
4
Developing New Animal Models for Biodefense Research
This chapter addresses the process and feasibility of developing new animal models for biodefense research. The previous chapter established that the ability of animal models to predict the human condition is not absolute and that collection of human clinical data is critical. The Committee on Animal Models for Assessing Countermeasures to Bioterrorism Agents concludes that focusing on the creation of new animal models—that is, continuing to rely exclusively on the use of animals for efficacy studies—is not warranted at this time. Although new models, such as hamsters, New World nonhuman primates, pigs, or bats, may be useful for basic research purposes, they will eventually encounter the same problems seen in the better-defined animal models currently in use.1 Instead, the Committee suggests that it is more useful to use different approaches that “support the qualification2 of animal models” (DHHS 2010, p 11) and increase understanding of how animal data may more consistently predict the human response as follows:
- Improve the quality and quantity of collected data about the natural history of the diseases studied and data from human patients in clinical settings.
- Continue and improve the acquisition of expanded data from phase 1 human safety trials.
- Maintain frequent interactions with the FDA.
- Focus on the reproducibility and scientific relevance of the compartmentalized3 animal model rather than on its validation.
- Develop strategies for interspecies comparisons, such as compartmentalization.
_________________________
1 As discussed in Chapter 2, current animal models do not adequately reflect the human condition; their validity cannot be easily evaluated due to paucity of human data; results from different species may not be comparable partly due to biocontainment constraints and partly due to methodological differences; and they are complex and expensive to develop.
2 The Animal Rule does not discuss the validation of animal models (FDA 2002).
3 Compartmentalization means to plan experiments that will yield information from components of the animal (organs, cells and systems) rather than data derived from the whole organism (for additional discussion see page 58).
USE OF HUMAN DATA TO IMPROVE THE VALUE OF ANIMAL MODELS
Postmarketing Clinical Studies
Before FDA approval, drugs and biological products must be tested in three phases of clinical trials to determine efficacy in humans. Increasing numbers of human volunteers will be used in the three phases to determine safety, and when feasible and ethical, under conditions that reflect the natural exposure or disease condition. Once the drug or biological product is approved, the FDA can require additional postmarketing clinical studies to determine whether there are safety issues that are revealed in larger populations than were tested in premarketing studies to ensure the quality and consistency of manufacturing and to gather more data on efficacy when the drug is used under normal clinical conditions. These postmarketing clinical studies are required for products that are approved under accelerated approval provisions. These provisions are for serious or life-threatening conditions; for marketed drugs approved in adults which have the potential for benefit in children; or more recently, for products that have been approved under the Animal Rule (21 CFR Parts 314 and 601 [2002]). In these cases, the FDA requires postmarketing commitments that outline the clinical studies that will be undertaken and the time frame under which they will be carried out. In addition, the agency can require postmarketing studies to determine whether there are reasons to withdraw the approval of the product; i.e., whether there is a known or potential serious risk related to the use of the drug. The FDA requires annual reporting of the status of the postmarketing studies until it determines that the commitments have been fulfilled (21 CFR Part 314.81(b)(2)(vii) [2011]).
For drugs and biologics approved under the Animal Rule, the FDA requires postmarketing studies or clinical trials to “verify and describe the drug’s [or biological product’s] clinical benefit and to assess its safety when used as indicated when these studies are feasible and ethical” (FDA 2002, p 37995, 37997). The FDA requires that applicants submit a plan or an approach, including the appropriate due diligence to identify opportunities to conduct these studies, and to carry out the clinical studies if and when it becomes feasible and ethical to do so. However, due to the nature of these products and the pathogens they are meant to counter, opportunities to conduct these clinical studies are rare and may only be possible under exposure to a chemical, biological, or nuclear threat. For some products, however, particularly for those used in the prevention or treatment of (emerging) infectious diseases, there may be particular environmental or natural conditions that provide an opportunity to collect human clinical efficacy data under natural exposure. In fact, the Animal Rule treats the use of a product approved under the rule in response to an exposure as a clinical trial from which essential data can be obtained (Walker and King 2011).
