
COMMERCIAL APPLICATIONS OF SYNTHETIC BIOLOGY
An Overview of Venture Capital
Venture capital is financial capital invested into high-potential companies. The role of venture capital is to support the entrepreneurial talent that takes basic science and breakthrough ideas to market by building companies. This risk capital ultimately supports some of the most innovative and promising companies—those that have gone on to change existing industries or create new ones altogether (Thompson Reuters, 2011).
Venture capital is a distinct asset class. Venture capital firms, which are professional, institutional capital managers, make investments by purchasing equity in a company. The stock acquired is an illiquid investment that requires the growth of the company for the investors to ultimately reap any potential return. It is this inability of venture capitalists to rapidly enter and exit investments, or “flip” them, that aligns their goals with those of the entrepreneurs. Venture capital is intrinsically a long-term investment (Thompson Reuters, 2011).
_______________
1 Affiliation: Flagship VentureLabs, Cambridge, MA, USA.
2 Key words: venture capital, biological engineering, synthetic biology, microbiome, diesel, photosynthesis, genome engineering.
Venture capitalists invest out of a fund, a vehicle that deploys capital on behalf of third-party investors. The investors in these funds, called limited partners, are often pension funds, foundations, corporations, endowments, and wealthy individuals, among others. Given the low liquidity associated with their investment into venture capital funds, limited partners expect large returns—better than those in the stock market—from the funds in which they invest. The funds represent a commitment of capital with a fixed life, typically 10 years. The general partner, a group of partners with fiduciary responsibility for the firm with the legal form of a partnership, manages the capital in the fund. The committed capital is called by the general partner from the limited partners to make a portfolio of investments. Ultimately, when investments mature and become liquid, the profits are shared, with the majority going back to the limited partners and the rest shared by the general partner.
Funding provided by venture capitalists typically takes the form of “rounds,” where a given amount of money is invested into a company at a valuation agreed upon between the management and the investors. Prior to an investment, the equity ownership is divided among the founders, management, and others. The valuation sets a share price against which the venture capital firm buys shares. At each round, the earlier investors and management team strive to increase the valuation for the subsequent round(s) of investment. The higher the valuation of a round, the less dilution (reduction in ownership) the existing shareholders take. While each round contemplates a share price that defines a paper value for an investor’s or an employee’s shares, little actual value is created. Only at a sale event or initial public offering do investors and the management team see a tangible financial return, which can take 5-8 years, if not longer.
Venture capital firms statistically see 100 business plans, take a deep look at 10 of these proposals, and invest in one. This process involves an assessment of the management team, the proposed business, its potential to exclude competition, the market being pursued, and how well the opportunity fits with the firm’s goals.
With an investment, a partner will typically get involved with a company by taking a seat on the board of directors, where he or she works closely with the management team on company strategy and growth. The venture capital industry plays an important role in the economy. Companies supported by early venture capital account for 21 percent of the U.S. gross domestic product by revenue, and 11 percent of private-sector jobs despite the fact that fewer than 1,000 new businesses get venture capital funding any given year (National Venture Capital Association, 2009).
A Brief History of the Venture Capital Industry
Venture capital is said to have originated in 1946 with the founding of the first two firms: American Research and Development Corporation (ARDC) and
J. H. Whitney & Company (Wilson, 1985). Georges Doriot, referred to as the “father of venture capitalism,” the former dean of Harvard Business School and founder of INSEAD, founded ARDC along with Karl Compton, the former president of MIT, as well as Ralph Flanders (Ante, 2008). ARDC sought to invest in businesses run by soldiers returning from World War II. The firm is most famous for investing $70,000 in Digital Equipment Corporation in 1957—a company that when it had its initial public offering in 1968 was valued at $355 million for a return to ARDC of over 1,200-fold. Employees of ARDC went on to found leading venture funds including Greylock Partners and Flagship Ventures, among others (Kirsner, 2008).
Two major government changes allowed venture capital to emerge as a fully fledged industry. First, the Small Business Investment Act of 1958 enabled the Small Business Administration to license Small Business Investment Companies to help finance and manage small entrepreneurial businesses. Second, in 1978, the Employee Retirement Income Security Act was altered to allow corporate pension funds to invest in venture capital. These two acts together supported the framework for venture capital and facilitated substantial investment in it.
Successes of the venture capital industry in the 1970s and 1980s, with companies including Digital Equipment Corporation, Apple, and Genentech resulting, led to rapid growth of the industry. With rapid growth came diminished returns. In the early 1990s the numbers of firms and managers shrank in response to declining investment performance. At the same time, the more successful firms retrenched, starting a wave of increased returns that began in 1995 and continued through the Internet bubble in 2000 (Metrick, 2007). Once again, with grossly increased returns, the investment into the sector and the number of funds skyrocketed. Beginning in March 2000, the NASDAQ crashed, and many funds suffered from a second contraction.
After the Internet bubble, the funds raised by venture firms shrank substantially. Amounts of committed capital increased through 2005 to a level much less than in 2000, and they remained flat until the economic meltdown in 2008. During the decade from 2000 to 2010, venture capital returns also fell dramatically to the point that the median 10-year return of all U.S. funds was less than the stock market (Thomson Reuters, 2011). These events resulted in another substantial contraction in the industry. The industry is currently responding to this most recent contraction. The number of funds has decreased as the average fund size has risen. This dynamic has caused venture funds to focus on either earlier-stage investments, later-stage investments (similar to private equity), or a combination. Other funds have started focusing on flipping assets by investing before or to induce specific value-creation events. This has created a new environment where only a small number of funds are focused on the earliest stage—that which venture capital is most associated with and most successful at—with several others focused on a more transactional business. This evolution is still in process, but it has been changing the nature of companies that receive investment as well.
At the earliest stage of investments, venture capitalists have returned to investing in outstanding teams and under the assumption that they can create great companies. A number of approaches have been taken to inspire innovation and support a new era of breakthrough companies. Various firms have taken different approaches. CMEA, for example, invests in proven entrepreneurs “prenapkin” (before the idea), on the belief that they will come up with ideas. Polaris’ Dogpatch Labs has created an environment where multiple entrepreneurs share a common environment and with light money attempt to prove out their concepts. Y Combinator gives entrepreneurs an education and a small amount of money to try out their ideas. Andreessen Horowitz has similarly created an infrastructure to support the earliest stages of companies and to allow them to focus their capital on the company. “Super Angels” such as Peter Thiel have also emerged to provide important early stage funding. Several companies produced from some of these efforts have emerged as important venture-backed companies. Flagship VentureLabs has created an internal infrastructure of serial entrepreneurs to co-iterate its own innovations and use that as a basis to build companies.
Flagship VentureLabs
Flagship VentureLabs was built with a focus on increasing the efficiencies of innovation and entrepreneurship. In the broadest context, both traditional entrepreneurship and venture capital have intrinsic benefits and inefficiencies. Entrepreneurs, for example, typically perform well when capital constrained, but, by the same token, avoid asking critical questions because if an undesirable answer results, they are unemployed. Venture capital has the advantage of large sample sizes and substantial funding, but it is limited in its investments to only those which it can see, and all of its investments must fundamentally go through a common set of efforts (i.e., financial infrastructure, legal, etc.). Fundamentally, Flagship VentureLabs removes the constraints from the typical elements of traditional ecosystems; that is, by harnessing the key constituents and requirements all under one roof, with the common goal of the betterment of humankind through innovation and entrepreneurship.
The focus of Flagship VentureLabs is to develop breakthrough technologies to match large unmet needs in life sciences and sustainability through the vehicle of startup companies. New companies come from a breakthrough innovation without a set utility or from work within Flagship VentureLabs identifying the intersection between the potential for technology solution and market pull. In the former case, a team is nucleated around the technology, including the inventor, to heavily iterate the concept and pressure test it against markets, intellectual property opportunities, team-building potential, and other features with the attempt at nonrationally identifying the “sweet spot.” In certain cases, this process results in the pseudolinear formation of a company focused on commercializing the technology. In others, however, through a progressive set of explorations,
the company may end up far from its origins, potentially not including the base technology. In the latter case, the defined intersection creates a hypothesis. If the hypothesis has already been manifest by others in a company or in academia (either singularly or through a combination of efforts), a simple investment may be warranted. In the absence of such a proof point, the concept then goes through heavy conceptual iteration with the attempt to prove the hypothesis wrong, and in the combination of not being able to make it fail and the generation of significant key stakeholder support (including industry and key opinion leaders), a company will be launched. Ultimately, this approach results in taking new ideas and forming companies several years before such an opportunity is likely to be compelling. The following discusses efforts in three such technology-based companies originating from Flagship VentureLabs covering both life sciences and sustainability.
Seres Therapeutics: Rethinking Drug Development
In an effort to reduce side effects, drug development has focused its efforts on target specificity, particularly on features including affinity, low off-target effects, pharmacokinetics, pharmacodynamics, and others. The Human Genome Project and systematic understandings of the functions of kinases have helped to drive this increasing target specificity. Nonetheless, the biology of diseases is complex and multi-factorial. Focusing drugs to single actors may reduce side effects but it also limits the spectrum of efficacy. The growing recognition of the nature of disease is driving the understanding of more complex biology and the development of drugs focused on the multitude of key factors.
One particular example is with the human microbiome. Microorganisms have long been thought of as independently functioning pathogens. Recently, however, the commensal and mutualistic natures of various microorganisms that inhabit the body have started to be characterized (Dethlefsen, 2011). The interactions between the multitude of organisms, as well as between the organisms and the host, play an important role in normal physiology broadly (Reid et al., 2011). Accordingly, disruptions in the microbiome, whether by antibiotics, diet, infection, or other means, can alter the microbiome and induce or simply increase the likelihood of a wide range of diseases, ranging from Clostridium difficile infection and inflammatory bowel disease to obesity and diabetes (Kau et al., 2011).
The complexity of the microbiome, including not only the interrelation between a number of species and the host but also the physical formation of the communities in specific niches (Rickard et al., 2003), is important to take into account when developing therapeutics aimed at diseases where the microbiome plays an important role. Seres Therapeutics was founded specifically to develop drugs based on the complexity of the microbiome. Probiotics and single biologics affect a limited scope of disease and, thus, have limited efficacy in complex diseases such as those involving the microbiome (Shen et al., 2009). By creat-
ing synthetic microbiomes aimed at disrupting pathogenic communities, Seres provides a therapeutics means by which a normal microbiome can be restored. Understanding biology and synthetically recapitulating conditions that can recover from a disease-associated insult enables a new class of therapeutics to be designed and developed that are focused on the etiology of underlying disease.
Sustainability
Persistently high fossil fuel prices, increasing dependency on foreign fuel supplies, and insecurity relating to the sources of petroleum have created substantial market pull for alternative solutions in the $6 trillion petrochemical industry. Outside of government-mandated markets, such as ethanol of late, fuels and chemicals are fungible products driven by price and purity, as well as supply and demand. Markets therefore require products with a known utility that meet certain industrial specifications while doing so at a competitive cost point. Consumers have not shown a willingness to pay for benefits such as greenhouse gas mitigation or domestic sourcing, so products made as alternatives must do so while competing head-to-head with the incumbents using the same metrics.
Fuels have traditionally originated from biology in some form or another. Fossil fuels are thought to ultimately derive from processing of ancient biomass through a process that takes millions to hundreds of millions of years. Historically, humans have also found faster cycle time sources of energy, namely the burning of trees for heat and energy, as well as the removal of spermaceti from whales as a source of wax. All of these resources have limited renewal potential and substantial environmental impact (Tertzakian, 2009). Given the central role biology has had in fossil fuels historically, it stands to reason that biology would be well positioned to be at the forefront of the future of sustainable fuels.
Biological engineering has evolved rapidly over the last 50-plus years. Breakthroughs in genomics research, increased genetic manipulation potential, and more complete knowledge of the inner workings of cells have set a stage for cells to be engineered to achieve desired functionalities. Moreover, the time from conception to proof of concept, and that from proof of concept to commercial viability, has reduced substantially. Historically, these periods have decreased threefold every 10 years. Given the technological potential enabled, the market needs can now drive the technological direction, thus leading toward an intersection between market pull and the potential for technology solution.
LS9: Ultraclean Renewable Diesel
In 2005, the U.S. government had built a robust market demand for ethanol by outlining a replacement timeline for methyl tert-butyl ether (MTBE), a fuel oxygenate that had been associated with groundwater contamination and potential increased cancer risks, with ethanol (U.S. Environmental Protection Agency,
2011a). Twelve billion gallons were mandated by 2012, effectively defining a market growth. This mandate was soon supplemented with the renewable fuel standard (RFS), and subsequently RFS2, ultimately requiring 36 billion gallons produced per year by 2022 (U.S. Environmental Protection Agency, 2011b). Corn ethanol was thus given ample runway to launch, and blenders were incorporating biologically based products into fuel nationwide. The intent of the MTBE replacement with ethanol, however, was replacing an oxygenate, not deeming ethanol a fuel. Nonetheless, outspoken investors were enthusiastically supportive of building the future of American renewable fuel on ethanol, asserting that it could be cheaper than and as efficient as petrochemically derived fuels (Khosla, 2006) despite the disadvantaged domestic cost structure and intrinsic lower energy density. The market was becoming well positioned for a viable alternative.
LS9 originated by asking the question, “If you could make any fuel from biology, what would you make?” The ideal fuel to be produced from biology would be diesel, given its high energy density and its use throughout the world as a primary transportation fuel. A market-acceptable biologically produced diesel must compete in a low-cost commodity market without subsidies, requiring an efficient biological pathway and process. Translating these needs into specific technological tasks required that the cell be engineerable to be feedstock agnostic (i.e., able to use any form of sugar), that the most efficient pathway of producing the product was available, that the product was to be made directly and secreted, and that it entailed both straightforward separations (a feature of the product) and no downstream processing (the final product is made by the cell).
Using the defined market constraints, various pathways to produce a straight-chain hydrocarbon were defined and evaluated. The fatty acid biosynthesis pathway not only has exceptionally high energy efficiency at over 90 percent but also produces a molecule that is chemically identical to diesel, requiring potentially fewer biological steps. Fatty acids are activated with coenzyme A (CoA) or acyl carrier protein (ACP) to make fatty acyl-CoA or fatty acyl-ACP (Zhang and Rock, 2008), which serve as the biological precursor products for fuel synthesis. These products are then modified to make a desired end product. The same pathways can be leveraged to make a series of other petrochemicals including fatty acid methyl esters, olefins, fatty alcohols, and others, in addition to alkanes (diesel) (Rude et al., 2011).
The product itself is insufficient for a commercial host and process. The identified market constraints require that the cell chassis has flexibility in feedstock, be optimized to maximize carbon flux to end product, and secrete the end product. Feedstock costs, driven by sugar prices, have risen dramatically over the past 6 years. Alternatives require the liberation of sugar from cellulosic biomass, which is done through exogenous enzymes at present. The expression of hemicellulases into the host already engineered to produce alkanes or other derivatives can enable consolidated bioprocessing, thereby reducing process costs (Magnuson et al., 1993). This is achievable, for example, with the endogenous production
of glycosyl hydrolases such as xylanase (Xsa) from Bacteroides ovatus and an endoxylanase catalytic domain (Xyn10B) from Clostridium stercorarium, which together hydrolyze hemicelluloses to xylose, which is usable in E. coli central metabolism (Adelsberger et al., 2004; Steen et al., 2010; Whitehead and Hespell, 1990). Optimizing the host requires focusing the flux of the sugar input through central metabolism to the product. Specifically, fadD and fadE knockouts block the first two steps of the ß-oxidation pathway, increasing end-product production three- to fourfold (Steen et al., 2010). Secreting the end product eliminates end-product inhibition and streamlines bioprocessing, thus increasing flux and reducing operating costs by supporting continuous operations (Berry, 2010). Expressing a leaderless version of TesA eliminates end-product inhibition, drives secretion, and notably also positively affects chain length with a natural preference for C14 fatty acids (Cho and Cronan, 1995; Jiang and Cronan, 1994; Steen et al., 2010).
Through this approach, an industrial chassis has been rationally developed to systematically meet commercial needs. By specifically including features necessary to ensure diverse feedstock utility, drop-in product synthesis, and lowest cost processing, LS9’s technology has been designed specifically to drive market pull.
Joule Unlimited: Renewable Solar Fuels
An intrinsic challenge of using a sugar-based feedstock is the price volatility associated with the commodity. Joule Unlimited was founded to develop a platform that could eliminate dependence on sugar feedstocks while still producing fuels in a way that meets market needs. A systematic exploration of sources of carbon that can be routed into central metabolism rapidly identifies photosynthesis, nature’s solution to carbon dioxide assimilation driven by solar energy, as a compelling, though insufficient, pathway. The Department of Energy’s Aquatic Species program, based on explorations of algal biofuels between 1976 and 1996, concluded that photosynthesis could support viable fuel processes, but it requires a set of key innovations to do so (Sheehan et al., 1998; Weyer et al., 2010; Zhu et al., 2008). At the outset of Joule Unlimited, the fundamental limitations of algal fuels were examined and coupled with market needs to design an entirely new and distinct approach, whose only similarity to algae was the use of photosynthesis.
A thorough exploration of market needs identified that an ideal solution would directly produce secreted fungible fuel directly from sunlight and carbon dioxide without a dependency on arable land, freshwater, or other costly reagents, while having a cost that could meet or beat fossil fuel equivalents in the absence of subsidies and at the same time scale modularly such that smaller-scale plants could be used to validate large-scale deployments. The simultaneous technological solution to meet all of these needs demands a genetically tractable cyanobacteria engineered to not need exogenous factors and to produce secreted fuel grown in a
modular bioreactor leveraging the two-dimensional scaling of light and incorporating fundamental process needs including proper mixing. A schematic of the Joule Unlimited approach, Helioculture™, is provided in Figure A1-1.
Cyanobacteria had previously been engineered to express recombinant proteins, but it had not been systematically engineered on a genome scale owing primarily to a lack of engineering tools (Alvey et al., 2011). A concerted effort using E. coli engineering over 50 years serves as a roadmap of the needs for genome engineering in cyanobacteria. Using these tools coupled with a systemic genome engineering effort allows one to overcome a theoretical maximum light use and net productivity for algal biofuels of ~2,000 gallons per acre per year by enabling photoautotrophs, for the first time, to function like industrialized heterotrophs, whose phases of growth and production are separated (Berry, 2010; Robertson et al., 2011). Systematic re-regulation of central metabolism directs 95 percent of photosynthetic activity to specific product synthesis versus up to 50 percent in un-engineered organisms, allowing for productivity ~95 percent of the light-exposed time through continuous operations versus substantial down time with batch processes, minimizing maintenance energy requirements, and limiting the energy lost to photorespiration.
At the same time, the engineered cells are grown in a reactor system designed to be low cost and linearly scalable. Low-cost product separation, cell mixing, and proper gas transfer to the cells are all incorporated into the reactor design. Coupled together, this systems approach allows for ~12 percent theoretical maximal photonic energy conversion versus 1.5 percent for traditional algal processes (Figure A1-2), which translates to unprecedented areal productivities of 25,000 gallons of ethanol per acre per year or 15,000 gallons of diesel per acre per year.
The high productivities achieved through the Joule Unlimited approach allow for cost points as low as $20/barrel for fungible diesel. Maximizing solar energy capture, carbon dioxide fixation as a replacement for sugar use, and organism productivity creates a system that can be market competitive while providing for the environmental benefits that have been sought in fossil fuel replacements. Specifically, Joule Unlimited’s technology eliminates the need for arable land, requires no freshwater, and reduces life-cycle greenhouse gases by over 90 percent by using carbon dioxide as a feedstock. By coupling the technical needs for a total solution with a market need, Joule Unlimited has uniquely developed a process that can produce a sugar-independent diesel in a highly scalable manner, overcoming the challenges of alternative approaches.
Conclusions
Systematic changes in venture capital have altered the entrepreneurial ecosystem. Flagship VentureLabs is pioneering a new approach of technology development through companies by building technologies that specifically address the intersection between the potential for technology solution with market pull
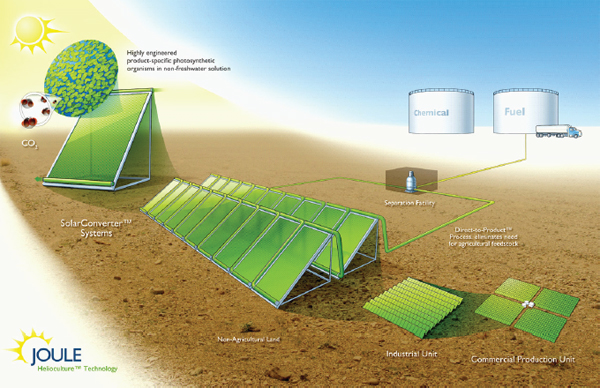
FIGURE A1-1 Schematic of the Joule Unlimited Helioculture systems approach. Specific cyanobacteria are engineered to convert sunlight, CO2, and nonfreshwater to diesel (or other fuels and chemicals). The engineered organisms are housed within a Solar Converter, a single reactor unit designed to interconnect with others and therefore scale linearly.
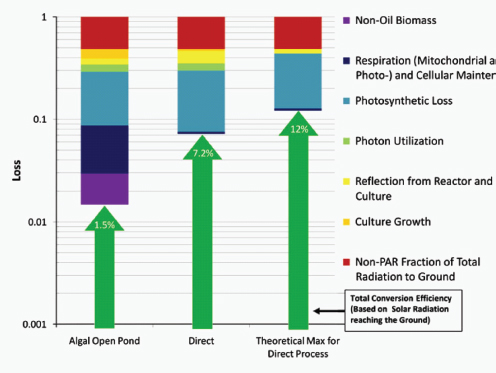
FIGURE A1-2 A summation of the accumulated photon losses for algal and direct fuel processes, as well as a theoretical maximum photonic energy conversion. The losses are shown through individual contributions accumulated serially and illustrated on a logarithmic scale, beginning with the percent of photosynthetically active radiation empirically measured at the ground. Total energy conversion efficiency as a function of the losses is indicated by the green arrows.
SOURCE: Adapted from Robertson et al. (2011).
driving invention and innovation toward market needs. The resultant companies are designed by exploring cutting-edge capabilities and iterating against market needs—not just from an evolutionary standpoint, but additionally identifying the true needs of an industry across multiple facets. Synthetic biology is a new and rapidly developing tool that has particular utility in meeting broad-based and distinct market needs, particularly through its ability to create functional modules in a cell-based system. By leveraging the potential of synthetic biology with market-driven needs, Flagship VentureLabs has been able to spearhead a set of breakthrough innovations in both life sciences and sustainability. This approach now takes market potential before traditional research approaches have made for a compelling and investable technology-driven opportunity and, through heavy iteration, can bring it to bear ahead of time through broad-based collaborations with industry and academia. This approach can be broadly leveraged to develop
a series of future breakthrough technologies in a variety of important market sectors.
References
Adelsberger, H., C. Hertel, E. Glawischnig, V. V. Zverlov, and W. H. Schwarz. 2004. Enzyme system of Clostrium stercorarium for hydrolysis of arabinoxylan: Reconstitution of the in vivo system from recombinant synzmes. Microbiology 150:2257-2266.
Alvey, R. M., A. Biswas, W. M. Schluchter, and D. A. Bryant. 2011. Attachment of noncognate chromophores to CpcA of Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002 by heterologous expression in Escherichia coli. Biochemistry 50:4890-4902.
Ante, S. E. 2008. The Birth of Venture Capital. Cambridge, MA: Harvard Business School Press.
Berry, D. A. 2010. Engineering organisms for industrial fuel production. Bioengineered Bugs 1: 303-308.
Cho, H., and J. E. Cronan, Jr. 1995. Defective export of a periplasmic enzyme disrupts regulation of fatty acid synthesis. World Journal of Biological Chemistry 270:4216-4219.
Dethlefsen, L., M. McFall-Ngai, and D. A. Relman. 2011. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811-818.
Jiang, P., and J. E. Cronan, Jr. 1994. Inhibition of fatty acid synthesis in Escherichia coli in the absence of phospholipid synthesis and release of inhibition by thioesterase action. Journal of Bacterialogy 176:2814-2821.
Kau, A. L., P. P. Ahern, N. W. Griffen, A. L. Goodman, and J. I. Gordon. 2011. Human nutrition, the gut microbiome and the immune system. Nature 474:327-336.
Khosla, V. 2006. Is ethanol controversial? Should it be? http://www.khoslaventures.com/khosla/papers.html (accessed June 8, 2011).
Kirsner, S. 2008. Venture capital’s grandfather. The Boston Globe, April 6.
Magnuson, K., S. Jackowski, C. O. Rock, and J. E. Cronan, Jr. 1993. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiology Reviews 57:522-542.
Metrick, A. 2007. Venture Capital and the Finance of Innovation. New York: John Wiley & Sons.
National Venture Capital Association. 2009. Venture Impact: The Economic Importance of Venture Backed Companies to the U.S. Economy. National Venture Capital Association: Arlington, VA.
Reid, G., J. A. Younes, H. C. Van der Mei, G. B. Gloor, R. Knight, and H. J. Busscher. 2011. Micro-biota restoration: Natural and supplemented recovery of human microbial communities. Nature Reviews Microbiology 9:27-38.
Rickard, A. H., P. Gilbert, N. J. High, P. E. Kolenbrander, and P. S. Handley. 2003. Bacterial coaggregation: An integral process in the development of multi-species biofilms. Trends in Microbiology 11:94-100.
Robertson, D. E., S. Jacobson, F. Morgan, D. Berry, G. M. Church, and N. B. Afeyan. 2011. A new dawn for industrial photosynthesis: An evaluation of photosynthetic efficiencies for production of liquid fuels. Photosynthesis Research 107:269-277.
Rude, M. A., T. S. Baron, S. Brubaker, M. Alibhai, S. B. Del Cardayre, and A. Schirmer. 2011. Terminal olefin (1-alkene) biosynthesis by a novel p450 fatty acid decarboxylase from Jeotgalicoccus species. Applied and Environmental Microbiology 77:1718-1727.
Sheehan, J., T. Dunahay, J. Benemann, and P. A. Roessler. 1998. A Look Back at the U.S. Department of Energy’s Aquatic Species Program: Biodiesel from Algae. U.S. Department of Energy, Office of Fuels Development: Closeout Report TP-580-241-24190.
Shen, J., H. Z. Ran, M. H. Yin, T. Z. Zhou, and D. S. Xiao. 2009. Meta-analysis: The effect and adverse events of lactobacilli versus placebo in maintenance therapy for Crohn disease. Internal Medical Journal 39:103-109.
Steen, E. J., Y. Kang, G. Bokinsky, Z. Hu, A. Schirmer, A. McClure, S. B. del Cardayre, and J. D. Keasling. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559-563.
Tertzakian, P. 2009. The End of Energy Obesity. New York: John Wiley & Sons.
Thomson Reuters. 2011. 2011 National Venture Capital Association Yearbook. New York: Thomson Reuters.
U.S. Environmental Protection Agency. 2011a. Methyl Tertiary Butyl Ether (MTBE). http://www.epa.gov/mtbe/faq.htm#concerns.
——. 2011b. Fuels and Fuel Additives. http://www.epa.gov/otaq/fuels/renewablefuels/index.htm
Weyer, K. M., D. R. Bush, A. Darzins, and B. D. Wilson. 2010. Theoretical maximum algal oil production. Bioenergy Research 3:204-213.
Whitehead, T. R., and R. B. Hespell. 1990. The genes for three xylan-degrading activities from Bacteroides ovatus are clustered in a 3.8-kilobase region. Journal of Bacteriology 172:2408-2412.
Wilson, J. W. 1985. The New Venturers: Inside the High-Stakes World of Venture Capital. Boston: Addison-Wesley.
Zhang, Y. M., and C. O. Rock. 2008. Membrane lipid homeostasis in bacteria. Nature Reviews Microbiology 6:222-233.
Zhu, X.-G., S. P. Long, and D. R. Ort. 2008. What is the maximum efficiency with which photosynthesis can convert solar energy into biomass? Current Opinion in Biotechnology 19:153-159.
SYNTHETIC BIOLOGY: APPLICATIONS COME OF AGE3
Ahmad S. Khalil4and James J. Collins4,5
Abstract
Synthetic biology is bringing together engineers and biologists to design and build novel biomolecular components, networks and pathways, and to use these constructs to rewire and reprogram organisms. These re-engineered organisms will change our lives over the coming years, leading to cheaper drugs, ‘green’ means to fuel our cars and targeted therapies for attacking ‘superbugs’ and diseases, such as cancer. The de novo engineering of genetic circuits, biological modules and synthetic pathways is beginning to address these crucial problems and is being used in related practical applications.
_______________
3 Originally printed as Khalil, A. S., and J. J. Collins. 2010. Synthetic biology: Applications come of age. Nature Reviews Genetics 11:367-379. Reprinted with kind permission from Nature Publishing Group.
4 Howard Hughes Medical Institute, Department of Biomedical Engineering, Center for BioDynamics and Center for Advanced Biotechnology, Boston University, Boston, Massachusetts 02215, USA.
5 Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts 02115, USA. Correspondence to J.J.C. e-mail: jcollins@bu.edu. doi:10.1038/nrg2775.
The circuit-like connectivity of biological parts and their ability to collectively process logical operations was first appreciated nearly 50 years ago (Monod and Jacob, 1961). This inspired attempts to describe biological regulation schemes with mathematical models (Glass and Kauffman, 1973; Savageau, 1974; Kauffman, 1974; Glass, 1975) and to apply electrical circuit analogies to biological pathways (McAdams and Arkin, 2000; McAdams and Shapiro, 1995). Meanwhile, breakthroughs in genomic research and genetic engineering (for example, recombinant DNA technology) were supplying the inventory and methods necessary to physically construct and assemble biomolecular parts. As a result, synthetic biology was born with the broad goal of engineering or ‘wiring’ biological circuitry — be it genetic, protein, viral, pathway or genomic — for manifesting logical forms of cellular control. Synthetic biology, equipped with the engineering-driven approaches of modularization, rationalization and modelling, has progressed rapidly and generated an ever-increasing suite of genetic devices and biological modules.
The successful design and construction of the first synthetic gene networks — the genetic toggle switch (Gardner et al., 2000) and the repressilator (Elowitz and Leibler, 2000) (Box A2-1) — showed that engineering-based methodology could indeed be used to build sophisticated, computing-like behaviour into biological systems. In these two cases, basic transcriptional regulatory elements were designed and assembled to realize the biological equivalents of electronic memory storage and timekeeping (Box A2-1). Within the framework provided by these two synthetic systems, biological circuits can be built from smaller, well-defined parts according to model blueprints. They can then be studied and tested in isolation, and their behaviour can be evaluated against model predictions of the system dynamics. This methodology has been applied to the synthetic construction of additional genetic switches (Gardner et al., 2000; Atkinson et al., 2003; Bayer and Smolke, 2005; Deans et al., 2007; Dueber et al., 2003; Friedland et al., 2009; Ham et al., 2006, 2008; Kramer and Fussenegger, 2005; Kramer et al., 2004), memory elements6 (Gardner et al., 2000; Friedland et al., 2009; Ham et al., 2006; Ajo-Franklin et al., 2005) and oscillators (Elowitz and Leibler, 2000; Atkinson et al., 2003; Fung et al., 2005; Stricker et al., 2008, Tigges et al., 2009; Danino et al., 2010), as well as to other electronics-inspired genetic devices, including pulse generators7 (Basu et al., 2004), digital logic gates8 (Anderson et al., 2007; Guet et al., 2002; Rackham and Chin, 2005; Rinaudo et al., 2007;
_______________
6 Memory elements – Devices used to store information about the current state of a system.
7 Pulse generators – Circuits or devices used to generate pulses. A biological pulse generator has been implemented in a multicellular bacterial system, in which receiver cells respond to a chemical signal with a transient burst of gene expression, the amplitude and duration of which depend on the distance from the sender cells.
8 Digital logic gates – A digital logic gate implements Boolean logic (such as AND, OR or NOT) on one or more logic inputs to produce a single logic output. Electronic logic gates are implemented using diodes and transistors and operate on input voltages or currents, whereas biological logic gates operate on cellular molecules (chemical or biological).
Stojanovic and Stefanovic, 2003; Win and Smolke, 2008), filters9 (Basu et al., 2005; Hooshangi et al., 2005; Sohka et al., 2009) and communication modules (Danino et al., 2010; Basu et al., 2005; Kobayashi et al., 2004; You et al., 2004).
Now, 10 years after the demonstration of synthetic biology’s inaugural devices (Gardner et al., 2000; Elowitz and Leibler, 2000), engineered biomolecular networks are beginning to move into the application stage and yield solutions to many complex societal problems. Although work remains to be done on elucidating biological design principles (Mukherji and van Oudenaarden, 2009), this foray into practical applications signals an exciting coming-of-age time for the field.
Here, we review the practical applications of synthetic biology in biosensing, therapeutics and the production of biofuels, pharmaceuticals and novel biomaterials. Many of the examples herein do not fit exclusively or neatly into only one of these three application categories; however, it is precisely this multivalent applicability that makes synthetic biology platforms so powerful and promising.
Biosensing
Cells have evolved a myriad of regulatory circuits — from transcriptional to post-translational — for sensing and responding to diverse and transient environmental signals. These circuits consist of exquisitely tailored sensitive elements that bind analytes and set signal-detection thresholds, and transducer modules that filter the signals and mobilize a cellular response (Box A2-2). The two basic sensing modules must be delicately balanced: this is achieved by programming modularity and specificity into biosensing circuits at the transcriptional, translational and post-translational levels, as described below.
Transcriptional biosensing. As the first dedicated phase of gene expression, transcription serves as one method by which cells mobilize a cellular response to an environmental perturbation. As such, the genes to be expressed, their promoters, rNA polymerase, transcription factors and other parts of the transcription machinery all serve as potential engineering components for transcriptional biosensors. Most synthetic designs have focused on the promoters and their associated transcription factors, given the abundance of known and characterized bacterial, archaeal and eukaryotic environment-responsive promoters, which include the well-known promoters of the Escherichia coli lac, tet and ara operons.
Both the sensory and transducer behaviours of a biosensor can be placed under synthetic control by directly engineering environment-responsive promoter sequences. In fact, this was the early design strategy adopted for establishing inducible expression systems (Brown et al., 1987; Deuschle et al., 1989; Hu and
_______________
9 Filters – Algorithms or devices for removing or enhancing parts or frequency components from a signal.
BOX A2-1
Early Synthetic Biology Designs: Switches and Oscillators
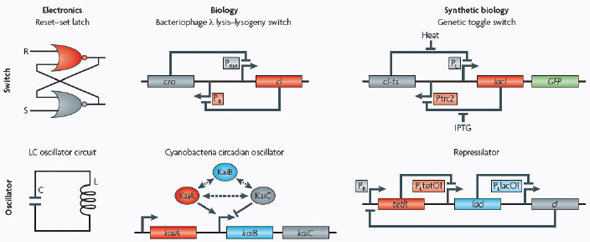
Switches and oscillators that occur in electronic systems are also seen in biology and have been engineered into synthetic biological systems.
Switches
In electronics, one of the most basic elements for storing memory is the reset–set (RS) latch based on logical NOR gates*. This device is bistable in that it possesses two stable states that can be toggled with the delivery of specified inputs. Upon removal of the input, the
circuit retains memory of its current state indefinitely. These forms of memory and state switching have important functions in biology, such as in the differentiation of cells from an initially undifferentiated state. One means by which cellular systems can achieve bistability is through genetic mutual repression. The natural PR–PRM genetic switch from bacteriophage λ, which uses this network architecture to govern the lysis–lysogeny decision, consists of two promoters that are each repressed by the gene product of the other (that is, by the Cro and CI repressor proteins). The genetic toggle switch (Gardner et al., 2000) constructed by our research group is a synthetically engineered version of this co-repressed gene regulation scheme. In one version of the genetic toggle, the PL promoter from λ phage was used to drive transcription of lacI, the product of which represses a second promoter, Ptrc2 (a lac promoter variant). Conversely, Ptrc2 drives expression of a gene (cI-ts) encoding the temperature-sensitive (ts) λ CI repressor protein, which inhibits the PL promoter. The activity of the circuit is monitored through the expression of a GFP promoter.
The system can be toggled in one direction with the exogenous addition of the chemical inducer isopropyl-β-d-thiogalactopyranoside (IPTG) or in the other direction with a transient increase in temperature. Importantly, upon removal of these exogenous signals, the system retains its current state, creating a cellular form of memory.
Oscillators
Timing mechanisms, much like memory, are fundamental to many electronic and biological systems. Electronic timekeeping can be achieved with basic oscillator circuits — such as the LC circuit (inductor L and capacitor C) — which act as resonators for producing periodic electronic signals. Biological timekeeping, which is widespread among living organisms (Dunlap, 1999), is achieved with circadian clocks and similar oscillator circuits, such as the one responsible for synchronizing the crucial processes of photosynthesis and nitrogen fixation in cyanobacteria. The circadian clock of cyanobacteria is based on, among other regulatory mechanisms, intertwined positive and negative feedback loops on the clock genes kaiA, kaiB and kaiC. Elowitz and Leibler constructed a synthetic genetic oscillator based not on clock genes but on standard transcriptional repressors (the repressilator) (Elowitz and Leibler, 2000). Here, a cyclic negative feedback loop composed of three promoter–gene pairs, in which the ‘first’ promoter in the cascade drives expression of the ‘second’ promoter’s repressor, and so on, was used to drive oscillatory output in gene expression.
——————
* NOR gate – A digital logic gate that implements logical NOR, or the negation of the OR operator. It produces a HIGH output (1) only if both inputs to the gate are LOW (0).
BOX A2-2
Synthetic Biosensors: Transcriptional and Translational Architectures and Examples
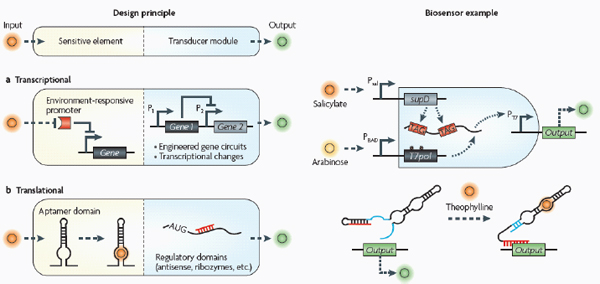
Biosensors consist of two basic modules (see the figure): sensitive elements for recognizing and binding analytes, and transducer modules for transmitting and reporting signals.
Transcriptional
Transcriptional biosensors (part a) are built by linking environment-responsive promoters to engineered gene circuits for programmed transcriptional changes. In the example shown, a transcriptional AND gate was designed to sense and report only the simultaneous presence of two environmental signals (for example, salicylate and arabinose) (Anderson et al., 2007). At one gate input, the researchers encoded an environment-responsive promoter (for example, PBAD) that activates transcription of a T7 RNA polymerase gene in response to a single environmental signal (for example, arabinose). The gene, however, carries internally encoded amber stop codons (red spiked circles) that function to block translation of its transcript. Activation of the second gate input is the key to unlocking translation; specifically, translation can be induced when a second promoter (for example, Psal) activates transcription of the supD amber suppressor tRNA in response to a second unique signal (for example, salicylate). In other words, only when the two environmental signals are simultaneously present can the T7 RNA polymerase be faithfully expressed and used to activate an output T7 promoter. This is an example of how sophisticated specificity can be programmed into a transducer module by creatively linking the sensory information of multiple sensitive elements. Furthermore, the design is transcriptionally modular in that different sets of environment-responsive promoters can be interfaced to the AND gate.
Translational
Translational biosensors (part b) are typically built by linking RNA aptamer domains to RNA regulatory domains. The example shown is an OFF ‘antiswitch’. Here, the small molecule theophylline is recognized and bound by the aptamer stem of the RNA biosensor. This causes a conformational change in the molecule that liberates the antisense domain from its sequestering stem loop and allows it to inhibit translation of an output reporter (Bayer and Smolke, 2005).
Davidson, 1987; Lutz and Bujard, 1997). By introducing, removing or modifying activator and repressor sites, a promoter’s sensitivity to a molecule can be tuned. Synthetic mammalian transactivation systems are generic versions of this strategy in which an environmentally sensitive transcription factor is fused to a mammalian transactivation domain to cause inducer-dependent changes in gene expression. Synthetic mammalian biosensors based on this scheme have been created for sensing signals such as antibiotics (Fussenegger et al., 2000; Gossen and Bujard, 1992; Weber et al., 2002), quorum-sensing molecules (Neddermann et al., 2003; Weber et al., 2003b), gases and metabolites (Malphettes et al., 2005; Mullick et al., 2006; Weber et al., 2006; Weber et al., 2004), and temperature changes (Boorsma et al., 2000; Weber et al., 2003a). Fussenegger and colleagues have even incorporated this transgene design into mammalian circuits, creating synthetic networks that are responsive to electrical signals (Weber et al., 2009).
Although the engineering of environment-responsive promoters has been valuable, additional control over modularity10 and specificity can be achieved by embedding environment-responsive promoters11 in engineered gene networks. Achieving true modularity with genetic parts is inherently difficult because of unintended interference among native and synthetic parts and therefore requires careful decoupling of functional modules. One such modular design strategy was used by Kobayashi et al. (2004) to develop whole-cell E. coli biosensors that respond to signals in a programmable fashion. In this design, a sensory module (that is, an environment-responsive promoter and associated transcription factor) was coupled to an engineered gene circuit that functions like a central processing unit. E. coli cells were programmed to respond to a deleterious endogenous input — specifically, DNA-damaging stimuli, such as ultraviolet radiation or mitomycin C. The gene circuit, which was chosen to be the toggle switch (Box A2-1), processes the incoming sensory information and flips from an ‘OFF’ to an ‘ON’ state when a signal threshold is exceeded. Because the biosensor has a decoupled, modular nature, it can be wired to any desired output, from the expression of a standard fluorescent reporter to the activation of natural phenotypes, such as biofilm formation (for example, through expression of traA) or cell suicide (for example, through expression of ccdB).
Sometimes a single signal may be too general to characterize or define an environment. For such situations, Anderson et al. (2007) devised a transcriptional AND gate that could be used to integrate multiple environmental signals into a single genetic circuit (Box A2-2), therefore programming the desired level of biosensing specificity. Genetic biosensors of this sort could be useful for communicating the state of a specific microenvironment (for example, in an industrial
_______________
10 Modularity – The capacity of a system or component to function independently of context.
11 Environment-responsive promoters – Promoters that directly transduce environmental signals (for example, heavy metal ions, hormones, chemicals or temperature) that are captured by their associated sensory transcription factors.
bioreactor) within a ‘sea’ of environmental conditions, such as temperature, metabolite levels or cell density.
Translational biosensing. RNA molecules have a diverse and important set of cellular functions (Eddy, 2001). Non-coding RNAs can splice and edit RNA, modify ribosomal RNA, catalyse biochemical reactions and regulate gene expression at the level of transcription or translation (Eddy, 2001; Doudna and Cech, 2002; Guerrier-Takada et al., 1983; Kruger et al., 1982). The regulatory subset of non-coding RNAs (Lee et al., 1993; Stougaard and Nordstrom, 1981; Wagner and Simons, 1994) is well-suited for rational design (Isaacs et al., 2006) and, in particular, for biosensing applications. Many regulatory RNA molecules are natural environmental sensors (Gelfand et al., 1999; Johansson et al., 2002; Lease and Belfort, 2000; Majdalani et al., 2002; Mandal et al., 2003; Mironov et al., 2002; Morita et al., 1999; Winkler et al., 2002; Winkler et al., 2004), and because of their ability to take on complex structures defined by their sequence, these molecules can mediate diverse modular functions across distinct sequence domains. Riboswitches (Winkler and Breaker, 2005), for instance, bind specific small-molecule ligands through aptamer12 domains and induce conformational changes in the 5′ UTR of their own mRNA, thereby regulating gene expression. Aptamer domains that are modelled after riboswitches are versatile and widely used sensitive elements for RNA-based biosensing. The choice and number of aptamer domains can provide control over specificity. Building an entire RNA-based biosensor typically requires coupling an aptamer domain (the sensitive element) with a post-transcriptional regulatory domain (the transducer module) on a modular RNA molecule scaffold.
Antisense RNAs13 (Wagner and Simons, 1994; Good, 2003) are one such class of natural regulatory RNAs that can control gene expression through posttranscriptional mechanisms. By linking a riboswitch aptamer to an antisense repressor on a single RNA molecule, Bayer and Smolke (2005) engineered transacting, ligand-responsive riboregulators14 of gene expression in Saccharomyces cerevisiae (Box A2-2). Binding of the aptamer to its ligand (for example, the small molecule theophylline) induces a conformational change in the RNA sensor that either sequesters the antisense domain in a stable stem loop (ON switch) or liberates it to inhibit translation of an output gene reporter (OFF switch). As a result of the cooperative dependence on both ligand and target mRNA, this biosensor shows binary-like switching at a threshold ligand concentration, similar
_______________
12 Aptamer – Oligonucleic acids that bind to a specific target molecule, such as a small molecule, protein or nucleic acid. Nucleic acid aptamers are typically developed through in vitro selection schemes but are also found naturally (for example, RNA aptamers in riboswitches).
13 Antisense RNAs – RNAs that bind segments of mRNA in trans to inhibit translation.
14 Riboregulators – Small regulatory RNAs that can activate or repress gene expression by binding segments of mRNA in trans. They are typically expressed in response to an environmental signalling event.
to the genetic toggle design. Importantly, this detection threshold can be adjusted by altering the RNA sequence and therefore the thermodynamic properties of the structure. In principle, the ‘antiswitch’ framework is modular; in other words, aptamers for different ligands and antisense stems targeting different downstream genes could be incorporated into the scaffold to create new sensors. In practice, developing new sensors by aptamer and antisense replacement often involves re-screening compatible secondary structures to create functioning switches. In the future, this platform could be combined with rapid, in vitro aptameric selection techniques (Cox et al., 2002; Ellington and Szostak, 1990; Hermann and Patel, 2000; Tuerk and Gold, 1990) for generating a suite of RNA biosensors that report on the levels of various mRNA species and metabolites in a cell. However, here it should also be noted that aptamers show specificity for a biased ligand space, and as a result aptamers for a target ligand cannot always be found.
