
3
Cutting-Edge Efforts to Advance MCM Regulatory Science
NONCLINICAL APPROACHES TO ASSESSING EFFICACY
A challenge facing developers of MCMs is how to increase the predictive value of nonclinical data, said panel moderator Lauren Black, senior scientific advisor at Charles River Laboratories. In the absence of clinical trials, nonclinical data can, for example, help define a human dose regimen and predict a reasonable likelihood of human efficacy. In addition to animal models, other nonclinical tools such asin silico biology and biomarkers can be employed to inform and advance MCM development.
In Silico Approaches to Efficacy Assessment of MCMs
A systems biology approach to health and disease acknowledges that there are likely complex molecular mechanisms, with groups of molecules, genes, proteins, and metabolites working in a coordinated fashion, that differ in healthy versus diseased states, explained Ramon Felciano, founder of Ingenuity Systems. These molecular mechanisms trigger higher-order cellular mechanisms and disease mechanisms that drive overall physiology (Figure 3-1). Technologies that have emerged over the past decade or so (e.g., genomics, proteomics, metabolomics) have generated a flood of new data, driving the need for new types of analytics such as in silico or computer modeling of biology. These new data enhance and align with existing knowledge of disease pathways and mechanisms from the literature.
A typical systems biology approach is philosophically data driven
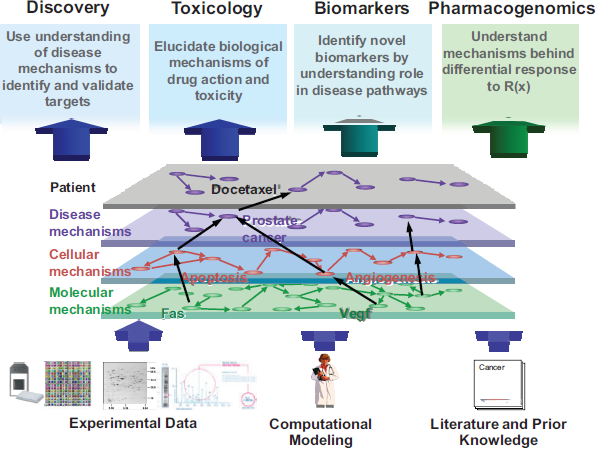
FIGURE 3-1 In silico modeling of disease mechanisms for drug development.
SOURCE: Ramon Felciano. 2011. Presentation at IOM workshop; AdvancingRegulatory Science for Medical Countermeasure Development.
and holistic, Felciano said, using computer-based tools and techniques to model and understand complex biological function. Experimental designs are typically comparative in nature (e.g., healthy versus disease, disease versus treatment, dose response). The complexity and volume of the data that are generated by these approaches typically require fairly sophisticated computational and statistical modeling for analysis and prediction. Research teams are often interdisciplinary by necessity, with therapeutic area researchers, computer scientists, statisticians, and others working together.
Primary benefits of this approach, Felciano said, include better understanding of disease progression, generation of novel hypotheses for therapeutic or diagnostic targets (i.e., biomarkers), and characterization of plausible mechanisms that correlate with these diagnostic and prognostic markers.
Compared with other therapeutic areas, there has been relatively little research done in the area of MCMs using a systems biology approach, Felciano noted. He cited one retrospective study of yellow fever vaccine that
demonstrates the potential ofin silico approaches to modeling. Querec and colleagues (2009) used a systems biology approach to identify early gene signatures that predicted immune response in humans to the yellow fever vaccine.
There are several challenges to using in silico techniques in MCM efficacy studies, Felciano said. As this is a new field, no dominant modeling formalisms have yet emerged, and there is a lot of new math being generated alongside the new data. Some of the “-omics” technologies are still relatively new, and there are issues to be addressed, for example: measurement accuracy and reproducibility, false positive results, and cost effectiveness. Felciano noted that FDA has a Voluntary Exploratory Data Submission (VXDS) program in which industry submits candidate datasets that FDA can use to evaluate the regulatory applicability of these new approaches. Other challenges are that systems biology experiments are complex and interdisciplinary, requiring substantial time, interdisciplinary expertise, and resources for analysis. Thus far, there are few successful applications of in silico techniques to infectious disease. In addition, there are few good predictive models to bridge animal data to humans.
To leverage in silico modeling for MCM development, Felciano said there is a need for more VXDS submissions for clinical infectious disease and MCM studies, with an emphasis on proposals including genomic markers of efficacy. Secondly, Felciano recommended a public “genome-to-phenome” database that characterizes, at a systems biology level, how existing animal models are representative of given target endpoints and underlying mechanisms. This would allow for assessment of concordance between existing “well-characterized animal models” and in silico links between molecular systems and animal study endpoints. Finally, Felciano recommended the collection and integration of quantitative data on human and animal model immunity in normal, vaccinated, and infected individuals. This would allow for analysis of efficacy and common markers of response for existing treatments between humans and animals, as well as between animal models.
In summary discussion, participants discussed clinical trial simulations, in which virtual patients are put through in silico trials, a process that allows a company to model a wide range of trial designs and analysis methods, with the goal of reaching programmatic decisions more quickly, more cheaply, and with greater certainty. To leverage in silico modeling, there is a need for more complete, shared databases of human and animal study data (e.g., genome-to-phenome information; data on human and animal model immunity in normal, vaccinated, and infected individuals).
It was also suggested that a Bayesian, model-based, predictive framework should be developed that would essentially create in silico animals and a virtual human. It was noted that such a project would require per-
missions, funding, and collaborations on the scale of IBM’s Watson project or the Manhattan Project.
Using Biomarkers to Connect Animal Systems with Clinical Efficacy
Measurements of biomarker molecules are intended to allow connection of physiological changes with changes in outcomes or risks, explained Leigh Anderson, founder and CEO of the Plasma Proteome Institute. Biomarkers measured in blood and tissue are generally proteins, measured by immunoassay, or mRNAs (ribonucleic acid), measured using microarrays. Anderson offered cardiac troponin as an example of a successful biomarker; an increase in this protein is indicative of a recent heart attack.
Candidate biomarkers can be identified via in silico modeling studies, experimental studies, and by analogy with other species. However, Anderson noted, establishing biomarker validity requires significant effort, and all methods of hypothesizing biomarkers are extremely failure prone (> 99 percent attrition).
There are 109 proteins for which tests have been approved or cleared by FDA, and 96 additional proteins that can be tested for using laboratory-developed tests (that have not been reviewed by FDA). Approval of new protein biomarkers occurs at a very slow rate, Anderson noted: about 1.5 new protein biomarkers per year over the last 15 years. This rate, he said, is insufficient to meet broad clinical needs, without even considering MCM development.
Part of this dearth of new biomarkers is caused by the lack of a real pipeline for systematic discovery, development, and marketing of biomarkers. There are technological issues, including the lack of reliable, high-throughput assays for most biomarker candidates and the slow pace of development of new protein assays. There are also challenges in accessing large, existing sample sets in which to test the clinical relevance of a biomarker. The basic understanding of the mechanism for cross-species extrapolation is also very poor.
An ideal biomarker measurement method would provide certainty as to analyte structure, Anderson said. It would include internal standards and would have a method of eliminating interferences. He noted that mass spectrometry allows for very high-specificity measurements of proteins, with quantitative accuracy and internal standards, and it is inherently multiplexable. These assays can be developed very quickly, and there is the potential that the improved science content could allow more rapid approval by FDA.
In conclusion, Anderson said the challenges of identifying biomarkers for development of MCMs are similar to those for biomarkers for general clinical use. He emphasized the fundamental need to develop a
biomarker pipeline capable of systematically addressing complex biology. Biomarkers of efficacy for MCMs must be established in advance for the species involved in MCM testing. This requires a systematic evaluation of candidate biomarker homologs across a range of species, something that has not been done thus far. Success of an MCM biomarker also relies heavily on parallel mechanisms of disease, treatment efficacy, and recovery across species. Anderson also recommended that biomarker measurement technology be based on a high-confidence, rapidly approvable analytical method.
It is now feasible, Anderson said, to make specific, FDA-approvable assays for all 20,000 human proteins. Despite statistical design challenges, it is near feasible, he suggested, to test all possible proteins as candidate biomarkers against a broad range of diseases. If this were done, it could establish broad parallelism between human and animal systems.