These clinical studies also offer the opportunity to evaluate the relevance and predictability of the animal models that were used in the approval process. These studies, therefore, represent important opportunities to refine the animal model systems, to evaluate the biomarkers identified and measured, and to correlate the clinical findings in humans to those seen in the animal models. By identifying and collecting data using the appropriate biomarkers4 and correlating clinical information to existing animal data, it should be possible to improve understanding of the relevance of the animal model to human clinical responses. Such data would be particularly informative to models developed with a product-neutral approach, which, by exhibiting a broader application profile, may be useful in “unknown-unknown” exigencies (see Chapter 2, p 30). Therefore, it would be useful for postmarketing
4 Biomarker is a biological characteristic that can be objectively measured and evaluated as an indicator of normal, pathogenic, or pharmacological responses or as the target of a therapeutic intervention. Biomarkers provide insight into disease progression, prognosis, and response to therapy.
commitments to include plans that provide data to evaluate the relevance of the animal model to the human condition.
Natural Disease Outbreaks
Many of the infectious disease threat agents that are the target of products developed under the Animal Rule occur naturally in many places of the world and may be responsible for outbreaks of disease in humans (Warfield et al. 2006). These disease outbreaks represent potential opportunities to collect prophylactic and therapeutic clinical data on the efficacy of drugs and biologics approved under the Animal Rule and to collect data to determine the relevance of the animal models used. Therefore, it is important to identify such potential opportunities and collaborations early in the clinical development process. For the countries where outbreaks are known to occur, the products approved under the Animal Rule also represent potentially important tools for public health officials to reduce morbidity and mortality in exposed populations, to protect and treat first responders during outbreaks, and to support ongoing surveillance and containment efforts.
For drugs and vaccines where natural and environmental exposure can occur, postmarketing commitments can be developed that include (1) the identification of potential sources of outbreaks in humans; (2) the opportunities for supporting first responders and surveillance and containment efforts; and (3) the development of a strategy to engage key partners from endemic country academic institutions, public health, and governmental agencies at an early stage of clinical development or at the time of approval.
Phase 1 Human Safety Trials
Safety evaluation and verification of clinical benefit of products approved under the Animal Rule is required and studied in human volunteers under the conditions described under 21 CFR Parts 312 and 320 [2010]. As mentioned in Chapter 2, about 20% of drugs fail due to safety concerns, many of which were either not observed or not predicted by animal trials. Even though safety trials are not a substitute for clinical trials with humans, the combined purpose of safety trials under the Animal Rule indicates that useful data can be collected to inform the efficacy of the product. For instance, if pharmacological responses were studied and biomarkers were developed in the animal models, biomarker levels in humans could be monitored and later correlated to the animal-based data.
Expanded data acquisition from these trials could be useful in multiple ways, especially if the duration of the trials were expanded and the trials structured to (1) mirror the anticipated treatment regimen in humans, and (2) reflect the heterogeneity of the general population.5 Furthermore, surrogate markers for efficacy can be tested in safety trials, such as finding the level and functionality of antibodies induced,6 the blood or tissue concentration of drugs relative to the critical bactericidal and antiviral concentrations, and the actual modification of immune responses.
_________________________
5 The FDA advisory committee tasked with the evaluation of the application of Human Genome Sciences for licensure of raxibacumab under the Animal Rule (see Chapter 3 for more details) requested that additional safety studies in humans include pediatric and elderly populations (FDA 2009b).
6 Data from human safety studies of raxibacumab show that blood concentrations of the antibody can be used as a surrogate endpoint predictive of clinical benefit (Migone et al. 2009).
INTERACTIONS WITH THE FOOD AND DRUG ADMINISTRATION
The Animal Rule regulatory pathway is relatively new to the FDA and the research community. As discussed in Chapter 3 only two drugs have been approved under this rule since 2002; both of them were repurposed for use as medical countermeasures. Most of the proposed approaches in this report are unlikely to rely on a precedent example from a marketed product. Thus, early interactions with the FDA are critical for products that will be considered under the Animal Rule regulatory pathway, as presented in Box 4-1.