Another method for transducing the sensory information captured by aptamer domains is to regulate translation through RNA self-cleavage (Winkler et al., 2004; Winkler and Breaker, 2005). RNA cleavage is catalysed by ribozymes, some of which naturally possess aptameric domains and are responsive to metabolites (Winkler et al., 2004). Yen et al. (2004) took advantage of this natural framework and encoded ligand-sensitive ribozymes in the mRNA sequences of reporter genes. In the absence of its cognate ligand, constitutive autocleavage of the reporter mRNA resulted in little or no signal. The RNA biosensor is flipped when the cognate ligand is present to inhibit the ribozyme’s activity. Similar to the ‘antiswitch’ framework (and with the same technical challenges), these engineered RNAs could potentially be used as endogenous sensors for reporting on a variety of intracellular species and metabolites.
Post-translational biosensing. Signal transduction pathways show vast diversity and complexity. Factors such as the nature of the molecular interactions, the number of interconnected proteins in a cascade and the use of spatial mechanisms dictate which signals are transmitted, whether a signal is amplified or attenuated and the dynamics of the response. Despite the multitude of factors and interacting components, signal transduction pathways are essentially hierarchical schemes based on sensitive elements and downstream transducer modules, and as such can be rationalized for engineering protein-based biosensors.
The primary sensitive element for most signal transduction pathways is the protein receptor. Whereas environment-responsive promoters and RNA aptamers are typically identified from nature or selected with high-throughput combinatorial methods, protein receptors can be designed de novo at the level of molecular interactions. For instance, Looger et al. (2003) devised a computational method for redesigning natural protein receptors to bind new target ligands. Starting with a ‘basis’ of five proteins from the E. coli periplasmic binding protein (PBP) superfamily, the researchers replaced each of the wild-type ligands with a new, non-native target ligand and then used an algorithm to combinatorially explore all
binding-pocket-residue mutations and ligand-docking configurations. This procedure was used to predict novel receptors for trinitrotoluene (TNT; a carcinogen and explosive), L-lactate (a medically-important metabolite) and serotonin (a chemical associated with psychiatric conditions). The predicted receptor designs were experimentally confirmed to be strong and specific in vitro sensors, as well as in vivo cell-based biosensors.
Protein receptors, such as the ones discussed above, are typically membrane-bound; they trigger protein signalling cascades that ultimately result in a cellular response. However, several synthetic methods can be used to transmit captured sensory information in a tunable and desirable manner. Skerker et al. (2008) rationally rewired the transmission of information through two-component systems15 by identifying rules governing the specificity of a histidine kinase to its cognate response regulator. Alternatively, engineered protein scaffolds can be designed to physically recruit pathway modulators and synthetically reshape the dynamical response behaviour of a system (Bashor et al., 2008) (Box A2-3). This constitutes a modular method for programming protein-based biosensors to have any desired response, including accelerated, delayed or ultrasensitive responses, to upstream signals.
Hybrid approaches. Combining synthetic transcriptional, translational and post-translational circuits into hybrid solutions and harnessing desired characteristics from each could lead to the creation of cell-based biosensors that are as robust as those of natural organisms. Using a synthetic hybrid approach, Voigt and colleagues (Levskaya et al., 2005; Levskaya et al., 2009; Tabor et al., 2009) developed E. coli-based optical sensors. A synthetic sensor kinase was engineered to allow cells to identify and report the presence of red light. As a result, a bacterial lawn of the engineered cells could faithfully ‘print’ a projected image in the biological equivalent of photographic film. Specifically, a membrane-bound photoreceptor from cyanobacteria was fused to an E. coli intracellular histidine kinase to induce light-dependent changes in gene expression (Levskaya et al., 2005) (Box A2-3). In a clever example of its use, the bacterial optical sensor was applied in image edge detection (Tabor et al., 2009). In this case, by wiring the optical sensor to transcriptional circuits that perform cell–cell communication (the quorum-sensing16 system from Vibrio fischeri) and logical functions (Box A2-3), the researchers programmed only the cells that receive light and directly neighbour cells that do not receive light to produce a pigment, allowing
_______________
15 Two-component systems – Among the simplest types of signal transduction pathways. In bacteria, they consist of two domains: a membrane-bound histidine kinase (sensitive element) that senses a specific environmental stimulus, and a cognate response regulator (transducer domain) that triggers a cellular response.
16 Quorum sensing – A cell-to-cell communication mechanism in many species of bacteria, whereby cells measure their local density (by the accumulation of a signalling molecule) and subsequently coordinate gene expression.
BOX A2-3
Synthetic Biosensors: Post-translational and Hybrid Architectures and Examples
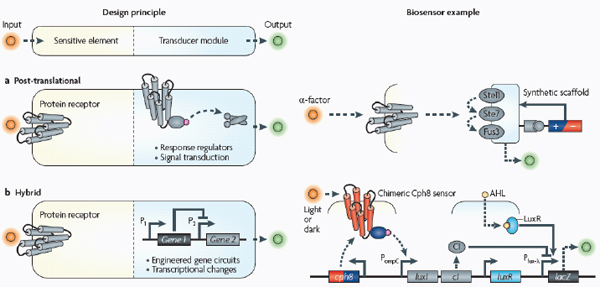
A basic biosensor has two modules (see the figure): the sensitive element recognizes and binds analytes, whereas the transducer module transmits and reports signals.
Post-translational
Post-translational biosensors (part a) consist of membrane-bound protein receptors that trigger signal transduction cascades through signalling proteins, such as response regulators of two-component systems. In the example shown, a synthetic protein scaffold was engineered to physically localize the pathway components of the yeast mitogen-activated protein kinase (MAPK) pathway, which here is being triggered by the mating α-factor (Bashor et al., 2008). By recruiting pathway positive and negative modulators (±) to the scaffold, the system can be tuned to enable desired responses to upstream signals (for example, accelerated, delayed or ultrasensitive responses).
Hybrid
The hybrid example (part b) shows a synthetic genetic edge detection circuit (Tabor et al., 2009). The sensitive element is a light–dark sensor, Cph8, made as a chimaera of the photoreceptor domain of the cyanobacteria phytochrome Cph1 and the kinase domain of Escherichia coli EnvZ. This synthetic sensor activates an engineered gene circuit that combines cell–cell communication (genes and promoters of the Lux operon) with a logical AND gate (Plux-λ) to trace the edges of an image. Specifically, the absence of light triggers Cph8 kinase activity, which correspondingly activates the ompC promoter. Cells not receiving light will therefore produce the cell–cell communication molecule 3-oxohexanoyl-homoserine lactone (AHL; yellow circle) through expression of its biosynthetic enzyme LuxI. In addition, these cells will produce the transcriptional repressor CI (grey oval). AHL binds to the constitutively expressed transcription factor LuxR (light blue oval) to activate expression from the Plux-λ promoter, which is simultaneously and dominantly repressed by CI. The result is that only cells that receive light (and therefore do not express the transcriptional repressor CI) and are nearby to AHL-producing dark cells will activate the final gate and produce pigment through β-galactosidase activity (encoded by lacZ).
the edges of a projected image to be traced. This work demonstrates that complex behaviour can emerge from properly wiring together smaller genetic programs, and that these programs can lead to unique real-world applications.
Therapeutics
Human health is afflicted by new and old foes, including emergent drug-resistant microbes, cancer and obesity. Meanwhile, progress in medicine is faced with challenges at each stage of the therapeutic spectrum, ranging from the drying up of pharmaceutical pipelines to limited global access to viable medicines. In a relatively short amount of time, synthetic biology has made promising strides in reshaping and streamlining this spectrum (Box A2-4). Indeed, the rational and model-guided construction of biological parts is enabling new therapeutic platforms, from the identification of disease mechanisms and drug targets to the production and delivery of small molecules.
Disease mechanism. An electrical engineer is likely to prototype portions of a circuit on a ‘breadboard’ before printing it as an entire integrated circuit. This allows for the rigorous testing of submodules in an isolated, well-characterized environment. Similarly, synthetic biology provides a framework for synthetically reconstructing natural biological systems to explore how pathological behaviours may emerge. This strategy was used to give mechanistic insights into a primary immunodeficiency, agammaglobulinaemia, in which patients cannot generate mature B cells and as a result are unable to properly fight infections (Ferrari et al., 2007). The researchers developed a synthetic testbed by systematically reconstructing the various components of the human B cell antigen receptor (BCr) signalling pathway in an orthogonal environment.17 This allowed them to identify network topology features that trigger BCR signalling and assembly. A rare mutation in the immunoglobulin-β-encoding gene was identified in one patient and introduced into the synthetic system, in which it was shown to abolish assembly of the BCR on the cell surface, thereby linking this faulty pathway component with disease onset. Pathogenic viral genomes can similarly be reconstructed for studying the molecular underpinnings of infectious disease pandemics. For instance, synthetic reconstruction of the severe acquired respiratory syndrome (SARS) coronavirus (Becker et al., 2008) and the 1918 Spanish influenza virus (Tumpey et al., 2005) helped to identify genetic mutations that may have conferred human tropism and increased virulence.
Drug-target identification. Building up synthetic pathways and systems from individual parts is one way of identifying disease mechanisms and therapeutic
_______________
17 Orthogonal environment – A cellular environment or host into which genetic material is transplanted to avoid undesired native host interference or regulation. Orthogonal hosts are often organisms with sufficient evolutionary distance from the native host.
targets. Another is to deploy synthetic biology devices to systematically probe the function of individual components of a natural pathway. Our group, for instance, has engineered modular riboregulators that can be used to tune the expression of a toxic protein or any gene in a biological network (Isaacs et al., 2004). To achieve post-transcriptional control over a target gene, the mRNA sequence of the riboregulator 5′ UTR is designed to form a hairpin structure that sequesters the ribosomal binding site (RBS) and prevents ribosome access to it. Translational repression of this cis-repressed mRNA can be alleviated by an independently regulated transactivating RNA that targets the stem-loop for unfolding. Engineered riboregulators have been used to tightly regulate the expression of CcdB, a toxic bacterial protein that inhibits DNA gyrase,18 to gain a better understanding of the sequence of events leading to induced bacterial cell death (Dwyer et al., 2007). These synthetic biology studies, in conjunction with systems biology studies of quinolones (antibiotics that inhibit gyrase) (Dwyer et al., 2007), led to the discovery that all major classes of bactericidal antibiotics induce a common cellular death pathway by stimulating oxidative damage (Kohanski et al., 2007, 2008). This work provided new insights into how bacteria respond to lethal stimuli and paved the way for the development of more effective antibacterial therapies.
Drug discovery. After a faulty pathway component or target is identified, whole-cell screening assays can be designed using synthetic biology strategies for drug discovery. As a demonstration of this approach, Fussenegger and colleagues (Weber et al., 2008) developed a synthetic platform for screening small molecules that could potentiate a Mycobacterium tuberculosis antibiotic (Box A2-4). ethionamide, currently the last line of defence in the treatment of multidrug-resistant tuberculosis, depends on activation by the M. tuberculosis enzyme EthA for efficacy. However, due to transcriptional repression of ethA by the protein EthR, ethionamide-based therapy is often rendered ineffective. To address this problem, the researchers designed a synthetic mammalian gene circuit that featured an EthR-based transactivator of a reporter gene and used it to screen for and identify EthR inhibitors that could abrogate resistance to ethionamide. Importantly, because the system is a cell-based assay, it intrinsically enriches for inhibitors that are non-toxic and membrane-permeable to mammalian cells, which are key drug criteria as M. tuberculosis is an intracellular pathogen. This framework, in which drug discovery is applied to whole cells that have been engineered with circuits that highlight a pathogenic mechanism, could be extended to other diseases and phenotypes.
_______________
18 DNA gyrase – A type II DNA topoisomerase that catalyses the ATP-dependent supercoiling of closed-circular dsDNA by strand breakage and rejoining reactions. Control of chromosomal topological transitions is essential for DNA replication and transcription in bacteria, making gyrase an effective target for antimicrobial agents (for example, the quinolone class of antibiotics).
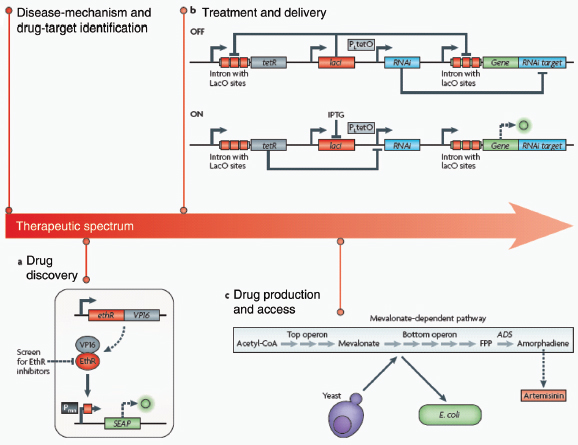
Drug discovery
Part a of the figure shows a synthetic mammalian gene circuit that enabled drug discovery for antituberculosis compounds (Weber et al., 2008). The antibiotic ethionamide is rendered cytotoxic to Mycobacterium tuberculosis by the enzyme EthA in infected cells. Because EthA is natively repressed by EthR, resistance to ethionamide treatment is common. In the gene circuit, a fusion of EthR and the mammalian transactivator VP16 binds a minimal promoter (Pmin) with a synthetic EthR operator site and activates expression of the reporter gene SEAP (human placental secreted alkaline phosphatase). This platform allows for the rapid screening of EthR inhibitors in mammalian cells.
Treatment and delivery
Part b shows a synthetic mammalian genetic switch for tight, tunable and reversible control of a desired gene for therapeutic or gene-delivery applications. In the OFF configuration (upper panel), expression of the gene of interest (green) is repressed at the levels of both transcription and translation. Constitutively expressed LacI repressor (red) binds to the lac operator sites in the transgene module of the gene of interest, therefore repressing its transcription. Any transcriptional leakage is repressed at the level of translation by an interfering RNA (blue), which targets the gene’s 3′ UTR. The system is switched ON (lower panel) by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG), which binds LacI repressor proteins and consequently relieves both forms of repression.
Drug production and access
The discovery of drugs does not always translate to the people who need them the most because drug production processes can be difficult and costly. Antibiotics are industrially produced from microbes and fungi, and are therefore widespread and cheap. Conversely, many other drugs are isolated from hosts that are not as amenable to large-scale production and are therefore costly and in short supply. Such drugs include the antimalaria drug artemisinin and the anticancer drug taxol. Fortunately, global access to drugs is being enabled by hybrid synthetic biology and metabolic engineering strategies for the microbial production of rare natural products. In the case of artemisinin (part c), there exist two biosynthetic pathways for the synthesis of the universal precursors to all isoprenoids, the large and diverse family of natural products of which artemisinin is a member. The native isoprenoid pathway found in Escherichia coli (the deoxyxylulose 5-phosphate (DXP) pathway) has been difficult to optimize, so instead researchers have synthetically constructed and tested the entire Saccharomyces cerevisiae mevalonate-dependent (MEV) pathway in E. coli in a piece-wise fashion (for example, by separating the ‘top’ and ‘bottom’ operons). The researchers initially used E. coli as a simple, orthogonal host platform to construct, debug and optimize the large metabolic pathway (Martin et al., 2003). They then linked the optimized heterologous pathway to a codon-optimized form of the plant terpene synthase ADS to funnel metabolic production to the specific terpene precursor to artemisinin. This work allowed them to build a full, optimized solution that could be ultimately and seamlessly deployed back into S. cerevisiae for cost-effective synthesis and purification of industrial quantities of the immediate drug precursor of artemisinin (Ro et al., 2006). FPP, farnesyl pyrophosphate.
Therapeutic treatment. Synthetic biology devices have additionally been developed to serve as therapies themselves. Entire engineered viruses and organisms can be programmed to target specific pathogenic agents and pathological mechanisms. For instance, in two separate studies (Lu and Collins, 2007, 2009) researchers used engineered bacteriophages to combat antibiotic-resistant bacteria by endowing them with genetic mechanisms that target and thwart bacterial mechanisms for evading antibiotic action. The first study was prompted by the observation that biofilms,19 in which bacteria are encapsulated in an extracellular matrix, have inherent resistance to antimicrobial therapies and are sources of persistent infections. To more effectively penetrate this protective environment, T7 phage was engineered to express the biofilm matrix-degrading enzyme dispersin B (DspB) upon infection (Lu and Collins, 2007). The two-pronged attack of T7 expressing DspB and phage-induced lysis fuelling the creation and spread of DspB resulted in the removal of 99.997% of the biofilm bacterial cells. In the second study (Lu and Collins, 2009), it was suggested that inhibition of certain bacterial genetic programs could improve the effectiveness of current antibiotic therapies. In this case, bacteriophages were deliberately designed to be non-lethal so as not to elicit resistance mechanisms; instead, a non-lytic M13 phage was used to suppress the bacterial SOS DNA-damage response by overexpression of its repressor, lexA3. The engineered bacteriophage significantly enhanced killing by three major classes of antibiotics in traditional cell culture and in E. coliinfected mice, potentiated killing of antibiotic-resistant bacteria and, importantly, reduced the incidence of cells with antibiotic-induced resistance.
Synthetically engineered viruses and organisms that are able to sense and link their therapeutic activity to pathological cues may be useful in the treatment of cancer, in which current therapies often indiscriminately attack tumours and normal tissues. For instance, adenoviruses were programmed to couple their replication to the state of the p53 pathway in human cells (Ramachandra et al., 2001). Normal p53 production would result in inhibition of a crucial viral replication component, whereas a defunct p53 pathway, which is characteristic of tumour cells, would allow viral replication and cell killing. In another demonstration of translational synthetic biology applied to cancer therapy, Voigt and colleagues (Anderson et al., 2006) developed cancer-targeting bacteria and linked their ability to invade the cancer cells to specific environmental signals. Constitutive expression of the heterologous invasin (inv) gene (from Yersinia pseudotuberculosis) can induce E. coli cells to invade both normal human cell lines and cancer cell lines. So, to preferentially invade cancer cells, the researchers placed inv under the control of transcriptional operons that are activated by environmental signals specific to the tumour microenvironment. These engineered bacteria could be made to carry or synthesize cancer therapies for the treatment of tumours.
_______________
19 Biofilms – Surface-associated communities of bacterial cells encapsulated in an extracellular polymeric substances (EPS) matrix. Biofilms are an antibiotic-resistant mode of microbial life found in natural and industrial settings.
Therapeutic delivery. In addition to engineered therapeutic organisms, synthetic circuits and pathways can be used for the controlled delivery of drugs as well as for gene and metabolic therapy. In some cases, sophisticated kinetic control over drug release in the body may yield therapeutic advantages and reduce undesired side effects. Most hormones in the body are released in time-dependent pulses. Glucocorticoid secretion, for instance, has a circadian and ultradian20 pattern of release, with important transcriptional consequences for glucocorticoid-responsive cells (Stavreva et al., 2009). Faithfully mimicking these patterns in the administration of synthetic hormones to patients with glucocorticoid-responsive diseases, such as rheumatoid arthritis, may decrease known side effects and improve therapeutic response (Stavreva et al, 2009). Periodic synthesis and release of biologic drugs can be autonomously achieved with synthetic oscillator circuits (Elowitz and Leibler, 2000; Atkinson et al., 2003; Fung et al., 2005; Stricker et al., 2008; Tigges et al., 2009) or programmed time-delay circuits (Weber et al., 2007). In other cases, one may wish to place a limit on the amount of drug released by programming the synthetic system to self-destruct after a defined number of cell cycles or drug release pulses. Our group has recently developed two variants of a synthetic gene counter (Friedland et al., 2009) that could be adapted for such an application.
Gene therapy is beginning to make some promising advances in clinical areas in which traditional drug therapy is ineffective, such as in the treatment of many hereditary and metabolic diseases. Synthetic circuits offer a more controlled approach to gene therapy, such as the ability to dynamically silence, activate and tune the expression of desired genes. In one such example (Deans et al., 2007), a genetic switch was developed in mammalian cells that couples transcriptional repressor proteins and an RNAi module for tight, tunable and reversible control over the expression of desired genes (Box A2-4). This system would be particularly useful in gene-silencing applications, as it was shown to yield >99% repression of a target gene.
Additionally, the construction of non-native pathways offers a unique and versatile approach to gene therapy, such as for the treatment of metabolic disorders. Operating at the interface of synthetic biology and metabolic engineering, Liao and colleagues (Dean et al., 2009) recently introduced the glyoxylate shunt pathway21 into mammalian liver cells and mice to explore its effects on fatty acid metabolism and, more broadly, on whole-body metabolism. Remarkably, the researchers found that when transplanted into mammals, the shunt actually increased fatty acid oxidation, evidently by creating an alternative cycle. Furthermore, mice expressing the shunt showed resistance to diet-induced obesity when
_______________
20 Ultradian – Periods of cycles that are repeated throughout a 24-hour circadian day.
21 Glyoxylate shunt pathway – A two-enzyme metabolic pathway unique to bacteria and plants that is activated when sugars are not readily available. This pathway diverts the tricarboxylic acid (TCA) cycle so that fatty acids are not completely oxidized and are instead converted into carbon energy sources.
placed on a high-fat diet, with corresponding decreases in total fat mass, plasma triglycerides and cholesterol levels. This work offers a new synthetic biology model for studying metabolic networks and disorders, and for developing treatments for the increasing problem of obesity.
Finally, the discovery of drugs and effective treatments may not quickly — or ever — translate to the people who need them the most because drug production processes can be difficult and costly. As discussed below, synthetic biology is allowing rare and costly drugs to be manufactured more cost-effectively (Box A2-4).
Biofuels, pharmaceuticals and biomaterials
Recent excitement surrounding the production of biofuels, pharmaceuticals and biomaterials from engineered microorganisms is matched by the challenges that loom in bringing these technologies to production scale and quality. The most widely used biofuel is ethanol produced from corn or sugar cane (Fortman et al., 2008); however, the heavy agricultural burden combined with the suboptimal fuel properties of ethanol make this approach to biofuels problematic and limited. Microorganisms engineered with optimized biosynthetic pathways to efficiently convert biomass into biofuels are an alternative and promising source of renewable energy. These strategies will succeed only if their production costs can be made to compete with, or even outcompete, current fuel production costs. Similarly, there are many drugs for which expensive production processes preclude their capacity for a wider therapeutic reach. New synthetic biology tools would also greatly advance the microbial production of biomaterials and the development of novel materials.
Constructing biosynthetic pathways. When engineering for biofuels, drugs or biomaterials, two of the first design decisions are choosing which biosynthetic pathway or pathways to focus on and which host organism to use. Typically, these decisions begin with the search for organisms that are innately capable of achieving some desired biosynthetic activity or phenotype (Alper and Stephanopoulos, 2009). For biofuel production, for instance, certain microorganisms have evolved to be proficient in converting lignocellulosic material to ethanol, biobutanol and other biofuels. These native isolates possess unique catabolic activity, heightened tolerances for toxic materials and a host of enzymes designed to break down the lignocellulosic components. Unfortunately, these highly desired properties exist in pathways that are tightly regulated according to the host’s evolved needs and therefore may not be suitable in their native state for production scale. A longstanding challenge in metabolic and genetic engineering is determining whether to improve the isolate host’s production capacity or whether to transplant the desired genes or pathways into an industrial model host, such as E. coli or S.
cerevisiae; these important considerations and trade-offs are reviewed elsewhere (Alper and Stephanopoulos, 2009).
The example of the microbial production of biobutanol, a higher energy density alternative to ethanol, provides a useful glimpse into these design trade-offs. Butanol is converted naturally from acetyl-CoA by Clostridium acetobutylicum (Jones and Woods, 1986). However, it is produced in low yields and as a mixture with acetone and ethanol, so substantial cellular engineering of a microorganism for which standard molecular biology techniques do not apply is needed to produce usable amounts of butanol (Tummala et al., 2003; Shao et al., 2007). Furthermore, importing the biosynthetic genes into an industrial microbial host can lead to metabolic imbalances (Inui et al., 2008). In an altogether different approach, Liao and colleagues (Atsumi et al., 2008) bypassed standard fermentation pathways and recognized that a broad set of the 2-keto acid intermediates of E. coli amino acid biosynthesis could be synthetically shunted to achieve high-yield production of butanol and other higher alcohols in two enzymatic steps.
Indeed, complementary to efforts in traditional metabolic and genetic engineering is the use of engineering principles for constructing functional, predictable and non-native biological pathways de novo to control and improve microbial production. In an exemplary illustration of this, Keasling and colleagues engineered the microbial production of precursors to the antimalarial drug artemisinin to industrial levels (Martin et al., 2003; Ro et al., 2006) (Box A2-4). There are now many such examples of the successful application of synthetic approaches to biosynthetic pathway construction — such approaches have been used in the microbial production of fatty-acid-derived fuels and chemicals (such as fatty esters, fatty alcohols and waxes) (Steen et al., 2010), methyl halide-derived fuels and chemicals (Bayer et al., 2009), polyketide synthases that make cholesterol-lowering drugs (Ma et al., 2009), and polyketides made from megaenzymes that are encoded by very large synthetic gene clusters (Kodumal et al., 2004).
Optimizing pathway flux. After biosynthetic pathways have been constructed, the expression levels of all of the components need to be orchestrated to optimize metabolic flux22 and achieve high product titres. A standard approach is to drive the expression of pathway components with strong and exogenously tunable promoters, such as the PLtet, PLlac, and PBAD promoters from the tet, lac and ara operons of E. coli, respectively. To this end, there are ongoing synthetic biology efforts to create and characterize more reusable, biological control elements based on promoters for predictably tuning expression levels (Alpert et al., 2005; Ellis et al., 2009). Further to this, synthetic biologists have devised a number of alternative methods for obtaining biological pathway balance, ranging from reconfiguring network connectivity to fine-tuning individual components. A richer
_______________
22 Metabolic flux – The rate of flow of metabolites through a metabolic pathway. The rate is regulated by the enzymes in the pathway.
discussion of these topics, including the fine-tuning of parts, the application of model-guided approaches and the development of next-generation interoperable parts, is presented elsewhere (Lu et al., 2009). In Box A2-5 and Box A2-6 we detail several synthetic biology strategies that specifically pertain to the optimization of metabolic pathway flux. These strategies range from those driven by evolutionary techniques, to those driven by rational design and in silico models, to those that combine both approaches.
Programming novel functionality and materials. Beyond facilitating metabolic tasks, synthetic systems can infuse novel functionality into engineered organisms for production purposes or for building new materials. Early work in the field laid the groundwork for constructing basic circuits that could sense and process signals, perform logic operations and actuate biological responses (Voigt, 2006). Wiring these modules together to bring about reliable, higher-order functionality is one of the next major goals of synthetic biology (Lu et al., 2009), and an important application of this objective is the layering of ‘smart’ control mechanisms over metabolic engineering. For instance, circuits designed to sense the bioreactor environment and shift metabolic phases accordingly would further improve biofuel production. Alternatively, autonomous timing circuits could be used to shut down metabolic processes after a prescribed duration of time. Biological timers of this sort have been developed using genetic toggle switches that were deliberately rendered imbalanced through model-guided promoter engineering (Ellis et al., 2009). These genetic timers were used to program the time-dependent flocculation23 of yeast cells to facilitate the separation of cells from, for instance, the alcohol produced in industrial fermentation processes.
Synthetic control systems can also be used to extract and purify the synthesized product. This is particularly important in the production of recombinant proteins, bioplastics and other large biomaterials, which can accumulate inside cells, cause the formation of inclusion bodies and become toxic to cells if they are present at high titres. To export recombinant spider-silk monomers, Widmaier et al. (2009) searched for a secretion system that would enable efficient and indiscriminate secretion of proteins through both bacterial membranes. The Salmonella type III secretion system (T3SS) not only fulfils these criteria but also possesses a natural regulatory scheme that ties expression of the protein to be secreted to the secretion capacity of the cell; as a result, the desired protein is only expressed when sufficient secretion complexes have been built. To obtain superior secretion rates of recombinant silk protein, the researchers needed only to engineer a control circuit that hitches the heterologous silk-protein-producing genes to the innate genetic machinery for environmental sensing and secretion commitment.
_______________
23 Flocculation – A specific form of cell aggregation in yeast triggered by certain environmental conditions, such as the absence of sugars. For example, flocculation occurs once the sugar in a beer brew has been fermented into ethanol.
Finally, there is an emerging branch of synthetic biology that seeks to program coordinated behaviour in populations of cells, which could lead to the fabrication of novel biomaterials for various applications. The engineering of synthetic multicellular systems is typically achieved with cell–cell communication and associated intracellular signal processing modules, as was elegantly used by Hasty and colleagues (Danino et al., 2010) to bring about synchronized oscillations in a population of bacterial cells. Weiss and colleagues (Basu et al., 2004, 2005) have similarly done pioneering work in building biomolecular signal-processing architectures that can filter communication signals originating from ‘sender’ cells. These systems, which can be programmed to form intricate multicellular patterns from a solid-phase cellular lawn, would aid the development of fabrication-free scaffolds for tissue engineering.
Future challenges and conclusions
The future of translational synthetic biology hinges on the development of reliable means for connecting smaller functional circuits to realize higher-order networks with predictable behaviours. In a previous article (Lu et al., 2009), we outlined four research efforts aimed at improving and accelerating the overall design cycle and allowing more seamless integration of biological circuitry (Box A2-7).
Beyond the challenge of improving the design cycle, applied synthetic biology would benefit from once again summoning the original inspiration of bio-computing. The ability to program higher-level decision-making into synthetic networks would yield more robust and dynamic organisms, including ones that can accomplish many tasks simultaneously. Furthermore, as adaptive and predictive behaviours are naturally present in all organisms (including microbes) (Mitchell et al., 2009; Tagkopoulos et al., 2008), synthetic learning networks built from genetic and biological parts (Fernando et al., 2009; Fritz et al., 2007) would infuse engineered organisms with more sophisticated automation for biosensing and related applications.
Finally, the majority of synthetic biology is currently practiced in microbes. However, many of the most pressing problems, and in particular those of human health, are inherently problems with mammalian systems. Therefore, a more concerted effort towards advancing mammalian synthetic biology will be crucial for next-generation therapeutic solutions, including the engineering of synthetic gene networks for stem-cell generation and differentiation.
By addressing such challenges, we will be limited not by the technicalities of construction or the robustness of synthetic gene networks but only by the imagination of researchers and the number of societal problems and applications that synthetic biology can resolve.
Evolutionary strategies
In the production of artemisinin precursors, the native Escherichia coli isoprenoid pathway (the deoxyxylulose 5-phosphate (DXP) pathway) was eschewed in favour of a heterologous pathway so as to circumvent the complex regulatory control imposed by the host (Box A2-4). In an alternative method of relieving regulatory control over the large number of DXP pathway components, Wang et al. (2009) diversified and, as a result, optimized the native DXP biosynthetic pathway in E. coli (see the figure, part a). The researchers developed a rapid, automated method for the in vivo directed evolution of pathways, which they termed multiplex automated genome engineering (MAGE). They then applied it to evolve the translational efficiencies of DXP pathway components to achieve maximal lycopene production. Specifically, cells were subjected to cycles of genetic modifications (through oligo-mediated allelic replacement) in an automated fashion to explore sufficient genomic diversity for optimizing biosynthetic pathways at laboratory timescales.
Rational design
At the other end of the spectrum are strategies that rely on quantitative models and blueprints for the rational design of optimized networks and pathways (part b). Typically, a component of interest (for example, an engineered promoter (P*) or ribosomal binding site (RBS*) sequence) will be built into a simple test network. The network and its input–output data will then be fed into a model, which attempts to determine a parameter set that optimally describes the component’s dynamics within the framework of the model. Finally, the optimized parameter set will be used to forward-engineer new networks and components. For example, stochastic biochemical models have been developed to capture the expression dynamics of synthetically engineered promoters; these models were subsequently used to predict the correct in vivo behaviour of different and more complex gene networks built from the modelled components (Blake et al., 2003; Guido et al., 2006). Similarly, at the level of translation, thermodynamic models that predict the relative translation initiation rates of proteins can be used to rationally forward-engineer synthetic RBS sequences to give desired expression levels (Salis et al., 2009). Such techniques harness modelled genetic parameters (transcriptional or translational) to predict the level of expression of proteins and enzymes in a network. DMAPP, dimethylallyl diphosphate; FPP, farnesyl diphosphate; G3P, glyceraldehyde 3-phosphate; IPP, isopentenyl diphosphate.
In a hybrid rational–combinatorial approach, Dueber et al. (2009) suggested that metabolic flux could be controlled by spatially recruiting the enzymes of a desired biosynthetic pathway using synthetic protein scaffolds. To construct the enzyme scaffolding, the researchers tethered protein–protein interaction domains (for example, GBD, SH3 and PDZ domains) from metazoan signalling proteins. These domains recognize and bind cognate peptides that were fused to the enzymes to be recruited (acetoacetyl-CoA thiolase (AtoB) from Escherichia coli and HMG-CoA (HMBS) synthase and HMG-CoA reductase (HMGR) from Saccharomyces cerevisiae). By varying the number of repeats of an interaction domain, the researchers could additionally control the stoichiometry of the enzymes recruited to the complex. Using the heterologous mevalonate-dependent (MEV) pathway in E. coli as a model, they combinatorially (albeit, at a substantially smaller scale) optimized the stoichiometry of the three enzymes responsible for producing mevalonate from acetyl-CoA. Finally, they showed that the optimized synthetic scaffold could substantially increase product titre while reducing the metabolic load on the host; in other words, their high product titres did not require the overexpression of biosynthetic enzymes in the cell.
BOX A2-7
Recommendations for Improving the Synthetic Biology Design Cycle
- Scaling up to larger and more complex biological systems while simultaneously minimizing interference among parts will require an expanded synthetic biology toolkit and, in particular, libraries of interoperable parts. Eukaryotic systems are fertile grounds for discovering such parts, as many synthetic biology devices are based on a small repertoire of bacterial and archaeal regulatory elements.
- Modelling and fine-tuning of synthetic networks should be emphasized, particularly as the network size and complexity increases. This will facilitate proper matching of input–output behaviours (that is, transfer functions) when distinct modules are connected.
- There is a need to develop new probes and high-throughput methods for the in vivo measurement of circuit dynamics to rapidly characterize parts and debug networks.
- Cellular testing platforms need to be developed to quicken the pace of identifying problematic network nodes and ease the failure-prone jumps associated with either building a more complex network or deploying a network in a more complex organism. These testing platforms could be cells engineered to have minimal genomes (Gibson et al., 2008; Glass et al., 2006; Lartigue et al., 2007; Lartigue et al., 2009) or lower model organisms that have been equipped with specific machinery from higher organisms.
Acknowledgements
We thank members of the Collins laboratory for helpful discussions and K. M. Flynn for help with artwork. We also thank the Howard Hughes Medical Institute and the US National Institutes of Health Director’s Pioneer Award Program for their financial support.
Competing Interests Statement
The authors declare competing financial interests: see Web version for details.
Databases
Entrez Gene: http://www.ncbi.nlm.nih.gov/gene ccdB | lexA3 | traA
OMIM: http://www.ncbi.nlm.nih.gov/omim agammaglobulinaemia
UniProtKB: http://www.uniprot.org CI | Cro | DspB | EnvZ | EthA | EthR
Further Information
James J. Collins’ homepage: http://www.bu.edu/abl
All links are active on the online PDF.
References
Ajo-Franklin, C. M. et al. Rational design of memory in eukaryotic cells. Genes Dev. 21, 2271–2276 (2007).
Alper, H. & Stephanopoulos, G. Engineering for biofuels: exploiting innate microbial capacity or importing biosynthetic potential? Nature Rev. Microbiol. 7, 715–723 (2009).
Alper, H., Fischer, C., Nevoigt, E. & Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc. Natl Acad. Sci. USA 102, 12678–12683 (2005).
Anderson, J. C., Clarke, E. J., Arkin, A. P. & Voigt, C. A. Environmentally controlled invasion of cancer cells by engineered bacteria. J. Mol. Biol. 355, 619–627 (2006).
This study provides a potential synthetic approach to cancer therapy. Bacteria were programmed to sense environmental cues of the tumour microenvironment and respond to them by invading malignant cells.
Anderson, J. C., Voigt, C. A. & Arkin, A. P. Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 3, 133 (2007).
Atkinson, M. R., Savageau, M. A., Myers, J. T. & Ninfa, A. J. Development of genetic circuitry exhibiting toggle switch or oscillatory behavior in Escherichia coli. Cell 113, 597–607 (2003).
Atsumi, S., Hanai, T. & Liao, J. C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451, 86–89 (2008).
Bashor, C. J., Helman, N. C., Yan, S. & Lim, W. A. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 319, 1539–1543 (2008).
Basu, S., Mehreja, R., Thiberge, S., Chen, M. T. & Weiss, R. Spatiotemporal control of gene expression with pulse-generating networks. Proc. Natl Acad. Sci. USA 101, 6355–6360 (2004).
Basu, S., Gerchman, Y., Collins, C. H., Arnold, F. H. & Weiss, R. A synthetic multicellular system for programmed pattern formation. Nature 434, 1130–1134 (2005).
An excellent demonstration of synthetic control over a population of cells. By splitting the V. fischeri quorum-sensing circuit between ‘sender’ and ‘receiver’ cells, bacteria were programmed to communicate to generate intricate two-dimensional patterns.
Bayer, T. S. & Smolke, C. D. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nature Biotech. 23, 337–343 (2005).
This paper establishes an RNA scaffold for the ligand-dependent ON–OFF switching of gene expression.
Bayer, T. S. et al. Synthesis of methyl halides from biomass using engineered microbes. J. Am. Chem. Soc. 131, 6508–6515 (2009).
Becker, M. M. et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl Acad. Sci. USA 105, 19944–19949 (2008).
Blake, W. J., Kærn, M., Cantor, C. R. & Collins, J. J. Noise in eukaryotic gene expression. Nature 422, 633–637 (2003).
Boorsma, M. et al. A temperature-regulated replicon-based DNA expression system. Nature Biotech. 18, 429–432 (2000).
Brown, M. et al. Lac repressor can regulate expression from a hybrid SV40 early promoter containing a lac operator in animal cells. Cell 49, 603–612 (1987).
Cox, J. C. et al. Automated selection of aptamers against protein targets translated in vitro: from gene to aptamer. Nucleic Acids Res. 30, e108 (2002).
Danino, T., Mondragon-Palomino, O., Tsimring, L. & Hasty, J. A synchronized quorum of genetic clocks. Nature 463, 326–330 (2010).
Dean, J. T. et al. Resistance to diet-induced obesity in mice with synthetic glyoxylate shunt. Cell Metab. 9, 525–536 (2009).
Deans, T. L., Cantor, C. R. & Collins, J. J. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 130, 363–372 (2007).
Deuschle, U. et al. Regulated expression of foreign genes in mammalian cells under the control of coliphage T3 RNA polymerase and lac repressor. Proc. Natl Acad. Sci. USA 86, 5400–5404 (1989).
Doudna, J. A. & Cech, T. R. The chemical repertoire of natural ribozymes. Nature 418, 222–228 (2002).
Dueber, J. E., Yeh, B. J., Chak, K. & Lim, W. A. Reprogramming control of an allosteric signaling switch through modular recombination. Science 301, 1904–1908 (2003).
Dueber, J. E. et al. Synthetic protein scaffolds provide modular control over metabolic flux. Nature Biotech. 27, 753–759 (2009).
The authors provide a modular framework for programming the input–output behavior of eukaryotic signalling-protein circuits, and construct synthetic switch proteins with a rich set of gating behaviours.
Dunlap, J. C. Molecular bases for circadian clocks. Cell 96, 271–290 (1999).
Dwyer, D. J., Kohanski, M. A., Hayete, B. & Collins, J. J. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 3, 91 (2007).
Eddy, S. R. Non-coding RNA genes and the modern RNA world. Nature Rev. Genet. 2, 919–929 (2001).
Ellington, A. D. & Szostak, J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990).
Ellis, T., Wang, X. & Collins, J. J. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nature Biotech. 27, 465–471 (2009).
Elowitz, M. B. & Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).24
Fernando, C. T. et al. Molecular circuits for associative learning in single-celled organisms. J. R. Soc. Interface 6, 463–469 (2009).
Ferrari, S. et al. Mutations of the Igβ gene cause agammaglobulinemia in man. J. Exp. Med. 204, 2047–2051 (2007).
Fortman, J. L. et al. Biofuel alternatives to ethanol: pumping the microbial well. Trends Biotechnol. 26, 375–381 (2008).
Friedland, A. E. et al. Synthetic gene networks that count. Science 324, 1199–1202 (2009).
Fritz, G., Buchler, N. E., Hwa, T. & Gerland, U. Designing sequential transcription logic: a simple genetic circuit for conditional memory. Syst. Synth. Biol. 1, 89–98 (2007).
Fung, E. et al. A synthetic gene-metabolic oscillator. Nature 435, 118–122 (2005).
Fussenegger, M. et al. Streptogramin-based gene regulation systems for mammalian cells. Nature Biotech. 18, 1203–1208 (2000).
Gardner, T. S., Cantor, C. R. & Collins, J. J. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342 (2000).24
Gelfand, M. S., Mironov, A. A., Jomantas, J., Kozlov, Y. I. & Perumov, D. A. A conserved RNA structure element involved in the regulation of bacterial riboflavin synthesis genes. Trends Genet. 15, 439–442 (1999).
Gibson, D. G. et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220 (2008).
Glass, J. I. et al. Essential genes of a minimal bacterium. Proc. Natl Acad. Sci. USA 103, 425–430 (2006).
Glass, L. Classification of biological networks by their qualitative dynamics. J. Theor. Biol. 54, 85–107 (1975).
Glass, L. & Kauffman, S. A. The logical analysis of continuous, non-linear biochemical control networks. J. Theor. Biol. 39, 103–129 (1973).
Good, L. Translation repression by antisense sequences. Cell. Mol. Life Sci. 60, 854–861 (2003).
Gossen, M. & Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA 89, 5547–5551 (1992).
_______________
24 [These references] describe synthetic biology’s first devices — the genetic toggle switch and the repressilator — and establish the engineering-based methodology for constructing sophisticated, dynamic behaviours in biological systems from simple regulatory elements.
Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N. & Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35, 849–857 (1983).
Guet, C. C., Elowitz, M. B., Hsing, W. & Leibler, S. Combinatorial synthesis of genetic networks. Science 296, 1466–1470 (2002).
Guido, N. J. et al. A bottom-up approach to gene regulation. Nature 439, 856–860 (2006).
Ham, T. S., Lee, S. K., Keasling, J. D. & Arkin, A. P. A tightly regulated inducible expression system utilizing the fim inversion recombination switch. Biotechnol. Bioeng. 94, 1–4 (2006).
Ham, T. S., Lee, S. K., Keasling, J. D. & Arkin, A. P. Design and construction of a double inversion recombination switch for heritable sequential genetic memory. PLoS ONE 3, e2815 (2008).
Hermann, T. & Patel, D. J. Adaptive recognition by nucleic acid aptamers. Science 287, 820–825 (2000).
Hooshangi, S., Thiberge, S. & Weiss, R. Ultrasensitivity and noise propagation in a synthetic transcriptional cascade. Proc. Natl Acad. Sci. USA 102, 3581–3586 (2005).
Hu, M. C. & Davidson, N. The inducible lac operator–repressor system is functional in mammalian cells. Cell 48, 555–566 (1987).
Inui, M. et al. Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl. Microbiol. Biotechnol. 77, 1305–1316 (2008).
Isaacs, F. J. et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nature Biotech. 22, 841–847 (2004).
Isaacs, F. J., Dwyer, D. J. & Collins, J. J. RNA synthetic biology. Nature Biotech. 24, 545–554 (2006).
Johansson, J. et al. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110, 551–561 (2002).
Jones, D. T. & Woods, D. R. Acetone-butanol fermentation revisited. Microbiol. Rev. 50, 484–524 (1986).
Kauffman, S. The large scale structure and dynamics of gene control circuits: an ensemble approach. J. Theor. Biol. 44, 167–190 (1974).
Kobayashi, H. et al. Programmable cells: interfacing natural and engineered gene networks. Proc. Natl Acad. Sci. USA 101, 8414–8419 (2004).
Kodumal, S. J. et al. Total synthesis of long DNA sequences: synthesis of a contiguous 32-kb polyketide synthase gene cluster. Proc. Natl Acad. Sci. USA 101, 15573–15578 (2004).
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A. & Collins, J. J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007).
Kohanski, M. A., Dwyer, D. J., Wierzbowski, J., Cottarel, G. & Collins, J. J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135, 679–690 (2008).
Kramer, B. P. & Fussenegger, M. Hysteresis in a synthetic mammalian gene network. Proc. Natl Acad. Sci. USA 102, 9517–9522 (2005).
Kramer, B. P. et al. An engineered epigenetic transgene switch in mammalian cells. Nature Biotech. 22, 867–870 (2004).
Kruger, K. et al. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 31, 147–157 (1982).
Lartigue, C. et al. Genome transplantation in bacteria: changing one species to another. Science 317, 632–638 (2007).
Lartigue, C. et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325, 1693–1696 (2009).
Lease, R. A. & Belfort, M. A trans-acting RNA as a control switch in Escherichia coli: DsrA modulates function by forming alternative structures. Proc. Natl Acad. Sci. USA 97, 9919–9924 (2000).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993).
Levskaya, A. et al. Synthetic biology: engineering Escherichia coli to see light. Nature 438, 441–442 (2005).
Levskaya, A., Weiner, O. D., Lim, W. A. & Voigt, C. A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009).
Looger, L. L., Dwyer, M. A., Smith, J. J. & Hellinga, H. W. Computational design of receptor and sensor proteins with novel functions. Nature 423, 185–190 (2003).
Lu, T. K. & Collins, J. J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl Acad. Sci. USA 104, 11197–11202 (2007).
——. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl Acad. Sci. USA 106, 4629–4634 (2009).25
Lu, T. K., Khalil, A. S. & Collins, J. J. Next-generation synthetic gene networks. Nature Biotech. 27, 1139–1150 (2009).
Lutz, R. & Bujard, H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1–I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210 (1997).
Ma, S. M. et al. Complete reconstitution of a highly reducing iterative polyketide synthase. Science 326, 589–592 (2009).