Marietta Anthony of the Critical Path Institute (C-Path) presented information about efforts of the Predictive Safety Testing Consortium (PSTC) to achieve FDA qualification of seven renal toxicity biomarkers. The specific context of use that FDA allowed was for drug induced kidney injury in GLP rat studies and to support clinical trials. She remarked that the next step for C-Path is to conduct human clinical studies to assess their seven renal biomarkers. If the data are found to be important, they will be submitted to FDA for qualification. Donna Mendrick of the National Center for Toxicological Research (NCTR) at FDA commented that translating biomarkers can be extremely challenging. Mendrick noted that for kidney biomarkers, the gold standard in animal studies is histopathology, while in the clinic, the gold standard is measurement of serum blood urea nitrogen (BUN) and creatinine, which become abnormal at a later stage in disease. Anthony noted that the seven renal biomarkers that were qualified reflect histopathology far more effectively than BUN. Vikram Patel of the Office of Testing and Research at FDA’s CDER reminded participants the ultimate proof of efficacy of an MCM only comes when it is used in humans. In that regard, having a biomarker is very important to help assess whether the MCM, or which of several MCMs, is effective in an emergency situation. He expressed concern that very little attention is being paid to development of biomarkers.
Animal Models of MCM Efficacy
Throughout the workshop a number of participants discussed limitations of animal models.
Michael Kurilla of NIAID set the stage by noting in his remarks in the session on enterprise stakeholder perspectives (Chapter 1) that animal models are critical to MCM development; however, most animal models
are not suitable for a number of potential reasons. Animal models are infection models, Kurilla reminded participants, not disease models, and some infectious diseases are uniquely human diseases (i.e., there may not be any appropriate animal model). In addition, pathogenesis differs among various species, animals may not fully model host defense responses, and the availability of species-specific reagents may preclude the ability to define correlates. Extensive pathogenesis and natural history studies are necessary to demonstrate the validity of a particular species to replicate a human disease. There are also feasibility issues with conducting pivotal efficacy studies in animal models, including the development of GLP animal models to support licensure.
Elizabeth Leffel of PharmAthene provided formal remarks about animal models, and a panel discussion ensued. In developing MCMs under the Animal Rule, stakeholders need to think of animal studies as the equivalent of traditional phase I to II clinical trials, said Leffel. Leffel emphasized that while aspects of animal models can be standardized, animals cannot be “validated,” just as we cannot validate humans in clinical trials. She also noted that both humans and animals are heterogeneous populations, and no model can be 100 percent predictive of what will happen in humans.
The primary regulatory science tool for animal models is, of course, the FDA Animal Rule. There is a relatively new draft guidance published to support the Animal Rule, entitled “Qualification Process for Drug Development Tools.” This guidance, Leffel clarified, is not a mechanism to discuss product-specific tools or assays; rather, it is meant to address how animal models can be applied broadly to more than one drug.
Leffel identified four key regulatory science needs relative to animal models. First, she said, the essence of the Animal Rule needs to be consistently defined to product sponsors. There are different interpretations across FDA divisions, she noted, and sometimes between reviewers within the same division, of how to apply the Animal Rule. Second, appropriate review of MCMs based on risk and benefit is needed. These are high-risk, life-threatening diseases, about which clinical knowledge is often limited. Third, Leffel noted, there is a need for precompetitive mechanisms to share basic animal model information quickly. This includes shared proof-of-concept studies to avoid duplication (e.g., for NIAID-sponsored studies, information on basic models for vaccine studies is available in cross-referenced master files for sponsors). Fourth, as noted by others, there must be ways to bridge nonclinical models to expected human outcomes, such as surrogate markers, correlates of protection, clinical observation in animals, and pathology.
Moving forward, the first priority, Leffel said, is to develop a strategic plan for applying the Animal Rule. She suggested:
- This includes finalizing the draft guidances to reflect current FDA thinking1 and then applying these standards consistently within and across divisions at FDA and across sponsors. Areas that could be standardized by disease should be identified, and those areas that cannot be should be recognized. The strategy should also include preparing the MCM enterprise to accept more risk, as well as adopting provisions to mitigate risk (by, for example, special licensing conditions such as restricted or conditional licenses).
- A second priority, Leffel said, should be to leverage existing initiatives or form new partnerships to enhance data sharing. There are a lot of partnerships already in existence, she noted, and we need to start using them more effectively. She cited the FDA-NIH regulatory science initiative as a potential opportunity to allow FDA to leverage scientific resources from NIH and further engage FDA scientists in professional development.
- Third, she suggested, licensure review could be expedited by engaging cross-functional expert teams early on. Specifically, Leffel noted, in addition to meetings between product sponsors and FDA, it might improve communication further if an FDA scientist could also be present at the regular meetings between product sponsors that have U.S. government contracts and the relevant funding agency, at least at significant time points or milestones.
- Public-private partnerships, such as early development partnerships between industry and DoD and NIH labs, could be effective, and cross-industry precompetitive collaboration models should be pursued. Leffel also suggested that the agency should initiate a risk communication strategy to the public and establish dedicated cross-divisional review teams to evaluate MCMs under the Animal Rule.
Animal Model Case Study and Discussion
Drusilla Burns from the Office of Vaccines Research and Review in FDA’s CBER offered as a case study the pathway to licensure for anthrax vaccines. Animal models were developed, she said, that were thought to be appropriately reflective of human disease. It was demonstrated that an immune marker, anthrax toxin neutralization antibodies, correlated with protection in the animals, and the protective level of antibody was
_____________
1 A participant from FDA clarified the status of the draft Animal Rule guidance. Following the public comment meeting in November 2010, the draft guidance is undergoing major revisions and, as such, will not be finalized but will be republished as a draft to allow for another comment period on the revised guidance.
identified. Further studies demonstrated that the assay that is used to measure these antibodies was species independent, allowing for bridging to humans (i.e., in a clinical trial, measuring the antibody levels in humans could be used to predict the potential efficacy in humans). While this may sound simple, Burns said, it was very resource intensive, involving convening a workshop, conducting a literature review and interviews with experts, and forming an interagency animal studies working group that called upon vaccine manufacturers, academicians, and government contractors as needed. She emphasized the importance of the scientific partnerships between FDA scientists and other government scientists or outside scientists, and the involvement of diverse disciplines.
Judy Hewitt, chief of the Biodefense Research Resources Section at NIAID, emphasized the importance of qualification of animal models in a product-neutral manner. Patel of FDA suggested having a control animal dataset in a national database to which sponsors could compare their animal test data. Leffel commented that organizations such as the Alliance for Biosecurity, a public-private partnership, have taken steps to pursue development of a shared database of anthrax animal model data; unfortunately, that effort was underfunded. She noted that BARDA has picked up some of this work in anthrax and is in the early stages of working with industry partners to conduct meta-analyses on contributed data. She emphasized that adequate funding is critical to the success of these types of initiatives.
In summary discussion, it was noted that there is a clear need for a better understanding of animal models and how to apply them in a variety of settings. One of the most significant challenges is the extrapolation of animal immunological and pathophysiological data to the human setting, and participants discussed the need for new approaches to bridge nonclinical models to expected human outcomes (e.g., surrogate markers, correlates of protection). A number of workshop participants noted that it is unlikely one species model will reflect human disease adequately, and a compartmentalization strategy, pooling data from several species models, was proposed. Workshop co-chair, Les Benet, Professor in the Department of Biopharmaceutical Sciences of the University of California, San Francisco, called attention to a series of five forthcoming papers, part of the PhRMA initiative on predicting models of efficacy, safety, and compound properties, that found that, for 108 new molecular entities where both human PK and animal data were available, the animal models were poor in predicting (Poulin et al., 2011a,b; Ring et al., 2011). There was also interest in setting up precompetitive mechanisms to share basic animal model information quickly (including proof of concept studies to avoid duplication).
Picking up on earlier discussions, Benet suggested that a retrospec-
tive look at historical animal data from approved vaccines, anti-infectives, and other products could help inform discussions about the Animal Rule. He proposed looking at the data from animal studies as if that were all that was available, and making a hypothetical approval decision under the Animal Rule criteria, and then comparing how well that correlates with what is known from the human clinical trials that the actual product approval was based on. In other words, asking “Using all of the predictive methodologies that we have available today, if we approved this product under Animal Rule, would we have made the ‘right’ decision?”