BOX 4-1
Formal Meetings with the FDA
Pre-investigational New Drug (IND) Application Meetings
Prior to the submission of an initial IND, the sponsor (applicant) can request a meeting with the FDA to review and reach agreement on the format of the IND (21 CFR § 312.82 [1998]), the phase 1 clinical protocol, the design of animal studies needed to support human clinical testing (e.g., protocols for toxicological studies), and chemistry, manufacturing, and controls (CMC) information. The CMC information includes a description of the product potency assay and early product stability assessment protocols. An important goal of a pre-IND meeting (regardless of the regulatory pathway) is to identify issues that may lead to a delay in the initiation of the phase 1 clinical trial, i.e., the “clinical hold” (21 CFR § 312.42 [2009]; FDA 1995, 2001). The overall plan for investigating the product should be considered in the context of the proposed clinical indications, including a proposed “indications and usage” section analogous to that seen on the package insert. Issues regarding the potential for fast-track designation may be discussed.
Early discussions regarding Animal Rule specific issues may also occur at the pre-IND meeting or separately. Examples of pertinent issues include the appropriateness of using the Animal Rule regulatory pathway for the specific clinical indications; pilot animal efficacy studies (protocols or data); and iterative determinations for the pivotal animal studies, such as the relevant dose level based on prior pilot animal and human studies.
End-of-Phase 1 (EOP 1) Meeting
A sponsor may request an EOP 1 meeting for a product with fast-track designation after completion of early phase 1 clinical studies to review the phase 1 data and reach agreement on clinical plans for the phase 2 program (21 CFR § 312.82(b) [1998]). Emerging data from pilot animal pharmacokinetic and efficacy studies, or animal study protocols, can also be discussed. For drugs for life-threatening diseases, the FDA will provide its best judgment, including whether pediatric studies will be required and, if so, whether their submission can be deferred until the product has been approved” for adults (21 CFR § 312.82(b) [2010]). Other issues can also be covered, e.g., emerging CMC and assay validation questions.
End-of-Phase 2 (EOP 2) and Prephase 3 Meeting
The purpose of this meeting is to review the phase 2 clinical data before proceeding to phase 3, to evaluate plans for the phase 3 clinical program and clinical protocols, and to identify any additional information necessary to support a marketing new drug application (NDA) or a biological license application (BLA) for the proposed clinical indications, e.g., unresolved CMC issues (21 CFR § 312.47(b)(1) [2002]). For products developed under the Animal Rule regulatory pathway, the phase 3 clinical studies may include the definitive clinical pharmacokinetic study (or immunogenicity study for vaccines) as well as larger well-controlled safety studies. The phase 3 equivalents for animal studies include animal pharmacokinetic and animal efficacy studies.
Specific Information on Pivotal Animal Studies (Phase 3)
When devising a development plan and protocols for animal studies, the applicant should recognize that certain principles of clinical trial design will also apply to the animal studies; therefore, multidisciplinary collaboration among research scientists, clinical trial experts and biostatisticians is highly recommended. The principles for adequate and well-controlled human efficacy trials should be applied (as appropriate) to the animal studies (described in 21 CFR § 314.126 [2010]). The animal study protocols and statistical analysis plan for the animal efficacy studies (including control groups, justification, and clear prospective
primary and secondary endpoints) are provided to the agency with sufficient time for FDA review and comment before conducting the study. The analysis of the primary efficacy endpoint will form the basis for approval.
Other studies in support of an NDA or BLA, such as the clinical or animal pharmacokinetic interaction studies (e.g., between the investigational drug and an approved drug that would be used concurrently), may be necessary.
“VALIDATION” OF ANIMAL MODELS FOR BIODEFENSE RESEARCH
A question that arises when applying the Animal Rule is whether or not an animal model is validated. Validation is a regulatory concept that describes the suitability of an analytical procedure for its intended purpose and includes such qualities as accuracy, precision, repeatability, intermediate precision, specificity, detection limit, quantitation limit, linearity, and range (ICH 2005).