Majdalani, N., Hernandez, D. & Gottesman, S. Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol. Microbiol. 46, 813–826 (2002).
Malphettes, L. et al. A novel mammalian expression system derived from components coordinating nicotine degradation in arthrobacter nicotinovorans pAO1. Nucleic Acids Res. 33, e107 (2005).
Mandal, M., Boese, B., Barrick, J. E., Winkler, W. C. & Breaker, R. R. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell 113, 577–586 (2003).
Martin, V. J., Pitera, D. J., Withers, S. T., Newman, J. D. & Keasling, J. D. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nature Biotech. 21, 796–802 (2003).
McAdams, H. H. & Arkin, A. Towards a circuit engineering discipline. Curr. Biol. 10, R318–R320 (2000).
McAdams, H. H. & Shapiro, L. Circuit simulation of genetic networks. Science 269, 650–656 (1995).
Mironov, A. S. et al. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 111, 747–756 (2002).
Mitchell, A. et al. Adaptive prediction of environmental changes by microorganisms. Nature 460, 220–224 (2009).
Monod, J. & Jacob, F. General conclusions: telenomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb. Symp. Quant. Biol. 26, 389–401 (1961).
Morita, M. T. et al. Translational induction of heat shock transcription factor σ32: evidence for a built-in RNA thermosensor. Genes Dev. 13, 655–665 (1999).
Mukherji, S. & van Oudenaarden, A. Synthetic biology: understanding biological design from synthetic circuits. Nature Rev. Genet. 10, 859–871 (2009).
Mullick, A. et al. The cumate gene-switch: a system for regulated expression in mammalian cells. BMC Biotechnol. 6, 43 (2006).
Neddermann, P. et al. A novel, inducible, eukaryotic gene expression system based on the quorum-sensing transcription factor TraR. EMBO Rep. 4, 159–165 (2003).
Rackham, O. & Chin, J. W. Cellular logic with orthogonal ribosomes. J. Am. Chem. Soc. 127, 17584–17585 (2005).
Ramachandra, M. et al. Re-engineering adenovirus regulatory pathways to enhance oncolytic specificity and efficacy. Nature Biotech. 19, 1035–1041 (2001).
Rinaudo, K. et al. A universal RNAi-based logic evaluator that operates in mammalian cells. Nature Biotech. 25, 795–801 (2007).
_______________
25 In [these references], engineered bacteriophages were used to deliver synthetic enzymes and perturb gene networks to combat antibiotic-resistant strains of bacteria.
Ro, D. K. et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943 (2006).26
Salis, H. M., Mirsky, E. A. & Voigt, C. A. Automated design of synthetic ribosome binding sites to control protein expression. Nature Biotech. 27, 946–950 (2009).
Savageau, M. A. Comparison of classical and autogenous systems of regulation in inducible operons. Nature 252, 546–549 (1974).
Shao, L. et al. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 17, 963–965 (2007).
Skerker, J. M. et al. Rewiring the specificity of two-component signal transduction systems. Cell 133, 1043–1054 (2008).
Sohka, T. et al. An externally tunable bacterial band-pass filter. Proc. Natl Acad. Sci. USA 106, 10135–10140 (2009).
Stavreva, D. A. et al. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nature Cell Biol. 11, 1093–1102 (2009).
Steen, E. J. et al. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463, 559–562 (2010).26
Stojanovic, M. N. & Stefanovic, D. A deoxyribozyme-based molecular automaton. Nature Biotech. 21, 1069–1074 (2003).
Stougaard, P., Molin, S. & Nordstrom, K. RNAs involved in copy-number control and incompatibility of plasmid R1. Proc. Natl Acad. Sci. USA 78, 6008–6012 (1981).
Stricker, J. et al. A fast, robust and tunable synthetic gene oscillator. Nature 456, 516–519 (2008).
Tabor, J. J. et al. A synthetic genetic edge detection program. Cell 137, 1272–1281 (2009).
An outstanding example of how smaller circuits can be combined to realize larger genetic programs with sophisticated and predictable behaviour. Here, logic gates and cell–cell communication modules were coupled to program a population of bacteria to sense and trace the edges of a projected image.
Tagkopoulos, I., Liu, Y. C. & Tavazoie, S. Predictive behavior within microbial genetic networks. Science 320, 1313–1317 (2008).
Tigges, M., Marquez-Lago, T. T., Stelling, J. & Fussenegger, M. A tunable synthetic mammalian oscillator. Nature 457, 309–312 (2009).
Tuerk, C. & Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990).
Tummala, S. B., Welker, N. E. & Papoutsakis, E. T. Design of antisense RNA constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum. J. Bacteriol. 185, 1923–1934 (2003).
Tumpey, T. M. et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310, 77–80 (2005).
Voigt, C. A. Genetic parts to program bacteria. Curr. Opin. Biotechnol. 17, 548–557 (2006).
Wagner, E. G. H. & Simons, R. W. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 48, 713–742 (1994).
Wang, H. H. et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898 (2009).
Provides a combinatorial/evolutionary approach to optimizing biosynthetic pathway components through rapid in vivo genome engineering by cycles of targeted genome modification and phenotype selection.
_______________
26 [These references] provide a paradigm for the application of synthetic biology to the construction and optimization of biosynthetic pathways for cost-effective and high-yield microbial production. In these papers, the authors demonstrate industrial production of the direct precursor to the antimalarial drug artemisinin as part of a broader effort to address worldwide shortages of rare drugs.
Weber, W. et al. Macrolide-based transgene control in mammalian cells and mice. Nature Biotech. 20, 901–907 (2002).
——. Conditional human VEGF-mediated vascularization in chicken embryos using a novel temperature-inducible gene regulation (TIGR) system. Nucleic Acids Res. 31, e69 (2003a).
——. Streptomyces-derived quorum-sensing systems engineered for adjustable transgene expression in mammalian cells and mice. Nucleic Acids Res. 31, e71 (2003b).
——. Gas-inducible transgene expression in mammalian cells and mice. Nature Biotech. 22, 1440–1444 (2004).
——. A synthetic time-delay circuit in mammalian cells and mice. Proc. Natl Acad. Sci. USA 104, 2643–2648 (2007).
——. A synthetic mammalian gene circuit reveals antituberculosis compounds. Proc. Natl Acad. Sci. USA 105, 9994–9998 (2008).
——. A synthetic mammalian electro-genetic transcription circuit. Nucleic Acids Res. 37, e33 (2009).
Weber, W., Link, N. & Fussenegger, M. A genetic redox sensor for mammalian cells. Metab. Eng. 8, 273–280 (2006).
Widmaier, D. M. et al. Engineering the Salmonella type III secretion system to export spider silk monomers. Mol. Syst. Biol. 5, 309 (2009).
Win, M. N. & Smolke, C. D. Higher-order cellular information processing with synthetic RNA devices. Science 322, 456–460 (2008).
Winkler, W., Nahvi, A. & Breaker, R. R. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419, 952–956 (2002).
Winkler, W. C., Nahvi, A., Roth, A., Collins, J. A. & Breaker, R. R. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428, 281–286 (2004).
Winkler, W. C. & Breaker, R. R. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 59, 487–517 (2005).
Yen, L. et al. Exogenous control of mammalian gene expression through modulation of RNA self-cleavage. Nature 431, 471–476 (2004).
You, L., Cox, R. S., Weiss, R. & Arnold, F. H. Programmed population control by cell–cell communication and regulated killing. Nature 428, 868–871 (2004).
THE GENOME AS THE UNIT OF ENGINEERING
Andrew D. Ellington and Jared Ellefson27
Introduction
Recent years have marked a dramatic increase of our capabilities to sequence and synthesize nucleic acids. Ten years ago the first human genome was sequenced, and at the time it was a monumental undertaking requiring billions of dollars in funding and legions of dedicated researchers. Today, the same genome sequence would cost a fraction of the price and soon personal genome sequencing will be commonplace. However, while our understanding of genomes has not yet
_______________
27 Center for Systems and Synthetic Biology, University of Texas at Austin.
caught up with our ability to generate data, there is no question that the technology revolution has transformed and will continue to drive the biological sciences, in particular the new integrative science known as systems biology.
The same revolution is occurring in DNA synthesis, as prices for DNA drop and technologies are developed for large-scale (and ever longer) syntheses. As with sequencing, the technology revolution will lead understanding, and it can be argued that, although we can synthesize a microbial genome (Gibson et al., 2010), we don’t necessarily understand how to design one. That’s fine; we can learn by doing, and as synthetic genomes become easier to synthesize, we will undoubtedly generate customized genomes that will test the simple hypothesis of whether they function as intended.
Together, these emerging issues are the essence of what can be seen as the holy grail of both systems and synthetic biology, the genome as the unit of engineering. Synthetic biology can, in many circumstances, be defined as the abstraction of biology to the point where it can be easily engineered. This is first and foremost an operational definition, not an intellectual one. Indeed, there hides in the background of synthetic biology the usually unstated hypothesis that biology was made to be engineered by engineers (a hypothesis we take issue with below). Parallels are often drawn between electrical engineering and synthetic biology, where genetic information like promoters and proteins can be compared to components of a circuit board such as transistors or capacitors. In the view of the new breed of synthetic biologists, standardized components can almost always be used to rationally design genetic elements to perform desired tasks. This view has much merit, since it has already yielded interesting products such as biofuels, pharmaceuticals, and biomaterials. For example, Bayer and Voigt coaxed yeast into making valuable methyl halides from biomass (Bayer et al., 2009), while the Keasling lab reengineered the common lab microbe, Escherichia coli, to produce large quantities of amorphadiene, a precursor to antimalarial and anticancer drugs (Martin et al., 2003). It is not unreasonable to suspect that synthetic circuits will be introduced in human hosts at some point; for example, Fussenegger has created a synthetic circuit composed of a bacterial uric acid sensor and a fungal urate oxidase (which converts uric acid into a more tolerable compound) that can be used to control uric acid levels in mammalian hosts and thus ameliorate chronic disease states such as gout (Kemmer et al., 2010).
However, it is nonetheless still the case that many of these systems must be optimized in order for them to function as intended, in large part because the constituent parts are neither truly modular nor is their function fully predictable in new contexts. The question thus becomes whether biology is really meant to be engineered the same way a circuit board is, whether engineers will learn to make biology a circuit board, or whether some composite view is more akin to reality. Or, stated another way, the question is whether genetic tinkering (which existed well before synthetic biology) has somehow entered a new, more robust phase or if it has just been relabeled.
Systems biology has taken a different approach to how to tinker with systems. This approach proceeds from an understanding of the system as a whole instead of as an amalgam of component parts. It is top down rather than bottom up. By integrating literally millions of points of data from genomes, transcriptomes, and proteomes across the phylogenetic spectra, systems biologists can draw remarkable conclusions, up to and including the identification of nonobvious and evolutionarily repurposed subsystems. It is this sort of understanding that allows us to realize that the same interactions that govern resistance to antifungals in yeast also govern blood vessel formation in higher organisms (McGary et al., 2010). While a comparable synthetic biology approach might be to repurpose a tractable signaling pathway for blood vessel formation, this approach would require extensive empirical testing. As technology continues to develop for high-throughput analysis of DNA, RNA, and proteins, systems biologists will have the benefit of several billion years of empirical testing and, thus, will hold the intellectual high ground for understanding how organisms truly work. Quantitative modeling of the connectedness of extant systems will, in the end, be more likely to allow us to build a functional genome from scratch than the untested engineering hypothesis that organisms should work like we want them to. In greater detail, this should inevitably lead to the following discussion.
Systems Biology Eats Synthetic Biology
The fiction of synthetic biology is that it is possible to engineer biological systems in modular, composable, scalable, and programmable ways using “parts” to build circuits and eventually systems (and ecosystems) (Bromley et al., 2008; Win et al., 2009). Both the field and the fiction have emerged largely not from scientific research but from the International Genetically Engineered Machines competition, iGEM, which is constrained to rely on “BioBricks” for the construction of student projects (Smolke, 2009). In fact, long before iGEM there were numerous biological engineers, and these biological engineers would, with different degrees of success, use genes (not then known as biological parts) to construct pathways (not then known as circuits) that had particular functions.
What has changed between the long period during which biological engineering has been maturing (indeed, one could argue that biological engineering in the form of selective breeding greatly predated man’s understanding of biology itself) and today to make biological engineering into synthetic biology, besides nomenclature? It could be argued that there are two important milestones: First, many researchers in other engineering disciplines were somehow shut out of the biology because it lacked an engineering flair and was more dissective than synthetic. In this view, the influx of engineers into biology is assisted by recasting biology in terms more familiar to engineers, and this is why we often see biological circuits represented (incorrectly, as I will argue later) as electronic circuits (Khalil and Collins, 2010).
Second, there is not just a quantitative but rather a qualitative or foundational difference in being able to make large amounts of DNA. The ability to remake a whole genome (Gibson et al., 2010) is so great relative to the ability to make a mutation in that genome (or in an episome sharing a cell with that genome) that there must now be a new engineering discipline that approaches genome construction in a wholly different way than traditional biological engineering would have been capable of doing. I think there is some merit to this explanation, although it is a bit like saying that genomics is different than genetics. While we would never argue that we do not have much greater understanding of molecular detail today than we did in Mendel’s day, our fantastic increase in knowledge does not invalidate the views of Mendel; rather, this knowledge merely adds depth to the fundamental concept of inheritance. Genomics further explains genetics; it does not remake it (although individual concepts in genetics, such as the role of environment in inheritance, have certainly been remade radically). Nonetheless, it is true that techniques for the rational manipulation of whole genomes will prove to be of enormous value into the future. And while many of us who wish to reconstruct living systems will continue to have “Venter envy,” a new generation of techniques such as multiplex automated genome engineering (Wang et al., 2009) are beginning to emerge that will make such manipulations possible even for individual investigators.
In the absence of the synthetic biology “revolution” (redefinition), how might biological engineers who were attempting to manipulate larger and larger amalgams of DNA have managed? Where would they have turned for knowledge and inspiration? I contend that it would not have been electrical engineering—the frequent muse to whom many who call themselves synthetic biologists appeal—but rather to systems biology. Systems biology attempts to understand the interrelationships between all of the molecular and cellular parts of system in a quantitative way, and it has as one of its ultimate goals the modeling of biological organisms down to the molecular level. If we had the same knowledge of organisms that we do of the hardware and software accompanying a computer, the entire organism would not just be a “chassis” for the installation of a synthetic circuit; it would be the unit of engineering itself. Fortunately for both traditional biological engineers and synthetic biologists, our understanding of systems has increased dramatically in recent years, and thus there will come a point where systems biology will not only completely inform biological engineering, but it will overtake the utility of orthogonal, add-on circuits that were largely meant to operate independently of the systems in which they were embedded. Systems biology will eat synthetic biology.
This change in perspective and approach will be absolutely essential as biological engineering moves forward. The utility of orthogonal synthetic circuits is limited for a variety of reasons. First, by failing to take into account the unity of the system, the predictability of circuits must be limited. This can best be seen by thinking about a very simple synthetic circuit that has been around for
a long time: a plasmid that is used for protein overproduction. You can in fact embed a plasmid in a strain and induce protein production. From this vantage, the synthetic circuit has worked. However, it is difficult if not impossible to predict protein yield at the outset with any degree of certainty. Different proteins will express to different extents, will form aggregates to different extents, and will hamper the growth of the organism to different extents, to mention just a few variables that ultimately affect yield. This variability has nothing to do with whether a given “part” has been well characterized or not, and everything to do with the interaction of the part with the system as a whole. Protein overexpression interfaces with the cell’s machinery for transcription, translation, and degradation in myriad ways, and in the absence of a complete understanding of how the circuit interfaces with the system it will be difficult to ultimately predict how the circuit will work. Outcomes will range from making huge amounts of a protein to killing the cell outright. Of course the circuit can always be tinkered with to make more protein, but that was true before it was called a circuit and was merely a plasmid. What has synthetic biology brought to the modular, composable, scalable, and programmable overexpression of proteins that was not previously known? Nothing, because the tinkering that is now possible following the invention of the discipline is the same tinkering that was possible prior to its invention. Collins has pointed out that really tinkering is all that is necessary for engineering to prosper in biology, and he is of course correct. However, understanding and progress in this area will come from an increasingly detailed understanding of systems biology and integrated models of metabolism.
In discussions with Erik Winfree, a more charitable interpretation of the coexplosion of both systems and synthetic biology emerges, which is that these are different means to the same end—a predictable biology. The more complex system, the organism, is at present less predictable, and thus nominally orthogonal subsystems as built by synthetic biologists may allow us to initially develop better models. As those models become more sophisticated, they will of necessity take into account the discoveries and models developed through the study of systems biology.
A second, more important objection is that, by treating systems as though they were mere amalgams of components, synthetic biology ignores what I would argue is the central tenet of biology: organisms evolve. Given that an orthogonal circuit is really just an abstraction, as such circuits draw upon the metabolic, transcription, translation, and other resources of a cell, the circuit must of necessity change the fitness of a cell. To the extent that any synthetic circuit is used in the context of an evolutionary machine—a replicating cell—the possibility exists that mutations will arise that change both the cell and its added circuitry, for better or worse. While one alternative is just to not have cells replicate and to let circuits execute in preset bags of enzymes, this denies one of the great possibilities inherent in biology: the ability to generate larger amounts of complex matter from simpler substrates. An understanding of how to engineer biological systems will therefore proceed in large measure from a better understanding of
the costs and benefits of circuits to the system as a whole, again a province of systems (and evolutionary) biology.
Remaking Organismal Operating Systems
It is with this somewhat different perspective that we can move forward from thinking about how to make orthogonal circuits to thinking about how to manipulate the operating systems of organisms. Ultimately, synthetic biology is not modular, composable, scalable, or programmable because the operating system of biology does not support these features. The operating system of biology is a kludge, the result of billions of years of happenstance and compromise, and it cannot at this juncture be remade by the simple expedient of saying it’s not so.
But this does not mean that the operating system cannot be remade.
In looking at the operating system of biology, there is really only one component that is remotely akin to the synthetic biology dream of being modular, composable, scalable, and programmable, and that is DNA (or, more generally, nucleic acids). The simplicity of Watson-Crick base pairing and its implementation in a regular biopolymer meets all of these requirements. Unfortunately, this simplicity is destroyed by translation, which turns a regular biopolymer into many irregular ones. Obviously, from the point of view of organismal fitness, this is a good thing.
Thus, the question becomes, how can we remake the operating system of an organism so that the features of DNA are the features of not just one component but of all the components? My answer to this question is a limited one, but I think it suggests new directions. It seemed to me that since it is possible to have the base-pairing properties of DNA, but with different backbones, such as those seen in RNA, locked nucleic acids (LNAs), and especially peptide nucleic acids (PNAs), that it should be possible to extend base pairing to other components in biological operating systems. In particular, it should be possible to allow proteins to base pair in much the same way that PNAs would.
To this end, my lab has embarked on a journey to expand the genetic code to include four monomers that have nucleobase amino acids with side chains that have pairing properties equivalent to G, A, T, and C. Given the exploits and insights of the Schultz lab and others, this is an engineering feat that should be possible. The punctuation of the genetic code, stop codons, can be reprogrammed to code for amino acids (although ultimately with a systems-level cost), and four base codons (and now even a four base codon–reading ribosome) allow a (still uncertain) expansion of the code and the production of genetically augmented proteins.
Imposing Rationality on Biology
Whereas computers were built from the bottom up, with all of the rationality that engineers could bring to bear on both the hardware and software, biology
and its operating systems were presented to us as a fait accompli, and thus have to be engineered from the top down, in a way that can best be described as irrational, despite the fiction of synthetic biology. Biological systems evolved as kludges, and are engineered as kludges. While the underpinnings of synthetic biology seem to be to ignore these kludges, the hope of systems biology is to understand these kludges well enough that engineering short circuits and patches seem seamless.
As a small step, I envision that the addition of nucleobase amino acids to the genetic codes will allow us to begin to engineer biology in a truly rational manner. In my vision, there will be several substantive benefits to this fundamental remaking of a biological operating system. First, engineering protein–protein interactions will become more predictable, as it may be possible to uniquely code for such interactions via strings of nucleobase amino acids. Second, the same benefit will obviously accrue for protein–nucleic acid interactions. Transcription factors and RNA-binding proteins will no longer have semiprogrammable interaction domains, such as zinc fingers, but will instead be fully programmable. Third, it should be possible to rationally design cellular interactions and architectures via membrane proteins that again are tailed with nucleobase amino acids. And finally, this fundamental change in the operating system of a biological system should greatly speed the course of evolution. This conclusion comes from thinking about how transcription factors evolve to acquire new specificities. In general, there are multiple amino acid changes that must occur in order to bind a new DNA sequence, and these changes are not one-to-one, since the “code” for protein–nucleic acid interactions is three-dimensional rather than linear. By switching out the three-dimensional code for a nominally linear one, the number of sequence changes that must be acquired to bind adjacent to a promoter (or to form a new protein–protein interaction, or to make a connection to a new cell) will be proportionately reduced, and thus the acceptance of these changes (evolution) can potentially occur much more quickly.
For the longer term, I think there is the possibility that the operating systems of organisms can be changed in an even more fundamental manner. Merely adding nucleobase amino acids to the repertoire of the genetic code simplifies interactions from three-dimensional and irrational to linear and rational, but it does nothing about the way in which the associated machinery actually performs computations. Signal-transduction pathways will still pass information from the outside world to the inside of cells via a series of just-so scaffolds and complex conformational changes that will remain subject to relatively irrational engineering (and, given how I have tried to define rationality at a global level, this statement takes nothing away from the extraordinary successes of Lim and others in engineering signal transduction) (Bashor et al., 2008).
Where, then, can we find a more rational operating system for biology? In parallel with the “revolution” of synthetic biology, a true revolution in nucleic acid programming has been brewing at Caltech, led by Erik Winfree, Niles
Pierce, Peng Yin, and others (Seelig et al., 2006; Yin et al., 2008; Zhang et al., 2007). These pioneers have taken the seminal work by Adleman (1994), showing that nucleic acids can be used for computation, and further transformed it, showing that nucleic acids can be computers. To my mind, the distinction is subtle but important. An algorithm can be embedded in nucleic acids, as Adleman showed, but nucleic acids may not be the best platform for executing that algorithm (electronics beats biology, at least in terms of speed, on most computations). When instead we attempt to identify how nucleic acids can best compute—how their intrinsic properties can be exploited to do computation—then we begin to make nucleic acid computers of the sort that have emerged from the Caltech researchers. These computers rely on a very simple reaction, strand exchange, in which one nucleic acid binds to and forms a duplex with another. Duplex formation can in turn lead to the displacement of a previous duplex or the alteration of a programmed secondary structure. The displacement can be transient, with new binding sites (so-called toeholds) being revealed and paving the way to the formation of additional and/or different duplexes. Overall, extraordinarily complex executable circuits can be built in a fully rational manner, and these circuits have now been adapted to a variety of interesting computations, from amplification to determining square roots. The comparison with synthetic biology is instructive: while Weiss has pointed out that it has proven difficult to make synthetic circuits with more than six or so layers (Purnick and Weiss, 2009), Winfree has now readily generated a DNA circuit with this many layers and the obvious capability to scale much farther (Qian and Winfree, 2011).
I therefore fantasize about a biological system that is based on a rational biological computer, akin to the strand exchange circuits. As tempting as it is to suggest we should tear down biology and start afresh, this is well beyond my capabilities, and thus even my fantasies must be instantiated incrementally. It is possible to envision how strand-exchange RNA circuits could be embedded in organisms. The outcome of such circuitry could be the production of particular miRNAs that would feed back on the system, in much the same way that Benenson and Weiss have nicely shown that logical miRNAs can be embedded in the current biological operating system (Rinaudo et al., 2007). In couple with a fully realized suite of genetically augmented proteins containing nucleobase amino acids, the possibilities for superimposing a rational operating system on the current irrational one is manifest (albeit still incremental).
In moving from current biological operating systems to a fanciful new operating system built on genetically augmented proteins and strand exchange reactions, I have always been sensitive to the nature of the operating system that we already have (even while attempting to remake it). This “feeling for the organism” (to steal a phrase from McClintock) is not stealth vitalism but rather a sense of how engineering must proceed from the materials at hand. In this regard, while engineers can certainly become biologists and vice versa, each brings their preconceptions to the table in a manner that should be explicitly recognized. From
the point of view of a biologist, biological circuits are utterly unlike electronic circuits, irrespective of the analogies that can be drawn (Simpson et al., 2009). Electronic circuits operate at light speed, with spatially defined interconnectivities. Biological circuits operate via chemical diffusion, with molecular recognition and reactivity defining interconnectivity. While software is independent of hardware, and while both types of circuits can make, say, an oscillator, that’s no reason to suspect that both types of circuits will be able to operate in the same time regimes or with the same fidelity, since these features will stem in part from capabilities of the materials from which they are constructed. Thus, it may not be unreasonable to suggest that in engineering biology we must build from the capabilities of the materials involved, rather than trying to superimpose a foreign mindset—the current and likely transient Zeitgeist of synthetic biology—on those materials.
References
Adleman, L. M. 1994. Molecular computation of solutions to combinatorial problems. Science 266:1021-1024.
Bashor, C. J., N. C. Helman, S. Yan, and W. A. Lim. 2008. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 319:1539-1543.
Bayer, T. S., D. M. Widmaier, K. Temme, E. A. Mirsky, D. V. Santi, and C. A. Voigt. 2009. Synthesis of methyl halides from biomass using engineered microbes. Journal of the American Chemical Society 131:6508-6515.
Bromley, E. H., K. Channon, E. Moutevelis, and D. N. Woolfson. 2008. Peptide and protein building blocks for synthetic biology: From programming biomolecules to self-organized biomolecular systems. ACS Chemical Biology 3:38-50.
Gibson, D. G., J. I. Glass, C. Lartigue, V. N. Noskov, R. Y. Chuang, M. A. Algire, G. A. Benders, M. G. Montague, L. Ma, M. M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N. Assad-Garcia, C. Andrews-Pfannkoch, E. A. Denisova, L. Young, Z. Q. Qi, T. H. Segall-Shapiro, C. H. Calvey, P. P. Parmar, C. A. Hutchison, 3rd, H. O. Smith, and J. C. Venter. 2010. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329:52-56.
Kemmer, C., M. Gitzinger, M. Daoud-El Baba, V. Djonov, J. Stelling, and M. Fussenegger. 2010. Self-sufficient control of urate homeostasis in mice by a synthetic circuit. Nature Biotechnology 28:355-360.
Khalil, A. S., and J. J. Collins. 2010. Synthetic biology: Applications come of age. Nature Reviews Genetics 11:367-379.
Martin, V. J., D. J. Pitera, S. T. Withers, J. D. Newman, and J. D. Keasling. 2003. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nature Biotechnology 21:796-802.
McGary, K. L., T. J. Park, J. O. Woods, H. J. Cha, J. B. Wallingford, and E. M. Marcotte. 2010. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proceedings of the National Academy of Sciences of the United States of America 107:6544-6549.
Purnick, P. E., and R. Weiss. 2009. The second wave of synthetic biology: From modules to systems. Nature Reviews Molecular Cell Biology 10:410-422.
Qian, L., and E. Winfree. 2011. Scaling up digital circuit computation with DNA strand displacement cascades. Science 332:1196-1201.
Rinaudo, K., L. Bleris, R. Maddamsetti, S. Subramanian, R. Weiss, and Y. Benenson. 2007. A universal RNAi-based logic evaluator that operates in mammalian cells. Nature Biotechnology 25:795-801.
Seelig, G., D. Soloveichik, D. Y. Zhang, and E. Winfree. 2006. Enzyme-free nucleic acid logic circuits. Science 314:1585-1588.
Simpson, Z. B., T. L. Tsai, N. Nguyen, X. Chen, and A. D. Ellington. 2009. Modelling amorphous computations with transcription networks. Journal of the Royal Society Interface 6(Suppl 4):S523-S533.
Smolke, C. D. 2009. Building outside of the box: iGEM and the BioBricks Foundation. Nature Biotechnology 27:1099-1102.
Wang, H. H., F. J. Isaacs, P. A. Carr, Z. Z. Sun, G. Xu, C. R. Forest, and G. M. Church. 2009. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894-898.
Win, M. N., J. C. Liang, and C. D. Smolke. 2009. Frameworks for programming biological function through RNA parts and devices. Chemical Biology 16:298-310.
Yin, P., H. M. Choi, C. R. Calvert, and N. A. Pierce. 2008. Programming biomolecular self-assembly pathways. Nature 451:318-322.
Zhang, D. Y., A. J. Turberfield, B. Yurke, and E. Winfree. 2007. Engineering entropy-driven reactions and networks catalyzed by DNA. Science 318:1121-1125.
SYNTHETIC BIOLOGY—A NEW GENERATION OF BIOFILM BIOSENSORS
James Chappell28and Paul S. Freemont27,29
Biofilms: A Survival Strategy
The notion that bacteria live an autonomous, independent, and planktonic lifestyle has been radically challenged with the realization of the abundance of bacterial communities known as biofilms (Hall-Stoodley et al., 2004). Biofilms are structured communities of adherent microorganisms encased in a self-produced complex extracellular polymeric substance (EPS) matrix as shown in Figure A4-1 (Costerton et al., 1999). The advent of microscopy techniques has revealed the complex nature of these structured communities, containing networks of channels for nutrient supply and organized three-dimensional structures. Moreover, bacteria within these biofilms are profoundly physiologically distinct from their planktonic counterparts, and they function in a coordinated manor as a cooperative consortium, more similar to that of multicellular organisms than a unicellular organism (Hall-Stoodley et al., 2004; Lindsay and von Holy, 2006; Parsek and Singh, 2003). The ability to form biofilms is almost ubiquitously found within the bacteria domain of life and indeed within many other classes of microorganisms, including fungi, yeasts, algae, and protoza (Van Houdt and Michiels, 2010). Evidence suggests that biofilm formation is an ancient ability,
_______________
28 Centre for Synthetic Biology and Innovation, Imperial College London, South Kensington Campus, London SW7 2AZ, United Kingdom.
29 Correspondence: e-mail: p.freemont@imperial.ac.uk, telephone: + 44 (0)2075945327.
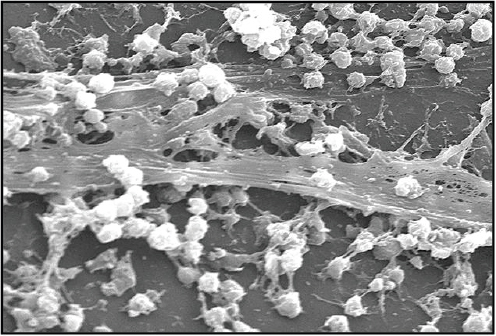
FIGURE A4-1 An electron micrograph of Staphylococcus aureus bacteria biofilms on the luminal surface of an indwelling catheter.
SOURCE: Image obtained from Public Health Image Library. Provided courtesy of R. Donlan, and J. Carr, Centers for Disease Control and Prevention. Photo by J. Carr (2005).
with biofilm microcolonies being identified in fossils from 3.3 to 3.4 billion years ago (Hall-Stoodley et al., 2004; Westall et al., 2001). Taken altogether, biofilm formation is an evolutionary conserved and widespread phenomenon.
The advantages of bacteria to abandon their independent lifestyle for that of a community is simply that it provides a more protective mode of growth. Biofilms allow bacteria to colonize and sustain favorable niches independent of larger environmental changes. For example, the EPS, a key component of biofilms, can serve as storage for nutrients and water, helping to stabilize local resources (Costerton et al., 1987; Donlan, 2002; Lindsay and von Holy, 2006). In addition, biofilms provide a defensive role, acting as a physical barrier from environmental dangers such as ultraviolet light, metal toxicity, acid exposure, dehydration, and even immune defenses such as phagocytosis (Donlan, 2002; Lindsay and von Holy, 2006; Parsek and Singh, 2003). These selective advantages of biofilm formation are obvious within the context of hostile and dynamic environments such as that of ancient Earth (Hall-Stoodley et al., 2004). Yet even in today’s relatively temperate environment, the formation of biofilms is still selectively advantageous and constitutes a major component of bacterial biomass in natural environments (Costerton et al., 1978, 1999; Lindsay and von Holy, 2006). Bacteria’s attempt
to survive by the formation of biofilms extends beyond just natural environments and provides bacteria protection in many man-made environments (Hall-Stoodley et al., 2004). However, the focus of this review is the presence of pathogenic bacterial biofilms within a clinical context.
Biofilms in a Clinical Context
It is only in recent years that the importance of biofilms in clinical settings as a source of pathogenic bacteria has been realised (Lindsay and von Holy, 2006). The persistence of biofilms in hospitals is undoubtedly a significant contribution to hospital-acquired infections or nosocomial infections (Costerton et al., 1999; Donlan and Costerton, 2002; Hall-Stoodley and Stoodley, 2009; Hall-Stoodley et al., 2004; Lindsay and von Holy, 2006). Indeed it has been estimated that biofilms contribute to 65 percent of nosocomial infections (Potera, 1999; Smith and Hunter, 2008). Table A4-1 shows the most frequent pathogenic strains associated with nosocomial infections, the most frequent and problematic being Pseudomonas aeruginosa, Staphylococcus aureus, and S. epidermidis (Costerton et al., 1999; Hall-Stoodley et al., 2004). Recent studies estimate that at any one time 9 percent of all in-patients in England and Wales (United Kingdom) suffer from hospital-acquired infections, resulting in approximately 5,000 deaths per year. The costs associated with these hospital-related infections for the U.K. National Health Service are estimated at £1 billion per year (Smith and Hunter, 2008).
TABLE A4-1 The Most Common Causes of Nosocomial Infections
| Nosocomial Infections | |
| ICU pneumonia | Gram-negative rods |
| Sutures | S. epidermidis and S. aureus |
| Exit sites | S. epidermidis and S. aureus |
| Arteriovenous shunts | S. epidermidis and S. aureus |
| Schleral buckles | Gram-positive cocci |
| Contact lens | P. aeruginosa and Gram-positive cocci |
| Urinary catheter cystitis | E. coli and other Gram-negative rods |
| Peritoneal dialysis (CAPD) peritonitis | A variety of bacteria and fungi |
| IUDs | Actinomyces israelii and many others |
| Endotacheal tubes | A variety of bacteria and fungi |
| Hickman catheters | S. epidermidis and Candida albicans |
| Central venous catheters | S. epidermidis and others |
| Mechanical heart valves | S. epidermidis and S. aureus |
| Vascular grafts | Gram-positive cocci |
| Biliary stent blockage | A variety of enteric bacteria and fungi |
| Orthopedic devices | S. epidermidis and S. aureus |
| Penile prostheses | S. epidermidis and S. aureus |
| SOURCE: Adapted from Costerton et al. (1999). | |
Although many nonpathogenic bacteria are able to form innocuous biofilms, it is the formation of pathogenic biofilms that is particularly important for contributing to the high level of hospital-acquired infections. It has been suggested that biofilm formation is analogous to a virulence factor, where biofilm formation increases the likelihood of a pathogen causing an infection.
Three general mechanisms have been proposed to explain the increase of virulence seen for pathogenic bacteria in a biofilm microenvironment (Hall-Stoodley and Stoodley, 2005). These are the survival and transmission of planktonic patho-
BOX A4-1
The Mechanisms of Biofilm-Associated Virulence
Survival and Transmission of Pathogenic Biofilms
Clinical environments such as hospitals are constantly exposed to a consortium of pathogenic bacteria. Many pathogenic bacteria can be easily eradicated when in planktonic mode of growth by biocidal agents. However, if these bacteria form biofilms, they will become significantly more resistant to biocidal agents. Established biofilms can tolerate antimicrobial agents at concentrations of 10-1,000 times that needed to kill genetically equivalent planktonic bacteria (Gander, 1996; Gilbert et al., 2002; O’Toole, 2002; Smith and Hunter, 2008). This high level of resistance makes eradication of pathogenic biofilms in hospitals largely unsuccessful. Indeed, studies have shown pathogenic biofilms to be present on a number of medical surfaces, including indwelling medical devices such as catheters and prostheses (Stickler, 2002), hospital water systems, stethoscopes (Guinto et al., 2002), soap dispensors (Brooks et al., 2002), and other hospital equipment.
The presence of these pathogenic biofilms is problematic because they act as nodes of infection, seeding the regular release of large numbers of pathogenic cells, potentially exposing patients to infectious doses of these organisms (Hall-Stoodley and Stoodley, 2005; Hall-Stoodley et al., 2004). Biofilms have been shown to regularly release cells either by exposure to external mechanical forces, such as by shear forces, or by internal biofilm processes including enzymatic degradation of matrix, release of EPS, and surface binding proteins (Hall-Stoodley et al., 2004; Stoodley et al., 2002). Taken together, the high level of resistance and regular release of pathogenic cells and the presence of pathogenic biofilms increase the potential of a patient being exposed to an infectious dose of pathogenic cells.
Heterogeneity and Selection of Virulent Phenotype
The complex nature of biofilms is that they provide heterogeneous microenvironments within the biofilms. These distinct microenvironments provide different selective pressures and are occupied by genetically distinct populations. Genetic screening of microenvironments has suggested that the biofilm mode of growth accelerates genetic variation in the biofilm populations (Boles et al., 2004; Kirisits et al., 2005; Stewart and Franklin, 2008). It has been shown that P. aeruginosa undergoes accelerated genetic
gens from biofilms, the phenotypic heterogeneity of biofilm populations and the potential evolution of infectious phenotypes, and the role of biofilm formation in the regulation of virulence mechanisms. Each of these mechanisms is discussed further in Box A4-1.
The seriousness of this problem has provided strong motivations for developing new antibiofilm therapies and detection methods. The development of new antibiofilm therapies is beyond the scope of this paper, although there are a number of extensive reviews (Lynch and Abbanat, 2010). Here we focus on cur-
diversification in the biofilm mode of growth. This mechanism was dependent on the recA gene, a key protein involved in DNA recombination, suggesting that recombination contributes to this variation (Boles et al., 2004). These genetic variations lead to more pathogenic variants; for example, hyperbiofilm formation and antibiotic-resistant phenotypes have been shown to develop in P. aeruginosa biofilm models (Drenkard and Ausubel, 2002; Parsek and Singh, 2003).
Virulence Products
Genes contributing to the virulence and pathogenicity of bacterial strains are commonly regulated by the cell-to-cell signaling mechanism known as quorum sensing. Quorum sensing is a mechanism whereby bacteria release and sense small signaling molecules, termed autoinducers. These autoinducers accumulate as a population increases, and, once a sufficient “quorum” (population) is reached, gene expression is induced in a coordinated manor across a population (Fuqua et al., 1994). The advantage of this cooperation is that it allows bacteria to act in concert, but only once a sufficient population has been reached. In terms of pathogenic bacteria, it allows bacterial densities to increase before expression of virulence factors and other pathogenic factors that might trigger immune responses. In P. aeruginosa, quorum sensing has been shown to regulate the expression of genes encoding virulence factors, antibiotic resistances, motility, siderophore production, adhesions, cytotoxtins, exotoxins, and type III secretion systems (Asad and Opal, 2008; Davies et al., 1998). Furthermore, the importance of quorum sensing for virulence has been confirmed in a number of animal models including mice, nematodes, and insects (Lesic et al., 2007; Mahajan-Miklos et al., 2000; Popat et al., 2008; Rumbaugh et al., 1999).
The relationship between quorum sensing and biofilms is one of positive feedback. It has also been shown that quorum sensing is required for biofilm formation. In P. aeruginosa it has been shown that mutants of quorum sensing transcription factors, LasR, produce flat, continuous, and sparse biofilms that had neither the density nor the complex channel structure seen in wild-type biofilms. In addition these mutant strain biofilms lose resistance to the biocide agent sodium dodecyl sulfate (SDS) (Davies et al., 1998). This evidence suggests a key role of quorum sensing in the formation of biofilms and regulation of virulence. Biofilms provide a niche that allows the cell population to increase and sufficient autoinducers to accumulate to thus trigger the expression of genes involved in virulence and pathogenicity (Asad and Opal, 2008; Hall-Stoodley and Stoodley, 2005).
rent methods for biofilm detection, as well as their limitations, and the prospect of synthetic biology to develop completely novel methods for the detection of pathogenic biofilms.
Current Methods of Biofilm Detection
The ability of biofilms to form on a variety of abiotic and biotic surfaces has resulted in colonization of many clinical environments. Detection of these biofilms has obvious advantages: to direct suitable sanitation protocols in biofilm niches, to indicate replacement of any contaminated medical devices such as indwelling catheters, and to direct suitable medication if patient infection ensues. It has now become common practice in hospitals to sample both environmental surfaces and patients, typically urine, for the presence of pathogenic biofilms. There are numerous methods used for detection, including conventional plate counting, phenotypic screens, microscopy methods, and genotypic methods. Two of the most established methods for biofilm detection used in laboratories and clinical settings are the genotypic and phenotypic methods that are described in more detail below.
Phenotypic methods rely on culturing bacterial samples and screening these cultures for the phenotype of biofilm producers. These methods rely on the hypothesis that biofilm formation is a marker of virulence. Three variants of phenotypic detection have been commonly used: the tissue culture plate, the tube method, and the Congo red agar (CRA) method. These methods rely on indirectly assessing biofilm producers by accumulation of biofilm components such as EPS, a polysaccharide involved in cell-cell adhesion and essential for biofilm formation.
Genotypic methods are based upon PCR amplification of specific genetic markers associated with pathogenic biofilms. The most notable example is the use of the ica cluster to identify biofilm-producing strains of Staphylococcus aureus and S. epidermidis. The ica cluster contains genes that are essential for these strains to produce EPS. In addition, PCR is commonly used to assess antibiotic resistance; for instance, the presence of the mecA gene encoding methicillin resistance can be used to identify strains of methicillin-resistant S. aureus.
Each of these methods has its own advantages and limitations. The major limitations of these are described below:
- Expertise and cost. Although techniques such as PCR and microscopy techniques have become commonplace in molecular biology laboratories, in hospitals, particularly in developing countries, access to the equipment and expertise required for analysis can be limited (Bose, 2009).
- In situ and point-of-care detection. Sampling of biofilms on hospital surfaces can be abrasive to attached cells and may result in damage to
-
cells, rendering them nonculturable (Lindsay and von Holy, 2006). In addition, sampling of indwelling medical devices such as catheters requires the removal of catheters, whether a biofilm is present or not. This can result in unnecessary stress for patients and wastefulness of such devices (Donlan and Costerton, 2002). Finally, the recovery efficiency of sampling methods commonly used is largely unknown, leaving the possibility of not detecting a representative population.
-
Reliability and reproducibility. The genotypic methods are generally considered more reliable and sensitive. However, they can be subject to false positives. This is due to the ica cluster not being absolutely required for biofilm formation. It has been shown that polysaccharide intercellular adhesin (PIA) synthesis from the ica operon alone is not sufficient to produce biofilms and that biofilms can form without producing PIA in staphylococci (Chokr et al., 2006).
For the phenotypic detection methods there is limited sensitivity, reliability, and reproducibility compared to genetic detection. There have been numerous independent studies comparing the different phenotypic methods, which have shown varying success rates using the different phenotypic methods. For example, studies assessing the sensitivities of the CRA method have shown a detection success ranging from 5.26 to 89 percent (Arciola et al., 2006; Bose, 2009; Knobloch et al., 2002; Mathur et al., 2006; Oliveira and Cunha, 2010). In addition, the phenotypic methods are commonly prone to false positives and nondetection of “weak” biofilm-forming strains.
- Time of response. The phenotypic detection methods rely upon the culturing of microorganisms collected from samples. These incubation periods are typically 24 to 48 hours and cultures are often analyzed offsite, increasing time for biofilms to develop (Bose and Ghosh, 2011).
- Informative. Neither the genotypic nor the phenotypic methods elicit an in situ biofilm structure. Although detection of the pathogenic biofilms indicates the potential for the presence of biofilms, it is not an accurate reflection of the actual microenvironment. This can lead to an inaccurate detection in the in situ environment and false diagnosis.
Whole-Cell Biosensors: Bridging the Detection Gap
As discussed above there is a strong motivation for the development of in situ detection methods with increased response time, specificity, and more informative outputs. A potential solution is the development of whole-cell biosensors, using genetically engineered biological cells to directly detect pathogenic biofilms. An essential ability for cells to function is the ability to sense and respond to a diverse range of environmental signals from environmental conditions such as temperature change to the detection of nutrients in the environment. This ability can be
broken down to two subfunctions, that of sensing and that of response (signal transduction). Whole-cell biosensors are essentially composed of the same two elements integrated into microorganisms: sensing elements detecting the analyte of interest and transducing elements that are usually coupled to the production of a detectable output, for example, expression of reporter proteins such as natural fluorescent proteins.
Some of the advantages of whole-cell biosensors are that they detect a variety of biological, organic, and nonorganic compounds. In addition, they can produce a number of detectable outputs that can be easily quantifiable, for example, fluorescent proteins measured by a fluorometer, dyes and pigments measured by spectroscopy and by eye, and electrochemical signals such as pH measured by litmus paper or a pH meter. These easily measured outputs require little expertise to use or to interpret. In addition, the timescale for biosensors is generally rapid, with signals being transduced in seconds to generate detectable signals within minutes to hours. In addition, compared to other in vitro biosensors like widely used enzymes or antibodies, whole-cell biosensors are easily produced by cell culture, in contrast to the costly and time-consuming purification of enzymes and antibodies.
Another advantage of whole-cell biosensors can be highlighted using the example of detecting pathogenic biofilms. Genotypic methods detect at a microscopic level the DNA genetic elements that are required for the formation of biofilms rather than the products of these genes, which can therefore lead to false positives. Phenotypic detection detects the products of biofilms but at a macroscopic level. Whole-cell biosensors offer a way to bridge the gap between these two methods, allowing sensing and evaluation at the level of microbial habitats. Whole-cell biosensors could allow the detection of new markers of biofilms to provide more effective detection methods. In recent years, there has been increased interest in the development of whole-cell biosensors, particularly within the field of synthetic biology. In the remainder of this review, the potential of synthetic biology for creating more effective biosensors for the detection of pathogenic biofilms is discussed.
Synthetic Biology: A New Generation of Biofilm Biosensors?