In discussion about this proposal, Robert M. Nelson, senior pediatric ethicist at FDA, noted a concern that most animal work is done for preclinical toxicology purposes, and there may not be a robust enough dataset around the appropriate animal model for this type of exercise. A participant from industry countered that they often conduct proof-of-concept efficacy studies in mice and rats prior to initiating phase II trials in humans. Ed Cox, Director of the Office of Antimicrobial Products within the Office of New Drugs of CDER, said to keep in mind there are different types of animal models, those intended to look at an activity (e.g., pharmacokinetic/pharmacodynamic[PK/PD]) and those that are intended to mirror the human condition (involving an actual tissue site where infection would occur and some of the local factors at that site). In addition, there are models of infection and models of disease. Participants also noted the challenge and the importance of comparing “apples to apples” when looking at historical data. Adding to the complexity is the fact that tests are done by different laboratories with different standards. Another participant suggested that an alternative approach could be to conduct a new animal study with a current, approved drug or vaccine, in an appropriate model, and base the predictions on that data.
Key Messages: Nonclinical Approaches to Assessing Efficacy
In Silico Approaches and Biomarkers
- Clinical trial simulations hold promise for modeling a wide range of trial designs and analysis methods and could facilitate reaching programmatic decisions more quickly, more cheaply, and with greater certainty.
- There is a need for a biomarker pipeline capable of systematically addressing complex biology. Efforts should include systematic evaluation of candidate biomarker homologs across a range of species.
- Big science” could be envisioned for new projects, such as:
- A Bayesian, model-based, predictive framework could be applied to create a “virtual human”; such a project would require momentum and collaboration on a large scale.
- Make specific assays for all 20,000 human proteins; statistical design challenges would need to be overcome.
Animal Models
- Building databases of existing animal models (genome to phenome) could allow for assessment of concordance between existing “well-characterized animal models” andin silico links between molecular systems and animal study endpoints.
- A control animal dataset in a national database would permit comparisons by sponsors of their animal test data.
- Scientific partnerships, including creation of an “ecosystem” of collaboration and a multidisciplinary approach, is important for addressing difficult regulatory science problems in assessing efficacy.
- Funding and substantial resources are essential to sustain interagency, public-private, and other enterprise partnerships and collaborations.
SAFETY AND REAL-TIME MONITORING
In a public health emergency, some of the MCMs used may be new molecular entities for which efficacy studies in humans were not done, and predeployment safety information is limited, said panel moderator, Carl Peck, of the University of California, San Francisco. He noted that once a new MCM is deployed, it will be especially important to monitor for side effects and to confirm effectiveness (so that use of an MCM that is not effective can be discontinued and further risk of adverse events reduced).
Toxicology Markers
Robert House, president of DynPort Vaccine Company, presented about toxicology markers from a vaccine development standpoint, noting that there are a variety of primary toxicological concerns. Local toxicity or “reactogenicity,” while not a main concern for small molecules, is a primary concern in developing vaccines. As with any drug, one must also be concerned with systemic toxicity. Toxicity testing is performed under GLP conditions to ensure the cleanest results, using GMP (or GMP-like) material, in a relevant animal model, House said. For vaccines, a standard toxicology profile must also include assessment of immunogenicity. Developmental toxicity and immunotoxicity are also assessed. Vaccine adjuvants must be tested as if they were a new chemical entity (and as such are tested twice, alone and as part of the vaccine). Other additives
TABLE 3-1 Prediction of Clinical Outcomes: Preclinical Toxicology Studies vs. Clinical Studies
| Parameter | Toxicology Studies | Clinical Studies |
| Survival | Yes | Yes |
| Pain upon injection | Difficult to assess | Yes |
| Fever | Dependent on animal model | Yes |
| Headache/malaise | No good animal models exist | Yes |
| Injection site reactions | Yes | Yes |
| Clinical signs | Yes | Yes |
| Body weights | Yes | Useful? |
| Clinical pathology | Yes | Yes/not usually |
| Necropsy, histopathology | Yes | Generally not |
| Antibodies | Yes | Yes |
| Immunotoxicity | Dependent on animal model | Yes/not usually done |
SOURCE: Robert House. 2011. Presentation at IOM workshop; Advancing Regulatory Science for Medical Countermeasure Development.
that go into vaccines, such as excipients or preservatives, must also be individually assessed for toxicity. Depending on how a vaccine is administered, it may also be necessary to assess the toxicology of the administration device.
Standard preclinical toxicological endpoints include body weights (as a measure of robust health); clinical observations (are the animals behaving normally); clinical pathology (including hematology, clinical chemistry, and other immunogenicity studies);anatomic pathology (including organ weights and histopathology to assess intended effect at the immune system target, as well as any effects at other points in the immune system); and local tolerance.
House compared preclinical toxicology studies to clinical studies in their ability to predict clinical outcomes (Table 3-1). He noted that several parameters (in italics)—pain upon injection, fever, headache and malaise, and injection site reactions—are often considered to be rather subjective and can be difficult assess in animal models.
Electronic Monitoring of Adverse Events
Kenneth Mandl of the Harvard Medical School Center for Biomedical Informatics characterized four main sources of clinical electronic health data:
- Reported data—Voluntary or mandated reporting of adverse events to FDA or disease outbreaks to CDC;
- Ambient data from the health system—Data that are produced through the routine processes of care;
- Meticulously collected data—Registries on a selected population of patients; and
- Patient-reported data—Consumer technologies, social networks, personal health records, and other technologies that can be accessed directly.
Mandl cited a study by Basch (2010) that highlights the value of patient-reported data. Individual contributions to drug safety data can include adverse effects, efficacy endpoints, adherence, satisfaction, and quality of life, as well as concomitant over-the-counter and complementary/alternative medicines.
A challenge to electronic data monitoring is that electronic medical records are managed by software that runs locally. Each one of these systems is different and often proprietary and unmodifiable. “The data are hard to get in and virtually impossible to get out,” Mandl said.
To help address this, Mandl and Kohane (2009) have proposed a platform approach to health data software design, for which many applications or “apps” could be developed (similar to the iPhone platform, Mandl explained). He noted that in such an environment, apps compete with each other in the apps store, so functionality and usability would be expected to improve continually and prices would be competitive as well. Mandl also referred workshop attendees to the “SMART Apps for Health” challenge on the challenge.gov website, a contest to develop apps that provide value to patients.
Mandl noted there has been over a decade of work on biosurveillance (real-time monitoring for detecting an emerging epidemic or an outbreak), and there are some fairly sophisticated systems and techniques that he suggested could be relevant to real-time safety surveillance of MCMs.
In closing, Mandl said that the Obama administration has made a $48 billion investment in health information technology for use across multiple sites, predominantly primary care facilities and hospitals, and he encouraged FDA to become very involved in the discussions of health information technology deployment.
Discussion
Richard Forshee of CBER explained that voluntary reporting of data (e.g., to the Vaccine Adverse Event Reporting System or VAERS) is a key component of FDA’s activities in near real-time safety surveillance in the
postmarket environment. An advantage of this passive reporting system, he said, is that it is fast. However, as it can be very labor intensive to sort through the many records in the system, the agency is developing text-mining systems that can narrow down the data to sets of adverse event reports that are most relevant for the medical officers to address.
Henry Francis, Deputy Office Director in the Office of Surveillance and Epidemiology of CDER, said that new approaches are needed in data collection (as noted, current systems are passive), quantitative analysis (FDA receives roughly 800,000 reports every year that are read by 43 people), qualitative assessment (e.g., a pharmacist may report that people are taking a medication incorrectly, such as chewing a Spiriva capsule instead of inhaling the contents), and reporting (to regulatory decision makers and the public).
Robert C. Nelson of Product Safety Assurance, Inc., recommended that the Postlicensure Rapid Immunization Safety Monitoring (PRISM) system that was developed to monitor the safety of the 2009 H1N1 influenza vaccine be institutionalized and used for all vaccines. He also noted that the FDA Adverse Event Reporting System (AERS) could be a starting point to help detect serious rare events associated with MCMs; however, monitoring the efficacy of the nonvaccine MCMs will be a significant challenge.
A participant drew attention to a collaboration between the Indiana University (IU) Medical Center, Eli Lilly, and the Regenstrief Institute for real-time adverse event monitoring. Data are collected at the point of care via a MedWatch-like form embedded into the electronic medical record system at IU. If a physician wants to report an adverse event, the form pops up in the system, and the completed form is simultaneously entered into the patient’s electronic medical record and forwarded to the manufacturer if there is a drug involved.