Such attributes do not apply to animal models and the Animal Rule does not mention “validated” animal studies. However, reproducibility and scientific relevance are essential qualities of successful animal models because they can lead to standardization of the model.7 The field of regulatory assessment (which includes drugs, chemicals, vaccines, cosmetics and pesticides), which depends both on animal testing and alternative methods to animal experiments, relies on a concept of validation that incorporates, among other factors, both reproducibility and scientific relevance, as depicted in Figure 4-1.
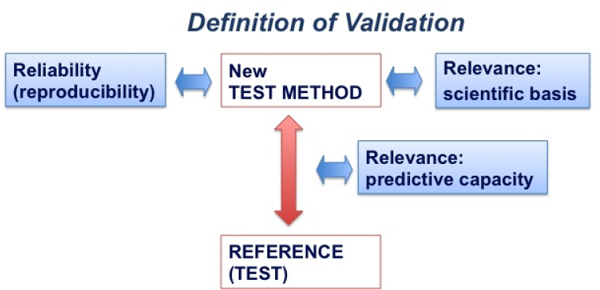
FIGURE 4-1 Definition of validation: “Validation is a process in which the scientific basis and reproducibility of a test system, and the predictive capacity of an associated prediction model, undergo independent assessment.” SOURCE: Adapted from Hartung 2007.
In this diagram the new test method (i.e., the animal model) is compared to the reference test. If no such reference test exists then a consensus standard, agreed upon by experts, is utilized, e.g., a number of positive and negative substances (Hoffmann et al. 2008). The results of the new method are
_________________________
7 The development of such a model by NIAID for inhalational anthrax is discussed in Appendix C.
then compared with the reference results. Three principal aspects of validity of the new method are examined: (1) reliability (i.e., reproducibility), (2) relevance of the model (i.e., its scientific basis), and (3) predictive capacity of the new method (i.e., the animal model) for the reference results. In addition, the new test is subjected to quality control (i.e., standardization).
In the case of animal models for countermeasures research there usually are no reference data available—that is, human pathological findings and clinical data are insufficient, there are no failed or successful drugs, and no reference test model has been established—with which to compare outcomes from the animal studies. Thus, a traditional validation assessment would not be possible.
Evaluation of the validity of a model is “an evolving process that is never completed because the models are always subject to further definitive reexaminations and revalidation as new technology becomes available” (Anderson and Tucker 2006). However, a model’s qualification (DHHS 2010) may be assessed by an approach that utilizes only some of the elements presented in Figure 4-1; the model’s reproducibility is coupled with an assessment of the scientific relevance of the model to the human condition that will include a robust analysis of modes of action in response to pathogens. Comparative analyses of outcomes from different animal species (see next section) may be an equally important source of information.
Taking into account the limitations of animal models outlined in Chapter 2 and the dearth of human reference data, the model’s reproducible response to a pathogen challenge across different laboratory settings and methods will help characterize or qualify the model rather than seek to validate it.8,9
COMPARATIVE BIOLOGY AND COMPARTMENTALIZATION IN ANIMAL MODEL DEVELOPMENT
An important problem facing animal modelers is that there is no roadmap for what makes an animal model good or bad. Historically, models were often deemed inferior because one protein in a critical pathway in one animal species was not identical to the corresponding pathway or protein seen in humans. A good example is found in the history of nitric oxide (NO), one of the primary effector molecules against a variety of intracellular pathogens (Chakravortty and Hensel 2003). Although murine macrophages readily produce NO, human cells often cannot (Yang et al. 2009). Consequently, the utility of murine models to investigate the role of NO was not obvious. However, those models could be quite useful in investigating most of the other aspects of host–intracellular pathogen interactions with the possible exception of the role of NO in humans. Thus, it would be more useful to compare the systems and pathways that lead up to a host response within a species, across species, and with humans rather than focusing on a single gene or protein or particular genes or proteins. This strategy is known as compartmentalization. If this strategy were applied to NO research, it would show that the mouse and human share most of the major pathways leading up to the production of interferon γ and subsequent macrophage activation except for the production of the final effector NO.
_________________________
8 A different set of criteria (Technology Readiness Levels) for medical countermeasures and detailed description of the development stages of animal models to support approval of countermeasures under the Animal Rule have been developed by the working groups of the Public Health Emergency Medical Countermeasures Enterprise for use across the public and private sector; https://www.medicalcountermeasures.gov/TRL_Page.aspx.