Synthetic biology is an application-driven field attempting to apply a rational engineering approach to the redesign of biological systems, to produce valuable and novel biological functions. Synthetic biology can be thought of as a natural evolution of biotechnology as opposed to being a separate field, although it does offer a novel and exciting approach. In the past 40 years biotechnology has undoubtedly produced many valuable biological products, yet current approaches tend to be lengthy due to the ad hoc nature of the approach. In addition, there is little lateral transfer of knowledge between projects, meaning knowledge gained in research and development in one area may not assist the post hoc development of projects in other, unrelated areas.
Synthetic biology aims to move away from this ad hoc design by providing a conceptual framework based on systematic design and rational engineering, so that redesigning biological systems would become more equivalent to that of redesigning a plane or a car. Indeed, it is the adoption of an engineering framework that has revolutionized the design and output of applications in the fields of mechanical, electrical, civil, and aeronautic engineering, and it is proposed that such an approach will do the same for biotechnology. The focus of this paper is to discuss the potential of whole-cell biosensors and the synthetic biology approach both in general and for the specific application of detection of pathogenic biofilms, the need of which was highlighted earlier.
Three major advantages of the synthetic biology approach for biosensor designs are highlighted: the advantages of the engineering approach, the development of novel sensing elements, and the complexity synthetic biology could enable.
Putting the Engineering into Genetic Engineering
Modern engineering relies on designing systems from catalogues of well-defined standard parts and higher-level devices that can be assembled into larger systems in a standardized manner. For electrical engineers, catalogues such as RS Components and Farnell provide well-defined parts and devices, each associated with data sheets of the functional characteristics. System designs can then be assessed by applying mathematical models in combination with known behavior characteristics, to computationally simulate their overall functions. This allows rapid assessment in silico of designs that meet the required specifications of design. Through iterations of this engineering approach or engineering cycle, efficient production of well-characterized and robust end products, from electronics to buildings, can be produced.
Synthetic biology is trying to adopt this engineering cycle and develop the tools required for high-throughput engineering of biological systems (Figure A4-2) (Gulati et al., 2009; Kitney et al., 2007). However, living systems are not computer chips and thus it is important to note that synthetic biology aims to provide a conceptual engineering approach that will allow biological engineering to be easier and more predictable. Living systems are intricate, formed from a complex network of interacting chemical components driven by the ability to self-replicate, adapt, and survive. Therefore, in living systems, context dependency and stochastic behavior are common features and therefore the engineering of predictable new cellular systems with defined functions is challenging. Nevertheless, the field of synthetic biology aims to provide a series of foundational technologies and a defined framework that will enable robust biological design and overcome these clear challenges. At present much emphasis in synthetic biology is focused on engineering single-cell organisms like Escherichia coli or yeast, given that our understanding of these organisms is more mature than for
FIGURE A4-2 The engineering cycle as an approach for synthetic biology. This adaption includes the four stages of defining specifications, design of biological devices, in silico modeling of the potential designs, and characterisation of performances.
multicellular systems. With this in mind, a synthetic biology engineering cycle is discussed below.
Engineering cycle
- Specification The first stage in the engineering cycle is to define the specifications required. For example, a previous whole-cell biosensor designed to detect arsenic in polluted water defined the detection range as defined by the World Health Organization recommendation of safe drinking water at 10 ppb arsenic. Other specifications can be tunable, for example, threshold, time of response, particular microbial organisms, or host chassis to be used.
- Design Once the specifications have been defined, genetic devices have to be designed to give the functions of interest to host cells, often microbes. For the design of a genetic device, the principle of abstraction is applied to help separate out unnecessary layers of complexity (Endy, 2005). At the most complex layer, the design of a biological device involves the writing of a DNA nucleotide sequence to give desired func-
-
tion when implanted in a cell. However, an abstract view can be taken and these sequences modularized into functional parts such as promoters, gene coding sequences, ribosome binding sites, and terminators. These modules of DNA allow the functional separation of biological parts (BioParts) and allow a higher level of design, where the underlying complexity of the DNA sequence can be hidden (Endy, 2005).
Collaborative and international efforts have been made to make open-source registries to document and physically store a huge variety of BioParts and devices, most notably being the BioBricks registry (http://partsregistry.org) and the International Open Facility Advancing Biotechnology (BIOFAB). Two important criteria of BioParts in these registries are the adherence of standards and the documentation of characterisation data. The BioBricks foundation has proposed a “request for comment” process, encouraging the synthetic biology community to list standards for physically joining DNA and protocols such as for characterisation of BioParts.
These catalogues are beginning to provide a huge toolbox of biological parts for the engineering of biological systems, from basic BioParts such as promoters, ribosome binding sites, genes, and terminators to more complex composite devices such as toggle switches, oscillators, logic gates, and cell-to-cell communication systems. Moreover, the standards required for submission to these registries ensure compatibility and interchangeability. For example, the BioBricks registry has defined standard restriction sites flanking all parts and devices, which has allowed standardized methods of assembly where any two parts can be combined by following use of a standardized protocol. Finally, the documentation of characterisation data allows transfer of knowledge and experience gained from separate projects to be used in the evaluation of potential designs (Arkin, 2008).
- Modeling After the design stage, in silico computational simulation of mathematical models representative of the genetic devices is performed. In the 1960s the mathematical logic in gene regulation of the lac operon was demonstrated (Andrianantoandro et al., 2006; Monod and Jacob, 1961). Since then, systems biology has transformed our understanding of biology using representative mathematical models based on quantitative data, most notably stochastic and ordinary differential equations. These modeling techniques, coupled with the use of experimentally defined characteristics defining model parameters, can predict the functional characteristics of a design without the need to undergo cloning or de novo synthesis, or experimental characterisation both of which increase overall time and cost of development. Ellis et al. (2009) demonstrated the use of computational modeling to predict the behavior of feedforward loops in yeast. To do so, a library of inducible promoters
-
with known minimum and maximum outputs (expression levels) were used in the device design. The characterisation data were incorporated into the modeling, the predictions of which were shown to accurately reflect the experimentally derived behaviors of these devices (Ellis et al., 2009).
-
Quality control The idea that a device’s behavior can be fully predicted by modeling the known characteristics of individual parts is obviously an oversimplification. It is inevitable that unexpected emergent properties will result when individual parts are put together that have been characterised in isolation and different contexts such as a switch in host chassis of E. coli to Bacillus subtilis (Arkin, 2008; Serrano, 2007). Unexpected interactions can occur from the context of the other BioParts in the device or even the host chassis. For example, in mRNA the untranslated regions of promoters can form secondary structures with ribosome binding sites of mRNA sequences modifying the function of this BioPart (Arkin, 2008).
These factors necessitate a quality control step, first to assess how well the device meets the specification but also to gain a greater understanding of the device being designed. Although approaches such as BioPart characterisation and modeling might not give an absolute prediction of device function as seen in other fields such as electronics, it nonetheless reduces the number of potential designs that need to be explored and therefore increases the overall efficiency.
Novel Biosensor Targets
As mentioned earlier, biosensors are composed of two subfunctions, that of sensing and that of signal transduction. The example of quorum sensing has already been discussed, where bacteria sense the local population densities based on the concentration of autoinducers and, once a threshold is reached, gene expression is induced. In this example, transcription factors act as the sensing elements binding to autoinducers and the quorum-sensing responsive promoters act as signal-transducer elements, defining the threshold of response and converting the autoinducer concentration to transcriptional output.
The bacterial toolbox of sensing elements is mostly mRNA and protein based, both of which can form complex tertiary structures able to bind a myriad of targets. Coupled to these sensing elements, the signal-transducing elements are generally transcriptional, translational, and posttranslational. Sensing modules and transducer modules can be separate biological components such as in quorum sensing, or within the same biological molecule such as riboswitches that contain an mRNA aptamer element able to sense and convert this sensing to translational output by sequestering or releasing a proximal ribosome binding site.
When considering the design of biosensors, it is essential to identify both
the sensing and the transducing elements. In general, synthetic biologists have employed three strategies to use these elements: first, to import exogenous sensing elements into new biological host chassis; second, to use and rewire existing endogenous sensing elements to detect new signals; and finally, in the production of novel elements not found in nature.
Three examples are described in Box A4-2 to explain each of these strategies. In addition, each example provides an illustration of biosensors functioning at the level of transcription, translation, and posttranslation.
Increasing Complexity
Within the field of synthetic biology, a variety of synthetic genetic devices have been developed. Many of these devices have been inspired by those found in electronics and include toggle switches (Atkinson et al., 2003; Bayer and Smolke, 2005; Deans et al., 2007; Dueber et al., 2003; Friedland et al., 2009; Gardner et al., 2000; Ham et al., 2006, 2008; Kramer and Fussenegger, 2005; Kramer et al., 2004), memory elements (Ajo-Franklin et al., 2007; Basu et al., 2004; Friedland et al., 2009; Ham et al., 2006, 2008), pulse generators (Basu et al., 2004), time-delayed circuits (Ellis et al., 2009; Weber et al., 2007), oscillators (Atkinson et al., 2003; Danino et al., 2010; Elowitz and Leibler, 2000; Fung et al., 2005; Stricker et al., 2008; Tigges et al., 2009), and logic gates (Anderson et al., 2007; Guet et al., 2002; Rackham and Chin, 2005; Rinaudo et al., 2007; Win and Smolke, 2008). These synthetic genetic devices are aiding the design of more complex biological systems the likes of which have not been previously seen. For the application of whole-cell biosensors, the implementation of these genetic devices could allow more complex and informative biosensors to be developed. For example, consider the use of logic gates for biosensor design. Logic gates are devices that perform a logical operation based on one or more inputs to produce a signal output. A frequently featured logic gate implemented in synthetic biology is the AND gate. An AND gate integrates two or more signals and will only give an output when all inputs are present. The principle and a biological example of an AND gate is shown in Figure A4-3.
The example of logic gates highlights the potential complexity of design that synthetic biology could offer for biosensor design. In general, multiple signal integration would allow fewer false positives by relying on more than one signal for detection and thus increasing the reliability. For example, the use of NOT gates could be used to remove known signals contributing to false positives. In addition, by being able to integrate multiple signals, logic gates could offer more complex and informative detection.
A more relevant illustration with regard to developing biosensors for detecting biofilms can be highlighted when we take into account mixed-species biofilms. Consider the case of biofilms on indwelling urinary catheters. A study of 106 biofilms found on catheters showed 14 species of bacteria to be commonly present (Macleod and Stickler, 2007; Stickler, 2008). Furthermore, although
BOX A4-2
Strategies and Examples of Biosensor Design
Transcriptional Control Using Exogenous Sensing Elements
Aleksic et al. (2007) engineered E. coli to detect toxic arsenic levels in drinking water, which today is a major pollutant of water in developing countries such as Bangladesh and West Bengal. To do so, exogenous transcription factors and promoters were cloned from B. subtilis into E. coli and coupled to the production of the enzyme urease, the production of which can be monitored by changes in pH. The arsR transcription factor and ars promoter were taken from B. subtilis (Sato and Kobayashi, 1998). In the absence of arsenic, the arsR represses transcription of the ars promoter. Upon binding of arsenic, the arsR repression is relieved, allowing the transcription of the chosen output. In this case a urease mRNA was the direct output that, once transcribed, is constitutively translated into the urease protein detectable by changes in pH.
The ability to import exogenous transcription factors and promoter regions between species of bacteria is relatively straightforward in theory because of the conserved mode of action of these transcription factors and conserved promoters. However, other sensing elements such as membrane protein receptors can be imported between strains, although, due to the varying nature of membranes in Gram-positive and Gram-negative species, it is only possible within these species groups. A major advantage of this approach is that it allows sensors found in strains that cannot easily be cultured to be incorporated into more commonly and well-defined strains such as E. coli. This is particularly advantageous when searching and identifying sensors from extremophiles that are often difficult to culture and would be impractical as whole-cell biosensors.
Translational Control Using De Novo Sensing Elements
Sinha et al. (2010) demonstrated that E. coli could be engineered to sense, follow, and destroy a commonly used herbicide, atrazine. To do so, de novo sensing elements had to be designed and constructed. They used the in vitro selection method, SELEX, to identify sequences of RNA that bound atrazine specifically and tightly. After identifying these RNA sequences, riboswitches were designed. Riboswitches are mRNA sequences that contain aptamer sequences that can sequester or release proximal
single-species biofilms were found, most were mixed-species biofilms—most frequently containing Enterococcus faecalis, Escherichia coli, Pseudomonas aeruginosa, and Proteus mirabilis. With the use of logic gates and multiple signal integration, synthetic biology could develop biosensors to respond not only to multiple targets but also with the ability to distinguish between species. This would allow general detection of biofilms as well as species-dependent detection that could aid correct treatment should the infection worsen.
ribosome binding site sequences upon binding or absence of a target. This allows sensing to be coupled to the translational control of any coding sequence placed downstream of the riboswitch. The atrizine riboswitch was designed so that, in the absence of atrazine, ribosome binding sites were sequestered by internal binding of proximal mRNA sequences, and in the presence and binding of atrixine, these ribosome binding sites were released, allowing translation of an output protein. In this example, the output protein was CheZ, a protein that controls motility in E. coli.
The design of de novo recognition elements allows the expansion of the natural “toolbox” of sensing elements available. New previously undetectable targets could be detected. This is particularly relevant for many of today’s environments, where the accumulation of man-made compounds and pollutants is relatively recent and for which nature has not evolved any response or sensing mechanisms.
Posttranslational Control Using Rewired Endogenous Sensing Elements
Baumgartner et al. (1994) rewired two transmembrane signaling pathways to create a hybrid signaling pathway. A hybrid transmembrane protein was created from the sensing domains of the Trg chemoreceptor and the transducing domains (kinase/phosphotase domains) of the EnvZ osomosensor. This engineered signaling pathway allowed the sensing of the sugar-occupied ribose-binding protein in the periplasmic domain and transduction of the EnvZ kinase and phosphatase domains, triggering a cascade of signaling proteins and ultimately expression of an OmpC-LacZ fusion protein. This hybrid signaling system was further engineered by Looger et al. (2003), who computationally designed artificial periplasmic binding proteins to sense novel targets such as TNT.
The potential advantage of posttranslational biosensors is that they offer a more rapid and precise biosensor. The transduction of signals by protein-protein interactions can be transduced in microseconds and are less affected by stochastic noise, a major cause of variation in transcriptional and translational biosensors (Legewie et al., 2008; Marchisio and Rudolf, 2011; Yu et al., 2008). In addition, although design principles of signal transduction are still being elicited, studies of natural pathways have shown that optimal design principles can produce high-fidelity signals, and characteristics such as latency phases after signaling can be produced (Colman-Lerner et al., 2005; Legewie et al., 2008).
Summary
Synthetic biology is a newly developing field that aims to provide an engineering framework for the predictable construction of new biological systems. The most immediate exemplars relate to biosensors where synthetic biology has already produced several living cell biosensors that act robustly and predictably. The application of synthetic biology design tools will enable the development of a new generation of biosensors to detect pathogenic biofilms. The implementation of these designs will have a profound impact in many areas including the control of hospital-acquired infections.
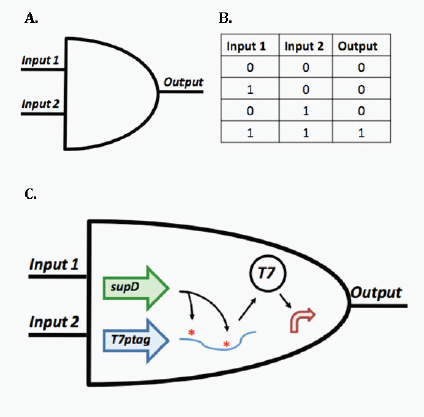
FIGURE A4-3 The principle and biological example of an AND gate (A) The symbolic representation of an AND gate and (B) a truth table describing the logic of an AND gate with regard to two inputs. Only when both inputs are present is an output produced. (C) An example of a biological AND gate previously described by Anderson et al. (2007). The AND gate is based around the interplay between two genes. One of these genes is the T7 RNA polymerase containing two amber stop codons (denoted by asterisks), meaning that translation of full-length T7 polymerase is inhibited (T7ptag). Only when the supD gene is transcribed are the amber stop codons translated into serines allowing full-length T7 polymerase to be translated and drive expression of the output from a T7 polymerase regulated promoter. This example represents an AND gate taking two transcriptional inputs and integrating these to a single transcriptional output.
References
Ajo-Franklin, C. M., D. A. Drubin, J. A. Eskin, E. P. Gee, D. Landgraf, I. Phillips, and P. A. Silver. 2007. Rational design of memory in eukaryotic cells. Genes & Development 21:2271-2276.
Aleksic, J., F. Bizzari, Y. Cai, B. Davidson, K. De Mora, S. Ivakhno, S. L. Seshasayee, J. Nicholson, J. Wilson, A. Elfick, C. French, L. Kozma-Bognar, H. Ma, and A. Millar. 2007. Development of a novel biosensor for the detection of arsenic in drinking water. IET Synthetic Biology 1(1-2):87-90.
Anderson, J. C., C. A. Voigt, and A. P. Arkin. 2007. Environmental signal integration by a modular AND gate. Molecular Systems Biology 3:133.
Andrianantoandro, E., S. Basu, D. K. Karig, and R. Weiss. 2006. Synthetic biology: New engineering rules for an emerging discipline. Molecular Systems Biology 2:2006.0028.
Arciola, C. R., D. Campoccia, L. Baldassarri, M. E. Donati, V. Pirini, S. Gamberini, and L. Montanaro. 2006. Detection of biofilm formation in Staphylococcus epidermidis from implant infections. Comparison of a PCR-method that recognizes the presence of ica genes with two classic phenotypic methods. Journal of Biomedical Materials Research Part A 76:425-430.
Arkin, A. 2008. Setting the standard in synthetic biology, Nature Biotechnology 26:771-774.
Asad, S., and S. M. Opal. 2008. Bench-to-bedside review: Quorum sensing and the role of cell-to-cell communication during invasive bacterial infection. Critical Care 12:236.
Atkinson, M. R., M. A. Savageau, J. T. Myers, and A. J. Ninfa. 2003. Development of genetic circuitry exhibiting toggle switch or oscillatory behavior in Escherichia coli. Cell 113:597-607.
Basu, S., R. Mehreja, S. Thiberge, M. T. Chen, and R. Weiss. 2004. Spatiotemporal control of gene expression with pulse-generating networks. Proceedings of the National Academy of Sciences of the United States of America 101:6355-6360.
Baumgartner, J. W., C. Kim, R. E. Brissette, M. Inouye, C. Park, and G. L. Hazelbauer. 1994. Trans-membrane signalling by a hybrid protein: Communication from the domain of chemoreceptor Trg that recognizes sugar-binding proteins to the kinase/phosphatase domain of osmosensor EnvZ. Journal of Bacteriology 176:1157-1163.
Bayer, T. S., and C. D. Smolke. 2005. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nature Biotechnology 23:337-343.
Boles, B. R., M. Thoendel, and P. K. Singh. 2004. Self-generated diversity produces “insurance effects” in biofilm communities. Proceedings of the National Academy of Sciences of the United States of America 101:16630-16635.
Bose, S. 2009. Detection of biofilm producing staphylococci: Need of the hour. Journal of Clinical and Diagnostic Research 3:1915-1920.
Bose, S., and A. K. Ghosh. 2011. Biofilms: A challenge to medical science. Journal of Clinical and Diagnostic Research 5:127-139.
Brooks, S. E., M. A. Walczak, R. Hameed, and P. Coonan. 2002. Chlorhexidine resistance in antibiotic-resistant bacteria isolated from the surfaces of dispensers of soap containing chlorhexidine. Infection Control & Hospital Epidemiology 23:692-695.
Chokr, A., D. Watier, H. Eleaume, B. Pangon, J. C. Ghnassia, D. Mack, and S. Jabbouri. 2006. Correlation between biofilm formation and production of polysaccharide intercellular adhesin in clinical isolates of coagulase-negative staphylococci. International Journal of Medical Microbiology 296:381-388.
Colman-Lerner, A., A. Gordon, E. Serra, T. Chin, O. Resnekov, D. Endy, C. G. Pesce, and R. Brent. 2005. Regulated cell-to-cell variation in a cell-fate decision system. Nature 437:699-706.
Costerton, J. W., G. G. Geesey, and K. J. Cheng. 1978. How bacteria stick. Scientific American 238:86-95.
Costerton, J. W., K. J. Cheng, G. G. Geesey, T. I. Ladd, J. C. Nickel, M. Dasgupta, and T. J. Marrie. 1987. Bacterial biofilms in nature and disease. Annual Review of Microbiology 41:435-464.
Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: A common cause of persistent infections. Science 284:1318-1322.
Danino, T., O. Mondragon-Palomino, L. Tsimring, and J. Hasty. 2010. A synchronized quorum of genetic clocks. Nature 463:326-330.
Davies, D. G., M. R. Parsek, J. P. Pearson, B. H. Iglewski, J. W. Costerton, and E. P. Greenberg. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295-298.
Deans, T. L., C. R. Cantor, and J. J. Collins. 2007. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 130:363-372.
Donlan, R. M. 2002. Biofilms: Microbial life on surfaces. Emerging Infectious Diseases 8:881-890.
Donlan, R. M., and J. W. Costerton. 2002. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clinical Microbiology Reviews 15:167-193.
Drenkard, E., and F. M. Ausubel. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740-743.
Dueber, J. E., B. J. Yeh, K. Chak, and W. A. Lim. 2003. Reprogramming control of an allosteric signaling switch through modular recombination. Science 301:1904-1908.
Ellis, T., X. Wang, and J. J. Collins. 2009. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nature Biotechnology 27:465-471.
Elowitz, M. B., and S. Leibler. 2000. A synthetic oscillatory network of transcriptional regulators. Nature 403:335-338.
Endy, D. 2005. Foundations for engineering biology. Nature 438:449-453.
Friedland, A. E., T. K. Lu, X. Wang, D. Shi, G. Church, and J. J. Collins. 2009. Synthetic gene networks that count. Science 324:1199-1202.
Fung, E., W. W. Wong, J. K. Suen, T. Bulter, S. G. Lee, and J. C. Liao. 2005. A synthetic gene-metabolic oscillator. Nature 435:118-122.
Fuqua, W. C., S. C. Winans, and E. P. Greenberg. 1994. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. Journal of Bacteriology 176: 269-275.
Gander, S. 1996. Bacterial biofilms: Resistance to antimicrobial agents. Journal of Antimicrobial Chemotherapy 37:1047-1050.
Gardner, T. S., C. R. Cantor, and J. J. Collins. 2000. Construction of a genetic toggle switch in Escherichia coli. Nature 403:339-342.
Gilbert, P., D. G. Allison, and A. J. McBain. 2002. Biofilms in vitro and in vivo: Do singular mechanisms imply cross-resistance? Journal of Applied Microbiology 92(Suppl):98S-110S.
Guet, C. C., M. B. Elowitz, W. Hsing, and S. Leibler. 2002. Combinatorial synthesis of genetic networks. Science 296:1466-1470.
Guinto, C. H., E. J. Bottone, J. T. Raffalli, M. A. Montecalvo, and G. P. Wormser. 2002. Evaluation of dedicated stethoscopes as a potential source of nosocomial pathogens. American Journal of Infection Control 30:499-502.
Gulati, S., V. Rouilly, X. Niu, J. Chappell, R. I. Kitney, J. B. Edel, P. S. Freemont, and A. J. deMello. 2009. Opportunities for microfluidic technologies in synthetic biology. Journal of the Royal Society Interface 6(Suppl 4):S493-S506.
Hall-Stoodley, L., and P. Stoodley. 2005. Biofilm formation and dispersal and the transmission of human pathogens. Trends in Microbiology 13:7-10.
——. 2009. Evolving concepts in biofilm infections. Cellular Microbiology 11:1034-1043.
Hall-Stoodley, L., J. W. Costerton, and P. Stoodley. 2004. Bacterial biofilms: From the natural environment to infectious diseases. Nature Reviews Microbiology 2:95-108.
Ham, T. S., S. K. Lee, J. D. Keasling, and A. P. Arkin. 2006. A tightly regulated inducible expression system utilizing the fim inversion recombination switch. Biotechnology and Bioengineering 94:1-4.
——. 2008. Design and construction of a double inversion recombination switch for heritable sequential genetic memory. PLoS One 3:e2815.
Jefferson, K. K. 2004. What drives bacteria to produce a biofilm? FEMS Microbiology Letters 236:163-173.
Kirisits, M. J., L. Prost, M. Starkey, and M. R. Parsek. 2005. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Applied and Environmental Microbiology 71:4809-4821.
Kitney, R. I., P. S. Freemont, and V. Rouilly. 2007. Engineering a molecular predation oscillator. IET Synthetic Biology 1:68-70.
Knobloch, J. K., M. A. Horstkotte, H. Rohde, and D. Mack. 2002. Evaluation of different detection methods of biofilm formation in Staphylococcus aureus. Medical Microbiology and Immunology 191:101-106.
Kramer, B. P., and M. Fussenegger. 2005. Hysteresis in a synthetic mammalian gene network. Proceedings of the National Academy of Sciences of the United States of America 102:9517-9522.
Kramer, B. P., A. U. Viretta, M. Daoud-El-Baba, D. Aubel, W. Weber, and M. Fussenegger. 2004. An engineered epigenetic transgene switch in mammalian cells. Nature Biotechnology 22:867-870.
Legewie, S., H. Herzel, H. V. Westerhoff, and N. Bluthgen. 2008. Recurrent design patterns in the feedback regulation of the mammalian signalling network. Molecular Systems Biology 4:190.
Lesic, B., F. Lepine, E. Deziel, J. Zhang, Q. Zhang, K. Padfield, M. H. Castonguay, S. Milot, S. Stachel, A. A. Tzika, R. G. Tompkins, and L. G. Rahme. 2007. Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathogens 3:1229-1239.
Lindsay, D., and A. von Holy. 2006. Bacterial biofilms within the clinical setting: What healthcare professionals should know. Journal of Hospital Infection 64:313-325.
Looger, L. L., M. A. Dwyer, J. J. Smith, and H. W. Hellinga. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423:185-190.
Lynch, A. S., and D. Abbanat. 2010. New antibiotic agents and approaches to treat biofilm-associated infections. Expert Opinion on Therapeutic Patents 20:1373-1387.
Macleod, S. M., and D. J. Stickler. 2007. Species interactions in mixed-community crystalline biofilms on urinary catheters. Journal of Medical Microbiology 56:1549-1557.
Mahajan-Miklos, S., L. G. Rahme, and F. M. Ausubel. 2000. Elucidating the molecular mechanisms of bacterial virulence using non-mammalian hosts. Molecular Microbiology 37:981-988.
Marchisio, M. A., and F. Rudolf. 2011. Synthetic biosensing systems. International Journal of Biochemistry & Cell Biology 43:310-319.
Mathur, T., S. Singhal, S. Khan, D. J. Upadhyay, T. Fatma, and A. Rattan. 2006. Detection of biofilm formation among the clinical isolates of staphylococci: An evaluation of three different screening methods. Indian Journal of Medical Microbiology 24:25-29.
Monod, J., and F. Jacob. 1961. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harbor Symposia on Quantitative Biology 26:389-401.
Oliveira, A., and M. de L. Cunha. 2010. Comparison of methods for the detection of biofilm production in coagulase-negative staphylococci. BMC Research Notes 3:260.
O’Toole, G. A. 2002. Microbiology: A resistance switch. Nature 416:695-696.
Parsek, M. R., and P. K. Singh. 2003. Bacterial biofilms: An emerging link to disease pathogenesis. Annual Review of Microbiology 57:677-701.
Popat, R., S. A. Crusz, and S. P. Diggle. 2008. The social behaviours of bacterial pathogens. British Medical Bulletin 87:63-75.
Potera, C. 1999. Forging a link between biofilms and disease. Science 283:1837, 1839.
Rackham, O., and J. W. Chin. 2005. Cellular logic with orthogonal ribosomes. Journal of the American Chemical Society 127:17584-17585.
Rinaudo, K., L. Bleris, R. Maddamsetti, S. Subramanian, R. Weiss, and Y. Benenson. 2007. A universal RNAi-based logic evaluator that operates in mammalian cells. Nature Biotechnology 25:795-801.
Rumbaugh, K. P., J. A. Griswold, B. H. Iglewski, and A. N. Hamood. 1999. Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in burn wound infections. Infection and Immunity 67:5854-5862.
Sato, T., and Y. Kobayashi. 1998. The ars operon in the skin element of Bacillus subtilis confers resistance to arsenate and arsenite. Journal of Bacteriology 180:1655-1661.
Serrano, L. 2007. Synthetic biology: Promises and challenges, Molecular Systems Biology 3:158.
Sinha, J., S. J. Reyes, and J. P. Gallivan. 2010. Reprogramming bacteria to seek and destroy an herbicide. Nature Chemical Biology 6:464-470.
Smith, K., and I. S. Hunter. 2008. Efficacy of common hospital biocides with biofilms of multi-drug resistant clinical isolates. Journal of Medical Microbiology 57:966-973.
Stewart, P. S., and M. J. Franklin. 2008. Physiological heterogeneity in biofilms. Nature Reviews Microbiology 6:199-210.
Stickler, D. J. 2002. Susceptibility of antibiotic-resistant Gram-negative bacteria to biocides: A perspective from the study of catheter biofilms. Journal of Applied Microbiology 92(Suppl): 163S-170S.
——. 2008. Bacterial biofilms in patients with indwelling urinary catheters. Nature Clinical Practice Urology 5:598-608.
Stoodley, P., R. Cargo, C. J. Rupp, S. Wilson, and I. Klapper. 2002. Biofilm material properties as related to shear-induced deformation and detachment phenomena. Journal of Industrial Microbiology and Biotechnology 29:361-367.
Stricker, J., S. Cookson, M. R. Bennett, W. H. Mather, L. S. Tsimring, and J. Hasty. 2008. A fast, robust and tunable synthetic gene oscillator. Nature 456:516-519.
Tigges, M., T. T. Marquez-Lago, J. Stelling, and M. Fussenegger. 2009. A tunable synthetic mammalian oscillator. Nature 457:309-312.
Van Houdt, R., and C. W. Michiels. 2010. Biofilm formation and the food industry, a focus on the bacterial outer surface. Journal of Applied Microbiology 109:1117-1131.
Weber, W., J. Stelling, M. Rimann, B. Keller, M. Daoud-El Baba, C. C. Weber, D. Aubel, and M. Fussenegger. 2007. A synthetic time-delay circuit in mammalian cells and mice. Proceedings of the National Academy of Sciences of the United States of America 104:2643-2648.
Westall, F., M. J. de Wit, J. Dann, S. van der Gaast, C. E. J. de Ronde, and D. Gerneke. 2001. Early Archean fossil bacteria and biofilms in hydrothermally-influenced sediments from the Barberton greenstone belt, South Africa. Precambrian Research 106:93-116.
Win, M. N., and C. D. Smolke. 2008. Higher-order cellular information processing with synthetic RNA devices. Science 322:456-460.
Yu, R. C., C. G. Pesce, A. Colman-Lerner, L. Lok, D. Pincus, E. Serra, M. Holl, K. Benjamin, A. Gordon, and R. Brent. 2008. Negative feedback that improves information transmission in yeast signalling. Nature 456:755-761.
SYNTHETIC BIOLOGY AND THE ART OF BIOSENSOR DESIGN
Christopher E. French,30Kim de Mora,31Nimisha Joshi,32Alistair Elfick,33James Haseloff,34and James Ajioka35
Introduction
The term “biosensor” refers to a wide variety of devices. The common element is that a biological component provides highly specific recognition of a certain target analyte, and this detection event is somehow transduced to give an easily detectable, quantifiable response, preferably one that can be easily converted to an electrical signal so that the result can be fed to an electronic device
_______________
30 School of Biological Sciences, University of Edinburgh.
31 School of Biomedical Sciences, University of Edinburgh.
32 School of Geosciences, University of Edinburgh.
33 School of Engineering & Electronics, University of Edinburgh.
34 Department of Plant Sciences, University of Cambridge.
35 Department of Pathology, University of Cambridge.
for signal processing, data storage, etc. The biological component in many biosensors is either an enzyme, as in the glucose-oxidase–based biosensors used for blood glucose monitoring, or an antibody, as in most optical biosensors. Another class of biosensor, sometimes also referred to as a bioreporter, uses living cells as a component. These cells detect the target analyte via some more-or-less specific receptor and generate a detectable response, most commonly by induction of a reporter gene. Many such devices have been reported in the scientific literature, with detection of mercury and arsenic in the environment being particularly common applications. However, very few such devices are commercially available, the best-known examples being the mutagen-detecting devices such as the SOS-Chromotest (Environmental Bio-Detection Products, Inc.) system. Here we discuss the reasons for this gap between promise and delivery, and ways in which the emerging discipline of synthetic biology may lead to a new generation of whole-cell biosensors.
Whole-Cell Biosensors
Whole-cell biosensors, or bioreporters, are living cells that indicate the presence of a target analyte. The most commercially successful by far are nonspecific toxicity sensors based on naturally occurring luminescent bacteria such as Vibrio harveyi, Vibrio (Photobacterium) fischeri, and Photobacterium phosphoreum. Examples include MicroTox (Strategic Diagnostics, SDIX) and BioTox (Aboatox). In these mainly marine organisms, above a certain population density, light is produced continuously by the action of bacterial luciferase (LuxAB), which oxidizes a long-chain aldehyde such as tetradecanal in the presence of FMNH2 and oxygen. Regeneration of the reduced flavin (catalyzed by LuxG) and the aldehyde substrate (catalyzed by LuxCDE) requires NADPH and ATP, so any toxic substance that interferes with metabolism will reduce light emission, which is easily detected using a luminometer.
However, these systems are nonspecific and are only useful for preliminary screening of environmental samples to determine whether or not a toxic substance is present. The potentially more useful class of bioreporter consists of genetically modified microorganisms in which the presence of a specific target analyte is linked to a detectable response. The genetic modification involved in these cases consists of linking the receptor for the target analyte to induction of an easily detectable reporter gene. Commonly used reporter genes are shown in Table A5-1. For recent reviews of such systems, see Belkin (2003), Daunert et al. (2000), Tecon and van der Meer (2008), and van der Meer and Belkin (2010).
Synthetic Biology and Whole-Cell Biosensors
Like “biosensor,” the term “synthetic biology” is widely used by different authors to mean different things. In this context, we are using it to refer to a
TABLE A5-1 Reporter Genes Commonly Used in Whole-Cell Biosensors
| Reporter | Characteristics |
| lacZ (ß-galactosidase) | Chromogenic (X-gal, o-nitrophenyl galactoside) and chemiluminescent substrates are available. In E. coli host strains with the lacZΔM15 mutation, only a small peptide representing the missing N-terminus, designated lacZ′α, is required. |
| luxAB (bacterial luciferase) | Blue bioluminescence in the presence of added substrate (a long-chain aldelhyde, usually decanal). |
| luxCDABE (bacterial luciferase) | As above; presence of luxCDE allows biosynthesis of the substrate so that it need not be added to the reaction. |
| Firefly or click-beetle luciferase | Bioluminescence in the presence of added substrate (D-luciferin). Quantum yield is higher than for bacterial luciferase, but the substrate is much more expensive. Luminescence is normally green, but color variants are now available. |
| Fluorescent proteins | Fluorescence when stimulated by ultraviolet or visible light. The original green fluorescent protein (GFP), still widely used, is stimulated best by ultraviolet; enhanced green fluorescent protein responds well to blue light, and numerous color variants are now available. |
systematic approach to rationalizing genetic modification to make it more like other engineering disciplines, in terms of the use of standardized parts that can be assembled in a modular way to make a variety of different constructs. Since whole-cell biosensors are intrinsically modular, consisting of a recognition element coupled to an arbitrarily chosen reporter, synthetic biology seems well suited to the development of such devices.
This approach to synthetic biology is associated particularly with MIT, BioBricks, the Registry of Standard Biological Parts,36 and iGEM (the International Genetically Engineered Machine competition).37 BioBricks (Knight, 2003) are a type of standardized biological “part,” consisting of pieces of DNA which conform to a certain standard (defined by a document known as RFC10, available
_______________
36 See http://partsregistry.org/Main_Page.
37 For more information, see the following: iGEM 2006, University of Edinburgh, http://parts.mit.edu/wiki/index.php/University_of_Edinburgh_2006; iGEM 2007, University of Cambridge, http://parts.mit.edu/iGEM07/index.php/Cambridge; iGEM 2007, University of Glasgow, http://parts.mit.edu/igem07/index.php/Glasgow; iGEM 2007, University of Science and Technology, China, http://parts.mit.edu/igem07/index.php/USTC; iGEM 2008, Harvard University, http://2008.igem.org/Team:Harvard; iGEM 2009, University of Cambridge, http://2009.igem.org/Team:Cambridge; iGEM 2010, Bristol Centre for Complexity Studies, http://2010.igem.org/Team:BCCS-Bristol; iGEM 2010, Imperial College, London, http://2010.igem.org/Team:Imperial_College_London; and iGEM 2010, Peking University, http://2010.igem.org/Team:Peking.
from the BioBricks Foundation38) specifying certain characteristics of the ends. Each BioBrick may be a protein-coding region, or some other component such as a promoter, ribosome binding site, transcription termination sequence, or any other piece of DNA that may be useful in making genetic constructs. The essential point of this format is that any BioBrick can, through a standardized procedure, be combined with any other BioBrick to form a new BioBrick, which can then be combined with any other BioBrick, and so on. In this way, quite large and complex constructs can be built up fairly quickly. The Registry of Standard Biological Parts, currently hosted at MIT, was established to store both the DNA of these parts and also associated information such as DNA sequence, performance characteristics, and user experience. The intention was, and is, that this library of BioBricks and associated information should become a valuable resource for synthetic biologists.
To demonstrate the potential of this approach to synthetic biology, the iGEM competition was established in 2005. Each year, interdisciplinary teams of undergraduates consisting of a mixture of biologists, engineers, and computer scientists compete over the summer vacation period to conceive, design, mathematically model, construct, and test novel genetically modified systems made using BioBricks from the Registry, as well as new BioBricks created specifically for the project. All new BioBricks made are deposited in the Registry and are available for use by future teams. iGEM projects, completed on a short timescale by undergraduate students, are generally not nearly as well characterized as systems reported in the peer-reviewed literature; however, they are often based on highly creative ideas and generally include a mathematical modeling component far in excess that usually found in biological publications. They can therefore be a very interesting way to follow possible future application areas in synthetic biology.
Since biosensors are conceptually simple devices with a clear real-world application, and many opportunities for elaboration in terms of novel input and output modalities, in vivo signal processing, and other aspects, they are a popular choice of project, and a number of interesting innovations have been reported as a result of iGEM projects. We refer to a number of these later in this report. All information relating to previous iGEM projects is available via the relevant websites.
Arsenic Biosensors
Arsenic is a particularly attractive target for whole-cell biosensors, in that it is a major groundwater contaminant in Bangladesh, West Bengal, and a number of other regions (Meharg, 2005; Smith et al., 2000). This only came to light in the 1980s, and it is a major and increasing public health issue. The problem initially arose when, to combat waterborne diarrheal diseases caused by consumption of contaminated surface water, nongovernmental organizations drilled some millions of tube wells to supply clean drinking water. Unexpectedly, it was
_______________
discovered some years later that many of these produced water with unacceptably high levels of arsenic. The current recommended World Health Organization (WHO) limit for drinking water is 10 ppb arsenic, while Bangladesh and some other countries maintain an earlier limit of 50 ppb, but many wells exceed this by a large margin. Chronic consumption of water with high arsenic concentrations leads to arsenicosis, resulting in skin lesions and various cancers. Thus, there is a clear and present need for cheap and simple tests for monitoring arsenic levels in drinking water. Current field test kits are based around the Gutzeit method, which involves reduction of arsenate to toxic arsine gas, and the detection of this as a color spot following reaction with mercuric salts. Such tests reportedly are unreliable at low but still significant arsenic concentrations, and disposal of mercuric salts poses its own environmental issues. Thus there is a clear potential niche for a simple arsenic biosensor device (Diesel et al., 2009; M. Owens, Engineers Without Borders, personal communication).
Most early arsenic biosensors were based on the arsenic detoxification operons of Staphylococcus plasmid pI258 and Escherichia coli plasmid R773. The former consists simply of the ars promoter controlling genes arsR, arsB, and arsC, encoding the repressor, arsenite efflux pump, and arsenate reductase, respectively, whereas the latter has a relatively complex structure, arsRDABC, with two separate repressors, ArsR and ArsD, which control the operon with different affinities (Oremland and Stolz, 2003). ArsD is also reported to act as a metallochaperone, carrying arsenite to the efflux pump formed by ArsAB (Lin et al., 2006). Later systems were based on the simpler E. coli chromosomal arsenic detoxification operon (Cai and DuBow, 1996; Diorio et al., 1995), which consists of the ars promoter followed by arsR, arsB, and arsC, and the similar operon of Bacillus subtilis (Sato and Kobayashi, 1998), which is discussed further below. In either case, the preparation of the biosensor organism is straightforward—the reporter gene is simply inserted adjacent to the controlled promoter so that induction of the promoter results in expression of the reporter gene, giving an easily detectable signal (usually a color change, luminescence, or fluorescence). A number of systems are reportedly at or near commercialization; for example, the Aboatox BioTox Heavy Metal Assay kits, developed at the University of Turku, use E. coli as host, with firefly luciferase as the reporter gene. Stocker et al. (2003) described production of a set of E. coli–based arsenic bioreporters using ß-galactosidase with a chromogenic substrate, bacterial luciferase, or green fluorescent protein (GFP) as reporter; one of these, based on the luciferase reporter gene, has been field tested in Vietnam (Trang et al., 2005) and was reported to give good results in comparison to chemical field tests. Whole-cell arsenic biosensors have been reviewed recently by Diesel et al. (2009).
The Edinburgh Arsenic Biosensor
One example which we consider in some detail is the first biosensor project to be submitted to iGEM: the arsenic biosensor submitted for iGEM 2006 by the
team from the University of Edinburgh, under the supervision of C. French and A. Elfick. (K. de Mora was a student member of this iGEM team.) The intention was to develop a device that would be suitable for field use in developing countries. It should therefore be cheap, simple to use, and should deliver an output that could be assessed by eye, without requiring expensive electrical equipment, but should also give a quantifiable response using cheap equipment where this was required. For this reason the standard reporters based on luminescence and fluorescence were not considered appropriate. Instead, it was decided that the output should be in the form of a pH change. This would give a bright and easily assessed visible response using a pH indicator chemical, and it could also give a quantitative electrical response using a cheap glass pH electrode or similar solid-state device (ISFET).
For practical reasons, it was decided to use a standard laboratory host strain of Escherichia coli. Such organisms are easy to manipulate, carry multiple disabling mutations that make them harmless to humans and unable to propagate in the environment, and grow well at temperatures up to 45°C. E. coli and related organisms naturally ferment a variety of sugars, including lactose, via the mixed acid pathway, resulting in the production of acetic, lactic, and succinic acids, which can rapidly lower the pH of the medium below 4.5. Many laboratory strains carry a deletion in the gene lacZ, encoding ß-galactosidase, the initial enzyme of lactose degradation, so that a nonfunctional truncated LacZ protein, missing the first few amino acids, is produced. This can be complemented by a short peptide known as the alpha peptide, consisting of the first 50 to 70 amino acids of LacZ. This is encoded by a short open reading frame known as lacZ′α. Thus, to generate an acid response, it was only necessary to use such a lactose-defective host strain, such as E. coli JM109, and place the lacZ′α gene under the control of the ars promoter in a standard Registry multicopy plasmid, pSB1A2. Thus, in the presence of arsenate or arsenite, expression of the LacZ alpha peptide would be induced, complementing the truncated LacZ and allowing rapid fermentation of lactose to acids, lowering the pH. To generate an alkaline response, the urease genes, ureABC, of Bacillus subtilis (Kim et al., 2005) were chosen. (Uropathogenic strains of E. coli also possess urease genes, but these are longer and more complex than those of B. subtilis.) Expression of these genes allows conversion of urea to ammonia and carbon dioxide and can raise the pH of the medium above 10. Both acid- and alkali-producing systems were tested and were found to work well (Aleksic et al., 2007).
The original design of the arsenic biosensor submitted for iGEM 2006 was a complex system, with a multistage output. This is discussed further below. However, the practical demonstration provided consisted only of the acid-generating system, which was found to give robust and reliable responses to arsenic concentrations as low as 2.5 ppb, with the time of pH change being related to the arsenic concentration in a simple and reproducible way (Aleksic et al., 2007; Joshi et al., 2009). This construct is available from the Registry of Standard Biological Parts (BBa_J33203), as are its components, the ars promoter and associated arsR gene
(BBa_J33201) and the lacZ′α reporter gene (BBa_J33202). Interestingly, during early testing to determine whether buffer ions expected to be present in groundwater might interfere with the pH response, we found that bicarbonate ions actually increase the sensitivity of the response, leading to induction at much lower arsenate levels (Joshi et al., 2009). The reason for this is not clear, but it may be due to altered speciation or uptake of arsenate (de Mora et al., 2011).
The original concept for this biosensor system involved use of a universal pH indicator solution which gives a strong color response—blue in alkaline conditions, green in neutral conditions, and red in acidic conditions—coupled with quantitation via a glass pH electrode. However, it became apparent that the red component of the universal pH indicator, as well as pure methyl red, were rapidly bleached in the presence of living cells under the conditions used. This was therefore replaced with bromothymol blue, which is blue under alkaline conditions and yellow under acid conditions, with pKa around 7.3 (Figure A5-1).
For quantitative monitoring of multiple samples simultaneously, as might be useful in a local or regional testing laboratory, an inexpensive system was developed based on the use of freeze-dried cells together with a webcam; following aseptic addition of groundwater samples to freeze-dried cells and sterile medium, the webcam would monitor the color of multiple tubes simultaneously, and software would extract the pixels representing the tubes and monitor the color of each over time. From these data, the time of color change could be extracted, and this was found to correlate well with arsenic levels in model groundwaters
FIGURE A5-1 Demonstration of the Edinburgh pH-based arsenic biosensor, Escherichia coli JM109/pSB1A2-BBa_J33203 with bromothymol blue as pH indicator, following static overnight incubation. From left to right: arsenic-free control; 5, 10, 25, 50, and 100 ppb arsenic as sodium arsenate; and cell-free control with 100 ppb arsenate. Note the increasing size of the cell pellet in tubes with increasing arsenic concentrations. Color change occurs more rapidly in samples with increasing arsenic concentration (not shown).