Mendrick cautioned that it can be very difficult to discriminate between drug-related postmarket adverse events and disease-related processes in humans.
Key Messages: Safety and Real-Time Monitoring
- Collaborative working groups should be convened to share data and experiences with respect to safety biomarkers.
- App” technology for surveillance, response, and adverse event monitoring holds promise.
When assessing microbial threats, strain identity is secondary to the organism’s resistance profile, asserted Kevin Judice, CEO of Achaogen, Inc. For example, the gram-positive bacteria, methicillin-resistant Staphylococcus aureus (MRSA) and Bacillus anthracis or anthrax, are similar at a genetic level and phenotypically. As such, an antimicrobial to treat MRSA will generally be effective against anthrax as well. If the microbial strain in question is, for example, susceptible to ciprofloxacin, there is generally no problem with treatment. However, strains that are resistant are essentially untreatable. The rise of ciprofloxicin resistance in E. coli over the past decade has been significant. Yersinia pestis (plague) is closely related to E. coli, and resistance genes can transfer naturally, resulting in ciprofloxacin-resistantY. pestis. So although this is a naturally occurring process in “civilian strains”(those found in nature), the result is a resistant strain of a biothreat agent.
Strategies to address drug-resistant civilian strains can suffice for MCMs as well, Judice said. The early development pathways are essentially identical (excluding any specialized microbiology, or specialized laboratories for handling threat strains). As later-stage clinical trials with threat agents are impossible, it is necessary to rely on civilian indications for clinical trials.
As an example, Judice discussed nosocomial pneumonia, which is a good proxy for plague. Judice provided background that an investigational Achaogen compound, ACHN-978, has been shown to be active against resistant strains of pseudomonas and acinetobacter, as well as Y. pestis and Francisella tularensis. To demonstrate the clinical utility of ACHN-978, the company is focusing on nosocomial pneumonia associated with resistant pseudomonas and acinetobacter.
Current guidance on trial design, however, calls for trials that Judice said are often too big or too complex for a small company to conduct. In a standard noninferiority clinical trial design, patients with suspected infection are typically randomized to control (standard of care) or investigational treatment arms before bacterial culture results are available (i.e., before the pathogen or its resistance profile is known). A subset of each group will have resistant pathogens; however, because randomization was done before their resistance profile was known, groups may be unbalanced with regard to patients with multidrug-resistant strains, making analysis challenging. If the intent is to test the investigational product for efficacy against resistant strains, this is not an optimal design. Judice suggested that, under current guidelines, such a trial would need roughly 2,500 patients, would cost around $300 million, and would take five to seven years to complete, which is generally beyond the resources or abilities of a small company.
To move forward, Judice said that industry and FDA must work together, focusing on reassessing benefits and risk. Industry, he said, needs to develop better molecules that solve real problems (e.g., antimicrobial resistance) and demonstrate benefits in the clinic (superiority versus resistant strains). FDA may need to accept more risk, he said, such as alternative noninferiority margins for a novel agent to treat life-threatening multidrug-resistant infections.
Judice emphasized the need to enroll more patients with resistant organisms into clinical trials and suggested a superiority trial design in which patients are enrolled but are not randomized until after culture and sensitivity results are known. To facilitate this, he added, rapid diagnostic tests are needed. Patients with resistant pathogens would then be randomized to the standard of care or investigational therapy.
In closing, Judice expressed concern that we are entering a “post-antibiotic era”for some types of infections. We must work together to solve these problems—better molecules, improved clinical and regulatory pathways—soon, he said.
Discussion
Many participants noted that there is a great need for new antibiotics for patients. Cox expanded on the concept of developing compounds for conventional disease that are also useful against a particular threat agent, often referred to as “dual use”drugs.2 While this may not be applicable in all settings, he said, studying a drug in a patient population with a common infectious disease provides an opportunity to collect safety, PK, and appropriate dosing data. It also provides an important opportunity to evaluate efficacy; often, for example, the pathology of the conventional disease may have similarities to the disease caused by the threat agent. In addition, if a product has a market for treatment of a conventional disease, it would generally be in production and available during a public health emergency.
Cox concurred that rapid diagnostics could play an important role in making clinical trials of antimicrobial drugs more efficient. Another challenge for antimicrobial clinical trials is that concomitant or prior antibacterial drug therapy cloud the assessment of the efficacy of the investigational drug. Resource barriers, as discussed by Judice, are also a concern,
_____________
2 Note that dual use is sometimes used to refer to the potential for technology to be both used for biomedical research and misused for hostile purposes (e.g., antibiotic-resistant strains developed to facilitate research on relevant antimicrobial therapies also being used as agents of bioterror). However, in this summary, dual use refers exclusively to a product and its potential to treat both conventional and bioterror-related diseases or conditions.
and Cox suggested that economic incentives may be helpful or necessary to offset some of the economic risk that product developers face.
Panel moderator Linda Miller, director of Clinical Microbiology at GSK, suggested the need for guidance that better addresses the issues of antimicrobial trials that were raised (e.g., size of trials, patient populations, appropriate margins for noninferiority trials, use of rapid diagnostics). She also suggested that approval based on pathogen-specific indications be considered.
Workshop co-chair John Rex, of AstraZeneca, raised a concern that the superiority trial approach may work well for the first new drug being compared to older drugs, but it could make it impossible to get further comparable new drugs approved as subsequently superior to that first new drug. Judice responded that a good, well-controlled superiority trial against classic drugs could then set the stage for better noninferiority trials down the road, with potentially more appropriate noninferiority margins. Cox agreed, noting that if a new drug that shows superiority is found to be safe and effective, then it would become the standard of care on which to base a noninferiority margin. It is important to have pathways available for noninferiority trials in conditions such as hospital-acquired pneumonia, ventilator-associated pneumonia, complicated urinary tract infection (UTI), complicated intra-abdominal infections, or community-acquired pneumonia.
A participant pointed out there is a rich trove of data housed at FDA from many years of clinical trials in which there were patients enrolled who had multidrug-resistant organisms and for whom the investigational treatment failed. He suggested there should be a way to de-identify and share those data for reference by researchers.
Key Messages: Antimicrobials
- There is a need to enroll more patients with resistant organisms into clinical trials through superiority trial design, with randomization after sensitivity results are available. Regulatory guidance should be developed to support this, addressing:
- Trial design issues such as trial size and appropriate margins for noninferiority trials.
- Use of rapid diagnostic tests.
- Consider making available data currently held at FDA on failure of investigational treatment in patients with multidrug-resistant organisms.
Vaccines
The current product development timeline for vaccines spans 10 years or longer and can cost $500 million or more, said Alan Magill, program manager at the Defense Advanced Research Projects Agency (DARPA). Vaccine development is also a very-high-risk endeavor.
Knowledge of a gene sequence of a recognized immunogen from a known pathogen (e.g., influenza hemagglutinin) does not guarantee an immunogenic vaccine candidate, Magill pointed out. He commented that vaccine discovery tends to be an empiric trial-and-error process, adding that we need a better understanding of how to design and build an immunogen or antigen that leads to protective antibodies. Toward this goal, the DARPA Protein Design Processes Program is developing tools for the design and synthesis of new functional proteins. Researchers ultimately hope to be able to design, within 24 hours after notification of a threat, a new complex protein that will inactivate the pathogenic organism.
Another issue for vaccine development is establishing immune correlates of protection, which Magill said are really biomarkers for efficacy (e.g., hemagglutinin-inhibition titers for influenza, neutralizing antibody titers for yellow fever). Identification and qualification of these biomarkers starts with collection of specimens and correlation of data to clinical outcomes in clinical trials.
With regard to animal models, the question is whether they are predictive of vaccine protection in humans. Animal models of vaccine protection are expensive and increasingly more difficult to do. Magill questioned the need for GLP toxicology studies in animals prior to phase I clinical vaccine studies, as in his experience they rarely identify problems. DARPA is currently using the Modular IMmune In Vitro Constructs (MIMIC, from Sanofi VaxDesign) system to determine human immune responses directly from an antigen in an assay plate. The ultimate goal would be for this system to eliminate the need for human testing in some settings. In reality, Magill said, it should assist in culling down selection of candidates; clinical trials will likely always be needed. Human tissue engineering is another area that DARPA is aggressively moving into as a potential tool for testing medicines in lieu of animal models (Figure 3-2).
