive manner.