SOURCE: C. French, unpublished.
and also in real arsenic-contaminated groundwater samples from Hungary (de Mora et al., 2011).
This system seems well suited for use in water quality laboratories at a local or regional level. However, our original aim was to develop a system that could be used by relatively unskilled users in the field, so that local people could easily monitor the quality of their own well water. This poses several further issues. One technical issue is that any contamination with lactose-degrading bacteria will lead to false-positive results. To avoid this, water samples must be sterilized prior to introduction into the test device. To overcome this problem, we envisage a disposable plastic device containing freeze-dried cells and medium components, with an integral sterile filter through which the water sample is introduced. A second potential complicating factor is the temperature dependence of the rapidity of color change. This requires further investigation. Storage lifetime under relevant conditions also requires further research. However, we are confident that these issues can be overcome, and that this system can form the basis of a simple, cheap, sensitive, and reliable field test for arsenic concentration in groundwater.
One nontechnical issue which must first be addressed is the regulatory and safety issues associated with use of live genetically modified bacteria outside of a contained laboratory context. As noted above, the host strains used are disabled and are unable to colonize humans or to propagate in the environment in competition with wild-type bacteria (Chart et al., 2001). Nevertheless, depending on the jurisdiction, there are many regulatory hurdles associated with the use of genetically modified mciroorganisms in poorly contained applications. This is discussed further below. In the meantime, we are focusing our attention on development of a device suitable for use in local or regional laboratories. A company, Lumin Sensors, has been formed to explore these possibilities (www.luminsensors.com).
In the remainder of this paper, we consider some of the ways synthetic biology can improve the performance of whole-cell biosensors and lead to the development of a new generation of devices with improved performance.
Better Biosensors Through Synthetic Biology
Alternative Host (Chassis) Organisms
Synthetic biology generally involves the introduction of a new genetic system into a host organism, which in the context of synthetic biology is often called a “chassis.” The great majority of whole-cell biosensors reported in the literature have used E. coli as a host organism, due to familiarity, ease of manipulation, and availability of a wide variety of vector systems and other tools. However, E. coli has a number of characteristics which may mean that it is not necessarily the ideal chassis for any given purpose. For example, as a Gram-negative bacterium, it produces and sheds lipopolysaccharide (endotoxin), a powerful activator of the innate immune system; hence, it is generally unsuitable for any in vivo uses. Its
outer membrane prevents large analytes such as peptides from approaching the cell membrane. Unlike many other bacteria, it generally does not secrete most proteins effectively into the medium, unless specific modifications are introduced to allow it. Most significantly in terms of biosensor applications, E. coli does not naturally produce dormant states such as spores, meaning that freeze drying is likely to be required for storage and distribution. While freeze drying of bacterial cells is a well-understood process, this adds an extra level of complexity and expense to the manufacturing process. Some other potential hosts have much better characteristics in this regard. Most attention has been paid to Bacillus subtilis, a low-GC Gram-positive soil bacterium. B. subtilis is used as a model Gram-positive bacterium; its physiology is therefore well studied, and a number of vector systems and other tools are available, though in both characteristics it lags well behind E. coli. In contrast to E. coli, B. subtilis is naturally competent during a certain stage of its life cycle, and, unlike E. coli, will happily take up large pieces of linear DNA and integrate them onto its chromosome by homologous recombination. Also, and more importantly, B. subtilis naturally forms a dormant resting state known as endospores (Errington, 2003). When conditions become unfavorable for growth, each cell undergoes an asymmetrical cell division resulting in a large “mother cell” and a small “forespore.” The mother cell engulfs the forespore and produces layers of protein coats to surround it, while the forespore produces small acid-soluble proteins and calcium dipicolinate, which act to protect its nucleic acids. When this process is complete, the mother cell lyses to release the mature endospore. The spores can simply be harvested and dried for storage and distribution. Endospores are extremely tolerant to heat, drying, and other stresses. There are well-attested reports of endospores surviving for hundreds and even thousands of years in dry conditions (Nicholson et al., 2000), as well as more controversial reports of survival for many millions of years in unusual contexts such as in the guts of insects preserved in amber (Cano and Borucki, 1995).
B. subtilis possesses an arsenic detoxification operon similar to that found on the E. coli chromosome (Sato and Kobayashi, 1998); thus, arsenic biosensors can be prepared in a similar way to that described above, either by adding the ars promoter and arsR regulatory gene to a reporter gene on a multicopy plasmid, or by introduction of such a reporter gene to the chromosome downstream of the ars promoter. As a demonstration of principle, we have constructed such a system, designated a “Bacillosensor,” using the plasmid vector pTG262 and the reporter gene xylE of Pseudomonas putida, which encodes catechol-2,3-dioxygenase. This enzyme acts on the cheap substrate catechol to produce a bright yellow compound, 2-hydroxy-cis,cis-muconic semialdehyde. This system was found to be sensitive to arsenic levels well below the WHO recommended limit of 10 ppb (Figure A5-2). Spores could be boiled for 2 minutes prior to use in the assay; this would not only kill competing organisms but also activate the spores for rapid germination. The ability to remove most contaminating organisms from
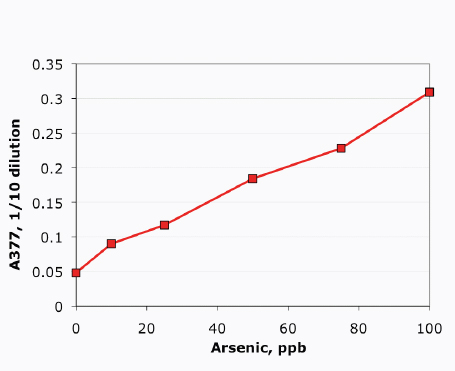
FIGURE A5-2 Detection of arsenic by B. subtilis 168/pTG262-arsR-xylE: absorbance at 377 nm vs. arsenic concentration (ppb arsenic as sodium arsenate). The vector and BioBrick components used to make this device (BBa_J33206, BBa_J33204) are available from the Registry of Standard Biological Parts. Conditions of the assay were as described by Joshi et al. (2009).
SOURCE: L. Montgomery and C. French, unpublished.
the sample simply by heat treatment offers a considerable potential advantage for field use of such devices, eliminating the need for filter sterilization or similar treatments.
Several reports in the literature have also described bioreporters based on B. subtilis and related organisms. Tauriainen et al. (1997, 1998) reported the use of B. subtilis as a host for firefly luciferase-based bioreporters for arsenic, antimony, cadmium, and lead but did not specifically describe the use of endospores in the assays. More recently, Date et al. (2007) reported the construction of bioreporters for arsenic and zinc based on endospores of B. subtilis and Bacillus megaterium, with ß-galactosidase plus a chemiluminescent substrate, or enhanced green fluorescent protein, as reporter genes. The genetically modified spores could be stored at room temperature for at least 6 months. The same authors later described incorporation of such endospore-based bioreporters into microfluidic devices (Date et al., 2010, discussed further below). Fantino et al. (2009) reported the construc-
tion of a device designated “Sposensor,” incorporating B. subtilis endospores engineered to produce ß-galactosidase in response to the target analyte. Systems responsive to zinc and bacitracin were demonstrated using a chromogenic substrate with spores dried on filter paper discs.
Another potentially interesting host is the yeast Saccharomyces cerevisiae. Again, this is a model organism, well studied, and for which numerous vector systems and other genetic modification tools are available. It can be stored and distributed in a “dry active” state (Baronian, 2003). However, as a eukaryote, S. cerevisiae has more complex regulatory systems than bacteria such as E. coli and B. subtilis, and to date there have been relatively few reports of its use as a host for bioreporter applications. One example is a nonspecific toxicity reporter described by Välimaa et al. (2008), using S. cerevisiae modified to produce firefly luciferase; as in the MicroTox system described above, toxic substances reduced the level of luminescence observed. S. cerevisiae has also been used as a platform for analyte-specific biosensors. For example, Leskinen et al. (2003) reported construction of a yeast-based bioreporter for copper ions, with firefly luciferase under control of the copper-responsive CUP1 promoter. This was used in environmental analysis for bioavailable copper (Peltola et al., 2005). Some further examples are described by Baronian (2003). With further development, analyte-specific yeast-based biosensors could be a useful addition to the biosensor toolkit.
Detection of Extracellular Analytes
In the examples discussed so far, the signal has been generated internally, either as a stress response (as in the case of the SOS-Chromotest) or else by binding of an intracellular protein, such as ArsR, to an analyte that has been internalized (arsenate is probably taken up in error by the phosphate uptake machinery). For medical applications, it would be advantageous to be able to detect and respond to analytes such as peptides, which do not naturally enter bacterial or fungal cells. However, bacteria are able to sense and respond to extracellular analytes via “two-component” systems. In these cases, one component is a sensor kinase that spans the cell membrane, and the second is a response regulator protein that binds DNA to activate or repress transcription from a given promoter. The extracellular analyte binds to the extracellular domain of the sensor kinase, and this increases or decreases the kinase activity of the intracellular domain. This alters the tendency of the kinase domain to phosphorylate the response regulator protein, which in turn alters its propensity to bind to and activate or repress the promoter(s) in question. Most bacteria possess multiple two-component sensor systems responding to a variety of different stimuli, including osmotic strength of the extracellular medium, extracellular phosphate levels, and the presence of various small molecules such as sugars and amino acids. One interesting subgroup consists of two-component sensor systems in Gram-positive bacteria such as Bacillus, Streptococcus, and Enterococcus, which sense and respond to the pres-
ence of short-peptide pheromones produced by other cells of the same species, a phenomenon analogous to the more familiar N-acyl homoserine lactone–based quorum-sensing systems of Gram-negative bacteria. One might imagine that such systems could be modified, perhaps by rational engineering or directed evolution, to respond instead to some peptide of analytical interest.
There are two interesting points regarding two-component sensor systems. One is that these systems show a surprising degree of modularity. A number of reports have described cases where the extracellular domain of one sensor kinase has been fused to the intracellular domain of another, giving a hybrid protein that responds to the normal stimulus of the first sensor kinase by activating the normal response regulator of the second. One well-known example is the report of Levskaya et al. (2005) describing fusion of the extracellular domain of the light-sensing domain of a cyanobacterial phytochrome, Cph1, to the intracellular domain of an E. coli osmotic stress sensor, EnvZ; the hybrid protein, Cph8, responded to red light by activating the response regulator, OmpR, which normally responds to EnvZ. When a promoter controlled by OmpR was fused to a pigment-producing gene, the resulting genetically modified cells responded to red light by producing a pigment, allowing “bacterial photographs” to be made by focusing images onto a plate of the bacteria (Levskaya et al., 2005). Another example is fusion of the extracellular domain of a chemotaxis receptor, Trg, which responds to the presence of ribose, among other chemoattractants, by controlling the cell’s motility apparatus, to the intracellular domain of EnvZ (Baumgartner et al., 1994). (In this case, the interaction is indirect: ribose binds to periplasmic ribose binding protein, which then interacts with the extracellular domain of Trg.) In the presence of the hybrid protein, designated Trz, an OmpR-controlled promoter (specifically, the ompC promoter) was found to respond to the presence of ribose. Recent structural studies have begun to offer some insight into the basis of communication between extracellular and intracellular domains in two-component sensor kinases (Casino et al., 2010), which may allow more rational engineering of such hybrid proteins.
The second point is that it is possible to engineer the recognition elements of such sensors to respond to nonnatural target molecules. For example, Looger et al. (2003) reported rational reengineering of the ribose binding protein, which binds ribose and interacts with the extracellular domain of the hybrid Trz sensor kinase mentioned above, so that it would respond to lactate or to 2,4,6-trinitro-toluene (TNT). In the presence of these molecules, OmpR-controlled promoters were reported to be activated. This opens the possibility that it might be possible to use such a platform as a “universal bioreporter” by generating a library of reengineered sensor kinases or associated binding proteins to respond to any analyte of interest. If such rational reengineering proved challenging in the general case, another interesting possibility would be to attempt fusion of the extracellular domain of such a sensor to some binding molecule such as a nanobody (variable region of a camelid heavy-chain-only antibody) or scFv fragment of an antibody,
so that any analyte to which an antibody could be raised could be detected by a bioreporter. Other protein-based scaffolds for specific recognition are also under development (Hosse et al., 2006; Nuttall and Walsh, 2008) and might also be suitable for such applications. Alternatively, extracellular domains able to bind a desired target analyte might be selected from a library of mutants by a technique such as phage display or cell surface display (Benhar, 2001); this might also enable simple screening for clones which were able not only to bind to the analyte of interest but also to generate the appropriate response on binding.
An interesting, but rather application-specific, approach to the detection of extracellular analytes via two-component sensing systems was proposed by the 2010 iGEM team of Imperial College, London. In this case, the objective was to detect cercaria (larvae) of the parasite Schistosoma in water. It was proposed that a protease produced by the parasite could be detected by cleavage of an engineered substrate to release the autoinducer peptide of Streptococcus pneumoniae; this peptide would then be detected by the two-component sensor system ComDE of S. pneumoniae expressed in B. subtilis. A similar system might be used to detect other proteases or hydrolytic enzymes of biological interest.
Another possible platform for detection of extracellular peptides would be the yeast mating peptide sensing system. In Saccharomyces cerevisiae, mating peptides (a and α) are detected by G-protein coupled receptors, Ste2 and Ste3, which initiate an intracellular signalling pathway (Bardwell, 2004; Naider and Becker, 2004). As noted above, S. cerevisiae is arguably an underexploited platform for biosensor development, with much potential for investigation. One interesting example of a yeast-based sensor system was reported by Chen and Weiss (2005). In this case, a cytokinin (plant hormone) was detected by yeast cells expressing Arabidopsis thaliana cytokinin receptor Atcre1, a two-component-type sensor kinase, which apparently is fortuitously able to interact with the endogenous yeast response regulator Ypd1 in the absence of Ypd1’s normal sensor kinase partner, Sln1 (Inoue et al., 2001). The engineered “receiver” cells expressed GFP from a Ypd1-activated promoter in the presence of cytokinin.
Animal cells have numerous and varied extracellular receptors but, with current techniques, probably lack the necessary robustness to be generally useful as a biosensor platform except possibly for specialized clinical uses.
Modulation of Sensitivity and Dynamic Range
The critical parameters in the performance of a sensor are the sensitivity (the lowest analyte concentration to which a detectable response is seen) and the dynamic range (in this context, the range of analyte concentrations over which the analyte concentration can be estimated based on the response; that is, between the sensitivity limit and the concentration at which the response saturates). In simple whole-cell biosensors such as those described above, a hyperbolic or sigmoidal response is seen, and it may be that the response curve initially obtained is not in
the desired range for the planned application. Obviously, one critical parameter is the affinity of the receptor for the analyte; however, in practice, many other factors are important, making analysis rather complicated (for a more detailed discussion of such issues, see van der Meer et al., 2004). For example, in the case of arsenic biosensors, arsenate is first taken up by the cell (probably mistakenly, via the phosphate uptake system). The characteristics of uptake determine the ratio between extracellular and intracellular arsenate concentrations. It then interacts with the ArsR repressor with a certain affinity, and interferes with the binding of the repressor to the ars promoter; both of these interactions have a characteristic affinity. Within a given cell, the numbers of arsenate anions, ArsR repressor molecules, and promoter sites are also all limited, so the relative numbers of these will also strongly affect the interaction. Finally, when the repressor is not bound to the promoter, and the promoter is thus “active,” RNA polymerase will bind the promoter with a certain affinity, and begin transcription with a certain efficiency. The actual level of arsenate repressor and reporter protein molecules generated will be strongly affected by the rate of messenger RNA synthesis, the rate of mRNA degradation, the affinity of ribosomes for the ribosome binding sites on the mRNA, and the degradation rate of the proteins. Thus, many steps intervene between the extracellular arsenate concentration and the level of the reporter protein. This means that, in practice, it is possible to modulate the sensitivity and dynamic range of the sensor without actually altering the receptor at all, simply by making minor changes to factors such as the strength of the ribosome binding sites.
This type of experiment is greatly facilitated by the modular, composable nature of BioBricks and the availability of a library of ribosome binding sites of different strengths in the Registry of Standard Biological Parts. To give one trivial example, we assembled an alternative version of the simple arsenic biosensor construct BBa_J33203, the differences being that the reporter gene was moved to a position between the promoter and the arsR gene encoding the repressor, and the native ribosome binding site of arsR was replaced by a strong synthetic ribosome binding site, BBa_J15001. The response characteristics of this modified construct were quite different from those of the original construct (Figure A5-3). While the mechanism of this was not investigated, one plausible explanation would be an increased level of expression of the ArsR repressor due to the stronger ribosome binding site.
More profound reorganizations of the arsenic recognition system have also been investigated. For example, a second copy of the ArsR-binding site was introduced between arsR and the reporter gene, with the aim of decreasing background expression in the absence of arsenic. This led to considerably improved induction characteristics (Stocker et al., 2003). It was further reported that modification of the activity or synthesis rate of the reporter enzyme (cytochrome c peroxidase or ß-galactosidase) led to strong changes in the system response to given arsenic concentrations (Wackwitz et al., 2008), allowing the generation of an array of
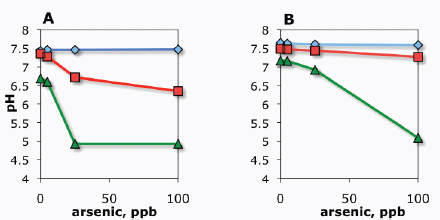
FIGURE A5-3 Altered response characteristics of a whole-cell arsenic biosensor through reassembly of the components. (A) Original Edinburgh arsenic biosensor, consisting of the E. coli chromosomal ars promoter and arsR gene (BBa_J33201) followed by lacZ′α (BBa_J33202). Both arsR and lacZ′α have their native ribosome binding site. (B) Reassembled operon consisting of ars promoter (BBa_J15301), strong synthetic ribosome binding site (BBa_J15001), lacZ′α coding sequence (BBa_J15103), ribosome binding site (BBa_J15001), and arsR coding sequence (BBa_J15101). Diamonds, time zero; squares, 6 hours; triangles, 24 hours. The vector was pSB1A2 and host was E. coli JM109 in all cases. Assay conditions were as described by Joshi et al. (2009). All components and assembled constructs are available from the Registry of Standard Biological Parts.
SOURCE: X. Wang and C. French, unpublished.
reporter strains with different response characteristics (discussed further below). In a similar investigation, using mercury biosensors, the 2010 Peking University iGEM team investigated the effects of placing the regulatory gene merR under the control of a variety of promoters of different strengths and found that this resulted in a wide variety of different sigmoidal response curves. A similar effect was achieved by screening a library of mutants with altered MerR-binding sites in the mercury-responsive promoter. Thus, a variety of simple modifications to the system can be used to achieve alterations in the sensitivity and dynamic range of such sensors. The composable nature of BioBricks, together with other assembly strategies used in synthetic biology, make it easy to generate and screen a large number of such systems to find a set with the desired characteristics.
It is also possible to use rational design principles to modify the dynamic range of a sensor, for example, by amplifying a weak transcriptional signal. One way to do this is through the use of genetic “amplifiers.” One set of such devices, submitted and tested by iGEM teams from the University of Cambridge in 2007 and 2009, consists of bacteriophage activator-promoter pairs. Rather than the
analyte-responsive promoter deriving the reporter gene directly, the promoter drives expression of an activator protein, which activates a second promoter, which controls the reporter gene. This gives a genetic “amplification” effect. A small library of cross-reactive activators and promoters allows a mix-and-match approach to select a pair that gives the desired response characteristics. In these projects, the promoters came from bacteriophages P2 (promoters PF, PO, PP, and PV) and P4 (promoters Psid and PLL), and the activators from bacteriophages P2 (Ogr), P4 (δ protein), PSP3 (Pag), and φR73 (δ protein) (Julien and Calendar, 1996). Fifteen promoter-activator combinations were characterized, allowing the biosensor designer to choose a pair with the desired response characteristics.
The availability of a number of similar biosensors with different response characteristics, as described above, allows the preparation of “bar graph”-like arrays of sensors to obtain a quantitative output over a wider range of analyte concentrations than any single sensor could achieve. This was proposed by Wackwitz et al. (2008), who used the term “traffic light biosensors” to describe such devices. In principle, such devices should not require calibration (van der Meer and Belkin, 2010).
Another issue with induction-based whole-cell biosensors is the time required for induction; few systems in the literature show detectable responses in less than 30 minutes or so, and several hours is more typical. For many applications, it would be advantageous to obtain a faster response. One ingenious approach to this problem was presented by the Imperial College iGEM team of 2010. In this case, the reporter enzyme is presynthesized in the cell in an inactive form. The analyte-responsive promoter drives expression of a protease, which cleaves and activates the reporter. Since each molecule of protease can rapidly activate multiple molecules of the reporter protein, this can potentially give a much faster response. In the case of the Imperial College iGEM project, the reporter, catechol-2,3-dioxygenase (XylE), was synthesised as an inactive fusion with GFP, joined by a linker which could be cleaved by site-specific TEV protease. Detection of the target analyte, in this case a peptide released by a protease of the parasite Schistosoma, led to induction of TEV protease expression and consequent cleavage of the linker, allowing rapid formation of active catechol-2,3-dioxygenase tetramers.
In Vivo Signal Processing and Multiplex Output
In addition to tuning of response characteristics, it is possible to introduce more complex forms of in vivo signal processing. One simple example is a genetic inverter, in which a promoter that would normally activate transcription is instead used to repress it. This is accomplished by having the analyte-responsive promoter drive production of a repressor, which represses expression from a second promoter driving the reporter gene. Three well-characterized repressor-promoter pairs are widely used in such systems: the LacI/lac promoter pair, bac-
teriophage λ cI/PL or PR pair, and the TetR-tet promoter pair (see, for example, Elowitz and Leibler, 2000). These repressor-promoter pairs are analogous to insulated wires used in an electronic circuit to communicate between different parts of the device. For complex devices, it would clearly be useful to have more than three such “wires” available. One interesting approach to this was reported by the USTC iGEM team of 2007 (following work described by Sartorius et al., 1989). In this case, mutations were made to the bases of the lac operator site involved in binding of the repressor LacI, and also to the amino acids of LacI involved in this binding. The libraries of mutant lac promoters and LacI repressors were then analyzed to determine which pairs interacted efficiently. From this experiment, multiple repressor-promoter pairs were chosen that did not crosstalk. Furthermore, these were used to generate biological equivalents of several logic gates. The simplest of these is the NOT gate, in which activation of one promoter leads to repression of another. This is simply achieved by having the first promoter drive expression of a repressor which represses the second, as described above. More complex gates include NAND and NOR. In the former case, binding of two different repressors is required to inactivate the second promoter; in the latter case, binding of either of two repressors is sufficient to achieve this effect. From combinations of such gates, more complex circuits can be assembled. Such logic systems can also be extended to systems consisting of several different types of engineered cell, with the cells communicating via quorum-sensing signals (Tamsir et al., 2010).
The term “traffic-light sensors,” discussed above, is also applied to a class of devices that have been proposed and modeled, but, so far as we know, never demonstrated in practice, in which discrete, different outputs are activated at different analyte levels by in vivo signal processing within a single bioreporter organism. The originally proposed iGEM 2006 Edinburgh arsenic biosensor fell into this category, giving an alkaline response at very low arsenate concentrations, a neutral pH at moderate arsenate concentrations, and an acidic response at dangerously high arsenate levels (Aleksic et al., 2007). This was to be achieved through the use of two separate repressors, with different affinities for arsenate, controlling two different reporters: urease for an alkaline response, and ß-galactosidase for the acidic response. Response of the high-affinity repressor-promoter pair was inverted via a repressor, so that the presence of a low concentration of arsenate led to production of a repressor that switched off production of urease, whereas higher levels of arsenate switched on production of ß-galactosidase. A similar arrangement of two different repressor systems can be used to generate a genetic “band detector,” which responds only to analyte concentrations within a certain concentration range.
Visual Outputs
While the majority of reported arsenic sensors use luminescence or fluorescence as output, it might be advantageous in some cases to have an output that can be easily detected by eye. The use of enzymes such as ß-galactosidase and catechol-2,3-dioxygenase, together with chromogenic substrates, offers one route to achieving this end. An alternative is the pH-based approach used in the Edinburgh arsenic biosensor described above; this is useful in that pH changes, together with standard pH indicators, give very strong and easily detected color changes, but they can also be quantified using an inexpensive pH electrode. In some cases it might be preferable to have cells produce an endogenous pigment in response to the target analyte. Several such examples have been reported. Fujimoto et al. (2006) described a system in which the photosynthetic bacterium Rhodovulum sulfidophilum was engineered to place the endogenous carotenoid pigment gene crtA under the control of the E. coli ars promoter, so that the presence of arsenite led to a change in cell pigmentation from yellow to red. Subsequently, Yoshida et al. (2008) reported a similar system based on Rhodopseudomonas palustris. The combinatorial nature of synthetic biology components, together with the possibility of multistage outputs discussed above, opens the possibility of systems in which a range of discrete colors is produced for different levels or combinations of analytes. To facilitate the construction of such devices, the University of Cambridge iGEM team, 2009, presented a set of modular BioBrick components that could generate a variety of carotenoid and indole-based pigments using different combinations of components from the carotenoid and violacein biosynthetic pathways (Figure A5-4). As with all of the iGEM entries discussed in this paper, these genetic modules are freely available from the Registry of Standard Biological Parts.

FIGURE A5-4 Escherichia coli cells producing a variety of pigments. From left to right, the first four tubes are derived from the carotenoid biosynthesis pathway, the last three from the violacein biosynthesis pathway. All of these pigment-producing pathways are available in BioBrick format from the Registry of Standard Biological Parts.
SOURCE: University of Cambridge, iGEM 2009.
Integration of Biological and Electronic Components
For data processing and storage, it is advantageous if biosensor outputs can easily be converted into electrical signals that can be read by a computer. A number of integrated biological-electrical devices have been reported. One example is the Bioluminescent Bioreporter Integrated Circuit (Nivens et al., 2004; Vijayaraghavan et al., 2007), in which luminescence emitted by bacterial luciferase is detected by a Complementary Metal-Oxide Semiconductor (CMOS) microluminometer incorporated with signal processing circuitry and other relevant components on a “biochip.” Several recent reports have described the integration of bioreporter cells into microfluidic devices. For example, Diesel et al. (2009) reported incorporation of an E. coli GFP-based arsenic bioreporter into a microfluidic device. Fluorescence from approximately 200 cells within the detection cavity was imaged using a camera. Date et al. (2010) reported the integration of Bacillus endospore-based biosensors for arsenic and zinc (described above) into a centrifugal microfluidic platform, in which pumping is provided by rotation of a disc-shaped substrate. Luminescence and fluorescence measurements were performed using separate laboratory instruments with fiber-optic probes held above the detection chambers of the microfluidic device. Storage and germination of endospores, and sensitive detection of the analytes, were reported.
Whereas these devices use “conventional outputs (fluorescence and luminescence), other reporter systems can give an electrical output directly, which may be more convenient for incorporation into simple devices for field use. One example is the Edinburgh arsenic biosensor described above. The pH response can easily be converted to a voltage signal using a standard glass pH electrode, or an equivalent solid-state device (ion selective field effect transistor). An alternative is the generation of an amperometric signal via a device similar to a microbial fuel cell. Bacteria respire by transferring electrons from a donor (such as a sugar) to an acceptor (such as oxygen). This electron transfer occurs at the cell membrane. Many bacteria can instead transfer electrons to or from an electrode, generating an electrical current; others can do so in the presence of electron shuttle molecules known as mediators (Lovley, 2006). An amperometric signal can be generated by placing synthesis of such a mediator, or of an essential component of the electron transfer apparatus, under the control of an analyte-responsive promoter. Both of these approaches have been described by iGEM teams. The iGEM 2007 entry from the University of Glasgow described a system for biosynthesis of pyocyanin, a redox-active metabolite of Pseudomonas aeruginosa, which could act as a mediator to transport electrons between the bacterial respiratory chain and an electrode. The iGEM 2008 entry from Harvard University took advantage of a naturally “electricigenic” bacterium, Shewanella oneidensis, by controlling synthesis of one of the outer membrane proteins, MtrB, required for efficient transfer of electrons to an electrode. Either of these approaches can allow controllable generation of an electrical current induced by the presence of a target analyte.
Regulatory Issues Related to Field Use of Whole-Cell Biosensors
The Edinburgh arsenic biosensor described above was designed to be used in the field in the form of a contained disposable device. Some other bioreporters have been designed for more direct use in the field. One of the earliest examples to be tested was that of Ripp et al. (2000), reportedly the first genetically modified microorganisms to be approved for field testing for a bioremediation application in the United States, in which a strain of Pseudomonas fluorescens modified to produce a luminescent signal in the presence of aromatic hydrocarbons was introduced into the soil in a contaminated site, and its persistence monitored over a 2-year period. While this organism was designed to persist in the environment for a prolonged period, there are other cases where the bioreporter only needs to survive for a few hours—just long enough to report on the conditions in which it finds itself. One well-known example is the Microbial Mine Detection System developed at Oak Ridge National Laboratories. This consisted of a strain of Pseudomonas modified to produce GFP in the presence of explosives leaking from land mines (discussed by Habib, 2007). The positions of the land mines could then be mapped using an ultraviolet lamp. This system was reportedly field tested with good results. Another example was reported by the iGEM team of the Bristol Centre for Complexity Studies (BCCS) in 2010. In this case, the bioreporter consisted of E. coli cells modified to indicate the presence of nitrate by producing a fluorescent response. These cells, encapsulated in a hydrophilic gel, were to be spread over the soil and left for several hours to express the fluorescent protein, after which the nitrate levels in the field could be mapped by a mobile device, providing information useful in agriculture.
On consideration of the examples described above, we can distinguish between four different cases:
- bioreporters designed to be used within the confines of a laboratory (e.g., the SOS-Chromotest);
- bioreporters designed to be used in a contained device, but outside of a laboratory (e.g., the Edinburgh arsenic biosensor);
- bioreporters designed to be exposed directly to the environment, but not to survive longer than required to report the levels of the target analyte (e.g., the MMDS and Bristol nitrate sensor); and
- bioreporters designed to survive and persist in the field for long periods (e.g., the bioremediation monitoring system of Ripp et al., 2000).
Unfortunately, current regulatory regimes in the United Kingdom and European Union do not appear to distinguish between cases 2, 3, and 4. Case 1 is unproblematic; such “contained use” applications are dealt with in the United Kingdom by the Health and Safety Executive under the Genetically Modified Organisms (Contained Use) Regulations 2000, whereas “uncontained uses” are dealt with by the Department for the Environment, Food and Rural Affairs un-
der the Genetically Modified Organisms (Deliberate Release) Regulations 2002 (which implement EU Directive 2001/18/EC). Whereas this is wholly appropriate for case 4, where such organisms are designed to persist in the environment, no provision seems to be made for cases 2 or 3, where organisms may be specifically designed not to survive in the environment, thereby minimizing any threat to ecosystems. This is a serious inhibiting factor in the use of genetically modified whole-cell biosensors or bioreporters outside of the laboratory, as expensive field trials with extensive postrelease monitoring are required. We feel that it would be useful if future legislation took this into account. In the United States, the situation is less clear to us; the relevant federal legislation appears to be the Toxic Substances Control Act (Sayre and Seidler, 2005). In the absence of specific legal and regulatory expertise, we will refrain from further comment. However, it is clear that until these regulatory issues are addressed, it will not be possible to use genetically modified bioreporter organisms on a large scale in the field, despite their obvious potential.
Conclusions
Sensitive and highly specific response to various molecules is one of the core functions of biological systems. As such, whole-cell biosensors offer a versatile and widely applicable method for detecting the presence of a wide range of analytes. The techniques of synthetic biology offer numerous possible improvements in terms of response tuning, in vivo signal processing, and direct interface with electronic devices for further signal processing and output. However, regulatory issues will need to be clarified before such devices can fulfill their true potential as highly sensitive, inexpensive sensors for field use.
Acknowledgments
The original Edinburgh arsenic biosensor concept was developed and tested by the 2006 University of Edinburgh iGEM team, consisting of Jelena Aleksic, Farid Bizzari, Yizhi Cai, Briony Davidson, Kim de Mora, Sergii Ivakhno, Judith Nicholson, Sreemati Lalgudi Seshasayee, and Jennifer Wilson. Valuable further contributions were made by Lucy Montgomery and Xiaonan Wang, formerly of the University of Edinburgh. Contaminated groundwater samples were provided by Dr. Balint L. Balint of the Department of Biochemistry and Molecular Biology, Medical and Health Science Centre, University of Debrecen.
Disclosures
The Edinburgh 2006 iGEM team was generously supported by the Gatsby Foundation, the Royal Commission for the Exhibition of 1851, and Synbiocomm, an initiative of the European Union. Funding for further development of the
Edinburgh arsenic biosensor was provided by the Biotechnology and Biological Sciences Research Council via the U.K. Synthetic Biology Standards Network, of which A. Elfick and J. Haseloff are codirectors.
References
Aleksic, J., F. Bizzari, Y. Z. Cai, B. Davidson, K. de Mora, S. Ivakhno, S. L. Seshasayee, J. Nicholson, J. Wilson, A. Elfick, C. E. French, L. Kozma-Bognar, H. W. Ma, and A. Millar. 2007. Development of a novel biosensor for detection of arsenic in drinking water. IET Synthetic Biology 1:87-90.
Bardwell, L. 2004. A walk-through of the yeast mating pheromone response pathway. Peptides 25(9):1465-1476.
Baronian, K. H. R. 2003. The use of yeast and moulds as sensing elements in biosensors. Biosensors and Bioelectronics 19:953-962.
Baumgartner, J. W., C. Kim, R. E. Brisette, M. Inoue, C. Park, and G. L. Hazelbauer. 1994. Trans-membrane signaling by a hybrid protein: Communication from the domain of chemoreceptor Trg that recognizes sugar-binding proteins to the kinase/phosphatase domain of osmosensor EnvZ. Journal of Bacteriology 176(4):1157-1163.
Belkin, S. 2003. Microbial whole-cell sensing systems of environmental pollutants. Current Opinion in Microbiology 6:206-212.
Benhar, I. 2001. Biotechnological applications of phage and cell display. Biotechnology Advances 19(1):1-33.
Cai, J., and M. S. DuBow. 1996. Expression of the Escherichia coli chromosomal ars operon. Canadian Journal of Microbiology 42:662-671.
Cano, R. J., and M. K. Borucki. 1995. Revival and identification of bacterial spores in 25 million year old to 40 million year old Dominican amber. Science 268(5213):1060-1064.
Casino, P., V. Rubio, and A. Marina. 2010. The mechanism of signal transduction by two-component systems. Current Opinion in Structural Biology 20:763-771.
Chart, H., H. R. Smith, R. M. La Ragione, and M. J. Woodward. 2001. An investigation into the pathogenic properties of Escherichia coli strains BLR, BL21, DH5α and EQ1. Journal of Applied Microbiology 89:1048-1058.
Chen, M. T., and R. Weiss. 2005. Artificial cell-cell communication in yeast Saccharomyces cerevisiae using signaling elements from Arabidopsis thaliana. Nature Biotechnology 23(12):1551-1555.
Date, A., P. Pasini, and S. Daunert. 2007. Construction of spores for portable bacterial whole-cell biosensing systems. Analytical Chemistry 79:9391-9397.
——. 2010. Integration of spore-based genetically engineered whole-cell sensing systems into portable centrifugal microfluidic platforms. Analytical and Bioanalytical Chemistry 398:349-356.
Daunert, S., G. Barret, J. S. Feliciano, R. S. Shetty, S. Shrestha, and W. Smith-Spencer. 2000. Genetically engineered whole-cell sensing systems: Coupling biological recognition with reporter genes. Chemical Reviews 100:2705-2738.
de Mora, K., N. Joshi, B. L. Balint, F. B. Ward, A. Elfick, and C. E. French. 2011. A pH-based biosensor for detection of arsenic in drinking water. Analytical and Bioanalytical Chemistry 400(4):1031-1039.
Diesel, E., M. Schreiber, and J. R. van der Meer. 2009. Development of bacteria-based bioassays for arsenic detection in natural waters. Analytical and Bioanalytical Chemistry 394:687-693.
Diorio, C., J. Cai, J. Marmor, R. Shinder, and M. S. DuBow. 1995. An Escherichia coli chromosomal ars operon homolog is functional in arsenic detoxification and is conserved in Gram negative bacteria. Journal of Bacteriology 177(8):2050-2056.
Elowitz, M. B., and S. Leibler. 2000. A synthetic oscillatory network of transcriptional regulators. Nature 403(6767):335-338.
Errington, J. 2003. Regulation of endospore formation in Bacillus subtilis. Nature Reviews Microbiology 1(2):117-126.
Fantino, J. R., F. Barras, and F. Denizot. 2009. Sposensor: A whole-cell bacterial biosensor that uses immobilized Bacillus subtilis spores and a one-step incubation/detection process. Journal of Molecular Microbiology and Biotechnology 17:90-95.
Fujimoto, H., M. Wakabayashi, H. Yamashiro, I. Maeda, K. Isoda, M. Kondoh, M. Kawase, H. Miyasaka, and K. Yagi. 2006. Whole cell arsenite biosensor using photosynthetic bacterium Rhodovulum sulfidophilum. Applied Microbiology and Biotechnology 73(2):332-338.
Habib, M. K. 2007. Controlled biological and biomimetic systems for landmine detection. Biosensors and Bioelectronics 23:1-18.
Hosse, R. J., A. Rothe, and B. E. Power. 2006. A new generation of protein display scaffolds for molecular recognition. Protein Science 15(1):14-27.
Inoue, T., M. Higuchi, Y. Hashimoto, M. Seki, M. Kobayashi, T. Kato, S. Tabata, K. Shinozaki, and T. Kakimoto. 2001. Identification of CRE1 as a cytokinin receptor from Arabidopsis. Nature 409(6823):1060-1063.
Joshi, N., X. Wang, L. Montgomery, A. Elfick, and C. E. French. 2009. Novel approaches to biosensors for detection of arsenic in drinking water. Desalination 248(1-3):517-523.
Julien, B., and R. Calendar. 1996. Bacteriophage PSP3 and φR73 activator proteins: Analysis of promoter specificities. Journal of Bacteriology 178(19):5668-5675.
Kim, J. K., S. B. Mulrooney, and R. P. Hausinger. 2005. Biosynthesis of active Bacillus subtilis urease in the absence of known urease accessory proteins. Journal of Bacteriology 187(20):7150-7154.
Knight, T. 2003. Idempotent vector design for standard assembly of Biobricks. http://web.mit.edu/synbio/release/docs/biobricks.pdf (accessed October 20, 2011).
Leskinen, P., M. Virta, and M. Karp. 2003. One-step measurement of firefly luciferase activity in yeast. Yeast 20(13):1109-1113.
Levskaya, A., A. A. Chevalier, J. J. Tabor, Z. Booth Simpson, L. A. Lavery, M. Levy, E. A. Davidson, A. Scouras, A. D. Ellington, E. M. Marcotte, and C. A. Voigt. 2005. Engineering Escherichia coli to see light. Nature 438:441-442.
Lin, Y.-F., A. R. Walmsley, and B. P. Rosen. 2006. An arsenic metallochaperone for an arsenic detoxification pump. Proceedings of the National Academy of Sciences of the United States of America 103(42):15617-15622.
Looger, L. L., M. A. Dwyer, J. J. Smith, and H. W. Hellinga. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423:185-190.
Lovley, D. 2006. Bug juice: Harvesting electricity with microorganisms. Nature Reviews Microbiology 4(7):497-508.
Meharg, A. 2005. Venomous Earth: How Arsenic Caused the World’s Worst Mass Poisoning. Basing-stoke, UK: Macmillan.
Naider, F., and J. M. Becker. 2004. The α-factor mating pheromone of Saccharomyces cerevisiae: A model for studying the interaction of peptide hormones and G protein-coupled receptors. Peptides 25(9):1441-1463.
Nicholson, W. L., N. Munakata, G. Horneck, H. J. Melosh, and P. Setlow. 2000. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiology and Molecular Biology Reviews 64(3):548-572.
Nivens, D. E., T. E. McKnight, S. A. Moser, S. J. Osbourn, M. L. Simpson, and G. S. Sayler. 2004. Bioluminescent bioreporter integrated circuits: Potentially small, rugged and inexpensive whole-cell biosensors for remote environmental monitoring. Journal of Applied Microbiology 96:33-46.
Nuttall, S. D., and R. B. Walsh. 2008. Display scaffolds: Protein engineering for novel therapeutics. Current Opinion in Pharmacology 8(5):609-615.
Oremland, R. S., and J. F. Stolz. 2003. The ecology of arsenic. Science 300(5621):939-944.
Peltola, P., A. Ivask, M. Astrom, and M. Virta. 2005. Lead and Cu in contaminated urban soils: Extraction with chemical reagents and bioluminescent bacteria and yeast. Science of the Total Environment 350(1-3):194-203.
Ripp, S., D. E. Nivens, Y. Ahn, C. Werner, J. Jarrell, J. P. Easter, C. D. Cox, R. S. Burlage, and G. S. Sayler. 2000. Controlled field release of a bioluminescent genetically engineered microorganism for bioremediation process monitoring and control. Environmental Science and Technology 34(5):846-853.
Sartorius, J., N. Lehming, B. Kisters, B. von Wilcken-Bergmann, and B. Müller-Hill. 1989. lac repressor mutants with double or triple exchanges in the recognition helix bind specifically to lac operator variants with multiple exchanges. EMBO Journal 8(4):1265-1270.
Sato, T., and Y. Kobayashi. 1998. The ars operon in the skin element of Bacillus subtilis confers resistance to arsenate and arsenite. Journal of Bacteriology 180(7):1655-1661.
Sayre, P., and R. J. Seidler. 2005. Application of GMOs in the US: EPA research and regulatory considerations related to soil systems. Plant and Soil 275(1-2):77-91.
Smith, A. H., E. O. Lingas, and M. Rahman. 2000. Contamination of drinking-water by arsenic in Bangladesh: A public health emergency. Bulletin of the World Health Organization 78(9): 1093-1103.
Stocker, J., D. Balluch, M. Gsell, H. Harms, J. Feliciano, S. Daunert, K. A. Malik, and J. R. van der Meer. 2003. Development of a set of simple bacterial biosensors for quantitative and rapid measurements of arsenite and arsenate in potable water. Environmental Science and Technology 37:4743-4750.
Tamsir, A., J. J. Tabor, and C. A. Voigt. 2010. Robust multicellular computing using genetically encoded NOR gates and chemical “wires.” Nature 469(7329):212-215.
Tauriainen, S., M. Kapr, W. Chang, and M. Virta. 1997. Recombinant luminescent bacteria for measuring bioavailable arsenite and antimonite. Applied and Environmental Microbiology 63(11):4456-4461.
——. 1998. Luminescent bacterial sensor for cadmium and lead. Biosensors and Bioelectronics 13:931-938.
Tecon, R., and J. R. van der Meer. 2008. Bacterial biosensors for measuring availability of environmental pollutants. Sensors 8(7):4062-4080.
Trang, P. T. K., M. Berg, P. H. Viet, N. V. Mui, and J. R. van der Meer. 2005. Bacterial bioassay for rapid and accurate analysis of arsenic in highly variable groundwater samples. Environmental Science and Technology 39:7625-7630.
Välimaa, A.-L., A. Kivistö, M. Virta, and M. Karp. 2008. Real-time monitoring of non-specific toxicity using a Saccharomyces cerevisiae reporter system. Sensors 8:6433-6447.
van der Meer, J. R., and S. Belkin. 2010. Where microbiology meets microengineering: Design and applications of reporter bacteria. Nature Reviews Microbiology 8:511-522.
van der Meer, J. R., D. Tropel, and M. Jaspers. 2004. Illuminating the detection chain of bacterial bioreporters. Environmental Microbiology 6(10):1005-1020.
Vijayaraghavan, R., S. K. Islam, M. Zhang, S. Ripp, S. Caylor, N. D. Bull, S. Moser, S. C. Terry, B. J. Blalock, and G. S. Sayler. 2007. Bioreporter bioluminescent integrated circuit for very low-level chemical sensing in both gas and liquid environments. Sensors and Actuators B 123:922-928.
Wackwitz, A., H. Harms, A. Chatzinotas, U. Breuer, C. Vogne, and J. R. van der Meer. 2008. Internal arsenite bioassay calibration using multiple reporter cell lines. Microbial Biotechnology 1:149-157.
Yoshida, K., K. Inoue, Y. Takahashi, S. Ueda, K. Isoda, K. Yagi, and I. Maeda. 2008. Novel carotenoid-based biosensor for simple visual detection of arsenite: Characterization and preliminary evaluation for environmental application. Applied and Environmental Microbiology 74:6730-6738.
SYSTEMS ANALYSIS OF ADAPTIVE IMMUNITY BY UTILIZATION OF HIGH-THROUGHPUT TECHNOLOGIES39
Sai T. Reddy40,41and George Georgiou40,41,42,43
A new generation of high-throughput technologies for quantitative and clonal analysis of adaptive immune responses have been developed. Functional analysis of lymphocyte populations has been accomplished via microfluidic assay systems. Additionally, lymphocyte receptor repertoires have been characterized on proteomic and genomic levels with multiplexed protein microarrays and high-throughput DNA sequencing. These tools are providing an unprecedented level of information depth on the distribution of adaptive immune cell (B and T cell) functionalities and repertoires, which develop upon activation following vaccination, pathogenic infection, or in disease states. These various high-throughput technologies have unlocked the potential to transform immunology into an information-rich science that will enable rapid expansion of the field of experimental systems immunology.
Introduction
Adaptive immunity plays an indispensible role in maintaining vertebrate host protection against a constant barrage of pathogens. Adaptive immunity is fundamentally reliant upon the generation of an incredible diversity of lymphocyte
_______________
39 Reprinted from Current Opinion in Biotechnology, 22/4, Reddy, S. T., and G. Georgiou, Systems analysis of adaptive immunity by utilization of high-throughput technologies, 584-589, 2011, with permission from Elsevier.