Magill commented that in biological manufacturing, regulatory requirements have led to a common notion that the “process is the product,” in that licensure requirements adhere to the “recipe” of how the product is manufactured. Conventional wisdom therefore holds that a product-specific facility is needed, which is very expensive, lengthy, and makes technology transfer particularly challenging. Magill described DARPA efforts to address this by creating “modular GMP” units that can be moved around (Figure 3-3). These mobile bioprocessing facilities
FIGURE 3-2 Human tissue engineering as a potential tool for testing medicines in lieu of animal models.
SOURCE: Alan Magill. 2011. Presentation at IOM workshop; Advancing Regulatory Science for Medical Countermeasure Development.

FIGURE 3-3 Mobile bioprocessing facilities (MBFs).
SOURCE: Alan Magill. 2011. Presentation at IOM workshop; Advancing Regulatory Science for Medical Countermeasure Development.
(MBFs) would have, for example, a protein-processing suite (or whatever is needed) and could result in reduced manufacturing costs and increased flexibility.
Another issue for consideration is antigens versus adjuvants versus vaccines. As subunit vaccines have not proven sufficiently immunogenic, various adjuvants have been used to try to boost immunogenicity and protection. Currently, FDA views approval of a vaccine as approval of a formulation (e.g., antigen and adjuvant). Magill suggested that there is a need for a qualification process for adjuvants, with generation of a drug master file where it would be possible to evaluate multiple antigens.
In closing points, Magill reminded participants that technology will not solve all of the problems of drug development and MCM development in particular. There is a need for better understanding of natural history and pathogenesis of disease and of immunology.
Adjuvants
Debbie Drane, senior vice president for Research and Development at CSL Biotherapies, highlighted some of the main problems faced in adjuvant development. Adjuvants are essentially platform technologies, she said, but they are not viewed that way by regulators (or product developers). Adjuvants themselves are not registered. There is a need to better understand the mechanisms of action of adjuvants. Another unknown is the long-term effects of strong immunopotentiators. There is a lack of preclinical biomarkers particularly for safety of adjuvants. There is also a lack of transparency around adjuvant research, as adjuvant development has largely been industry based.
Drane offered the CSL adjuvant, ISCOMATRIX, as a case study in adjuvant development and evaluation. ISCOMATRIX is a proprietary, saponin-based adjuvant in which ISCOPREP saponin is complexed cholesterol and phospholipids. CSL sought to understand the mode of action (immunogenicity and safety) and the mechanism of action. Animal models were used to evaluate immunogenicity. Different adjuvants have effects on different arms of the immune system, she noted; for example, water-in-oil adjuvants induce antibody responses but are less effective in inducing T-cell responses. Animal models were also used to provide information on the kinetics and potential mechanisms of the response. It can be very useful, Drane said, to understand which immune cells the vaccine formulation is targeting. In vitro studies in human immune cells are done to help link human response to the animal models. Another approach is using human cell lines and in vitro biomarkers (e.g., induction of pro-inflammatory cytokines) to predict potential safety signals associated with adjuvants. Gene profiling can also aid understanding of the tolerability of vaccines.
Safety of an adjuvant is key, Drane emphasized. CSL and other partners are working to establish an integrated database of adjuvant-related safety information from both nonclinical and clinical studies.
In summary, Drane said that vaccine developers must share and evaluate knowledge, and she suggested establishing an adjuvant advisory group that includes FDA, industry, and academia to convene workshops around a variety of adjuvant-specific issues such as safety biomarkers, perceived biothreats, and adjuvant combinations. Drane concurred with Magill that there is a need for a different approach to licensing adjuvants for MCMs, such as broad licensure for adjuvants that could then be used with virtually any MCM vaccine.
Discussion
Hana Golding, chief of the Laboratory of Retrovirus Research at CBER, said that one of the key strengths of FDA scientists is versatility—specifically, the ability of scientists to move from one pathogen to another, from one type of research to another, to address developing diseases. FDA scientists are also in a position to proactively identify gaps that will need to be addressed for new products to move into humans. Golding offered several examples of FDA scientific advances that were shared with MCM developers, such as the development of a high-throughput vaccinia virus neutralization assay, which the agency published and shared with multiple manufacturers of vaccinia immunoglobulins and new vaccines. She also highlighted several ongoing areas of research, such as faster or alternative approaches to assessing vaccine potency and new methodologies to evaluate tumorigenic cells to be used as cell substrates in lieu of egg-based vaccine processes (e.g., for tumorigenicity, oncogenicity, unknown adventitious agents). Basil Golding, director of the Division of Hematology at CBER, added that an advantage of FDA research and development is that the agency scientists are then very familiar with the parameters and pitfalls of various assays, and can provide valuable technical assistance to sponsors. David Frucht, chief of the Laboratory of Cell Biology at CDER, also agreed, noting that promoting research (both regulatory and product related) at FDA not only helps overcome development hurdles, but it establishes the subject matter experts at FDA that can provide rational and expedited reviews prior to and during emergencies.
With regard to vaccine development, Frucht emphasized that vaccine potency is a critical product quality characteristic, and the potency assay used should reflect the presumed mechanism of action of the product in humans. This is especially relevant, he said, when the Animal Rule is being used to establish clinical efficacy. He advocated for continued research into the most biologically relevant bioassays and expansion of
existing programs. Frucht also said that more in-depth knowledge of product quality early in the development cycle could help accelerate availability of MCMs for potential use under an EUA. The manufacturer should have an in-depth understanding of the critical process parameters and the manufacturing process early on in development. If done late in the development cycle, changes to manufacturing (e.g., scale up) and changes in the producer cell type can affect product quality, resulting in delays.
Alan Shaw, chief scientific officer for Vaxinnate, reinforced Magill’s point that one of the major problems of viral vaccine development is the protracted time frame. To help speed development, Vaxinnate is using a platform approach in which the company is inserting antigens onto flagellin (a toll-like receptor agonist). This, he explained, renders highly visible a protein or an antigen that is otherwise basically invisible to the immune system. This technique can be used for a variety of targets. Vaxinnate’s primary focus is influenza, but Shaw noted that they are also applying this approach to flaviviruses (e.g., West Nile virus, Japanese encephalitis, yellow fever, hepatitis C virus). There are other relevant platforms that could be applied, and Shaw suggested a platform consortium be formed. He also suggested a viral structure consortium that would develop common databases of structural information characterizing the different classes of virus that are likely to emerge as human pathogens.
Ed Nuzum of NIAID noted that there are numerous challenges for MCM sponsors beyond the Animal Rule: process development, manufacturing, product characterization, and potency assays are just examples. Despite FDA’s willingness to communicate early and often with sponsors, in practice it often does not happen for various reasons. He expressed concern that FDA’s action teams, discussed earlier by Luciana Borio of FDA, address higher-level, crosscutting, cross-center issues but not the product-specific concerns that sponsors may have. Nuzum supported the idea of working groups or “product acceleration teams” to enhance communication between sponsors and FDA, especially for companies that lack adequate expertise.
Key Messages: Vaccines and Adjuvants
- Manufacturing and other process changes should be reviewed to determine what process requirements are necessary, and creative solutions such as MBFs should be investigated.
- Vaccine potency assays should be further studied to ensure they are biologically relevant and reflect the presumed mechanism of action in humans.
- There is a need for a defined regulatory pathway or qualification process for vaccine adjuvants; broad licensure for adjuvants that can be used with virtually any MCM vaccine should be considered.
- An adjuvant advisory group, including FDA, industry, and academia, could be formed to convene workshops on adjuvant-specific issues such as safety biomarkers and adjuvant combinations.
- Consortia groups should be developed to address issues such as:
- platform technology, and
- viral structure, to develop common databases of structural information characterizing virus classes likely to emerge as pathogens.
SYNTHETIC AND COMPUTATIONAL BIOLOGY AND PLATFORM TECHNOLOGIES
Synthetic Biology
John Glass, senior scientist at the J. Craig Venter Institute, defined the emerging field of synthetic biology as the production of biological life, or essential components of living systems, by synthesis, to make new organisms with extraordinary properties. What makes synthetic biology possible is the fact that genes can be synthesized using the four bases that make up DNA. This capacity to synthesize DNA is changing the field of biological research, he said.