9 In its Draft Guidance to Industry the FDA has provided a list of the “essential data elements of an animal model.” As the document clearly states, however, “These elements serve as a guide. They may be modified or revised…” (FDA 2009, p 16-17).
The basic premise of compartmentalization is that each species is made up of a variety of physiological compartments that contribute to the host response to an infectious agent: One compartment is the innate immune response, a second compartment is the acquired immune response, a third includes the determinants of the pathogen’s tropism for that species, and so on. Pathophysiological data from humans can be used to identify the specific compartments of the animal models that are most relevant to the human condition and that can be the focus of research protocols and data evaluation. The possibility that these compartments overlap among several animal models and are not mutually exclusive would suggest a consistent response among different species. This response can be extrapolated more confidently to the human condition. Some of these overlapping compartmental responses may even occur in species in which important deviations from the human responses have been observed.
It is necessary to develop compartmentalization strategies and algorithms for comparing infectious disease models across several species to increase the likelihood of extrapolating the product effect in humans. This type of strategy may relieve the pressure to find one or two optimal models that can be used to address all aspects of the extrapolation and to focus all efforts and resources.10 For example, because susceptibility to anthrax is variable among animal species based on the role of toxin or capsule (see Chapter 2, pages 29-30), a synergistic strategy has been proposed to increase the relevance of anthrax animal models in the holistic modeling of the human disease (Goossens 2009). In variola studies, which are constrained by the species-specific tropism of individual poxviruses (McFadden 2005), data from multiple species that respond to different poxviruses (cowpoxvirus in murine species and monkeypoxvirus in nonhuman primates) can be combined. Otherwise, a suboptimal nonhuman primate model of variola that is challenged with massive nonphysiological doses of the virus must be relied upon (Jahrling et al. 2004).
Another advantage of the compartmentalization approach is the potential repurposing of animal models to discreet components of countermeasure development for which they may be better suited. For example, the results from the use of antitoxin therapeutics translate well from the murine model infected with the anthrax Sterne strain (Chapter 2, page 29) to the rabbit model of inhalational anthrax (Loving et al. 2009). This result suggests that, in some cases, the model selection for the development of countermeasures could be guided by the specific experimental question or the target. In other words, in the compartmentalization approach, the animal model is specialized (i.e., made to fit) to only one pathophysiology component, the toxin, so that collected information is specific to the host–toxin interaction and not to the whole organism and host–pathogen interaction.
OPTIMIZING CURRENT ANIMAL MODELS
The production of relevant and effective medical countermeasures to biothreats would greatly improve if the focus were shifted from developing new animal models to improving the extrapolation of animal data from the current models to the human response. This shift can be accomplished by cultivating alternative or underutilized sources of information, such as the data collected from current models and human populations.
_________________________
10 An example of the difficulty of developing optimal animal models in different species is presented in Appendix C in the section 4.1 Anthrax.
The Committee recommends that the TMT adopt the following strategies to improve the usefulness of current animal models:
- To control interspecies variability and improve the comparativeness of infectious disease models across different species, adopt the concept of compartmentalization. As each species is made up of a variety of physiological compartments that contribute to the host response to an infectious agent, compartmentalization is a strategy to compare the systems and pathways that lead up to the host response within a species, across species, and with humans rather than focusing on a single gene or protein or particular genes or proteins.
- To support the qualification of animal models as an alternative to validation, establish the compartmentalized model’s scientific relevance and reproducibility across different methods and laboratories. These comparative datasets may subsequently be used to define appropriate criteria to characterize or qualify vs. validate the animal model.
The Committee also recommends the following to improve the usefulness of the information derived from human populations:
- To address the dearth of data from human populations, expand the collection of data from patients in outbreak zones and from postmarketing studies. In addition, expand the acquisition of data from phase 1 safety trials by (1) increasing the duration of these trials; (2) diversifying the enrolled subjects to mirror the general population; and (3) including the anticipated treatment in the field as part of the trial protocol.