40 Department of Chemical Engineering, University of Texas at Austin, Austin, TX 78712, USA.
41 Department of Biomedical Engineering, University of Texas at Austin, Austin, TX 78712, USA.
42 Institute for Cellular and Molecular Biology, University of Texas at Austin, Austin, TX 78712, USA.
43 Section of Molecular Genetics and Microbiology, University of Texas at Austin, Austin, TX 78712, USA.
Corresponding author: Georgiou, George (gg@che.utexas.edu)
Current Opinion in Biotechnology 2011, 22:1–6
This review comes from a themed issue on systems biology
Edited by Roy Kishony and Vassily Hatzimanikatis
0958-1669/$ – see front matter
# 2011 Elsevier Ltd. All rights reserved.
DOI 10.1016/j.copbio.2011.04.015
receptors (B cell receptors, or BCRs; and T cell receptors, or TCRs); furthermore, the development of lymphocytes subsets, which display a plethora of functions, act synergistically to counteract pathogenic invasion. Receptor diversity in lymphocyte populations is generated by the genetic recombination of noncontiguous germline elements: variable (V), diversity (D), and joining (J) gene segments. Primary and secondary diversification via germline and junctional recombination, combinatorial pairing, and somatic hypermutation (for B cells only) combine to generate theoretical diversities of human antibodies and TCRs of ~1013 and ~1018, respectively (Janeway, 2005), far outnumbering the physiological number of lymphocytes in an average human (~1010). In addition to molecular diversity, there is an assortment of lymphocyte cellular subclasses (e.g., CD4+ and CD8+ T cells, Tregs, Th17 cells) and differentiation states (e.g., naïve, memory, and plasma B cells) that synergize to produce a multiprong attack on pathogens, leading to infection resolution and long-term immunological memory. Finally, the anatomical compartments (e.g., lymph nodes, blood, bone marrow) in which lymphocyte activation, homing, and trafficking take place add to the complex interconnected nature of adaptive immunity.
Conventional approaches for measuring adaptive immune responses have so far relied on cursory biological measurements, such as antibody titers to an antigen or lymphocyte assays that measure a single (ELISPOT) or multiple cytokines (intracellular staining, ICS) expressed by individual cells (Janetzki et al., 2004; Papagno et al., 2007). However, these techniques are surrogate measures and provide only a partial picture of the state of the immune system; for example, antibody titers do not provide specific molecular information on the monoclonal antibody components that comprise polyclonal responses in serum or secretory fluids. Likewise, ELISPOT or ICS assays can interrogate only a subset of lymphocytes based on functional properties; furthermore, they are sample-destructive thus precluding the simultaneous cloning and analysis of the respective BCR and TCR repertoires. Very importantly, existing assays typically capture the mean of a particular response or cellular phenotype and afford limited quantification. The understanding of immunological processes at greater resolution is not only important from a scientific point of view but can have major ramifications for human health. Detailed and quantitative deconvolution of the humoral immune response can assist in human immunotherapy by the identification of neutralizing or cytotoxic antibodies or of antigens overexpressed in a disease state (Gnjatic et al., 2010; Law et al., 2008; Yu et al., 2008). High-resolution information on adaptive immune responses to immunization may also greatly aid in assessing the efficacy of experimental vaccines (Germain et al., 2010; Querec et al., 2009), a process that currently relies on longitudinal studies of neutralizing antibody titers as the primary readout. In this review, we highlight recent advances in the high-throughput analysis of functional properties, proteomic interactions, and genomic repertoires of lymphocyte populations following an adaptive immune response (Table A6-1).
| High-throughput technology | Applications | Data Scale | Refs. |
| Single B cell-cloning | Antibody discovery; repertoire analysis | ~102 | Tiller et al., 2008; Smith et al., 2009; Wrammert et al., 2008; Meijer et al., 2006; Poulsen et al., 2007; Wiberg et al., 2006 |
| Microengraved devices | Single cell functional analysis, antibody and TCR specificity; antibody discovery; T cell cytokine reactivity; transcriptional profiling | ~105 | Tokimitsu et al., 2007; Jin et al., 2009; Love et al., 2006; Ogunniyi et al., 2009; Story et al., 2008; Song et al., 2010; Han et al., 2010; Flatz et al., 2011; Kwong et al., 2009 |
| Protein microarrays | Antigen discovery from pathogens, cancer and autoimmune disease; serological antibody profiling; vaccine development | ~103–104 | Davies et al., 2005; Felgner et al., 2009; Kunnath-Velayudhan et al., 2010; Robinson et al., 2002; Gnjatic et al., 2009; Reddy et al., 2011; Legutki et al., 2010 |
| Next generation DNA sequencing | Antibody and TCR repertoire analysis, immunological biomarker discovery, clinical monitoring, antibody library quality control, monoclonal antibody discovery | ~106–107 | Weinstein et al., 2009; Jiang et al., 2011; Boyd et al., 2009; Freeman et al., 2009; Robins et al., 2009, 2010; Warren et al., forthcoming; Glanville et al., 2009; Ge et al., 2010; Ravn et al., 2010; Reddy et al., 2010 |
Single Cell Analysis of Lymphocyte Populations
Single cell PCR-cloning of immunoglobulin genes
Until recently, the determination of the surface-anchored immunoglobulins/antibodies (BCRs) expressed by animal or human cells has relied on B cell immortalization techniques such as hybridoma technology or viral transduction (Becker et al., 2010; Kwakkenbos et al., 2009; Lanzavecchia and Sallusto, 2009); however, these techniques are compatible with only some stages of B cell maturation and furthermore, due to their low efficiencies (merely 1–3% of B cells interrogated) they are unable to generate a comprehensive picture of humoral repertoires. For example, terminally differentiated antibody-secreting plasma cells are not amenable to immortalization. Consequently, the antibodies isolated by immortalization of memory B cells may neither correspond to nor exhibit the same therapeutic potency as the population of circulating antibodies.
These problems can be addressed by single-cell cloning. Briefly, a desired B cell population is plated at limiting dilution in microtiter well plates and
then the variable heavy (VH) and light (VL) chains of surface immunoglobulins (BCRs) or of secreted antibodies from antibody secreting cells are amplified by PCR using primer sets. The VH and VL genes are then inserted into suitable vectors for expression from heterologous hosts, resulting in recombinant antibody fragments (e.g., scFv, FAB) or full-length IgGs. Subsequent antigen-specificity and binding affinity of antibodies are determined by standard ELISA techniques (Tiller et al., 2008). In one notable example, flow cytometry was used to isolate human plasmablasts from the peripheral blood of patients immunized with the influenza vaccine. The cells were sorted into 96 well plates at limiting dilution to yield approximately one cell per well. Nested RT-PCR and cDNA amplification of VL and VH genes was performed to amplify V genes in wells occupied by cells, followed by cloning into eukaryotic expression vectors (Smith et al., 2009). This approach was used to study the dynamics and antigen specificity of transient plasmablasts that arise three to seven days after vaccination and decay within a month. It was observed that at the peak of the response, nearly 80% of the plasmablasts expressed influenza-specific antibodies and the respective antibody repertoires were pauciclonal, as they were dominated by only a few sequences (Wrammert et al., 2008). Alternative techniques have been developed that rely on single B-cell cloning by linking VL and VH genes during RT-PCR followed by subsequent expression and screening in bacterial systems. For example, peripheral blood plasma cells were isolated from patients receiving tetanustoxiod (TT) immunizations; single-cell PCR and screening was performed which was then followed by extensive bioinformatic analysis of VL and VH pairing (Meijer et al., 2006). In another study, Andersen and co-workers isolated tetanus toxoid-specific antibodies from immunized patients and showed by biochemical characterization that they display affinity and kinetic binding constants close to the proposed limits for protective immunity (Poulsen et al., 2007). These single-cell cloning methods have been used by the company Symphogen (Denmark) to develop recombinant polyclonal antibodies, potentially valuable as human therapeutics. Multiple neutralizing antibodies were expressed under carefully controlled bioprocess conditions to produce a ‘recombinant polyclonal’ mixture that mimics the diversity, specificity, and binding affinity of natural polyclonal human immune responses (Wiberg et al., 2006).
Single cell microarrays for functional profiling of lymphocytes
Recently, large-scale analysis of lymphocyte populations has been enabled by methods in soft lithography and microengraving. These techniques have generated lymphocyte functional profiles. For example, microfluidic devices were deployed for the large-scale detection of antigen-specific B lymphocytes at the single-cell level and the subsequent cloning of their respective V genes. Kishi and co-workers used deep reactive ion etching on silicon to produce chips containing arrays of 10 mm diameter and 20 mm deep wells to trap single B lympho-
cytes at a density >200,000 cells per chip. The well surfaces were coated with anti-immunoglobulin antibodies, which allowed for capture of antibodies from antibody-secreting cells isolated from mouse or human B cells (Tajiri et al., 2007; Tokimitsu et al., 2007). Next, antigen was added to discover cells producing specific antibodies, which was followed by single-cell RT-PCR and cloning into a eukaryotic system for the generation of recombinant monoclonal antibodies (Jin et al., 2009). Alternatively, Love et al. used photolithography and intaglio printing to generate microarrays from poly(dimethylosiloxane). The dimensions of the arrays were compatible for imaging on a microscope slide, therefore allowing fluorescence-based imaging of B lymphocytes expressing antigen-specific antibodies (Love et al., 2006; Ogunniyi et al., 2009). Microengraved devices were used to assay the molecular profile of individual B cells isolated from mice following immunization; multiparametric analysis was performed to determine antibody specificities, isotypes, and apparent affinities (Story et al., 2008). The high throughput afforded by the microwell chip arrays described above notwithstanding, these methods rely on single-cell PCR and thus suffer from practical cloning limitations, therefore making it challenging to obtain a complete picture of the repertoire.
In addition to B cells, the functional profiles of T cells can also be measured by single cell microarray technologies (Song et al., 2010). Multidimensional activation profiles were generated from human peripheral blood T cells; cytokine secretion rates from individual cells were measured with very high sensitivity (0.5–4 molecules/s) (Han et al., 2010). In another recent study, single cell gene expression profiling was performed on CD8 T cell populations following various prime-boost HIV DNA vaccine schemes in mice. Multiparameter transcriptional analysis from single T cells was used to measure mRNA expression of 91 different genes related to phenotype, regulation, survival, and function, allowing discrimination of central memory and effector memory T cell subsets; furthermore providing quantitative parameters associated with vaccine efficacy and protective immunity (Flatz et al., 2011). The Heath group has developed a system for detecting antigen-specific T cells using nucleic acid cell sorting. Peptide-major histocompatibility complex tetramers (p/MHC) were site-specifically attached to DNA oligomers, which were annealed to complimentary strands printed on a glass slide. These p/MHC microarrays allowed for multiplexed sorting of antigen-specific T cells from heterogenous cell suspensions; this system can be exploited for clinical lymphocyte detection, such as detection of T cells from cancer patients (Kwong et al., 2009). These examples illustrate that single-cell methods are rapidly expanding the ability to generate high-throughput data for quantitative molecular analysis and functional profiling of adaptive immune responses.
Microarrays for Studying Protein–Protein Interactions in the Adaptive Immune Response
Systems-level analysis of gene expression using DNA microarrays has had a tremendous impact on cellular and molecular biology; the potential now exists
for proteomic microarrays to have a similar influence on immunological research (Michaud et al., 2003; Prechl et al., 2010). A substantial amount of work has been done to apply multiplexed high-throughput protein microarrays for the serological profiling and analysis of antibody responses; this research has ranged from infectious disease to cancer to autoimmune disease (Kingsmore, 2006).
Technologies for high-throughput printing of pathogen proteomes (with each protein expressed individually) onto protein microarrays have been developed. A typical approach consists of high-throughput PCR amplification of each predicted open-reading frame, which is followed by in vivo homologous recombination into a T7 expression vector. An Escherichia coli-based cell-free in vitro expression system is used to generate crude lysates possessing recombinant proteins, which are then printed onto nitrocellulose membranes resulting in a pathogen proteome microarray. In one example, the 185 viral proteins that comprise the vaccinia virus proteome were expressed individually and used to prepare protein arrays, which then were used to analyse the specificity of serum antibodies from vaccinia virus-vaccinated subjects; this allowed for high-throughput evaluation of humoral immunity (Davies et al., 2005). In an even more impressive study, protein microarrays comprising the 1205 proteins encoded by Burkholderia pseudomallei were constructed and probed with sera from infected and healthy patients. Such antibody profiling studies can reveal immunodominant antigens, which can be exploited as biomarkers for diagnostic purposes or to guide vaccine development (Felgner et al., 2009). Utilizing this same protein microarray methodology, it was found that in tuberculosis patients, circulating antibodies react with 10% of the Myobacterium tuberculosis proteome. Further analysis identified that during active infection the humoral response was highly biased towards only 0.5% of the proteome and particularly towards extracellular proteins, suggesting that the humoral immune response to M. tuberculosis correlates with the evolution of the infection (Kunnath-Velayudhan et al., 2010).
In addition to pathogenic proteins, autologous cellular antigens expressed by malignant cells or in autoimmune disorders can also elicit robust antibody responses. These autoantibodies serve as potential immunological biomarkers valuable for diagnosis and possibly for treatment. In an early study, a protein array was constructed by the immobilization of 196 different human proteins onto a glass slide and used for the detection of autoantibodies (Robinson et al., 2002). More recently, Old and co-workers constructed an array of 329 cancer antigens that was used to detect antibodies produced in cancer patients but not in healthy volunteers (Gnjatic et al., 2009). In a follow-up study, they expanded their analysis and used an array consisting of >8000 human proteins to probe the antibody specificities in sera from over 150 individuals, including healthy volunteers and ovarian and pancreatic cancer patients; this led to the identification of autoantigen signatures for these two malignancies (Gnjatic et al., 2010). In a recent study, a combinatorial library of unnatural synthetic molecules was used to screen for ligands reactive with antibodies abundant in serum (Reddy et al., 2011). The underlying concept of this study was that synthetic molecules act as a ‘shape library’, such that they are outside of the chemical space range occupied
by natural biomolecules. Microarrays of peptoids (N-substituted oligoglycines) were used to identify reactive IgGs in a mouse model of multiple sclerosis and in human patients with Alzheimer’s disease, thus demonstrating the potential to identify immunological serum antibody biomarkers without previous knowledge of antigen. In a similar approach, a microarray consisting of 10,000 random peptides was used to determine the serological antibody binding profile in mice and humans immunized with influenza. Although peptides were random sequences, this analysis still allowed for discrimination of pre-immune and post-immune samples, suggesting a novel platform for the discovery of immunological signatures present in antibody responses (Legutki et al., 2010).
High-Throughput DNA Sequencing of Adaptive Immune Repertoires
The emergence of ‘next generation’ high-throughput DNA sequencing platforms has been applied to the analysis of immune repertoires. The sequencing of immune repertoires at unprecedented levels of depth has opened up a realm of possibilities; these include answering basic questions on repertoire diversity and selection, monitoring repertoire diversity for clinical applications, and advancing strategies in monoclonal antibody discovery and engineering. The challenge of sequencing variable regions of immune receptors is that there is significant redundancy in the sequences encoding the four framework regions but extensive diversity in the three hypervariable complementarity determining regions (CDRs). As a result, sequencing platforms such as SOLiD (Applied Biosystems) that generate short reads (<100 bp) cannot be used to assemble the complete variable genes. Therefore, typically, the 454 (Roche) technology which yields reads >350 bp has been used to sequence the entire variable genes but at a relatively lower throughput (up to 106 reads per plate). Alternatively, the Illumina technology which currently has a maximum read length of ~200 bp (using paired-end sequencing) has been used to sequence the most hypervariable domain of the V gene (CDR3) at high depth (>106) (Fischer, 2011).
In one of the first examples, Quake and co-workers used 454 GS-FLX to generate 640 million bp of antibody V gene sequences from zebrafish, thus providing the first ever glimpse of the complete antibody diversity in an organism. Zebra-fish were shown to utilize between 50% and 86% of all possible germline VDJ combinations. Additionally convergence was observed such that several fish had identical antibody sequences (Weinstein et al., 2009). In a follow-up work, Jiang et al. showed that in early stages of zebrafish development, the VDJ repertoire is highly stereotypical, but becomes more diversified as it matures, suggesting both deterministic and stochastic elements shaped the antibody repertoire (Jiang et al., 2011). Boyd, Fire, and colleagues recently explored the application of antibody repertoire sequencing for clinical studies. In this work, the B lymphocyte clonality of VH genes was determined from the blood of healthy patients and from various tissues (e.g., bone marrow, lymph nodes) of patients with haematological
malignancies. This study demonstrated that high-resolution analysis of immunoglobulin repertoires and their clonality offers a potential strategy for diagnosis and therapeutic monitoring in cancer (Boyd et al., 2009).
Similarly the human TCR repertoire has been analyzed by Illumina-based high-throughput sequencing; high-resolution analyses were performed on CDR3 length distribution frequency and germline usage in TCRb chains from peripheral blood T cells (Freeman et al., 2009; Robins et al., 2009). In an expanded study, the TCRb CDR3 regions from naïve and memory CD8+ T cells isolated from seven patients were sequenced revealing a strong preferential usage of certain V–J combinations, with a bias towards a reduction in nontemplated nucleotide insertions. A computational analysis indicated a substantial overlap in the CD8+ CDR3 repertoire between any two individuals, approaching ~7000 fold greater than what would be expected based on the theoretical diversity of the TCR repertoire (Robins et al., 2010). In another similar study, the TCRb CDR3 regions from three different individuals were sequenced; comparisons between these repertoires (at the amino acid level) also revealed substantial overlap (e.g., two donors had 14.4% common sequences) (Warren et al., forthcoming). These studies suggest that overlap may be indicative of CDR3 TCRs that share specificity to common pathogens; the presence of such sequences could potentially be used as biomarkers for detection and diagnosis in infectious and autoimmune disease.
High-throughput DNA sequencing and analysis of immune repertoires has also started to make an impact on the field of monoclonal antibody discovery and engineering. For example, the antibody repertoire diversity in a phage display human combinatorial library was precisely quantified using 454 sequencing and bioinformatic analysis; it was found that nearly all germline families were present and that a substantial amount of diversity was generated by somatic mutations in CDR1 and 2 (Glanville et al., 2009). Similarly, a synthetic antibody library constructed by polynucleotide annealing and extension (amplification, PCR-free) was subjected to deep-sequencing for accurate determination of diversity and quality control (Ge et al., 2010). The Illumina platform was used to sequence CDR3 regions from an antibody fragment phage display library following rounds of antigen panning; this sequence-based approach allowed for identification and rescue of clones that were lost by subsequent rounds of screening (Ravn et al., 2010). In addition to in vitro libraries, high-throughput sequencing has been utilized for sequencing of antibody repertoires in immunized animals. Recently, 454 sequencing was performed on the antibody repertoires derived from bone marrow plasma cells of protein-immunized mice; it was found that VH and VL repertoires were highly polarized with the most abundant sequences representing 1–10% of the entire repertoire (Reddy et al., 2010). The most abundant VH and VL genes were paired simply by relative frequency and then constructed by high-throughput automated gene synthesis. Following recombinant expression, it was found that >75% (21/27) of antibodies were antigen-specific, representing the first ever example of monoclonal antibody discovery without screening.
Conclusions
The development of high-throughput technologies has led to a substantial amount of progress in the understanding of adaptive immune responses, which has allowed for a shift towards information-rich, systems-based approaches to immunological research (Table A6-1). Techniques that utilize single-cell analysis have generated high content genetic and functional data from various lymphocyte populations in mice and humans. Additionally, protein microarray technologies have been extensively used for humoral serological profiling and identification of antigens from a variety of pathogens and disease states. Finally, high-throughput DNA sequencing of adaptive immune repertoires has facilitated substantial advances in our understanding of immunological diversity and its subsequent exploitation for clinical and technological applications. These are only the beginning stages; the next steps will be to leverage these high-throughput technologies for the integration of biophysical, biochemical, genomic, and proteomic data sets, which will require the incorporation of statistical and mathematical modelling. This rapidly expanding field of experimental systems immunology will enable a more comprehensive understanding of adaptive immunity.
Acknowledgements
We thank Gregory C. Ippolito for helpful discussions and review of our manuscript. This work was funded by grants from the Cancer Prevention Research Institute of Texas (CPRIT) and Defense Advanced Research Projects Agency (DARPA).
References
Becker PD, Legrand N, van Geelen CMM, Noerder M, Huntington ND, Lim A, Yasuda E, Diehl SA, Scheeren FA, Ott M et al.: Generation of human antigen-specific monoclonal IgM antibodies using vaccinated ‘human immune system’ mice. PLoS ONE 2010, 5:e13137.
Boyd SD, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, Jones CD, Simen BB, Hanczaruk B, Nguyen KD et al.: Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Trans Med 2009, 1:12ra23.
Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y, Romero KM, Nguyen TT, Kalantari-Dehaghi M, Crotty S et al.: Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci U S A 2005, 102:547-552.
Felgner PL, Kayala MA, Vigil A, Burk C, Nakajima-Sasaki R, Pablo J, Molina DM, Hirst S, Chew JSW, Wang D et al.: A Burkholderia pseudomallei protein microarray reveals serodiagnostic and cross-reactive antigens. Proc Natl Acad Sci U S A 2009, 106:13499-13504.
Fischer N: Sequencing antibody repertoires: the next generation. mAbs 2011, 3:17-20.
Flatz L, Roychoudhuri R, Honda M, Filali-Mouhim A, Goulet J-P, Kettaf N, Lin M, Roederer M, Haddad EK, Sékaly RP et al.: Single-cell gene-expression profiling reveals qualitatively distinct CD8 T cells elicited by different gene-based vaccines. Proc Natl Acad Sci U S A 2011, 108:5724-5729.
Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA: Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res 2009, 19:1817-1824.
Ge X, Mazor Y, Hunicke-Smith SP, Ellington AD, Georgiou G: Rapid construction and characterization of synthetic antibody libraries without DNA amplification. Biotechnol Bioeng 2010, 106:347-357.
Germain RN, Meier-Schellersheim M, Nita-Lazar A, Fraser IDC: Systems biology in immunology: a computational modeling perspective. Annu Rev Immunol 2010, 29:527-585.
Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GMR et al.: Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A 2009, 106:20216-20221.
Gnjatic S, Wheeler C, Ebner M, Ritter E, Murray A, Altorki NK, Ferrara CA, Hepburne-Scott H, Joyce S, Koopman J et al.: Seromic analysis of antibody responses in non-small cell lung cancer patients and healthy donors using conformational protein arrays. J Immunol Methods 2009, 341:50-58.
Gnjatic S, Ritter E, Büchler MW, Giese NA, Brors B, Frei C, Murray A, Halama N, Zörnig I, Chen Y-T et al.: Seromic profiling of ovarian and pancreatic cancer. Proc Natl Acad Sci U S A 2010, 107:5088-5093.
Han Q, Bradshaw EM, Nilsson B, Hafler DA, Love JC: Multidimensional analysis of the frequencies and rates of cytokine secretion from single cells by quantitative microengraving. Lab Chip 2010, 10:1391-1400.
Janetzki S, Schaed S, Blachere NEB, Ben-Porat L, Houghton AN, Panageas KS: Evaluation of Elispot assays: influence of method and operator on variability of results. J Immunol Methods 2004, 291:175-183.
Janeway C: Immunobiology. Garland Pub.; 2005.
Jiang N, Weinstein JA, Penland L, White RA, Fisher DS, Quake SR: Determinism and stochasticity during maturation of the zebrafish antibody repertoire. Proc Natl Acad Sci U S A 2011.
Jin A, Ozawa T, Tajiri K, Obata T, Kondo S, Kinoshita K, Kadowaki S, Takahashi K, Sugiyama T, Kishi H et al.: A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat Med 2009, 15:1088-1092.
Kingsmore SF: Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nat Rev Drug Discov 2006, 5:310-320.
Kunnath-Velayudhan S, Salamon H, Wang H-Y, Davidow AL, Molina DM, Huynh VT, Cirillo DM, Michel G, Talbot EA, Perkins MD et al.: Dynamic antibody responses to the Mycobacterium tuberculosis proteome. Proc Natl Acad Sci U S A 2010, 107:14703-14708.
Kwakkenbos MJ, Diehl SA, Yasuda E, Bakker AQ, van Geelen CMM, Lukens MV, van Bleek GM, Widjojoatmodjo MN, Bogers WMJM, Mei H et al.: Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nat Med 2009, 16:123-128.
Kwong GA, Radu CG, Hwang K, Shu CJ, Ma C, Koya RC, Comin-Anduix B, Hadrup SR, Bailey RC, Witte ON et al.: Modular nucleic acid assembled p/MHC microarrays for multiplexed sorting of antigen-specific T cells. J Am Chem Soc 2009, 131:9695-9703.
Lanzavecchia A, Sallusto F: Human B cell memory. Curr Opin Immunol 2009, 21:298-304.
Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI et al.: Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 2008, 14:25-27.
Legutki JB, Magee DM, Stafford P, Johnston SA: A general method for characterization of humoral immunity induced by a vaccine or infection. Vaccine 2010, 28:4529-4537.
Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL: A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat Biotechnol 2006, 24:703-707.
Meijer P-J, Andersen PS, Haahr Hansen M, Steinaa L, Jensen A, Lantto J, Oleksiewicz MB, Tengbjerg K, Poulsen TR, Coljee VW et al.: Isolation of human antibody repertoires with preservation of the natural heavy and light chain pairing. J Mol Biol 2006, 358:764-772.
Michaud GA, Salcius M, Zhou F, Bangham R, Bonin J, Guo H, Snyder M, Predki PF, Schweitzer BI: Analyzing antibody specificity with whole proteome microarrays. Nat Biotechnol 2003, 21:1509-1512.
Ogunniyi AO, Story CM, Papa E, Guillen E, Love JC: Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat Protoc 2009, 4:767-782.
Papagno L, Almeida JR, Nemes E, Autran B, Appay V: Cell permeabilization for the assessment of T lymphocyte polyfunctional capacity. J Immunol Methods 2007, 328:182-188.
Poulsen TR, Meijer P-J, Jensen A, Nielsen LS, Andersen PS: Kinetic, affinity, and diversity limits of human polyclonal antibody responses against tetanus toxoid. J Immunol (Baltimore, MD) 2007, 179:3841-3850.
Prechl J, Papp K, Erdei A: Antigen microarrays: descriptive chemistry or functional immunomics? Trends Immunol 2010, 31:133-137.
Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B et al.: Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 2009, 10:116-125.
Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P, Magistrelli G, Farinelli L, Kosco-Vilbois MH, Fischer N: By-passing in vitro screening—next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res 2010, 38:e193.
Reddy MM, Wilson R, Wilson J, Connell S, Gocke A, Hynan L, German D, Kodadek T: Identification of candidate IgG biomarkers for Alzheimer’s disease via combinatorial library screening. Cell 2011, 144:132-142.
Reddy ST, Ge X, Miklos AE, Hughes RA, Kang SH, Hoi KH, Chrysostomou C, Hunicke-Smith SP, Iverson BL, Tucker PW et al.: Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat Biotechnol 2010, 28:965-969.
Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS: Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009, 114:4099-4107.
Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH: Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med 2010, 2:47ra64.
Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, Fong D, Genovese MC, de Vegvar HEN et al.: Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med 2002, 8:295-301.
Smith K, Garman L, Wrammert J, Zheng N-Y, Capra JD, Ahmed R, Wilson PC: Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc 2009, 4:372-384.
Song Q, Han Q, Bradshaw EM, Kent SC, Raddassi K, Nilsson B, Nepom GT, Hafler DA, Love JC: On-chip activation and subsequent detection of individual antigen-specific T cells. Anal Chem 2010, 82:473-477.
Story CM, Papa E, Hu C-CA, Ronan JL, Herlihy K, Ploegh HL, Love JC: Profiling antibody responses by multiparametric analysis of primary B cells. Proc Natl Acad Sci U S A 2008, 105:17902-17907.
Tajiri K, Kishi H, Tokimitsu Y, Kondo S, Ozawa T, Kinoshita K, Jin A, Kadowaki S, Sugiyama T, Muraguchi A: Cell-microarray analysis of antigen-specific B-cells: single cell analysis of antigen receptor expression and specificity. Cytometry 2007, 71:961-967.
Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H: Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods 2008, 329:112-124.
Tokimitsu Y, Kishi H, Kondo S, Honda R, Tajiri K, Motoki K, Ozawa T, Kadowaki S, Obata T, Fujiki S et al.: Single lymphocyte analysis with a microwell array chip. Cytometry 2007, 71:1003-1010.
Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, Webb JR, Holt RA: Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. Advanced Online Publication.
Weinstein JA, Jiang N, White RA, Fisher DS, Quake SR: High-throughput sequencing of the zebrafish antibody repertoire. Science (New York, NY) 2009, 324:807-810.
Wiberg FC, Rasmussen SK, Frandsen TP, Rasmussen LK, Tengbjerg K, Coljee VW, Sharon J, Yang CY, Bregenholt S, Nielsen LS et al.: Production of target-specific recombinant human polyclonal antibodies in mammalian cells. Biotechnol Bioeng 2006, 94:396-405.
Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng N-Y, Mays I, Garman L, Helms C et al.: Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453:667-671.
Yu X, Tsibane T, McGraw PA, House FS, Keefer CJ, Hicar MD, Tumpey TM, Pappas C, Perrone LA, Martinez O et al.: Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 2008, 455:532-536.
THE NEW SCIENCE OF SOCIOMICROBIOLOGY AND THE REALM OF SYNTHETIC AND SYSTEMS ECOLOGY
E. Peter Greenberg44
The Emergence of Sociomicrobiology as an Interest Area in Microbiology
Up until the later part of the past century, microbiologists believed that with rare exceptions bacteria did not practice sociality; this was in spite of the fact that growing bacteria as colonies on agar plates was and still remains a cornerstone technology for the discipline. Just the terminology “colonial” or “colony growth” implies some sort of social interactions. But with the few exceptional scientists working on myxobacteria, a special group of social bacteria, the eyes of microbiology did not see social interactions among individual bacteria. The longstanding view that bacteria were by-and-large asocial creatures began to crumble in the 1990s with rapid developments in the areas of quorum sensing and biofilm research. Now it seems apparent that the same selective pressures that led to the evolution of sociality in animals are forces for evolution of sociality in bacteria, and we see and appreciate social behavior in bacteria (Camilli and Bassler, 2006; Parsek and Greenberg, 2005). I have no proof but I believe that contributing to our past resistance to sociomicrobiology is the fact that bacteria are at once single cells and single individuals. They have served biology as wonderful models for cellular activities. We have not focused as much on the bacterial cell as an individual member of a species as we have on their existence as single-celled organisms.
My laboratory has focused on three areas of sociomicrobiology. The first is quorum sensing, which can be defined as a cell-to-cell communication system that enables individuals to sense the local population density and regulate expression of specific genes in response to population density. This is the primary topic
_______________
44 Department of Microbiology, University of Washington School of Medicine.
of this paper. The second is biofilm biology. Biofilms are organized assemblages of bacteria embedded in a self-produced extracellular matrix. As a consequence of the biofilm lifestyle, the assembled bacteria exist in a heterogeneous environment and bacteria in different regions of a biofilm have different functions, which confer different traits to the group. As a consequence of the biofilm lifestyle, at least a subpopulation of the assemblage is protected from environmental stresses and in fact biofilm infections are difficult or impossible to cure by antibiotic treatment (Costerton et al., 1999). Because biofilm infections are very prevalent medical problems there is considerable interest in developing novel therapies that are effective in killing biofilm bacteria. To name just a few, infections of any implanted medical device are biofilm infections, cystic fibrosis lung infections are biofilm infections, heart valve infections are biofilms, and at least some chronic middle ear infections are biofilms (Costerton et al., 1999). Third, we have also worked on conspecific territoriality exhibited by swarms of bacteria moving over surfaces (Gibbs and Greenberg, 2011; Gibbs et al., 2008).
The emergence of sociomicrobiology as a new subdiscipline is important for several reasons. First, there is an idea that we might be able to develop novel antivirulence therapeutics, which target quorum sensing in bacterial species that control virulence gene expression by cell-to-cell signaling. In fact there are many investigators working to identify potent quorum-sensing and biofilm inhibitors, and we ourselves have undertaken such programs together with collaborators in the pharmaceutical industry (Banin et al., 2008; Muh et al., 2006a,b), but this is not the primary motivation of our work. This is in part because it is my belief that we do not yet understand enough about the biology of quorum sensing to predict how, when, or where its inhibition might be of therapeutic value. Second, it is now clear that if one strives to really understand bacteria, social aspects of their biology cannot be ignored. Third, now that we understand that bacteria are social and we understand sociality at an unprecedented level of molecular detail in bacteria like Pseudomonas aeruginosa, these model organisms have become wonderful tools to study fundamental questions about the costs and benefits of cooperation, the selective pressures that lead to cooperative behaviors, and the advantages of controlling cooperative behaviors by communication. Finally, our detailed understanding of quorum-sensing signal synthesis and signal reception, as is discussed below, has led to the widespread use of quorum-sensing regulatory circuits in synthetic biology (Marguet et al., 2010, and references therein).
Quorum Sensing in Proteobacteria
In the late 1960s and early 1970s there was a very modest literature describing pheromone production and activity in bacteria (Eberhard, 1972; Tomasz, 1965). It was not until the 1980s that work on quorum sensing in marine luminescent bacteria led to the idea that these sorts of gene regulatory activities function as intercellular communication systems that coordinate group activities.
Not until the 1990s did we begin to understand the prevalence of quorum sensing in bacteria. Although there are many different types of bacterial cell-to-cell signaling systems considered as quorum-sensing systems, our work focuses on acyl-homoserine lactone quorum-sensing systems prevalent but not universal among the Proteobacteria. Our original model system was a marine bacterium, Vibrio fischeri, which controls a small set of about 25 or fewer genes, including genes for light production, by a transcriptional activator called LuxR, and 3-oxo-hexanoyl-homoserine lactone (3OC6-HSL), which is produced by the luxI gene product (Antunes et al., 2007; Engebrecht et al., 1983). This quorum-sensing system allows V. fischeri to discriminate between its free-living low-population-density lifestyle and its high-density host-associated lifestyle (Figure A7-1). It exists in the light organs of specific marine animals where it produces light, which serves the mutualistic symbiosis. Of note, 3OC6-HSL moves in and out of cells by passive diffusion. In this way the environmental signal concentration is a reflection of cell density. The luxI gene itself is controlled by quorum sensing—it is activated by LuxR and 3OC6-HSL. In terms of biology, this provides hysteresis to the system. The population density required to activate quorum-controlled genes is much higher than the density required to shut down an activated system.
We later turned our attention to the opportunistic pathogen P. aeruginosa. We learned that there were two acyl-HSL circuits, the C4-HSL-RhlI-RhlR circuit and the 3OC12-HSL-LasI-LasR circuits, which together are required for activation of about 325 genes (Figure A7-2) (Pearson et al., 1994, 1995; Schuster et al., 2003). Other investigators showed that at least under certain experimental conditions quorum-sensing mutants were impaired in virulence (Pearson et al., 2000; Tang et al., 1996), and thus the belief was that, as for V. fischeri quorum sensing, P. aeruginosa quorum sensing allowed discrimination between host and free-living states. However, it is not clear from the evidence that this is in fact the case. Among the 300-plus quorum-controlled genes, those coding for extracellular products like exoenzymes or coding for functions required for the production of exoproducts like hydrogen cyanide or pyocyanin are grossly overrepresented.
In a social context these sorts of extracellular products can be considered public goods or resources produced by individuals and shared by all members of the group. Thus we believe quorum sensing serves to coordinate cooperative behaviors. Investigators have devised experiments where growth of P. aeruginosa requires quorum sensing. This can be accomplished by providing protein as the sole source of carbon and energy. Growth requires quorum-sensing-induced production of the exoprotease elastase. In this setting, LasR quorum-sensing mutants will emerge and become a stable, significant minority of the overall population. These cheaters or freeloaders do not bear the cost of contributing to the public good (elastase in this case), but they benefit from use of the public goods (Sandoz et al., 2007). It is obvious that light production by V. fischeri is a shared behavior and we believe this also represents quorum control of cooperation. As far as I am aware, there are no biological systems sensitive enough to detect the light pro-
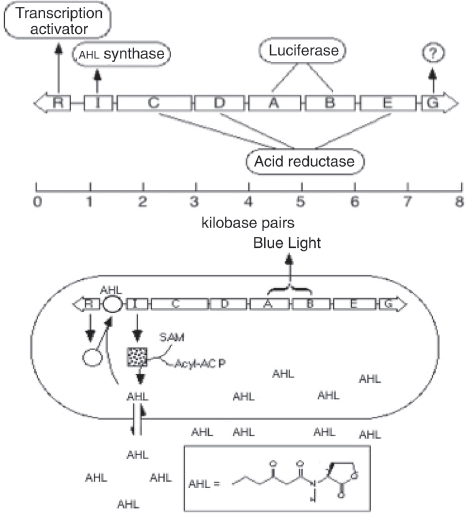
FIGURE A7-1 Quorum sensing in Vibrio fischeri. (Top) The lux gene cluster. The luxR gene encodes a 3OC6-HSL (AHL)-dependent transcriptional activator of the luxI-G operon. The luxI product is the AHL synthase; luxC, D, and E form a complex responsible for generation of one of the substrates for the luciferase reaction, luxA and B encode the two subunits of luciferase, and the function of luxG remains unknown. (Bottom) Cartoon of a V. fischeri cell producing the diffusible AHL signal. At low cell densities the luminescence operon is transcribed at a basal level. At high cell densities the AHL signal can reach a sufficient concentration and bind to the cellular LuxR protein, which will then activate transcription of the luminescence operon. The substrates for AHL synthesis are shown, as is the molecule. Note the positive autoregulation of luxI, a common feature of R-I circuits.
FIGURE A7-2 Diagram of the acyl-HSL quorum-sensing regulatory circuit in P. aeruginosa. The lasI and R genes are linked on the chromosome, as are the rhlR and I genes. The qscR gene is not linked tightly to any other quorum-sensing regulator. LasI-R sit atop a quorum-sensing cascade. The rhl quorum-sensing system is among the functions controlled by LasI-R and transcription of qscR is positively autoregulated but requires production of 3OC12-HSL by LasI. Pink ball, 3OC12-HSL; orange ball, C4-HSL.
duced by one or a few V. fischeri cells (maximum emission is about 1,000 photons per second per cell). But the light produced by large groups of cells can easily be observed. For example, we can see the light produced by colonies of V. fischeri growing on a Petri plate. So here are two examples where it seems apparent that quorum-sensing functions control and coordinate cooperative behaviors. Is this universal for LuxR-LuxI-acyl-HSL-type systems? Of course we do not know the answer to this question and one might imagine that such regulatory elements have been adapted by different bacteria for different purposes.
As a general principle, the LuxR-LuxI type of regulatory circuits appears to represent elements of true communication. That is, the signal and the signal receptor are coevolved (Keller and Surette, 2006). One finds that a disturbing anthropomorphic trend has crept into the field and the term “bacterial language” is sometimes used to describe quorum-sensing systems. I do not believe any linguist would find evidence for syntax and grammar in the bacterial world.
With the discovery that quorum-sensing-controlled virulence of bacteria like P. aeruginosa, the number of scientists studying acyl-HSL signaling grew exponentially. Generally speaking, studies of quorum sensing and control of social activity in bacteria are important for the several reasons listed earlier in this paper. We might be able to develop novel antivirulence therapeutics that target
quorum sensing in bacteria like P. aeruginosa. Sociality is an important aspect of bacterial biology. Bacteria are now viewed as experimental models for fundamental studies about the costs and benefits of cooperation, the selective pressures that lead to cooperative behaviors, and the advantages of controlling cooperative behaviors by communication. In addition, our detailed understanding of signal synthesis by Lux-type proteins and signal reception by LuxR-type transcription factors has led to their widespread use in the area of synthetic biology (Marguet et al., 2010). This was a largely unintended consequence of our work, and I believe there are three reasons for the popularity of these systems in building synthetic regulatory circuits. First, the acyl-HSLs are diffusible signaling ligands (Kaplan and Greenberg, 1985). Thus, the complication of transport systems for the ligand need not concern the synthetic biologist. Second, the V. fischeri system (Engebrecht et al., 1983), the P. aeruginosa systems, and many other acyl-HSL quorum-sensing circuits have positive autoregulatory loops. Signal production proceeds at a low level until it has accumulated sufficiently to activate quorum-controlled genes, one of which is the gene for its own synthesis. The signal concentration then increases very rapidly. Work in the area of synthetic biology has focused on this phenomenon as a method to reduce noise in regulatory circuits or build gene expression oscillators. Finally, we have identified many different systems and we know of dozens of acyl-HSL signals—the signal specificity of a given LasR homolog resides in the nature of the acyl side group. Thus, multiple systems can be strung together in a single bacterium with each controlling, for example, expression of fluorescent proteins that emit at different wavelengths. Examples showing the diversity of acyl-HSL signals identified in different bacterial species are shown in Figure A7-3.
New Twists to the Acyl-Homoserine Lactone Signaling Story
The P. aeruginosa quorum-sensing circuitry described in Figure A7-2 shows, in addition to LasR-LasI and RhlR-RhlI, a LuxR homolog, QscR. There is no cognate acyl-HSL synthase gene for QscR and it has been termed an orphan quorum-sensing signal receptor (Chugani et al., 2001). These orphans, which have also been called solos (Subramoni and Venturi, 2009), are quite commonly found in sequenced proteobacterial genomes. We know that QscR responds to the LasI-produced 3OC12-HSL, and it controls a set of genes that partially overlaps with the genes controlled by LasR and RhlR (Lee et al., 2006; Lequette et al., 2006). There has been recent interest in studying orphans (Subramoni and Venturi, 2009). There are orphan LuxR homologs in Salmonella and in pathogenic E. coli, neither of which have any luxI homologs. Evidence indicates that the E. coli and Salmonella LuxR orphans respond to acyl-HSLs made by other bacterial species in mixed microbial communities (Soares and Ahmer, 2011; Sperandio, 2010).
Most of the acyl-HSL signals that have been identified are fatty acyl-HSLs with fatty acyl groups of varying carbon length and with a limited number of
FIGURE A7-3 Some examples of acyl-HSL quorum-sensing signals. From top to bottom; the P. aeruginosa RhlI signal butanoyl-HSL, the V. fischeri LuxI signal, 3-oxo-hexanoyl-HSL, The B. mallei BmaI signal, 3-hydroxy-decanoyl-HSL, and the Rhodopseudomonas palustris RpaI signal, para-coumaryl-HSL.
substitutions on the fatty acyl carbon chain. We recently described examples of two bacteria that use LuxI-LuxR homologs to produce and respond to aryl-HSLs. Both are photosynthetic; Rhodopseudomonas palustris uses p-coumaroyl-HSL as a quorum-sensing signal and photosynthetic baradyrhizobia uses cinnamoyl-HSL (Ahlgren et al., 2011; Schaefer et al., 2008). These discoveries broaden the possibilities in terms of signal molecules. Furthermore, the photosynthetic bradyrhizobia produce nanomolar amounts of cinnamoyl-HSL and respond to picomolar
amounts. This is of interest because the fatty acyl-HSL systems that have been described involve responses to nanomolar signal levels and the signals are usually produced at levels in the micromolar range. The bradyrhizobium system is shifted to much lower levels for production and detection. The significance of the low production and ultrasensitivity is not clearly understood. However, it is clear that, by assuming acyl-HSLs will be produced at higher levels, we likely have overlooked at least some acyl-HSL signaling systems.
Concluding Comments
This paper seeks to introduce an emerging field, sociomicrobiology, and to point out opportunities for synthetic biology and therapeutic development in the area of sociomicrobiology. The paper emphasizes quorum sensing in Proteobacteria and provides examples where quorum sensing serves to allow individuals of a species to communicate. This rudimentary form of chemical communication serves to coordinate certain cooperative behaviors. Whether this is a universal characteristic of acyl-HSL signaling systems remains to be seen. There are also a variety of other small-molecule signals produced by bacteria and many of these are considered quorum-sensing signals (Camilli and Bassler, 2006). If and how these intercellular signals are controlling cooperation also remains to be seen. Finally, this Forum has covered topics related to synthetic and systems biology, and opportunities in the area of human health and disease. We need to remain cognizant that bacterial cells are individual organisms involved in complex social interactions. So not only do we need to think about systems and synthetic biology from a cellular perspective, but we need to think about systems and synthetic ecology too.
References
Ahlgren, N. A., C. S. Harwood, A.L. Schaefer, E. Giraud, and E. P. Greenberg. 2011. Aryl-homoserine lactone quorum sensing in stem-nodulating photosynthetic bradyrhizobia. Proceedings of the National Academy of Sciences of the United States of America 108:7183-7188.
Antunes, L. C., A. L. Schaefer, R. B. R. Ferreira, N. Qin, A. M. Stevens, E. G. Ruby, and E. P. Greenberg. 2007. Transcriptome analysis of the Vibrio fischeri LuxR-LuxI regulon. Journal of Bacteriology 189:8387-8391.
Banin, E., A. Lozinski, et al. 2008. The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proceedings of the National Academy of Sciences of the United States of America 105:16761-16766.
Camilli, A., and B. L. Bassler 2006. Bacterial small-molecule signaling pathways. Science 311: 1113-1116.
Chugani, S. A., M. Whiteley, K. M. Lee, D. D’Argenio, C. Manoil, and E. P. Greenberg. 2001. QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America 98:2752-2757.
Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: A common cause of persistent infections. Science 284:1318-1322.
Eberhard, A. 1972. Inhibition and activation of bacterial luciferase synthesis. Journal of Bacteriology 109:1101-1105.
Engebrecht, J., K. H. Nealson, and M. Silverman. 1983. Bacterial bioluminescence: Isolation and genetic analysis of functions from Vibrio fischeri. Cell 32:773-781.
Gibbs, K. A., and E. P. Greenberg. 2011. Territoriality in Proteus: Advertisement and aggression. Chemical Reviews 111(1):188-194.