Glass described six examples of how synthetic biology can be used to advance MCM development. First, synthetic bacterial cells can be used to produce live attenuated vaccines (or the equivalent). The first bacterial cell with a chemically synthesized genome was Mycoplasma mycoides. This was the result of two technologies that were developed at the Venter Institute: the “Gibson Assembly Method” of rapidly and efficiently assembling oligonucleotides by overlapping synthetic DNA molecules into larger molecules, and genome transplantation, the capacity to transfer a whole, naked bacterial genome from one organism into another so the recipient cell is converted into the same organism as the donor cell (Figure 3-4).
With this approach, for example, one could synthesize pathogenic bacteria devoid of all virulence factors, which could then potentially be used as live attenuated vaccines, Glass said. The transplanted genomes could contain only those genes necessary to generate an immune response and keep the cell alive. Adjuvants could be built into the organisms, and if desired, suicide genes could be inserted so that the organism could only survive for a specified number of generations. Another example would be the synthesis of gram-negative bacteria that produce outer membrane
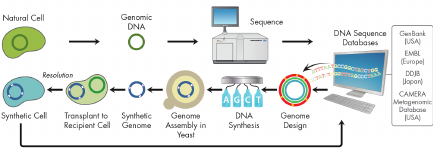
FIGURE 3-4 Synthesis of bacterial cells.
SOURCE: John Glass. 2011. Presentation at IOM workshop; Advancing Regulatory Science for Medical Countermeasure Development.
vesicles containing immunogenic proteins from a variety of pathogenic bacteria, kind of an “omnibus vaccine,” Glass said. This allows for immunization with membrane-bound proteins without exposing people to live cells.
Second, synthetic biology offers the potential to make influenza virus vaccine production better and faster. Using reverse genetics to synthesize hemagglutinin and neuraminidase can decrease the time needed to achieve a pure culture of vaccine seed stock from 35 days to 7 days (Figure 3-5). However, Glass said, while the technology to produce influenza virus vaccines has advanced greatly over the last 20 years, the regulatory processes for licensing new pandemic and seasonal influenza virus vaccine have not progressed in concert.
Other examples of potential uses of synthetic biology for MCM development are:
- The synthesis of a bacteriophage for use as an antibacterial therapeutic.
- Microbial manufacturing of drugs currently obtained from scarce natural sources (e.g., the antimalarial drug, artemisinin, originally extracted from Chinese wormwood bark, is now synthetically engineered).
- Synthesizing antigens for use in diagnostic assays and evaluation of therapeutics and vaccines (e.g., expressing protein epitopes from four different Borrelia burgdorfigenes on one recombinant protein to make an effective B. burgdorfi diagnostic).
- Finally, synthetic biology can be used to develop new tools and approaches to discover novel therapeutics.
FIGURE 3-5 Accelerating seasonal influenza virus vaccine development.
NOTES: HA, hemagglutinin; JCVI, J. Craig Venter Institute; NA, neuraminidase; WHO, World Health Organization.
SOURCE: John Glass. 2011. Presentation at IOM workshop; Advancing Regulatory Science for Medical Countermeasure Development.
In closing, Glass said that fast, accurate DNA synthesis of genes and genomes can eliminate the time and effort needed to obtain a natural DNA template for known pathogens or unknown or newly described infectious agents. Accurate, inexpensive synthetic DNA makes it possible to create many new therapeutic and diagnostic tools that were previously impossible or impractical to build. Engineering of microbes with extraordinary properties will soon be done in days or weeks.
However, Glass predicted, this accelerated pace of new biotechnology development may swamp FDA capacity to evaluate what he said would be “an avalanche of new drugs, vaccines, therapeutic, and diagnostic assays.” To help advance development, Glass suggested that older licensed compounds, vaccines, and assays that are made using new synthetic biology tools should be considered equivalent to their predecessors.
Computational and Systems Biology
Modern day virologists and immunologists have access to substantial technology, information, and computational infrastructure, said Michael Katze, Professor of Microbiology at the University of Washington, noting,
however, that even though the tools are there, scientists are ill equipped and ill trained to deal with the data. In addition, it is still not fully understood how a virus kills a cell, or how host attributes contribute to response. For example, when using model systems, if key findings do not align, is that a problem with the model or is it related to other attributes of the host?
Handling next generation sequencing, microarray, proteomics, and other “-omics” data requires development of a sophisticated information technology infrastructure, which, Katze pointed out, is expensive and complex. These new tools and technologies are important to virologists because they aid the study of the global impact of virus infection on host gene expression; and they can be used to discover cellular regulatory pathways targeted by viruses, identify new cellular targets for antiviral therapies, and develop new vaccines. Katze proposed an integrated approach to infectious disease, combining traditional histopathological, virological, and biochemical approaches with functional genomics, proteomics, and computational biology.
As an example, Katze described a systems biology approach to studying the 2009 H1N1 pandemic across species. He noted that H1N1 does not generally kill swine, and perhaps understanding why pigs do not die will provide a better understanding about host defense against influenza. Studies of the transcription of immune-related genes across mice, macaques, and pigs suggested that the nature of the immune response in each species may be quite different. Overall functional analysis showed significant alteration in immune response genes in all three species. Although the numbers of genes changing was similar, the precise genes changing were very different.
Katze suggested that applying systems biology to vaccine development could potentially help detect early signatures of efficacy and offer information about why a vaccine fails. Once host factors of virulence and pathogenesis are identified, systems biology could be applied to drug development, for example, to “score” drugs based on response to treatment, or to better understand mechanisms of action (including off-target effects of drug treatment and toxicity) through gene expression studies.
Computational biology will be key to modeling and predicting host response, Katze said, adding, however, that several challenges exist. As the landscape for certain high-throughput technologies is still being defined, there is a need to be able to accommodate constant evolution. Cost is an issue, as is communication; in other words, there is a need to connect the right people (computational scientists, mathematicians, biologists) as well as teach a new generation of scientists a new vocabulary and a new way of viewing science. To begin to address these challenges, Katze recommended:
- Early integration of high-throughput data collection in drug and vaccine development as a mechanism to understanding global impact, off-target effects, and biomarkers for efficacy.
- In silico screening for drug-drug interactions and as a tool for novel drug discovery.
- Increased interdisciplinary crosstalk between computational scientists and bench scientists to define standards for study designs.
In discussion, a participant raised the issue of training the next generation of the workforce to advance regulatory science outside the context of a particular product. Katze noted that universities have started offering interdisciplinary programs in computational biology where previously there was very little interaction between computer science and biology. A participant from FDA added that the agency has been putting resources into science computing capacity and is training agency reviewers and researchers to be able to use them.
Platform Technology
As an example of the use of platform technology to advance MCM development, Patrick Iversen, senior vice president of research and innovation at AVI BioPharma, described his company’s approach for the rapid development RNA-based therapeutics. AVI’s platform is based on the development of translation-suppressing oligomers that target single-stranded RNA (which could be from a host cell or from the pathogen), preventing the assembly of the ribosomal complex on the mRNA transcript, thereby preventing the production of a specific protein.
AVI has developed a predictable way of designing the oligomers, which makes the platform very flexible and allows for very rapid response. They have defined both the optimal position in the transcript, and the optimal length of the oligomer, and are developing a database of oligomers for a growing list of viral and bacterial targets and host genes. This knowledge base, Iversen predicted, would allow AVI to develop a putative solution to a new threat in a matter of hours.
Iversen noted that AVI currently has open INDs for oligomers for Ebola and Marburg viruses. Studies in mice, guinea pigs, and nonhuman primates have shown significant protection (i.e., survival). Crossover studies confirmed the specificity of the oligomers for the intended target (i.e., the Ebola virus oligomer was not effective against Marburg virus, and vice versa). Other endpoints investigated included dose-dependent survival increases, reduction in clinical signs, reduction in viremia, increase in platelet count, and improvement in both hepatic and renal markers of toxicity.
In closing, Iverson raised several questions regarding animal studies and human safety testing. For animal models, he asked, how should a viral challenge strain be chosen? For example, would it be better to use Marburg Angola or Marburg Musoke? Quasi-species characterization could reveal that there are elements or portions of both viruses in every outbreak. And the next outbreak will be a new quasispecies. “Deep sequencing” technology, he suggested, could provide insight into how to choose challenge strains.