Anderson J, Tucker K. 2006. Development and validation of animal models. In: Swearengen JR, ed. Biodefense: Research Methodology and Animal Models. Boca Raton: CRC Press. p 41-60.
Chakravortty D, Hensel M. 2003. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect 5(7):621-627.
DHHS [U.S. Department of Health and Human Services]. 2010. The Public Health Emergency Medical Countermeasures Enterprise Review: Transforming the Enterprise to Meet Long-Range National Needs. Assistant Secretary for Preparedness and Response, U.S. Department of Health and Human Services. Available online (https://www.medicalcountermeasures.gov/documents/MCMReviewFinalcover508.pdf), accessed May 2011.
FDA [Food and Drug Administration]. 1995. FDA Guidance for Industry. Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-Derived Products. Center for Drug Evaluation, Center for Biologics Evaluation and Research, Food and Drug Administration. Available online (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071597.pdf), accessed March 2011.
FDA. 2001. FDA Guidance for Industry. IND Meetings for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information. Available online (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070568.pdf), accessed March 2011).
FDA. 2002. New Drug and Biological Drug Products; Evidence Needed to Demonstrate Effectiveness of New Drugs When Human Efficacy Studies Are Not Ethical or Feasible. Fed Regist 67(105):37988-37998.
FDA. 2009a. Draft Guidance for Industry on Animal Models-Esential Elements to Address Efficacy Under the Animal Rule. Notice. Fed. Regist 74(12)3610-3611. Available online
(http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078923.pdf), accessed May 2011.
FDA. 2009b. Summary Minutes of the Anti-infective Drugs Advisory Committee Meeting, October 27, 2009. Center for Drug Evaluation and Research, Food and Drug Administration. Available online (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AntiInfectiveDrugsAdvisoryCommittee/UCM196436.pdf), accessed September 23, 2011.
Goossens PL. 2009. Animal models of anthrax: The Quest for the Holy Grail. Mol Aspects Med 30(6):467-480.
Hartung T. 2007. Food for thought … on validation. ALTEX 24(2):67-73.
Hoffmann S, Edler L, Gardner I, Gribaldo L, Hartung T, Klein C, Liebsch M, Sauerland S, Schechtman L, Stammati A, Nikolaidis E. 2008. Points of reference in validation: The report and recommendations of ECVAM Workshop 66. Altern Lab Anim 36(3):343-352.
ICH [International Conference on Harminization of technical Requirements for Registration of Pharmaceuticals for Human Use]. 2005. ICH Harmonized Tripartite Guideline. Validation of Analytical Procedures:Text and Methodology Q2(R1). Available online (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf), assessed September 2011.
Jahrling PB, Hensley LE, Martinez MJ, LeDuc JW, Rubins KH, Relman DA, Huggins JW. 2004. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc Natl Acad Sci USA 101(42):15196-15200.
Loving CL, Khurana T, Osorio M, Lee GM, Kelly VK, Stibitz S, Merkel TJ. 2009. Role of anthrax toxins in dissemination, disease progression, and induction of protective adaptive immunity in the mouse aerosol challenge model. Infect Immun. 77(1):255–265.
McFadden G. 2005. Poxvirus tropism. Nat Rev Microbiol 3(3):201-213.
Migone TS, Subramanian GM, Zhong J, Healey LM, Corey A, Devalaraja M, Lo L, Ullrich S, Zimmerman J, Chen A, Lewis M, Meister G, Gillum K, Sanford D, Mott J, Bolmer SD. 2009. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med 361(2):135-144.
Walker RL, King NMP. 2011. Biodefense research and the U.S. regulatory structure. Whither nonhuman primate moral standing? Kennedy Inst Ethics J 2193):277-310.
Warfield KL, Jaax NK, Deal EM, Swenson DL, Larsen T, Bavari S. 2006. Viral hemorrhagic fevers. In: Swearengen JR, ed. Biodefense: Research Methodology and Animal Models. Boca Raton: CRC Press. p 227-257.
Yang CS, Yuk JM, Jo EK. 2009. The role of nitric oxide in mycobacterial infections. Immune Netw 9(2):46-52.