Gibbs, K. A., M. L. Urbanowski, and E. P. Greenberg. 2008. Genetic determinants of self identity and social recognition in bacteria. Science 321:256-259.
Kaplan, H. B., and E. P. Greenberg. 1985. Diffusion of autoinducer is involved in regulation of the Vibrio fischeri luminescence system. Journal of Bacteriology 163:1210-1214.
Keller, L., and M. G. Surette. 2006. Communication in bacteria: An ecological and evolutionary perspective. Nature Reviews Microbiology 4:249-258.
Lee, J. H., Y. Lequette, and E. P. Greenberg. 2006. Activity of purified QscR, a Pseudomonas aeruginosa orphan quorum-sensing transcription factor. Molecular Microbiology 59:602-609.
Lequette, Y., J. H. Lee, F. Ledgham, A. Lazdunski, and E. P. Greenberg. 2006. A distinct QscR regulon in the Pseudomonas aeruginosa quorum-sensing circuit. Journal of Bacteriology 188: 3365-3370.
Marguet, P., Y. Tanouchi, E. Spitz, C. Smith, and L. C. You. 2010. Oscillations by minimal bacterial suicide circuits reveal hidden facets of host-circuit physiology. PLoS One 5:e11909.
Muh, U., B. J. Hare, B. A. Duerkop, M. Schuster, B. L. Hanzelka, R. Heim, E. R. Olson, and E. P. Greenberg. 2006a. A structurally unrelated mimic of a Pseudomonas aeruginosa acyl-homoserine lactone quorum-sensing signal. Proceedings of the National Academy of Sciences of the United States of America 103:16948-16952.
Muh, U., M. Schuster, R. Heim, A. Singh, E. R. Olson, and E. P. Greenberg. 2006b. Novel Pseudomonas aeruginosa quorum-sensing inhibitors identified in an ultra-high-throughput screen. Antimicrobial Agents and Chemotherapy 50:3674-3679.
Parsek, M. R., and E. P. Greenberg. 2005. Sociomicrobiology: The connections between quorum sensing and biofilms. Trends in Microbiology 13:27-33.
Pearson, J. P., K. M. Gray, L. Passador, K. D. Tucker, A. Eberhard, B. H. Iglewski, and E. P. Greenberg. 1994. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proceedings of the National Academy of Sciences of the United States of America 91:197-201.
Pearson, J. P., L. Passador, B. H. Iglewski, and E. P. Greenberg. 1995. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America 92:1490-1494.
Pearson, J. P., M. Feldman, B. H. Iglewski., and A. Prince. 2000. Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infection and Immunity 68:4331-4334.
Sandoz, K. M., S. M. Mitzimberg, and M. Schuster. 2007. Social cheating in Pseudomonas aeruginosa quorum sensing. Proceedings of the National Academy of Sciences of the United States of America 104:15876-15881.
Schaefer, A. L., E. P. Greenberg, C. M. Oliver, Y. Oda, J. J. Huang, G. Bittan-Banin, C. M. Peres, S. Schmidt, K. Juhaszova, J. R. Sufrin, and C. S. Harwood. 2008. A new class of homoserine lactone quorum-sensing signals. Nature 454:595-599.
Schuster, M., C. P. Lostroh, T. Ogi, and E. P. Greenberg. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: A transcriptome analysis. Journal of Bacteriology 185:2066-2079.
Soares, J. A., and B. M. Ahmer. 2011. Detection of acyl-homoserine lactones by Escherichia and Salmonella. Current Opinions in Microbiology 14:188-193.
Sperandio, V. 2010. SdiA sensing of acyl-homoserine lactones by enterohemorrhagic E. coli (EHEC) serotype O157:H7 in the bovine rumen. Gut Microbes 1:432-435.
Subramoni, S., and V. Venturi. 2009. LuxR-family ‘solos’: Bachelor sensors/regulators of signalling molecules. Microbiology 155:1377-1385.
Tang, H. B., E. DiMango, R. Bryan, M. Gambello, B. H. Iglewski, J. B. Goldberg, and A. Prince. 1996. Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infection and Immunity 64:37-43.
Tomasz, A. 1965. Control of the competent state in Pneumococcus by a hormone-like cell product: An example for a new type of regulatory mechanism in bacteria. Nature 208:155-159.
CREATION OF A BACTERIAL CELL CONTROLLED BY A CHEMICALLY SYNTHESIZED GENOME45
Daniel G. Gibson,46John I. Glass,46Carole Lartigue,46Vladimir N. Noskov,46Ray-Yuan Chuang,46Mikkel A. Algire,46Gwynedd A. Benders,47Michael G. Montague,46Li Ma,46Monzia M. Moodie,46Chuck Merryman,46Sanjay Vashee,46Radha Krishnakumar,46Nacyra Assad-Garcia,46Cynthia Andrews-Pfannkoch,46Evgeniya A. Denisova,46Lei Young,46Zhi-Qing Qi,46Thomas H. Segall-Shapiro,46Christopher H. Calvey,46Prashanth P. Parmar,46Clyde A. Hutchison III,47Hamilton O. Smith,47J. Craig Venter46,47,48
We report the design, synthesis, and assembly of the 1.08–mega–base pair Mycoplasma mycoides JCVI-syn1.0 genome starting from digitized genome sequence information and its transplantation into a M. capricolum recipient cell to create new M. mycoides cells that are controlled only by the
_______________
45 From Daniel G. Gibson, et al. 2010. Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science 329:52-56. Reprinted with permission from AAAS.
46 The J. Craig Venter Institute, 9704 Medical Center Drive, Rockville, MD 20850, USA.
47 The J. Craig Venter Institute, 10355 Science Center Drive, San Diego, CA 92121, USA.
48 To whom correspondence should be addressed. E-mail: jcventer@jcvi.org
We thank Synthetic Genomics, Inc. for generous funding of this work. We thank J. B. Hostetler, D. Radune, N. B. Fedorova, M. D. Kim, B. J. Szczypinski, I. K. Singh, J. R. Miller, S. Kaushal, R. M. Friedman, and J. Mulligan for their contributions to this work. Electron micrographs were generously provided by T. Deerinck and M. Ellisman of the National Center for Microscopy and Imaging Research at the University of California at San Diego. J.C.V. is chief executive officer and co-chief scientific officer of SGI. H.O.S. is co-chief scientific officer and on the Board of Directors of SGI. C.A.H. is chairman of the SGI Scientific Advisory Board. All three of these authors and JCVI hold SGI stock. JCVI has filed patent applications on some of the techniques described in this paper.
Supporting Online Material www.sciencemag.org/cgi/content/full/science.1190719/DC1
Materials and Methods
Figs. S1 to S6
Tables S1 to S7
References
9 April 2010; accepted 13 May 2010
Published online 20 May 2010; 10.1126/science.1190719.
Include this information when citing this paper.
synthetic chromosome. The only DNA in the cells is the designed synthetic DNA sequence, including “watermark” sequences and other designed gene deletions and polymorphisms, and mutations acquired during the building process. The new cells have expected phenotypic properties and are capable of continuous self-replication.
In 1977, Sanger and colleagues determined the complete genetic sequence of phage ϕX174 (Sanger et al., 1977), the first DNA genome to be completely sequenced. Eighteen years later, in 1995, our team was able to read the first complete genetic sequence of a self-replicating bacterium, Haemophilus influenzae (Fleischmann et al., 1995). Reading the genetic sequence of a wide range of species has increased exponentially from these early studies. The ability to rapidly digitize genomic information has increased by more than eight orders of magnitude over the past 25 years (Venter, 2010). Efforts to understand all this new genomic information have spawned numerous new computational and experimental paradigms, yet our genomic knowledge remains very limited. No single cellular system has all of its genes understood in terms of their biological roles. Even in simple bacterial cells, do the chromosomes contain the entire genetic repertoire? If so, can a complete genetic system be reproduced by chemical synthesis starting with only the digitized DNA sequence contained in a computer?
Our interest in synthesis of large DNA molecules and chromosomes grew out of our efforts over the past 15 years to build a minimal cell that contains only essential genes. This work was inaugurated in 1995 when we sequenced the genome of Mycoplasma genitalium, a bacterium with the smallest complement of genes of any known organism capable of independent growth in the laboratory. More than 100 of the 485 protein-coding genes of M. genitalium are dispensable when disrupted one at a time (Hutchison et al., 1999; Glass et al., 2006; Smith et al., 2008). We developed a strategy for assembling viral-sized pieces to produce large DNA molecules that enabled us to assemble a synthetic M. genitalium genome in four stages from chemically synthesized DNA cassettes averaging about 6 kb in size. This was accomplished through a combination of in vitro enzymatic methods and in vivo recombination in Saccharomyces cerevisiae. The whole synthetic genome [582,970 base pairs (bp)] was stably grown as a yeast centromeric plasmid (YCp) (Gibson et al., 2008b).
Several hurdles were overcome in transplanting and expressing a chemically synthesized chromosome in a recipient cell. We needed to improve methods for extracting intact chromosomes from yeast. We also needed to learn how to transplant these genomes into a recipient bacterial cell to establish a cell controlled only by a synthetic genome. Because M. genitalium has an extremely slow growth rate, we turned to two faster-growing mycoplasma species, M. mycoides subspecies capri (GM12) as donor, and M. capricolum subspecies capricolum (CK) as recipient.
To establish conditions and procedures for transplanting the synthetic ge-
nome out of yeast, we developed methods for cloning entire bacterial chromosomes as centromeric plasmids in yeast, including a native M. mycoides genome (Lartigue et al., 2009; Benders et al., 2010). However, initial attempts to extract the M. mycoides genome from yeast and transplant it into M. capricolum failed. We discovered that the donor and recipient mycoplasmas share a common restriction system. The donor genome was methylated in the native M. mycoides cells and was therefore protected against restriction during the transplantation from a native donor cell (Lartigue et al., 2007). However, the bacterial genomes grown in yeast are unmethylated and so are not protected from the single restriction system of the recipient cell. We overcame this restriction barrier by methylating the donor DNA with purified methylases or crude M. mycoides or M. capricolum extracts, or by simply disrupting the recipient cell’s restriction system (Lartigue et al., 2009).
We now have combined all of our previously established procedures and report the synthesis, assembly, cloning, and successful transplantation of the 1.08-Mbp M. mycoides JCVI-syn1.0 genome, to create a new cell controlled by this synthetic genome.
Synthetic Genome Design
Design of the M. mycoides JCVI-syn1.0 genome was based on the highly accurate finished genome sequences of two laboratory strains of M. mycoides subspecies capri GM12 (Lartigue et al., 2009; Benders et al., 2010).49 One was the genome donor used by Lartigue et al. [GenBank accession CP001621] (2007). The other was a strain created by transplantation of a genome that had been cloned and engineered in yeast, YCpMmyc1.1-ΔtypeIIIres [GenBank accession CP001668] (Lartigue et al., 2009). This project was critically dependent on the accuracy of these sequences. Although we believe that both finished M. mycoides genome sequences are reliable, there are 95 sites at which they differ. We began to design the synthetic genome before both sequences were finished. Consequently, most of the cassettes were designed and synthesized based on the CP001621 sequence.49 When it was finished, we chose the sequence of the genome successfully transplanted from yeast (CP001668) as our design reference (except that we kept the intact typeIIIres gene). All differences that appeared biologically significant between CP001668 and previously synthesized cassettes were corrected to match it exactly.49 Sequence differences between our synthetic cassettes and CP001668 that occurred at 19 sites appeared harmless and so were not corrected. These provide 19 polymorphic differences between our synthetic genome (JCVI-syn1.0) and the natural (nonsynthetic) genome (YCpMmyc1.1) that we have cloned in yeast and use as a standard for genome transplantation from yeast (Lartigue et al., 2009). To further differentiate between the synthetic
_______________
49 Supporting material is available on Science Online.
genome and the natural one, we designed four watermark sequences (fig. S1) to replace one or more cassettes in regions experimentally demonstrated [watermarks 1 (1246 bp) and 2 (1081 bp)] or predicted [watermarks 3 (1109 bp) and 4 (1222 bp)] to not interfere with cell viability. These watermark sequences encode unique identifiers while limiting their translation into peptides. Table S1 lists the differences between the synthetic genome and this natural standard. Figure S2 shows a map of the M. mycoides JCVI-syn1.0 genome. Cassette and assembly intermediate boundaries, watermarks, deletions, insertions, and genes of the M. mycoides JCVI syn1.0 are shown in fig. S2, and the sequence of the transplanted mycoplasma clone sMmYCp235-1 has been submitted to GenBank (accession CP002027).
Synthetic Genome Assembly Strategy
The designed cassettes were generally 1080 bp with 80-bp overlaps to adjacent cassettes.50 They were all produced by assembly of chemically synthesized oligonucleotides by Blue Heron (Bothell, Washington). Each cassette was individually synthesized and sequence-verified by the manufacturer. To aid in the building process, DNA cassettes and assembly intermediates were designed to contain Not I restriction sites at their termini and recombined in the presence of vector elements to allow for growth and selection in yeast (Gibson et al., 2008b).50 A hierarchical strategy was designed to assemble the genome in three stages by transformation and homologous recombination in yeast from 1078 1-kb cassettes (Fig. A8-1) (Gibson, 2009; Gibson et al., 2008a).
Assembly of 10-kb synthetic intermediates
In the first stage, cassettes and a vector were recombined in yeast and transferred to Escherichia coli.50 Plasmid DNA was then isolated from individual E. coli clones and digested to screen for cells containing a vector with an assembled 10-kb insert. One successful 10-kb assembly is represented (Fig. A8-2A). In general, at least one 10-kb assembled fragment could be obtained by screening 10 yeast clones. However, the rate of success varied from 10 to 100%. All of the first-stage intermediates were sequenced. Nineteen out of 111 assemblies contained errors. Alternate clones were selected, sequence-verified, and moved on to the next assembly stage.50
Assembly of 100-kb synthetic intermediates
The pooled 10-kb assemblies and their respective cloning vectors were transformed into yeast as above to produce 100-kb assembly intermediates.50
_______________
50 Supporting material is available on Science Online.
FIGURE A8-1 The assembly of a synthetic M. mycoides genome in yeast. A synthetic M. mycoides genome was assembled from 1078 overlapping DNA cassettes in three steps. In the first step, 1080-bp cassettes (orange arrows), produced from overlapping synthetic oligonucleotides, were recombined in sets of 10 to produce 109 ~10-kb assemblies (blue arrows). These were then recombined in sets of 10 to produce 11 ~100-kb assemblies (green arrows). In the final stage of assembly, these 11 fragments were recombined into the complete genome (red circle). With the exception of two constructs that were enzymatically pieced together in vitro (Gibson et al., 2009) (white arrows), assemblies were carried out by in vivo homologous recombination in yeast. Major variations from the natural genome are shown as yellow circles. These include four watermarked regions (WM1 to WM4), a 4-kb region that was intentionally deleted (94D), and elements for growth in yeast and genome transplantation. In addition, there are 20 locations with nucleotide polymorphisms (asterisks). Coordinates of the genome are relative to the first nucleotide of the natural M. mycoides sequence. The designed sequence is 1,077,947 bp. The locations of the Asc I and BssH II restriction sites are shown. Cassettes 1 and 800-810 were unnecessary and removed from the assembly strategy. (Supporting material is available on Science Online.) Cassette 2 overlaps cassette 1104, and cassette 799 overlaps cassette 811.
FIGURE A8-2 Analysis of the assembly intermediates. (A) Not I and Sbf I double restriction digestion analysis of assembly 341-350 purified from E. coli. These restriction enzymes release the vector fragments (5.5 and 3.4 kb) from the 10-kb insert. Insert DNA was separated from the vector DNA on a 0.8% E-gel (Invitrogen). M indicates the 1-kb DNA ladder (New England Biolabs; NEB). (B) Analysis of assembly 501-600 purified from yeast. The 105-kb circles (100-kb insert plus 5-kb vector) were separated from the linear yeast chromosomal DNA on a 1% agarose gel by applying 4.5 V/cm for 3 hours. S indicates the BAC-Tracker supercoiled DNA ladder (Epicentre). (C) Not I restriction digestion analysis of the 11 ~100-kb assemblies purified from yeast. These DNA fragments were analyzed by FIGE on a 1% agarose gel. The expected insert size for each assembly is indicated. λ indicates the lambda ladder (NEB). (D) Analysis of the 11 pooled assemblies shown in (C) following topological trapping of the circular DNA and Not I digestion. One-fortieth of the DNA used to transform yeast is represented.
Our results indicated that these products cannot be stably maintained in E. coli, so recombined DNA had to be extracted from yeast. Multiplex polymerase chain reaction (PCR) was performed on selected yeast clones (fig. S3 and table S2). Because every 10-kb assembly intermediate was represented by a primer pair in this analysis, the presence of all amplicons would suggest an assembled 100-kb
intermediate. In general, 25% or more of the clones screened contained all of the amplicons expected for a complete assembly. One of these clones was selected for further screening. Circular plasmid DNA was extracted and sized on an agarose gel alongside a supercoiled marker. Successful second-stage assemblies with the vector sequence are ~105 kb in length (Fig. A8-2B). When all amplicons were produced following multiplex PCR, a second-stage assembly intermediate of the correct size was usually produced. In some cases, however, small deletions occurred. In other instances, multiple 10-kb fragments were assembled, which produced a larger second-stage assembly intermediate. Fortunately, these differences could easily be detected on an agarose gel before complete genome assembly.
Complete genome assembly
In preparation for the final stage of assembly, it was necessary to isolate microgram quantities of each of the 11 second-stage assemblies.51 As reported (Devenish and Newlon, 1982), circular plasmids the size of our second-stage assemblies could be isolated from yeast spheroplasts after an alkaline-lysis procedure. To further purify the 11 assembly intermediates, they were treated with exonuclease and passed through an anion-exchange column. A small fraction of the total plasmid DNA (1/100) was digested with Not I and analyzed by field-inversion gel electrophoresis (FIGE) (Fig. A8-2C). This method produced ~1 mg of each assembly per 400 ml of yeast culture (~1011 cells).
The method above does not completely remove all of the linear yeast chromosomal DNA, which we found could substantially decrease the yeast transformation and assembly efficiency. To further enrich for the 11 circular assembly intermediates, ~200 ng samples of each assembly were pooled and mixed with molten agarose. As the agarose solidifies, the fibers thread through and topologically “trap” circular DNA (Dean et al., 1973). Untrapped linear DNA can then be separated out of the agarose plug by electrophoresis, thus enriching for the trapped circular molecules. The 11 circular assembly intermediates were digested with Not I so that the inserts could be released. Subsequently, the fragments were extracted from the agarose plug, analyzed by FIGE (Fig. A8-2D), and transformed into yeast spheroplasts.51 In this third and final stage of assembly, an additional vector sequence was not required because the yeast cloning elements were already present in assembly 811-900.
To screen for a complete genome, multiplex PCR was carried out with 11 primer pairs, designed to span each of the 11 100-kb assembly junctions (table S3). Of 48 colonies screened, DNA extracted from one clone (sMmYCp235) produced all 11 amplicons. PCR of the wild-type positive control (YCpMmyc1.1) produced an indistinguishable set of 11 amplicons (Fig. A8-3A). To further demonstrate the complete assembly of a synthetic M. mycoides genome, intact DNA
_______________
51 Supporting material is available on Science Online.
FIGURE A8-3 Characterization of the synthetic genome isolated from yeast. (A) Yeast clones containing a completely assembled synthetic genome were screened by multiplex PCR with a primer set that produces 11 amplicons; one at each of the 11 assembly junctions. Yeast clone sMmYCp235 (235) produced the 11 PCR products expected for a complete genome assembly. For comparison, the natural genome extracted from yeast (WT, wild type) was also analyzed. PCR products were separated on a 2% E-gel (Invitrogen). L indicates the 100-bp ladder (NEB). (B) The sizes of the expected Asc I and BssH II restriction fragments for natural (WT) and synthetic (Syn235) M. mycoides genomes. (C) Natural (WT) and synthetic (235) M. mycoides genomes were isolated from yeast in agarose plugs. In addition, DNA was purified from the host strain alone (H). Agarose plugs were digested with Asc I or BssH II, and fragments were separated by clamped homogeneous electrical field (CHEF) gel electrophoresis. Restriction fragments corresponding to the correct sizes are indicated by the fragment numbers shown in (B).
was isolated from yeast in agarose plugs and subjected to two restriction analyses: Asc I and BssH II.52 Because these restriction sites are present in three of the four watermark sequences, this choice of digestion produces restriction patterns that are distinct from that of the natural M. mycoides genome (Figs. A8-1 and A8-3B). The sMmYCp235 clone produced the restriction pattern expected for a completely assembled synthetic genome (Fig. A8-3C).
Synthetic Genome Transplantation
Additional agarose plugs used in the gel analysis above (Fig. A8-3C) were also used in genome transplantation experiments.52 Intact synthetic M. mycoides
_______________
52 Supporting material is available on Science Online.
genomes from the sMmYCp235 yeast clone were transplanted into restriction-minus M. capricolum recipient cells, as described (Lartigue et al., 2009). Results were scored by selecting for growth of blue colonies on SP4 medium containing tetracycline and X-gal at 37°C. Genomes isolated from this yeast clone produced 5 to 15 tetracycline-resistant blue colonies per agarose plug, a number comparable to that produced by the YCpMmyc1.1 control. Recovery of colonies in all transplantation experiments was dependent on the presence of both M. capricolum recipient cells and an M. mycoides genome.
Semisynthetic Genome Assembly and Transplantation
To aid in testing the functionality of each 100-kb synthetic segment, semisynthetic genomes were constructed and transplanted. By mixing natural pieces with synthetic ones, the successful construction of each synthetic 100-kb assembly could be verified without having to sequence these intermediates. We cloned 11 overlapping natural 100-kb assemblies in yeast by using a previously described method (Leem et al., 2003). In 11 parallel reactions, yeast cells were cotransformed with fragmented M. mycoides genomic DNA (YCpMmyc1.1) that averaged ~100 kb in length and a PCR-amplified vector designed to overlap the ends of the 100-kb inserts. To maintain the appropriate overlaps so that natural and synthetic fragments could be recombined, the PCR-amplified vectors were produced via primers with the same 40-bp overlaps used to clone the 100-kb synthetic assemblies. The semisynthetic genomes that were constructed contained between 2 and 10 of the 11 100-kb synthetic subassemblies (Table A8-1). The
TABLE A8-1 Genomes that have been assembled from 11 pieces and successfully transplanted. Assembly 2-100, 1; assembly 101-200, 2; assembly 201-300, 3; assembly 301-400, 4; assembly 401-500, 5; assembly 501-600, 6; assembly 601-700, 7; assembly 701-799, 8; assembly 811-900, 9; assembly 901-1000, 10; assembly 1001-1104, 11. WM, watermarked assembly.
| Genome assembly | Synthetic fragments | Natural fragments |
| Reconstituted natural genome | None | 1–11 |
| 2/11 semisynthetic genome with one watermark | 5 WM, 10 | 1–4, 6–9, 11 |
| 8/11 semisynthetic genome without watermarks | 1–4, 6–8, 11 | 5, 9, 10 |
| 9/11 semisynthetic genome without watermarks | 1–4, 6–8, 10–11 | 5, 9 |
| 9/11 semisynthetic genome with three watermarks | 1, 2 WM, 3 WM, 4, 6, 7 WM, 8, 10–11 | 5, 9 |
| 10/11 semisynthetic genome with three watermarks | 1, 2 WM, 3 WM, 4, 5 WM, 6, 7 WM, 8, 10–11 | 9 |
| 11/11 synthetic genome, 811-820 correction of dnaA | 1, 2 WM, 3 WM, 4, 5 WM, 6, 7 WM, 8, 9–11 | None |
| 11/11 synthetic genome, 811-900 correction of dnaA | 1, 2 WM, 3 WM, 4, 5 WM, 6, 7 WM, 8, 9–11 | None |
production of viable colonies produced after transplantation confirmed that the synthetic fraction of each genome contained no lethal mutations. Only one of the 100-kb subassemblies, 811-900, was not viable.
Initially, an error-containing 811-820 clone was used to produce a synthetic genome that did not transplant. This was expected because the error was a single–base pair deletion that creates a frameshift in dnaA, an essential gene for chromosomal replication. We were previously unaware of this mutation. By using a semisynthetic genome construction strategy, we pinpointed 811-900 as the source for failed synthetic transplantation experiments. Thus, we began to reassemble an error-free 811-900 assembly, which was used to produce the sMmYCp235 yeast strain. The dnaA-mutated genome differs by only one nucleotide from the synthetic genome in sMmYCp235. This genome served as a negative control in our transplantation experiments. The dnaA mutation was also repaired at the 811-900 level by genome engineering in yeast (Noskov et al., 2010). A repaired 811-900 assembly was used in a final-stage assembly to produce a yeast clone with a repaired genome. This yeast clone is named sMmYCP142 and could be transplanted. A complete list of genomes that have been assembled from 11 pieces and successfully transplanted is provided in Table A8-1.
Characterization of the Synthetic Transplants
To rapidly distinguish the synthetic transplants from M. capricolum or natural M. mycoides, two analyses were performed. First, four primer pairs that are specific to each of the four watermarks were designed such that they produce four amplicons in a single multiplex PCR reaction (table S4). All four amplicons were produced by transplants generated from sMmYCp235, but not YCpMmyc1.1 (Fig. A8-4A). Second, the gel analysis with Asc I and BssH II, described above (Fig. A8-3C), was performed. The restriction pattern obtained was consistent with a transplant produced from a synthetic M. mycoides genome (Fig. A8-4B).
A single transplant originating from the sMmYCp235 synthetic genome was sequenced. We refer to this strain as M. mycoides JCVIsyn1.0. The sequence matched the intended design with the exception of the known polymorphisms, eight new single-nucleotide polymorphisms, an E. coli transposon insertion, and an 85-bp duplication (table S1). The transposon insertion exactly matches the size and sequence of IS1, a transposon in E. coli. It is likely that IS1 infected the 10-kb subassembly following its transfer to E. coli. The IS1 insert is flanked by direct repeats of M. mycoides sequence, suggesting that it was inserted by a transposition mechanism. The 85-bp duplication is a result of a nonhomologous end joining event, which was not detected in our sequence analysis at the 10-kb stage. These two insertions disrupt two genes that are evidently nonessential. We did not find any sequences in the synthetic genome that could be identified as belonging to M. capricolum. This indicates that there was a complete replacement of the M. capricolum genome by our synthetic genome during the trans-
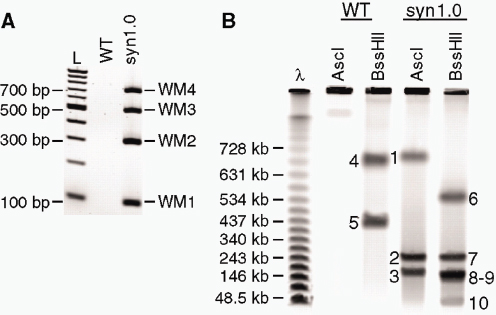
FIGURE A8-4 Characterization of the transplants. (A) Transplants containing a synthetic genome were screened by multiplex PCR with a primer set that produces four amplicons, one internal to each of the four watermarks. One transplant (syn1.0) originating from yeast clone sMmYCp235 was analyzed alongside a natural, nonsynthetic genome (WT) transplanted out of yeast. The transplant containing the synthetic genome produced the four PCR products, whereas the WT genome did not produce any. PCR products were separated on a 2% E-gel (Invitrogen). (B) Natural (WT) and synthetic (syn1.0) M. mycoides genomes were isolated from M. mycoides transplants in agarose plugs. Agarose plugs were digested with Asc I or BssH II and fragments were separated by CHEF gel electrophoresis. Restriction fragments corresponding to the correct sizes are indicated by the fragment numbers shown in Fig. A8-3B.
plant process. The cells with only the synthetic genome are self-replicating and capable of logarithmic growth. Scanning and transmission electron micrographs (EMs) of M. mycoides JCVI-syn1.0 cells show small, ovoid cells surrounded by cytoplasmic membranes (Fig. A8-5, C to F). Proteomic analysis of M. mycoides JCVI-syn1.0 and the wild-type control (YCpMmyc1.1) by two-dimensional gel electrophoresis revealed almost identical patterns of protein spots (fig. S4) that differed from those previously reported for M. capricolum (Lartigue et al., 2007). Fourteen genes are deleted or disrupted in the M. mycoides JCVI-syn1.0 genome; however, the rate of appearance of colonies on agar plates and the colony morphology are similar (compare Fig. A8-5, A and B). We did observe slight differences in the growth rates in a color-changing unit assay, with the JCVI-syn1.0 transplants growing slightly faster than the MmcyYCp1.1 control strain (fig. S6).
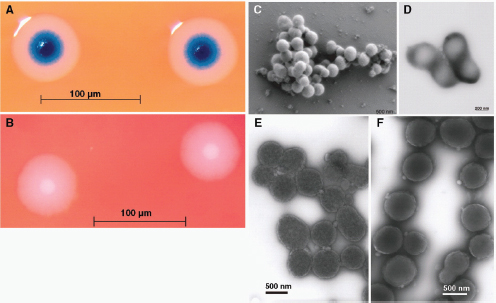
FIGURE A8-5 Images of M. mycoides JCVI-syn1.0 and WT M. mycoides. To compare the phenotype of the JCVI-syn1.0 and non-YCp WT strains, we examined colony morphology by plating cells on SP4 agar plates containing X-gal. Three days after plating, the JCVI-syn1.0 colonies are blue because the cells contain the lacZ gene and express β-lactosidase, which converts the X-gal to a blue compound (A). The WT cells do not contain lacZ and remain white (B). Both cell types have the fried egg colony morphology characteristic of most mycoplasmas. EMs were made of the JCVI-syn1.0 isolate using two methods. (C) For scanning EM, samples were postfixed in osmium tetroxide, dehydrated and critical point dried with CO2, and visualized with a Hitachi SU6600 SEM at 2.0 keV. (D) Negatively stained transmission EMs of dividing cells with 1% uranyl acetate on pure carbon substrate visualized using JEOL 1200EX CTEM at 80 keV. To examine cell morphology, we compared uranyl acetate–stained EMs of M. mycoides JCVI-syn1.0 cells (E) with EMs of WT cells made in 2006 that were stained with ammonium molybdate (F). Both cell types show the same ovoid morphology and general appearance. EMs were provided by T. Deerinck and M. Ellisman of the National Center for Microscopy and Imaging Research at the University of California at San Diego.
Discussion
In 1995, the quality standard for sequencing was considered to be one error in 10,000 bp, and the sequencing of a microbial genome required months. Today, the accuracy is substantially higher. Genome coverage of 30 to 50× is not unusual, and sequencing only requires a few days. However, obtaining an error-free genome that could be transplanted into a recipient cell to create a new cell controlled only by the synthetic genome was complicated and required many quality-control steps. Our success was thwarted for many weeks by a single–base
pair deletion in the essential gene dnaA. One wrong base out of more than 1 million in an essential gene rendered the genome inactive, whereas major genome insertions and deletions in nonessential parts of the genome had no observable effect on viability. The demonstration that our synthetic genome gives rise to transplants with the characteristics of M. mycoides cells implies that the DNA sequence on which it is based is accurate enough to specify a living cell with the appropriate properties.
Our synthetic genomic approach stands in sharp contrast to various other approaches to genome engineering that modify natural genomes by introducing multiple insertions, substitutions, or deletions (Itaya et al., 2005; Itaya, 1995; Mizoguchi et al., 2007; Chun et al., 2007; Wang et al., 2009). This work provides a proof of principle for producing cells based on computer-designed genome sequences. DNA sequencing of a cellular genome allows storage of the genetic instructions for life as a digital file.
The synthetic genome described here has only limited modifications from the naturally occurring M. mycoides genome. However, the approach we have developed should be applicable to the synthesis and transplantation of more novel genomes as genome design progresses (Khalil and Collins, 2010). We refer to such a cell controlled by a genome assembled from chemically synthesized pieces of DNA as a “synthetic cell,” even though the cytoplasm of the recipient cell is not synthetic. Phenotypic effects of the recipient cytoplasm are diluted with protein turnover and as cells carrying only the transplanted genome replicate. Following transplantation and replication on a plate to form a colony (>30 divisions or >109-fold dilution), progeny will not contain any protein molecules that were present in the original recipient cell (Lartigue et al., 2007).53 This was previously demonstrated when we first described genome transplantation (Lartigue et al., 2007). The properties of the cells controlled by the assembled genome are expected to be the same as if the whole cell had been produced synthetically (the DNA software builds its own hardware).
The ability to produce synthetic cells renders it essential for researchers making synthetic DNA constructs and cells to clearly watermark their work to distinguish it from naturally occurring DNA and cells. We have watermarked the synthetic chromosome in this and our previous study (Gibson, et al., 2008b).
If the methods described here can be generalized, design, synthesis, assembly, and transplantation of synthetic chromosomes will no longer be a barrier to
_______________
53 A mycoplasma cell, with a mass of about 10–13 g, contains fewer than 106 molecules of protein. (If it contains 20% protein, this is equivalent to 2 × 10–14 g of protein per cell. At a molecular mass of 120 daltons per amino acid residue, each cell contains (2 × 10–14)/120 = 1.7 × 10–16 mol of peptide residues. This is equivalent to (1.7 × 10–16) × (6 × 1023) = 1 × 108 residues per cell. If the average size of a protein is 300 residues, then a cell contains about 3 × 105 protein molecules.) After 20 cell divisions the number of progeny exceeds the total number of protein molecules in the recipient cell. So, following transplantation and replication to form a colony on a plate, most cells will contain no protein molecules that were present in the original recipient cell.
the progress of synthetic biology. We expect that the cost of DNA synthesis will follow what has happened with DNA sequencing and continue to exponentially decrease. Lower synthesis costs combined with automation will enable broad applications for synthetic genomics.
We have been driving the ethical discussion concerning synthetic life from the earliest stages of this work (Cho et al., 1999; Garfinkel et al., 2007). As synthetic genomic applications expand, we anticipate that this work will continue to raise philosophical issues that have broad societal and ethical implications. We encourage the continued discourse.
References
G. A. Benders et al., Nucleic Acids Res. 38, 2558 (2010).
M. K. Cho, D. Magnus, A. L. Caplan, D. McGee, Science 286, 2087, 2089 (1999).
J. Y. Chun et al., Nucleic Acids Res. 35, e40 (2007).
W. W. Dean, B. M. Dancis, C. A. Thomas Jr., Anal. Biochem. 56, 417 (1973).
R. J. Devenish, C. S. Newlon, Gene 18, 277 (1982).
R. D. Fleischmann et al., Science 269, 496 (1995).
M. S. Garfinkel, D. Endy, G. L. Epstein, R. M. Friedman, Biosecur. Bioterror. 5, 359 (2007).
D. G. Gibson et al., Proc. Natl. Acad. Sci. U.S.A. 105, 20404 (2008a).
——, Science 319, 1215 (2008b).
——, Nat. Methods 6, 343 (2009a).
D. G. Gibson, Nucleic Acids Res. 37, 6984 (2009b).
J. I. Glass et al., Proc. Natl. Acad. Sci. U.S.A. 103, 425 (2006).
C. A. Hutchison III et al., Science 286, 2165 (1999).
M. Itaya, FEBS Lett. 362, 257 (1995).
M. Itaya, K. Tsuge, M. Koizumi, K. Fujita, Proc. Natl. Acad. Sci. U.S.A. 102, 15971 (2005).
A. S. Khalil, J. J. Collins, Nat. Rev. Genet. 11, 367 (2010).
C. Lartigue et al., Science 317, 632 (2007).
——, Science 325, 1693 (2009).
S.-H. Leem et al., Nucleic Acids Res. 31, e29 (2003).
H. Mizoguchi, H. Mori, T. Fujio, Biotechnol. Appl. Biochem. 46, 157 (2007).
V. N. Noskov, T. H. Segall-Shapiro, R. Y. Chuang, Nucleic Acids Res. 38, 2570 (2010).
F. Sanger et al., Nature 265, 687 (1977).
H. O. Smith, J. I. Glass, C. A. Hutchison III, J. C. Venter, in Accessing Uncultivated Microorganisms: From the Environment to Organisms and Genomes and Back, K. Zengler, Ed. (American Society for Microbiology, Washington, DC, 2008), p. 320.
J. C. Venter, Nature 464, 676 (2010).
H. H. Wang et al., Nature 460, 894 (2009).
SYNTHETIC BIOLOGY “FROM SCRATCH”
Gerald F. Joyce54
The Darwinian Basis of Life
The previous presentations in this session described a mix of top-down and bottom-up approaches to synthetic biology. This presentation is intended to take us all the way down to the very bottom as we discuss synthetic biology “from scratch,” that is, life from scratch. Life is something we all know when we see it, right? Consider the giant bacterium Titanospirillum velox, which we all would agree is alive. Yet in a recent publication by Richard Hoover that appeared in the online Journal of Cosmology (Hoover, 2011), he describes what he believes is an extraterrestrial analog of Titanospirillum, found within a certain class of carbonaceous meteorites. Much of the popular media fell for it, abetted by a Fox News report showing a micrograph of Titanospirillum, rather than the mineralic artifacts within the meteorite that most experts agree have nothing to do with life.
But surely the experts must know life when they see it, right? And therefore the experts must know how to define life. Indeed some scientists have advanced a definition, or at least a working definition, of life. Some, including me, have pointed to the so-called NASA working definition of life, which is a self-sustained chemical system that is capable of undergoing Darwinian evolution (Joyce, 1994). In several of the presentations at this workshop, evolution has been referred to as the distinguishing, if not defining, feature of life. The Darwinian paradigm is the only one we know that explains how biological complexity can sustain itself against the vagaries of a changing environment. Darwinian evolution has a rigorous scientific foundation, but the word “life” is not a scientific term. As Andrew Ellington would say, it is a term for poets and philosophers, and scientists, let alone a government agency such as NASA, should not be in the business of trying to define it.
Without becoming bogged down in definitions, we can agree that life is all about Darwinian evolution, and that scientists understand what Darwinian evolution is all about. The key principles of Darwinian evolution are, first, heritable variation of form and function among a population of individuals; second, competition for finite resources by those individuals; and third, preferential reproduction of variants that operate most effectively in the competitive environment. In The Origin of Species, Charles Darwin made the observation: “Owing to this struggle for life, any variation…if it be in any degree profitable to an individual of any species, in its infinitely complex relations to other organic beings and to external nature…will generally be inherited by its offspring” (Darwin, 1859;
_______________
54 The Scripps Research Institute.
italics added). If life has a slogan, if not a formal definition, it would be inherit profitable variation. I, for one, would be comfortable abiding by this slogan.
In considering synthetic biology from scratch, the focus is on the evolution of functional molecules, rather than organisms. The principles of directed molecular evolution are the same as those for the Darwinian evolution of organisms, again captured by the slogan inherit profitable variation. In chemical terms, Darwinian evolution involves three processes: (1) reproduction of information-carrying molecules (inherit), (2) selection of molecules that meet some fitness criteria (profitable), and (3) maintenance of chemical diversity among the population of molecules (variation).
Over the past two decades, the technology of direct molecular evolution has become very powerful, but also routine. There are many methods for introducing molecular variation, both for generating initial combinatorial libraries and for maintaining variation in a population. Individuals that meet some fitness test can be separated based on a high-throughput screen or a selection procedure. Through screening, one or more highly advantageous variants can be identified and subsequently mutagenized to provide a second-generation combinatorial library. This process of iterative high-throughput screening can be a powerful discovery tool, although it does not fully capture the power of Darwinian evolution, which requires that the population as a whole be subject to repeated rounds of selection and randomization. Maintaining a diverse population is key to exploring the fitness landscape because it allows both dominant and subdominant individuals to give rise to novel variants. The subdominant individuals, following the acquisition of a few beneficial mutations, may yield descendants that are more advantageous compared to the previously dominant individuals. Ideally, the loss of variation due to selection should be compensated by the introduction of novel variation throughout the course of evolution.
There are many methods for selecting profitable molecules. For example, molecules can be selected based on their specific chromatographic mobility, their ability to withstand exposure to some physical condition, their ability to bind to a target ligand (aptamers), or their ability to catalyze a particular chemical transformation (enzymes). More complex selection criteria can be imposed, such as a combination of both positive and negative selection, conditional selection, and the selection for multiple attributes.
Finally, there are various methods for reproducing the profitable molecules in order to bring about the inheritance of selectively advantageous traits. If the selected molecules are DNA or RNA, then it is straightforward to achieve their amplification by using the appropriate polymerase enzyme(s), resulting in large numbers of progeny. If the selected molecules are proteins, which cannot be amplified directly, then one must amplify nucleic acid molecules that encode and are physically linked to the corresponding proteins. Techniques such as phage display, ribosome display, and compartmentalized self-replication make it possible to amplify an ensemble of genes that encode a corresponding set of proteins. The same principle of genetic encoding can be used to amplify other
informational macromolecules, such as peptide analogs, polysaccharides, and even multicomponent organic molecules.
All of the amplification methods discussed above are not part of a self-sustained evolving system because they rely on informational macromolecules that are not themselves subject to evolution within the system. Protein polymerases, phage particles, and ribosomes all are the products of Darwinian evolution in biology. They are employed as operators in laboratory evolution systems, but their information content is not subject to evolution within those systems.
The experimental pursuit of life from scratch began in 1953 with the work of Stanley Miller, then a graduate student at the University of Chicago, who sought to cook up a prebiotic soup from entirely abiotic ingredients (Miller, 1953). None of those ingredients had information content derived from Darwinian processes. It has been almost 60 years since Miller’s classic experiments, and life has yet to be produced starting from a prebiotic soup. However, considerable progress has been made toward synthesizing life using other methods, and it now appears that the production of life from scratch will be achieved in the near future. Some research efforts along these lines have attempted, as Miller did, to recapitulate the historical origins of life on Earth. Other efforts take inspiration from the origins of life on Earth, but aim for a second origin that would occur under decidedly artificial laboratory conditions. Considerable attention has been directed toward the critical role that RNA is thought to have played in the early history of life on Earth, during an era referred to as the “RNA world” (Atkins et al., 2011). RNA continues to play a central role in contemporary biology, which further motivates the goal of constructing RNA-based life from scratch.
Self-Sustained Darwinian Evolution
An explicit aim of my own research program is to construct a system of RNA molecules that undergo self-sustained Darwinian evolution. In fact this goal was recently achieved, although the system still lacks the complexity and inventiveness of what one might regard as life. The self-sustained evolving system employs populations of RNA enzymes that catalyze the RNA-templated joining of RNA substrates. The enzymes contain ~55 essential nucleotides and can be made to join pairs of RNA substrates of almost any sequence (Rogers and Joyce, 2001). If the substrates, once joined, form additional copies of the enzymes, then self-replication can be achieved. The newly formed enzymes behave similarly, resulting in exponential growth (Paul and Joyce, 2002). However, the process cannot be sustained indefinitely and is informationally restricted by the requirement that the original and newly formed enzymes must have the same sequence.
An improved version of the replication system employs two different RNA enzymes that catalyze each other’s synthesis, enabling their cross-replication and sustained exponential growth (Kim and Joyce, 2004; Lincoln and Joyce, 2009). Each enzyme of the cross-replicating pair contains two substrate-binding domains that recognize corresponding oligonucleotide substrates through Watson-Crick pairing.
During cross-replication, the “Watson” enzyme joins two pieces of RNA to form the “Crick” enzyme, while the “Crick” enzyme joins two pieces of RNA to form the “Watson” enzyme. Information is passed back and forth between these two enzymes in the form of particular sequences within the two substrate-binding domains.
Following optimization of the cross-replication system, it now is possible to achieve 100-fold amplification in just a few hours at a constant temperature and in the absence of any biological materials (Lincoln and Joyce, 2009). The only informational macromolecules in the system are the enzymes and their components, which themselves are subject to Darwinian evolution within the system. The only other components are MgCl2, a buffer to maintain pH, and H2O. Evolution can occur because there are many potential variants of the cross-replicating enzymes that must compete for a finite supply of substrates and can undergo mutation through recombination of the two substrate-binding domains.
Beginning with a small seed of the cross-replicating enzymes, amplification occurs with exponential growth, limited only by the amount of substrates that are available. The amplification profile follows the logistic growth equation [enzyme]t = a / (1 + be–ct), where a is the maximum extent of amplification, b is the degree of sigmoidicity, and c is the exponential growth rate. This equation also describes population growth for biological organisms constrained by the carrying capacity of their local environment.
Cross-replication of the RNA enzymes can be sustained indefinitely by continuing to supply the necessary substrates. This is most conveniently achieved through a serial transfer procedure, whereby a small aliquot is taken from a spent reaction mixture and transferred to a new reaction vessel that contains a fresh supply of substrates. The new reaction mixture contains only those enzymes that were carried over in the aliquot, and these enzymes immediately resume exponential amplification in the new mixture. Within a period of 24 hours, an overall amplification factor of >109 can be achieved (Lincoln and Joyce, 2009).
Self-sustained exponential amplification provides the growth engine for Darwinian evolution, but it is the diversity of enzymes in the population, their differential reproductive fitness, and their capacity for mutation that provides the opportunity to evolve macromolecular information. Profitable variation in the system emerges through particular combinations of substrates that form cross-replicators with high reproductive fitness. The genetic basis for this variation is represented by the two “loci”—the two substrate-binding domains—that can exist as any of a large number of possible “alleles.” Each allele can be made to encode a different corresponding phenotypic trait, embodied by the functional domain that is physically linked to that allele. If there are n potential variants of the first allele and m potential variants of the second allele, then the combinatorial complexity of the system is n × m.
As an example, a population of cross-replicating enzymes was constructed with 12 different alleles at each of the two loci, providing a combinatorial complexity of 12 × 12 = 144. Each variant allele was linked to a different form of the catalytic center of the enzyme, which resulted in differential reproductive fitness
for various combinations of alleles at the two loci (Lincoln and Joyce, 2009). The evolution process was seeded with 12 different cross-replicating pairs that, due to mutation, could give rise to any of the other 132 combinations. Evolution was allowed to proceed in a self-sustained manner for 100 hours, with an overall amplification factor of 1025. During this time the starting 12 replicators decreased in abundance as novel variants arose and came to dominate the population. Three of these novel variants together accounted for about half of the population members after 100 hours. The basis for their selective advantage was shown to be their relatively fast amplification rate and their propensity to cross-mutate to form additional copies of each other.