Iversen also questioned whether the use of healthy volunteers for safety assessment is necessary for MCM development. He noted that in normal healthy volunteers, the dose limiting toxicity may fall below the anticipated therapeutic dose. How should that limitation be interpreted; what distance between anticipated therapeutic benefit and dose limiting toxicity will be tolerable? Also, how should the size of the required human safety database be calculated? He asked, if these MCMs will never be used unless there is an outbreak, and will be used only used under an EUA, is a human safety database needed?
Discussion
William Fogler, senior director of portfolio planning and analysis at Intrexon Corporation, pointed out that the need for rapid response generally occurs under worst-case scenarios, often in association with compromised infrastructure. While these synthetic, computational, and platform technologies offer tremendous promise to respond rapidly to a pathogenic threat, they must be scalable and deliverable under such a scenario. He suggested that there are additional technologies that exist in terms of generating DNA vaccines, in which modular components can be predesigned, stored, and ready to assemble on short notice. Other modules could be devised in which immune-enhancing agents could be quickly assembled. These modules in the structure of a DNA vaccine can be under the control of inducible promoters, so that following injection of the vaccine, an activating ligand (e.g., a small molecule) would be taken orally to “turn the vaccine on,” and upon removal of the ligand, it would be “turned off.” This also offers the possibility of a needle-free vaccine-boosting mechanism, Fogler said.
Mendrick said that researchers at FDA are looking at these emerging technologies and are trying to anticipate and solve some of the questions that may arise. For example, NCTR has a nanotoxicology core facility that is looking at genetic toxicity assays to evaluate the carcinogenicity of nanoparticles.
Harvey Rubin, executive director of the Institute for Strategic Threat Analysis and Response at the University of Pennsylvania, emphasized
that computational biology is not simple mathematics. The scale of computational biology spans angstroms to kilometers, and nanoseconds to millennia, he said. The processes are very complicated, and include, for example, deterministic, stochastic, continuous, discrete, or hybrid processes. With regard to organization, the system could be structured, unstructured, or homogeneous. There are complexities and interdependencies that make modeling biological systems especially difficult, Rubin said. Motivations to do complicated mathematical modeling include the need to predict something (e.g., protein structure, epidemiologic patterns), to design something (e.g., new molecular structures, new controllers and regulators, new phenotypes), or to interpret something (e.g., data, patterns).
Rubin highlighted several research priorities that can help populate some of these mathematical models:
- There are many model-specific questions that need to be answered, such as what are the effects of interventions on infectivity, and what are the effects of disease and interventions on immunocompromised hosts?
- There is also general research needed on organizational structures, risk communication strategies, interdependencies (e.g., how the environment, economics, or politics impact the model), and health impact information.
- Also to be resolved is who should be funding this work—NIH, the National Science Foundation, DARPA, FDA, or industry.
Significant resources are dedicated to identifying and characterizing an emerging biological threat, said Daniel Wattendorf, program manager in the Defense Sciences Office of DARPA, but rarely is there subsequent broad distribution of new diagnostic assays for the identified threat to point-of-care settings. In cases where the decision to quarantine or treat is time sensitive, the turnaround time to ship samples to a reference laboratory is prohibitive.
Wattendorf cited several barriers to more rapidly fielding diagnostics for emerging threats. In some cases, the diagnostics platforms have not been made suitable for use in distributed settings. As an example, Wattendorf pointed out that PCR has been in use since 1983, yet no PCR-based diagnostic test is approved for a physician office setting. Additionally, if diagnostic tests are not already in place before an emergency, it is very difficult to get physicians to employ them in a crisis if they do not have prior experience with the test or have not been shown evidence of
utility. In the absence of specific diagnostic tests for emerging threats, there is interest in developing panels of early detection biomarkers that could detect a host immune response before an individual begins to exhibit symptoms of a disease.
Sample collection is another challenge for diagnostic testing. Current biospecimen collection generally involves collection of wet samples, such as through test tubes, which requires that the patient have access to medical personnel (e.g., a phlebotomist) who can collect the sample, and which also may require cold storage. There is also the option of taking dried blood spots on filter paper, but according to Wattendorf such samples have limited use. In this regard, Wattendorf suggested that a role for regulatory science would be the development of new formats for simple, self-collected biospecimens, formats that would be optimized for specimen source (blood, urine, etc.) and analyte class (specific proteins, types of RNA, etc.), and would be stable during storage to facilitate functional assays.
Wattendorf also noted that currently, teams of experts travel to a site, collect samples, and return to CDC or the DoD to run tests and identify the new threat. He suggested that, instead of moving the sample, it could be possible to move the data electronically. The use of highly multiplexed platforms could facilitate local testing, and the data could then be sent to a central facility for analysis. This would be faster and would provide distributed diagnostic capability where there is unmet need.
In summary, Wattendorf listed several questions for discussion:
- Can universal sample storage formats be developed for dried or near-dried self-collected biospecimens that show equivalence to fresh samples?
- Can highly multiplexed protein or molecular diagnostic platforms be developed that are suitable for use in a physician office base setting, from which data could then be sent for interpretation by highly trained laboratorians at a remote site?
- Are measurements of immune or metabolic status useful in the absence of a diagnostic test for a specific pathogen? If so, what should be measured? Could it be measured at the point of care? And, as it is not specific to a given disease, what would be the regulatory pathway?
In the panel discussion, Charles Daitch, CEO of Akonni Biosystems, said that from a technical perspective, the capability to communicate from remote sites to a central facility already exist, and it would be straightforward to develop and implement ways to communicate using either raw or processed data. Sally Hojvat of CDRH concurred and suggested that
this would be covered under existing regulations that address electronic records and the transfer of data from an instrument at a clinical site to a central facility for analysis (21 CFR 11). She cautioned that it would be necessary to demonstrate the accuracy and traceability of the results of a test performed remotely by an unqualified individual.
Panel moderator Bruce Burlington, an independent consultant, questioned how it could be determined that an immune status test was relevant for many different illnesses. Would test developers need to undertake a variety of disease challenges? Hojvat responded that it could be considered more of a prognostic type of marker, and such data would be one way FDA could begin to assess the test. With regard to its commercial value, Daitch said that the market for such a test is not obvious. A test that predicts, based on immune status, that someone is in the early stages of an infectious disease might be useful, for example, for astronauts about to go on the space shuttle or for troops about to be deployed, he said. Burlington added that it could also be used in an epidemic for health care workers or other first responders.
Participants discussed the potential for commercial assays on multiplex platforms to be used as epidemiological surveillance tools (as opposed to diagnostic tests where results are reported back to the patient). Hojvat suggested that companies could aid the surveillance effort by developing cassettes for biothreats for their multiplex systems. Daitch and David Ecker, founder of Ibis Biosciences, agreed it would be possible, but noted that key challenges would be validation of the test for broad groups of organisms and ensuring that data could be transferred over a secure network to somebody who has the capability to interpret the data correctly.
Participants also discussed the concept of an evolving label. Performance characteristics of a diagnostic test need to be defined in terms of sensitivity and specificity, but a challenge is how to present that information in the label when the background prevalence of what is being tested for is almost zero. It would be helpful if, as the threat emerges, new information and data based on use could be made available rapidly. Hojvat responded that FDA has the technology to do that, and there is an ongoing electronic labeling project.
In summary discussion, participants observed that it is important to remember that diagnostics are also MCMs. Several options for more efficient use of diagnostics were suggested, including the development of new formats for collection, transport, and stable storage of biospecimens, and the development of highly multiplexed testing platforms for local site use, with data then sent electronically to experts at a central facility for analysis. It was also noted that rapid diagnostics could improve the
efficiency of antimicrobial trials, allowing for enrichment of the population with patients infected with resistant organisms.
The goal of clinical trial simulation in drug development programs is to reach a decision faster, cheaper, and with greater certainty, explained Stephen Ruberg, distinguished research fellow and scientific leader in advanced analytics at Eli Lilly and Company. Companies seek to “kill” ineffective or unsafe investigational drugs sooner and advance potentially effective drugs as quickly as possible and at the lowest cost possible. Clinical trial simulation allows for examination of a broad range of clinical trial designs, decision rules, and analysis methods. In simulations, models can be used to create virtual patients that are then randomly selected for inclusion into in silico clinical trials using sophisticated software tools. These models for virtual patients can be PK/PD models, empirical statistical models of response over doses and time, or mechanistic disease models. Known design and analysis parameters can be controlled (e.g., sample size, number of doses or visits, analysis strategies for testing hypotheses or estimating key drug effect parameters), and a range of possibilities for unknown parameters and factors that cannot be controlled can be assessed (e.g., drug effect, true dose response curve, adverse event rate, placebo response, dropout rate). Dozens of combinations of factors are typically evaluated with the goal of selecting the design and analysis parameters that will minimize false positive and false negative findings in the drug development program.
From a regulatory science perspective, Ruberg said, this will require training of FDA staff on the use of simulation tools, some of which are becoming commercially available. FDA statistical and medical reviewers will have to understand and accept modeling simulation as a tool for study design. Simulation trial designs generated may not look like classic trial designs or may not have theoretically or mathematically described properties, he said. This is of particular concern when designing phase III trials due to the need to control the type I error (false positive) rate at 0.05. As this cannot always be done analytically, Ruberg asked whether FDA will accept simulated results in lieu of analytical proof. He noted that the FDA draft guidance on adaptive designs3 is a substantial step forward in helping the industry understand how best to move forward with innovative trial designs. Another topic for consideration is the simulation of the
_____________
3 See Guidance for Industry Adaptive Design Clinical Trials for Drugs and Biologics (Draft Guidance) http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm201790.pdf (accessed June 9, 2011).
sequence of clinical trials spanning an entire clinical drug development program, which, Ruberg said, companies could realistically be doing in the next couple of years.
A goal in drug development is to use as much data as possible—current or historical—to make decisions on drug safety and efficacy. Current practice in the vast majority of phase III clinical programs is for each clinical trial to stand on its own as an independent piece of evidence in the evaluation of a drug’s effect. This is afrequentist statistical approach. Eli Lilly, Ruberg said, is currently implementingBayesian methods for phase I and phase II trial design and analysis. There are many ways in which Bayesian methods can be used in clinical drug development. One example presented by Ruberg is a Bayesian augmented control design, in which control group data from the current prospective study is supplemented with historical control data. This allows for smaller trials (saving both time and resources) and for more enrolled patients to be allocated to treatment groups.
While the use of Bayesian statistical methods is a technical topic, Ruberg opined that the largest barriers to implementation are social. There will need to be changes in philosophy and mind-set within some FDA centers and other regulatory agencies around the world. There are also legitimate scientific debates relative to the choice of historical data to include in analyses and how to weigh those data relative to data generated from a new study, he added. From a regulatory science perspective, Ruberg said that the use of Bayesian approaches for phase III confirmatory trials would require in-depth sponsor-agency discussions at the end-of-phase-II meeting or sooner.
Important to the use of Bayesian approaches is the development of a comprehensive data element dictionary. Data element standards allow for more efficient collection of data and routine use of standardized software. More importantly, common data element standards allow for the simple, rapid integration of data from multiple sources, facilitating more comprehensive statistical analysis in order to draw the best scientific conclusions possible. Such a dictionary should be maintained by a central authoritative group, Ruberg said, and must be free, broadly accessible in electronic form, and downloadable for use within IT systems. He acknowledged the various ongoing standardization efforts (e.g., CDISC, HL7),4 but said that
_____________
4 The mission of the global, nonprofit, multidisciplinary Clinical Data Interchange Standards Consortium (CDISC) is to “develop and support global, platform-independent data standards that enable information system interoperability to improve medical research and related areas of health care.” See http://www.cdisc.org/. Health Level Seven International (HL7) is a nonprofit “standards-developing organization dedicated to providing a comprehensive framework and related standards for the exchange, integration, sharing, and retrieval of electronic health information.” See http://www.hl7.org/ (accessed June 9, 2011).
data element standards needs to go deeper in terms of specificity, broader in terms of accommodation of all therapeutic areas and measurements, and faster in terms of development and deployment.
In closing, Ruberg offered several ideas to advance the use of trial simulation and Bayesian statistics, and the standardization of data elements:
- For study design, Ruberg suggested adaptive/pooled studies as a way to more rapidly and uniformly test compounds. Such studies use a single trial design with a common control group that allows companies to insert their drug into a perpetually ongoing trial (such as the I-SPY 2 breast cancer clinical trial).
- Ruberg also directed participants to the Drug Information Association (DIA) Working Group on Bayesian Methods. Ruberg noted that Bayesian approaches were discussed in a recent National Research Council (NRC) report on how to handle missing data in clinical trials (NRC, 2010), and he suggested that the National Academies conduct a study to evaluate the use of Bayesian methods in clinical trials, with particular emphasis on phase III confirmatory trials.
In panel discussion, there was much discussion about the use of Bayesian statistical methods for analysis of clinical trials. Jeffrey Wetherington of GSK said that his company has made significant use of Bayesian methods for phase II proof-of-concept studies and dose-ranging studies, and he estimated that use of these methods has saved the company nearly $15 million on study costs over the past year. Similar to Eli Lilly, Wetherington said, GSK uses augmented control groups, decreasing study sample sizes by several hundred people. Bayesian methods provide very interpretable results, he said.
Estelle Russek-Cohen, acting director of the Division of Biostatistics at CBER, noted that CDRH frequently uses Bayesian analysis in the context of device modification submissions. She added that CBER has seen submissions with Bayesian and adaptive designs, primarily in phase I and II studies, many of which have been oncology studies. For phase II studies, a variety of skill sets are needed when considering the benefits and risks of the analysis approaches (e.g., medical officers, statisticians). Russek-Cohen said a concern with historical controls is how far back to go if the standard of care is changing. A question for consideration is whether, in the context of MCMs, there is a real and compelling need for these approaches.
Panel moderator Burlington noted that the toxicology community routinely uses historical controls, pooling data from control animals from many experiments. Russek-Cohen responded that pooling of historical control data is used for safety assessment as there is often not enough
power in individual studies, but it has not yet been done for efficacy studies, in part because FDA statutes call for adequate and well-controlled trials. In a phase II environment, it makes sense for industry to find ways to pool control information across companies pursuing similar projects.
Wetherington said that drugs such as anti-infectives can have small niche markets and limited profit margins. Using Bayesian-type designs for phase III studies, especially when comparing the novel agent to a well-characterized standard of care, could save time and money, and get products to patients more quickly.
Participants discussed the potential for use of simulation and Bayesian approaches as the basis for approval of an MCM in anticipation of future use. Goodman responded that part of the FDA MCM initiative is to consider novel approaches, and the agency is open to these possibilities. He encouraged developers of MCMs to discuss this with their FDA review team as part of their product development planning.
Burlington questioned whether FDA could mandate or incentivize companies to submit their data in conformation with data element standards. Russek-Cohen responded that implementing standards is part of the broader FDA initiative. Ruberg suggested that the National Library of Medicine or FDA could take the lead on pushing forward with data element standards. A participant noted that CDISC is approaching standards development disease by disease. In response, Ruberg suggested that there could be a working group of experts in MCMs to define what generally needs to be measured and start discussing standard data elements.
In summary discussion, the statistics of diagnostics and dealing with false positives was also considered. A participant said that CDRH has asked developers of new multiplex diagnostic assays to offer ideas about how to handle false positives, for example if three or four positives were found where one was expected. A participant said to keep in mind the primary question the assay is answering: Is it diagnosing an individual or determining if there is an outbreak? It was noted that there is an interagency meeting being planned on this issue.
Several workshop presenters and discussants noted that Bayesian statistical methodology can be used for both study design (e.g., supplementing the control group data with historical control data) and analysis (of both actual and simulated trials). Workshop participants offered suggestions for themes and future directions with respect to statistical methodologies and data analysis. The following individual suggestions were made:
- Training in Bayesian approaches and causal mechanisms of actions will be needed for both scientists and the public.
- The use of Bayesian approaches would be enhanced by the development of common data element standards (e.g., to facilitate pooling of data across studies).
- Christian Macedonia, medical sciences advisor to Admiral Mullen, the chair of the Joint Chiefs of Staff, raised the idea of electronically tagging every piece of information obtained in biomedical research (e.g., date, time, group, unique animal identification, institution) so data from large multicenter trials could be traced back, even years later, for further analysis. He likened this to the way electronic data are broken into packets and tagged for transfer across computer networks, to be reassembled at the other end.
- There was also interest in platform approaches to health data software design, for which many applications or “apps” could be developed. These could be used for collection, management, and analysis of electronic health data, specifically for monitoring of adverse events.
This page intentionally left blank.




