Toward Inventive Evolution
The capacity for the invention of novel function within the context of Darwinian evolution depends on both the genetic complexity of the system and the functional richness of the corresponding phenotypes. A population of 144 replicators is represented by only ~7 bits of genetic information. This is much less than the genetic complexity of even the simplest biological systems, which have an information content of 2 bits per base pair of genetic material. In principle, the synthetic RNA-based evolving system could have an information content of 30 bits, considering the 15 total base pairs within the two genetic loci. This would provide a molecular diversity of 415 = 109. However, it would not be possible to manage such high diversity because of the vast number of substrate molecules that would need to be present in the reaction mixture. These would slow replication as each enzyme must find its cognate substrates from among the complex mixture.
Current research efforts in our laboratory aim to maximize the genetic complexity of the self-sustained evolving system within the practical limits of both generating and harvesting molecular diversity. We constructed a population of cross-replicating enzymes with 64 different alleles for each of the two genetic loci, providing a combinatorial complexity of 64 × 64 = 4,096. In this test population, each variant allele was linked to the same functional sequence. This was done to assess the extent to which differences in genotype alone would result in differential fitness, something that should be minimized to allow the broadest exploration for novel phenotypes. Starting with this library, 106-fold selective amplification was carried out; then individuals were cloned from the population and sequenced. Indeed two sources of genotype-related bias were identified, and these biases were eliminated from subsequent populations that were constructed.
Although the replicative function is the most important aspect of phenotype, replication can be made contingent on other functions so that fitness reflects the ability to execute those other functions. There is a stem-loop region of the RNA enzyme that supports the structure of the catalytic center and is generic in sequence, so long as it forms a stable secondary structure. This stem loop can be replaced by a ligand-binding (aptamer) domain, configured so that in the absence
of the ligand the domain is unstructured, whereas in the presence of the ligand the domain adopts a folded state that supports the active structure of the enzyme. In this way replication can be made contingent upon recognition of the target ligand (Lam and Joyce, 2009).
As two examples, the supporting stem loop of the enzyme was replaced by an aptamer that recognizes either theophylline or FMN (Lam and Joyce, 2009). In the absence of the ligand there is no replication, but in the presence of the ligand there is sustained exponential growth. Furthermore, the exponential growth rate depends on the concentration of the ligand relative to the Kd (equilibrium binding constant) of the ligand-binding domain. This method for quantitative, ligand-dependent exponential amplification may have applications in biosensing and molecular diagnostics. It is analogous to quantitative PCR for the measurement of nucleic acid targets, but it operates at a constant temperature and can be generalized to non–nucleic acid targets, including proteins, drugs, and metabolites that can be recognized by an aptamer (Lam and Joyce, 2011).
The self-sustained evolution system could, in principle, be used to discover novel aptamers. A genetically encoded random-sequence domain could be placed adjacent to the catalytic center such that ligand recognition would result in selective amplification of the functional molecules. This would require sufficient genetic complexity to encode a population containing enough variants to include molecules with the desired function. Our most recently constructed populations of cross-replicating enzymes have 256 different alleles for each of the two loci, providing a combinatorial complexity of 256 × 256 = 65,536. This still is likely to be insufficient to derive novel aptamers within the context of self-sustained evolution, unless the target ligands are compounds that have a strong propensity to bind to RNA.
The populations with a combinatorial complexity of 256 × 256 were constructed by two different methods. The first involved serial production of all 256 + 256 = 512 allelic variants, synthesizing individual DNA templates to link each genotype-phenotype combination, then transcribing the templates to generate the corresponding RNAs. The advantage of this approach is that each variant is stored in a separate location, allowing one to prepare custom sublibraries. The disadvantage is that, at cost of synthesis of ~$10 per variant, the method cannot be extended to much more complex populations. The second approach for constructing the populations involved a novel split-and-pool method that makes it possible to synthesize the entire population in parallel. The parallel method enables the construction of populations with as many as 109 different members, although, as discussed above, it would be difficult to manage such high-diversity populations throughout the course of a self-sustained evolution experiment.
With increasing population complexity it becomes important to consider the nature of the code that relates particular genetic sequences to their corresponding functional sequences. This code can be chosen arbitrarily, but evolutionary optimization likely will benefit if more closely related genetic sequences correspond to more closely related functional sequences. The code need not have a collinear
3:1 relationship, as is the case for the genetic code in biology which relates trinucleotide codons within mRNA to individual amino acids within proteins. Furthermore, the same code need not apply for all genotype positions, although this simplification clearly has great selective advantage for natural biology. In synthetic biology, the experimenter must decide what genetic code to employ based on theoretical and practical considerations.
In preparing the populations of 256 × 256 combinatorial complexity, two different “sparse” codes were implemented, whereby each nucleotide within the genotype region encodes one or more nucleotides within the phenotype region. For the serially constructed population, each of four genetic nucleotides encodes three noncontiguous nucleotides in the functional region, with a different codon relationship for each genetic position. For the parallel library, each of four genetic nucleotides encodes either one or two contiguous nucleotides in the functional region, again with a different codon relationship for each genetic position. Experimental studies are under way to assess the operational characteristics of these different codes.
When practicing synthetic biology from scratch the experimenter makes the rules and allows Darwinian evolution to play the game. The capacity of the system to invent novel function is the most important measure of its robustness. Inventiveness should be as broad as possible so that the system can adapt to unanticipated changes in its environment. Life on Earth, although vulnerable to extreme changes of environmental conditions, has demonstrated extraordinary resiliency and inventiveness in adapting to highly disparate niches. Perhaps the most significant invention of life is a genetic system that has an extensible capacity for inventiveness, something that likely will not be achieved soon for synthetic biological systems. However, once informational macromolecules are given the opportunity to inherit profitable variation through self-sustained Darwinian evolution, they just may take on a life of their own.
Acknowledgments
Jeff Rogers developed the RNA enzyme that provides the basis for replication, Natasha Paul provided the first demonstration of a self-replicating RNA enzyme, Dong-Eun Kim converted the system to a cross-replication format, Tracey Lincoln first achieved self-sustained Darwinian evolution of RNA enzymes, Bianca Lam made replication contingent upon recognition of a target ligand, and Michael Robertson and Jonathan Sczepanski constructed increasingly complex populations of the cross-replicating RNA enzymes. This work was supported by grants from the National Aeronautics and Space Administration (NNX10AQ91G), the National Institutes of Health (GM065130), the National Science Foundation (MCB-0948161), and the Defense Advanced Research Projects Agency (DSO BAA-09-63).
References
Atkins, J. F., R. F. Gesteland, and T. R. Cech (eds). 2011. RNA Worlds: From Life’s Origins to Diversity in Gene Regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Darwin, C. R. 1859. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. London: John Murray. P. 61.
Hoover, R. B. 2011. Fossils of cyanobacteria in CI1 carbonaceous meteorites. Journal of Cosmology 13, published online March 2011.
Joyce, G. F. 1994. Foreword. In Origins of Life: The Central Concepts, edited by D. W. Deamer and G. R. Fleischacker. Boston: Jones and Bartlett. Pp. xi-xii.
Kim, D. E., and G. F. Joyce. 2004. Cross-catalytic replication of an RNA ligase ribozyme. Chemistry and Biology 11:1505-1512.
Lincoln, T. A., and G. F. Joyce. 2009. Self-sustained replication of an RNA enzyme. Science 323: 1229-1232.
Lam, B. J., and G. F. Joyce. 2009. Autocatalytic aptazymes enable ligand-dependent exponential amplification of RNA. Nature Biotechnology 27:288-292.
——. 2011. An isothermal system that couples ligand-dependent catalysis to ligand-independent exponential amplification. Journal of the American Chemical Society 133:3191-3197.
Miller, S. L. 1953. A production of amino acids under possible primitive Earth conditions. Science 117:528-529.
Paul, N., and G. F. Joyce. 2002. A self-replicating ligase ribozyme. Proceedings of the National Academy of Sciences of the United States of America 99:12733-12740.
Rogers, J., and G. F. Joyce. 2001. The effect of cytidine on the structure and function of an RNA ligase ribozyme. RNA 7:395-404.
MANUFACTURING MOLECULES THROUGH METABOLIC ENGINEERING55
Metabolic engineering has the potential to produce from simple, readily available, inexpensive starting materials a large number of chemicals that are currently derived from nonrenewable resources or limited natural resources. Microbial production of natural products has been achieved by transferring product-specific enzymes or entire metabolic pathways from rare or genetically intractable organisms to those that can be readily engineered, and production of unnatural specialty chemicals, bulk chemicals,
_______________
55 From Keasling, J. D. 2010. Manufacturing molecules through metabolic engineering. Science 330 (6009): 1355-1358. Reprinted with permission from AAAS.
56 Joint BioEnergy Institute, 5885 Hollis Street, Emeryville, CA 94608, USA.
57 Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
58 Synthetic Biology Engineering Research Center, Department of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA.
E-mail: keasling@berkeley.edu
and fuels has been enabled by combining enzymes or pathways from different hosts into a single microorganism and by engineering enzymes to have new function. Whereas existing production routes use well-known, safe, industrial microorganisms, future production schemes may include designer cells that are tailor-made for the desired chemical and production process. In any future, metabolic engineering will soon rival and potentially eclipse synthetic organic chemistry.
The term “metabolic engineering” was coined in the late 1980s–early 1990s (Bailey, 1991). Since that time, the range of chemicals that can be produced has expanded substantially, in part due to notable advances in fields adjacent to metabolic engineering: DNA sequencing efforts have revealed new metabolic reactions and variants of enzymes from many different organisms; extensive databases of gene expression, metabolic reactions, and enzyme structures allow one to query for desired reactions and design or evolve novel enzymes for reactions that do not exist; new genetic tools enable more precise control over metabolic pathways; new analytical tools enable the metabolic engineer to track RNA, protein, and metabolites in a cell to identify pathway bottlenecks; and detailed models of biology aid in the design of enzymes and metabolic pathways. Yet even with these substantial developments, microbial catalysts are not as malleable as those in synthetic organic chemistry, and metabolic engineers must weigh many trade-offs in the development of microbial catalysts: (i) cost and availability of starting materials (e.g., carbon substrates); (ii) metabolic route and corresponding genes encoding the enzymes in the pathway to produce the desired product; (iii) most appropriate microbial host; (iv) robust and responsive genetic control system for the desired pathways and chosen host; (v) methods for debugging and debottlenecking the constructed pathway; and (vi) ways to maximize yields, titers, and productivities (Fig. A10-1). Unfortunately, these design decisions cannot be made independently of each other: Genes cannot be expressed, nor will the resulting enzymes function, in every host; products or metabolic intermediates may be toxic to one host but not another host; different hosts have different levels of sophistication of genetic tools available; and processing conditions (e.g., growth, production, product separation and purification) are not compatible with all hosts. Even with these many challenges, metabolic engineering has been successful for many applications, and with continued developments more applications will be possible.
Starting Materials, Products, and Metabolic Routes
One area where metabolic engineering has a sizable advantage over synthetic organic chemistry is in the production of natural products, particularly active pharmaceutical ingredients (APIs), some of which are too complex to be chemically synthesized and yet have a value that justifies the cost of developing a
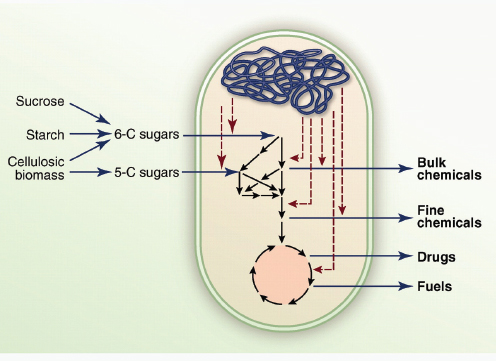
FIGURE A10-1 Conversion of sugars to chemicals by means of microbial catalysts.
genetically engineered microorganism. The cost of starting materials is generally a small fraction of their cost, and relatively little starting material is necessary so availability is not an issue. Most APIs fall into three classes of natural products, and many of the biosynthetic pathways for their precursors have been reconstituted in heterologous hosts.
Alkaloids are nitrogen-containing, low molecular weight compounds found primarily in and derived from plants and widely used as drugs. Two recent studies conclude that the large group of benzyl isoquinoline alkaloids (BIAs) will one day be producible in Escherichia coli and Saccharomyces cerevisiae (Hawkins and Smolke, 2008). Unfortunately, the BIAs are only one of four major alkaloid groups, all of which are produced through different pathways. As the metabolic pathways for other alkaloids are discovered in their natural producers, many more of these valuable molecules could be produced microbially.
Polyketides and nonribosomal peptides (NRPs) have found broad use as APIs, veterinary agents, and agrochemicals. Naturally occurring polyketides and NRPs are produced by a number of bacteria and fungi using large, modular enzymes. Their titers and yields in the native producers have been improved through traditional strain engineering and advanced metabolic engineering. More recently, some of the most valuable molecules have been produced with engineered industrial hosts (Pfeifer et al., 2001). Recombination of various synthase modules
allows one to produce a nearly infinite range of chemicals (Menzella et al., 2005; Siewers et al., 2010), opening up the possibility that they may one day be used to produce fine and bulk chemicals. Isoprenoids have found use as fragrances and essential oils, nutraceuticals, and pharmaceuticals. Many isoprenoids have been produced microbially, including carotenoids and various plant-derived terpenes (Martin et al., 2003; Ro et al., 2006; Leonard et al., 2010), taking advantage of terpene synthases to form the most complicated part of the molecules and hydroxylases to introduce hydroxyl group that can be subsequently functionalized chemically or biologically (Ro et al., 2006; Chang et al., 2007). Isoprenoids are one of the few classes of natural products where there are alternative precursor production pathways. An example of using metabolic engineering and synthetic chemistry together to produce an API is the semisynthesis of the antimalarial drug artemisinin with S. cerevisiae engineered to produce artemisinic acid, the most complex part of the molecule, and synthetic chemistry to produce artemisinin from the microbially sourced artemisinic acid (Martin et al., 2003; Ro et al., 2006; Chang et al., 2007). Beyond producing natural products, laboratory evolution or rational engineering of terpene cyclases, terpene hydroxylases, and a host of other terpene-functionalizing enzymes (Leonard et al., 2010; Yoshikuni et al., 2006; Schmidt-Dannert et al., 2000; Dietrich et al., 2009) and combinatorial expression of these evolved enzymes in a heterologous host will enable the production of unnatural terpenes, some of which might be more effective than the natural product for the treatment of human disease.
Although individual metabolic pathways have been developed to produce natural products derived from a single pathway, there is an opportunity to synthesize multisubstituent APIs (e.g., Taxol) or other molecules from the products of multiple biosynthetic pathways. This will require simultaneous expression of multiple precursor pathways in a single microorganism, as well as “ligases” that can assemble multiple substituents together into a single molecule. The benefit would be the synthesis of complicated molecules that might not otherwise be produced.
Although not as valuable as pharmaceuticals, many fine chemicals have been produced economically from natural and engineered microorganisms, including amino acids, organic acids, vitamins, flavors, fragrances, and nutraceuticals. For fine chemicals, profit margins are generally much lower than for APIs and may be affected by substrate availability and cost. Some of these molecules are sufficiently complicated that they cannot be produced economically by any route other than biological production, whereas others have chemical routes. For some important products (fragrances, flavors, amino acids), heterologous hosts have been engineered to enhance their production. Yet we have barely begun to investigate what will be possible to produce.
In contrast, bulk chemicals such as solvents and polymer precursors are rarely produced from microorganisms, because they can be produced inexpensively from petroleum by chemical catalysis. Due to fluctuations in petroleum prices and recognition of dwindling reserves, trade imbalances, and political considerations, it is now possible to consider production of these inexpensive
chemicals from low-cost starting materials such as starch, sucrose, or cellulosic biomass (e.g., agricultural and forest waste, dedicated energy crops, etc.) with a microbial catalyst. For example, 1,3-propanediol (1,3-PDE), a useful intermediate in the synthesis of polyurethanes and polyesters, is now being produced from glucose by E. coli engineered with genes from Klebsiella pneumoniae and S. cerevisiae (Nakamura and Whited, 2003). There is an opportunity to produce many other bulk chemicals (e.g., polymer precursors) by using metabolically engineered cells, but the key will be to produce the exact molecule needed for existing products rather than something “similar but green” that will require extensive product testing before it can be used.
By far the highest-volume (and lowest-margin) application for engineered metabolism is the production of transportation fuels. For many of the same reasons that it is desirable to produce petroleum-derived chemicals using biological systems, it is desirable to produce transportation fuels from readily available, inexpensive, renewable sources of carbon. There is a long history of using microorganisms to produce alcohols, primarily ethanol and butanol. Although much of the work on these alcohols was done by traditional strain mutagenesis and selection, more recent work focused on engineering yeasts and bacteria to produce ethanol or butanol from a variety of sugars while eliminating routes to side products and improving the tolerance of the host to the alcohol (Steen et al., 2008). Larger, branched-chain alcohols can be produced by way of the Ehrlich pathway. By incorporating broad substrate-range 2-keto acid decarboxylases and alcohol dehydrogenases, several microbes have now been engineered to produce these fuels (Donaldson et al., 2007; Atsumi et al., 2008). These alcohols are generally considered better fuels than ethanol and butanol and can also be used to produce a variety of commodity chemicals.
Recent advances in metabolic pathway and protein engineering have made it possible to engineer microorganisms to produce hydrocarbons with properties similar or identical to those of petroleum-derived fuels and thus compatible with our existing transportation infrastructure. Linear hydrocarbons (alkanes, alkenes, and esters) typical of diesel and jet fuel can be produced by way of the fatty acid biosynthetic pathway (Steen et al., 2010; Beller et al., 2010; Schirmer et al., 2010). For diesel in cold weather and jet fuel at high altitudes, branches in the chain are beneficial—regularly branched and cyclic hydrocarbons of different sizes with diverse structural and chemical properties can be produced via the isoprenoid biosynthetic pathway (Withers et al., 2007; Renninger and McPhee, 2008). Both the fatty acid–derived and the isoprenoid-derived fuels diffuse (or are pumped) out of the engineered cells and phase separate in the fermentation, making purification simple and reducing fuel cost.
Although the pathways described above produce a wide range of fuel-like molecules, there are many other molecules that one might want to produce, such as short, highly branched hydrocarbons (e.g., 2,2,4-trimethyl pentane or isooctane) that would be excellent substitutes for petroleum-derived gasoline. Additionally, most petroleum fuels are mixtures of large numbers of components
that together create the many important properties of the fuels. It should be possible to engineer single microbes or microbial consortia to produce a mixture of fuels from one of the biosynthetic pathways or from multiple biosynthetic pathways. Indeed, some enzymes produce mixtures of products from a single precursor—maybe these enzymes could be tuned to produce a fuel mixture ideal for a particular engine type or climate.
To make these new fuels economically viable, we must tap into inexpensive carbon sources (namely, sugars from cellulosic biomass). Given the variety of sugars in cellulosic biomass, the fuel producer must be able to consume both five- and six-carbon sugars. Because many yeasts do not consume five-carbon sugars, recent developments in engineering yeast to catabolize these sugars will make production of these fuels more economically viable (Wisselink et al., 2009). Engineering fuel-producing microorganisms to secrete cellulases and hemicellulases to depolymerize these sugar polymers into sugars before uptake and conversion into fuels has the potential to substantially reduce the cost of producing the fuel.
Hosts and Expression Systems
From the applications cited above, it should be evident that the product, starting materials, and production process all affect host choice. Some of the most important qualities one must consider when choosing a host are whether the desired metabolic pathway exists or can be reconstituted in that host; if the host can survive (and thrive) under the desired process conditions (e.g., ambient versus extremes of temperature, pH, ionic strength, etc.); if the host is genetically stable (both with the introduced pathway and not susceptible to phage attack); and if good genetic tools are available to manipulate the host. Widely used, heterologous hosts include E. coli, S. cerevisiae, Bacillus subtilis, and Streptomyces coelicolor, to name a few. Although E. coli and S. cerevisiae excel in the genetic tools available, E. coli has the disadvantage of being susceptible to phage attack. And while B. subtilis and S. coelicolor have the ability to easily express polyketide synthases, they have fewer genetic tools available than either S. cerevisiae or E. coli. Although minimal, bacterial hosts may have scientific interest (Gibson et al., 2010), minimal hosts that require addition of many nutrients or that cannot cope with stresses in processing will probably not find a niche in industrial chemical or fuel production where cost is critical. Thus, it is essential to have genetic tools for existing industrial hosts that can grow on simple, inexpensive carbon sources and salts or on an inexpensive, undefined medium with minimal additions (Wang et al., 2009; Pósfai et al., 2006).
The key issue necessitating good genetic tools is the introduction of foreign genes encoding the metabolic pathway and control over their expression to maximize yields and titers. The genes encoding the transformational enzymes in metabolically engineered cells do not need to be highly expressed, but must be produced in catalytic amounts sufficient to adequately transform the metabolic
intermediates into the desired products at a sufficient rate. Expression of the desired genes at too high a level will rob the cell of metabolites that might otherwise be used to produce the desired molecule of interest, particularly important for production of low-margin chemicals, while underexpressed genes will create pathway bottlenecks. Furthermore, because intermediates of a foreign metabolic pathway can be toxic to the heterologous host (Martin et al., 2003), which results in decreased production of the desired final compound, it is essential that the relative levels of the enzymes be coordinated.
Central to any genetic manipulation is the vector used to carry and/or harbor the transforming DNA in the host. Important features of the cloning vector include segregational stability, minimal and consistent copy number in all cells of a culture, and the ability to replicate and express large sequences of DNA. There is growing recognition that one or only a few copies of a gene are needed, particularly for metabolic engineering applications. With the ability to vary promoter (Jensen and Hammer, 1998) and ribosome binding strength (Salis et al., 2009), as well as the stabilities of the mRNA (Smolke et al., 2000) and the resulting protein produced from it, there are many factors other than copy number that can be manipulated to alter enzyme production.
Promoters play an essential role in controlling biosynthetic pathways. Inducible promoters are one of the easiest and most effective ways to regulate gene expression, but it is essential that the promoter be induced consistently in all cells of a culture (Khlebnikov et al., 2001). Constitutive promoters (Jensen and Hammer, 1998) and promoters that respond to a change in growth condition or to an important intermediary metabolite (Farmer and Liao, 2000) allow for inexpensive, inducer-free gene expression, which is particularly important where cost is an issue (Fig. A10-2). Although there are many inducible promoters for
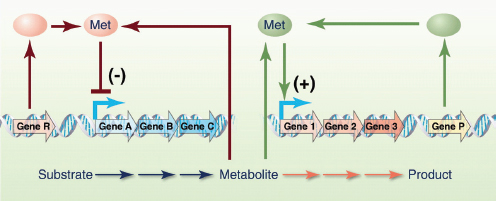
FIGURE A10-2 Use of synthetic regulators to modulate metabolic pathways that have a toxic intermediate. Regulatory proteins or RNAs bind the toxic metabolite and down-regulate the biosynthetic pathway and up-regulate the consumption pathway.
bacteria, the small number of inducible promoters for yeast and other potential industrial hosts makes regulation of metabolic pathways in those organisms more challenging than in bacteria.
Because production of complicated molecules often requires several enzymes, it is desirable to coordinate expression of the genes encoding these enzymes to prevent accumulation of toxic intermediates and bottlenecks in biosynthetic pathways. There are many ways to coordinate expression of multiple genes, such as using a non-native RNA polymerase or transcription factor to induce multiple promoters (Alper and Stephanopoulos, 2007); grouping multiple, related genes into operons; varying the ribosome binding strength for the enzymes encoded in the operon (Salis et al., 2009); or controlling segmental mRNA stability of each coding region to regulate the amount of each enzyme produced (Pfleger et al., 2006). One of the limitations to expressing multiple genes in yeast is the lack of internal ribosomal entry sequences (IRESs) that are available for higher eukaryotes. The development of yeast IRESs would allow one to express genes encoding metabolic pathways without the need for a promoter for each gene.
Debottlenecking, Debugging, and Process Optimizing
Even with a kit full of tools, building a biosynthetic pathway is made difficult without accurate blueprints. In almost all areas of engineering, there are models and simulation tools that allow one to predict which components to assemble to obtain a larger system with a desired function or characteristic. Similar biological design tools are in their infancy. However, metabolic models that incorporate cell composition and gene regulation have become relatively predictive and may one day be used to design metabolic pathways and predict the level of gene expression needed to achieve a particular flux through a reaction or pathway (Edwards et al., 2001).
Regardless of how sophisticated the design tools and how good the blueprint, there will always be “bugs” in the engineered system. Analogous to software debuggers that allow one to find and fix errors in computer code, the development of similar tools for biological debugging would reduce development times for optimizing engineered cells. For the development of microbial chemical factories, functional genomics can serve in the role of debugging routines (Park et al., 2007), because imbalances in a metabolic pathway often elicit a stress response in central metabolism (due to protein overproduction or accumulation of toxic intermediates or end products) (Martin et al., 2003; Kizer et al., 2008). Information from one or more of these techniques can be used to diagnose the problem and modify expression of genes in the metabolic pathway or in the host to improve titer and/or productivity.
Many desirable chemicals will be toxic to the producer, particularly at the high titers needed for industrial-scale production. Taking advantage of the cell’s
native stress response pathways can be an effective way to alleviate part or all of the toxicity (Alper et al., 2006). Even better, transporters could be used to pump the desired product outside the cell, reducing intracellular toxicity and purifying the product from the thousands of contaminating intracellular metabolites (Dunlop et al., 2010).
Designer Cells for Designer Chemicals
One can envision a future when a microorganism is tailor-made for production of a specific chemical from a specific starting material, much like chemical engineers build refineries and other chemical factories from unit operations (Fig. A10-3). The chemical and physical characteristics of the product and starting materials would be considered in the design of the organism to minimize both production and purification costs (e.g., operating the engineered cell at the boiling point of volatile, toxic products to drive production and reduce product toxicity). The cell envelope would be designed to be resistant to the specific desired chemical, and the cell wall would be designed to make the organism tolerant to industrial processing conditions. Specific, engineered transporters would be incorporated into the membrane to pump the desired product out of the cell and keep it out and to import the desired starting material. The biosynthetic pathway would be constructed from a parts registry containing all known enzymes by means of retrosynthesis software (Prather and Martin, 2008), and done so to maximize yield and minimize the time required to grow the organism and produce the desired chemical from the desired starting material. In the event that an enzyme does not exist for a particular reaction or set of reactions, one would use computer-aided design (CAD) software to design the desired enzyme (Siegel et al., 2010).
Once the cell has been designed in the computer, the genetic control system would be designed to control expression of all the genes at the correct time and at the appropriate levels. Redundancies in the genetic control system would be engineered to ensure that design parameters are maintained regardless of the transient changes the cell encounters during the production process. Simulations and scenario planning would test various designs, including genetic control system failure. Safety for the plant operators and the environment would be an essential design criterion. When the genetic controls were fully designed and tested, the chromosome(s) would be designed and constructed. Gene location, modularity, and ease of construction are but a few of the important considerations in designing the chromosome. The chromosome would be ordered from a commercial DNA manufacturer. Depending on the state of the technology at the time, the chromosome would arrive in pieces and be assembled in the constructed envelope or would be completely assembled at the factory and sent to another location to be introduced into the ghost cell. One can even envision a day when cell manufacturing is done by different companies, each specializing in certain aspects of
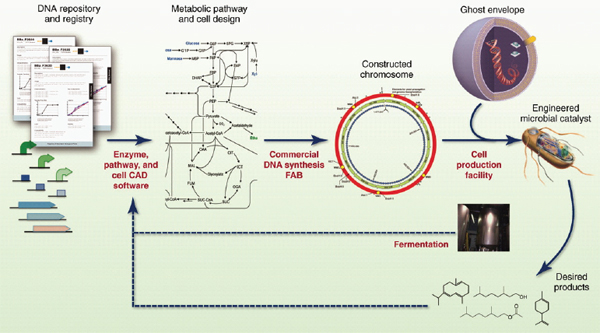
FIGURE A10-3 The future of engineered biocatalysts, enzymes, and genetic controls are designed from characteristic of parts (enzymes, promoters, etc.) by means of pathway and enzyme CAD software. The chromosomes encoding those elements are synthesized at a FAB and incorporated into a ghost envelope to obtain the new catalyst. The design of the engineered catalyst is influenced by the desired product and the production process.
the synthesis—one company constructs the chromosome, one company builds the membrane and cell wall (the “bag”), one company fills the bag with the basic molecules needed to boot up the cell.
Until this future arrives, manufacturing of molecules will be done with well-known, safe, industrial microorganisms that have tractable genetic systems. Continued development of tools for existing, safe, industrial hosts, cloning and expressing genes encoding precursor production pathways, and the creation of novel enzymes that catalyze unnatural reactions will be necessary to expand the range of products that can be produced from biological systems. When more of these tools are available, metabolic engineering should be just as powerful as synthetic chemistry, and together the two disciplines can greatly expand the number of products available from renewable resources.
References and Notes59
H. Alper, G. Stephanopoulos, Metab. Eng. 9, 258 (2007).
H. Alper, J. Moxley, E. Nevoigt, G. R. Fink, G. Stephanopoulos, Science 314, 1565 (2006).
S. Atsumi, T. Hanai, J. C. Liao, Nature 451, 86 (2008).
J. E. Bailey, Science 252, 1668 (1991).
H. R. Beller, E.-B. Goh, J. D. Keasling, Appl. Environ. Microbiol. 76, 1212 (2010).
M. C. Chang, R. A. Eachus, W. Trieu, D. K. Ro, J. D. Keasling, Nat. Chem. Biol. 3, 274 (2007).
J. A. Dietrich et al., ACS Chem. Biol. 4, 261 (2009).
G. K. Donaldson, A. C. Eliot, D. Flint, L. A. Maggio-Hall, V. Nagarajan, U.S. Patent 20070092957 (2007).
M. J. Dunlop, J. D. Keasling, A. Mukhopadhyay, Syst. Synth. Biol. 4, 95 (2010).
J. S. Edwards, R. U. Ibarra, B. O. Palsson, Nat. Biotechnol. 19, 125 (2001).
W. R. Farmer, J. C. Liao, Nat. Biotechnol. 18, 533 (2000).
D. G. Gibson et al., Science 329, 52 (2010).
K. M. Hawkins, C. D. Smolke, Nat. Chem. Biol. 4, 564 (2008).
P. R. Jensen, K. Hammer, Appl. Environ. Microbiol. 64, 82 (1998).
A. Khlebnikov, K. A. Datsenko, T. Skaug, B. L. Wanner, J. D. Keasling, Microbiology 147, 3241 (2001).
L. Kizer, D. J. Pitera, B. F. Pfleger, J. D. Keasling, Appl. Environ. Microbiol. 74, 3229 (2008).
E. Leonard et al., Proc. Natl. Acad. Sci. U.S.A. 107, 13654 (2010).
V. J. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman, J. D. Keasling, Nat. Biotechnol. 21, 796 (2003).
H. G. Menzella et al., Nat. Biotechnol. 23, 1171 (2005).
C. E. Nakamura, G. M. Whited, Curr. Opin. Biotechnol. 14, 454 (2003).
J. H. Park, K. H. Lee, T. Y. Kim, S. Y. Lee, Proc. Natl. Acad. Sci. U.S.A. 104, 7797 (2007).
B.A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane, C. Khosla, Science 291, 1790 (2001).
B. F. Pfleger, D. J. Pitera, C. D. Smolke, J. D. Keasling, Nat. Biotechnol. 24, 1027 (2006).
G. Pósfai et al., Science 312, 1044 (2006).
K. L. Prather, C. H. Martin, Curr. Opin. Biotechnol. 19, 468 (2008).
_______________
59 This work was supported in part by the Synthetic Biology Engineering Research Center, which is funded by National Science Foundation Award No. 0540879 and by the Joint BioEnergy Institute, which is funded by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231. Competing financial interests: The author is a founder of and owns equity in Amyris and LS9.
N. S. Renninger, D. J. McPhee, World Patent 200804555 (2008).
D. K. Ro et al., Nature 440, 940 (2006).
H. M. Salis, E. A. Mirsky, C. A. Voigt, Nat. Biotechnol. 27, 946 (2009).
A. Schirmer, M. A. Rude, X. Li, E. Popova, S. B. del Cardayre, Science 329, 559 (2010).
C. Schmidt-Dannert, D. Umeno, F. H. Arnold, Nat. Biotechnol. 18, 750 (2000).
J. B. Siegel et al., Science 329, 309 (2010).
V. Siewers, R. San-Bento, J. Nielsen, Biotechnol. Bioeng. 106, 841 (2010).
C. D. Smolke, T. A. Carrier, J. D. Keasling, Appl. Environ. Microbiol. 66, 5399 (2000).
E. J. Steen et al., Microb. Cell Fact. 7, 36 (2008).
——, Nature 463, 559 (2010).
H. H. Wang et al., Nature 460, 894 (2009).
H. W. Wisselink, M. J. Toirkens, Q. Wu, J. T. Pronk, A. J. van Maris, Appl. Environ. Microbiol. 75, 907 (2009).
S. T. Withers, S. S. Gottlieb, B. Lieu, J. D. Newman, J. D. Keasling, Appl. Environ. Microbiol. 73, 6277 (2007).
Y. Yoshikuni, T. E. Ferrin, J. D. Keasling, Nature 440, 1078 (2006).
NOVEL APPROACHES TO COMBAT BIOFILM DRUG TOLERANCE
Kim Lewis60
The Nature of the Threat
It is a given that new antibiotics are needed to combat drug-resistant pathogens. However, this is only part of the need—we actually never had antibiotics capable of eradicating an infection. Currently used antibiotics have been developed against rapidly growing bacteria, and most of them have no activity against stationary-state organisms, and none are effective against dormant persister cells. The relative effectiveness of antibiotics in treating disease is largely a result of a cooperation with the immune system, which mops up after antibiotics eliminate the bulk of a growing population. But the deficiency of existing antibiotics against supposedly drug-susceptible pathogens is becoming increasingly apparent with the rise of immunocompromised patients (HIV infected, undergoing chemotherapy) and the wide use of indwelling devices (catheters, prostheses, and heart valves), where the pathogen forms biofilms protecting cells from the components of the immune system. The ineffectiveness of the immune system leads to chronic diseases, which make up approximately half of all infectious disease cases in the developed world. The main culprit responsible for tolerance of pathogens to antibiotics are specialized survivors, persister cells (Lewis, 2007, 2010), which we examine in the following section.
_______________
60 Antimicrobial Discovery Center and the Department of Biology, Northeastern University, Boston, MA, USA. E-mail: k.lewis@neu.edu, http://www.biology.neu.edu/faculty03/lewis03.html.
Persisters
Persisters represent a small subpopulation of cells that spontaneously go into a dormant, nondividing state. When a population is treated with a bactericidal antibiotic, regular cells die, while persisters survive (Figure A11-1). In order to kill, antibiotics require active targets, which explains the tolerance of persisters. Taking samples and plating them for colony counts over time from a culture treated with antibiotic produces a biphasic pattern, with a distinct plateau of surviving persisters. By contrast, resistance mechanisms prevent antibiotics from binding to their targets (Figure A11-2).
Infectious disease is often untreatable, even when caused by a pathogen that is not resistant to antibiotics. This is the essential paradox of chronic infections. In most cases, chronic infections are accompanied by the formation of biofilms, which seems to point to the source of the problem (Costerton et al., 1999; Del Pozo and Patel, 2007). Biofilms have been linked to dental disease, endocarditis, cystitis, urinary tract infection, deep-seated infections, indwelling device and catheter infections, and the incurable disease of cystic fibrosis. In the case of indwelling devices such as prostheses and heart valves, reoperation is the method of choice for treating the infection. Biofilms do not generally restrict penetration of antibiotics (Walters et al., 2003), but they do form a barrier for the larger com-
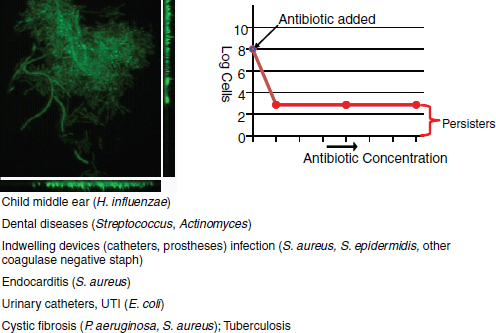
FIGURE A11-1 Persisters and biofilms. Dose-dependent killing with a bactericidal antibiotic reveals a small subpopulation of tolerant cells, persisters, that are formed within a biofilm.
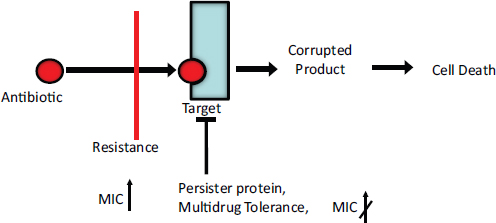
FIGURE A11-2 Resistance and tolerance. Bactericidal antibiotics kill cells by forcing the active target to produce corrupted products. Persister proteins act by blocking the target, so no corrupted product can be produced. By contrast, all resistance mechanisms prevent the antibiotic from binding to the target.
ponents of the immune system (Jesaitis et al., 2003; Leid et al., 2002; Vuong et al., 2004). The presence of biofilm-specific resistance mechanisms was suggested to account for the recalcitrance of infectious diseases (Stewart and Costerton, 2001). However, the bulk of cells in the biofilm are actually highly susceptible to killing by antibiotics; only a small fraction of persisters remains alive (Spoering and Lewis, 2001). Based on these findings, we proposed a simple model of a relapsing chronic infection: antibiotics kill the majority of cells, and the immune system eliminates both regular cells and persisters from the bloodstream (Lewis, 2001) (Figure A11-3). The only remaining live cells are then persisters in the biofilm. Once the level of antibiotic drops, persisters repopulate the biofilm, and the infection relapses. While this is a plausible model, it is not the only one. A simpler possibility is that antibiotics fail to effectively reach at least some cells in vivo, resulting in a relapsing infection.
Establishing potential causality between persisters and therapy failure is not trivial, since these cells form a small subpopulation with a temporary phenotype, which precludes introducing them into an animal model of infection. We reasoned that causality can be tested based on what we know about selection for high persister (hip) mutants in vitro. Periodic application of high doses of bactericidal antibiotics leads to the selection of strains that produce increased levels of persisters (Moyed and Bertrand, 1983; Wolfson et al., 1990). This is precisely what happens in the course of treating chronic infections: the patient is periodically exposed to high doses of antibiotics, which may select for hip mutants. But hip mutants would only gain advantage if the drugs effectively reach, and kill, the regular cells of the pathogen.
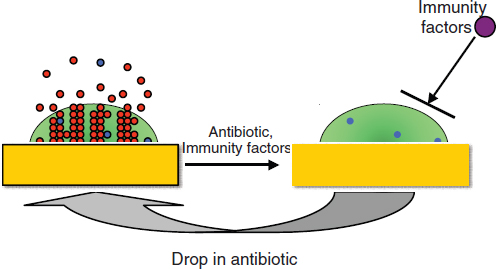
FIGURE A11-3 A model of a relapsing biofilm infection. Regular cells (red) and persisters (blue) form in the biofilm and are shed off into surrounding tissue and the bloodstream. Antibiotics kill regular cells, and the immune system eliminates escaping persisters. The matrix protects persisters from the immune system, and when the concentration of the antibiotic drops, they repopulate the biofilm, causing a relapse.
Patients with cystic fibrosis (CF) are treated for decades for an incurable Pseudomonas aeruginosa infection to which they eventually succumb (Gibson et al., 2003). The periodic application of high doses of antibiotics provides some relief by decreasing the pathogen burden, but it does not clear the infection. If hip strains of pathogens were selected in vivo, they would most likely be present in a CF patient. We took advantage of a set of longitudinal P. aeruginosa isolates from a single patient, collected over the course of many years (Smith et al., 2006). Testing persister levels by monitoring survival after challenge with a high dose of ofloxacin showed a dramatic, 100-fold increase in surviving cells in the last four isolates (Mulcahy et al., 2010). Testing paired strains from additional patients showed that, in most cases, there was a considerable increase in persister levels in the late isolate from a patient. Interestingly, most of the hip isolates had no increase in minimum inhibitory concentration (MIC) compared to their clonal parent strain to ofloxacin, carbenicillin, and tobramycin, suggesting that classical acquired resistance plays little or no role in the recalcitrance of CF infection. These experiments directly link persisters to the clinical manifestation of the disease and suggest that persisters are responsible for the therapy failure of chronic CF infection. But why have the hip mutants with their striking survival phenotype evaded detection for such a long time?
The main focus of research in antimicrobials has been on drug resistance, and the basic starting experiment is to test a clinical isolate for its ability to grow in
the presence of elevated levels of different antibiotics and to record any increases in the MIC. This is also the standard test employed by clinical microbiology laboratories. The hip mutants are of course missed by this test, which explains why they had remained undetected in spite of a major effort aimed at understanding pathogen survival to antimicrobial chemotherapy. Given that hip mutants are the likely main culprit responsible for morbidity and mortality of the CF infection, it makes sense to test for their presence. Testing for persister levels is not that much more difficult as compared to a MIC test.
Is selection for hip mutants a general feature of chronic infections? We recently examined patients with chronic oral thrush caused by Candida albicans (LaFleur et al., 2010). These were cancer patients undergoing chemotherapy, and suppression of the immune system caused the fungal infection. In patients where the disease did not resolve, the C. albicans isolates were almost invariably hip mutants, as compared to patients where the disease cleared within 3 weeks of treatment with chlorhexidine. The eukaryotic C. albicans forms persisters (Al-Dhaheri and Douglas, 2008; Harrison et al., 2007; LaFleur et al., 2006) through mechanisms that are probably analogous, rather than homologous, to that of their bacterial counterparts. Given the similar lifestyles of the unrelated P. aeruginosa and C. albicans, we may expect that the survival advantage of a hip mutation is universal. Just as multidrug resistance has become the prevalent danger in acute infections, multidrug tolerance of persisters and hip mutants may be the main, but largely overlooked, culprit of chronic infectious disease.
Biofilms apparently serve as a protective habitat for persisters (Harrison et al., 2005a,b, 2009; LaFleur et al., 2006; Spoering and Lewis, 2001), allowing them to evade the immune response. However, a more general paradigm is that persisters will be critical for pathogens to survive antimicrobial chemotherapy whenever the immune response is limited. Such cases would include disseminating infections in immunocompromised patients undergoing cancer chemotherapy or infected with HIV. Persisters are also likely to play an important role in immunocompetent individuals in cases where the pathogen is located at sites poorly accessible by components of the immune system. These include the central nervous system, where pathogens cause debilitating meningitis and brain abscesses (Honda and Warren, 2009), and the gastrointestinal tract, where hard-to-eradicate Helicobacter pylori causes gastroduodenal ulcers and gastric carcinoma (Peterson et al., 2000). Tuberculosis is perhaps the most prominent case of a chronic infection by a pathogen evading the immune system. The acute infection may resolve spontaneously or as a result of antimicrobial therapy, but the pathogen often remains in a “latent” form (Barry et al., 2009). It is estimated that one of every three people carry latent Mycobacterium tuberculosis, and 10 percent of carriers develop an acute infection at some stage in their lives. One simple possibility is that persisters are equivalent to the latent form of the pathogen. We recently isolated persisters from a growing culture of M. tuberculosis following lysis with d-cycloserine and found that these cells exhibit a general
shutdown of biosynthetic processes, a hallmark of dormancy (Keren et al., 2011). The M. tuberculosis persisters also had several toxin-antitoxin modules overexpressed. These findings will enable the use of the “persister signature” to establish the equivalence of these cells and the dormant form of the pathogen in vivo.
The above analysis underscores the significance of drug tolerance as a barrier to effective antimicrobial chemotherapy. Given its significance—roughly half of all cases of infection (Figure A11-4)—the number of studies dedicated to tolerance is tiny compared to the number of publications on resistance. The difficulty in pinpointing the mechanism of biofilm recalcitrance and the formidable barriers to studying persister cells account for the lack of parity between these two comparably significant fields. Hopefully a better balance will be achieved, and the following discussion summarizes recent advances in understanding the mechanism of tolerance.
Persisters were initially discovered in 1944, but the mechanism of their formation eluded us for a very long time. Only recently has the molecular mechanism of dormancy begun to emerge.
The most straightforward approach to finding an underlying mechanism of a complex function is by screening a library of transposon insertion mutants. This produces a set of candidate genes, and subsequent analysis leads to a pathway and a mechanism. This is indeed how the basic mechanisms of sporulation, flagellation, chemotaxis, virulence, and many other functions have been established.
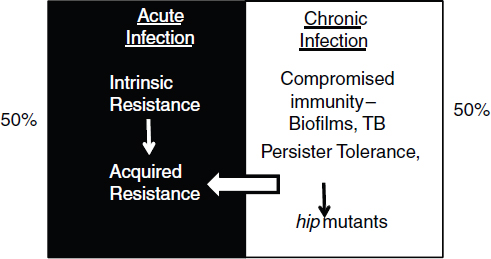
FIGURE A11-4 The two faces of recalcitrance. Drug resistance plays an important role in recalcitrance of acute infections, while drug tolerance is largely responsible for failures of chemotherapy in chronic infections. Tolerance allows the pathogen to survive for lengthy periods of time, and this relapsing infection increases the probability of producing classical resistant cells.
However, screening a Tn insertion library of Escherichia coli for an ability to tolerate high doses of antibiotics produced no mutants completely lacking persisters (Hu and Coates, 2005; Spoering, 2006). With the development of the complete, ordered E. coli gene knockout library by the Mori group (Baba et al., 2006) (the Keio collection), it seemed reasonable to revisit the screening approach. Indeed, there always remains a possibility that transposons missed a critical gene, or the library was not large enough. The use of the Keio collection largely resolves this uncertainty.
This advanced screen (Hansen et al., 2008), similarly to previous efforts, did not produce a single mutant lacking persisters, suggesting a high degree of redundancy. The screen did identify a number of interesting genes, with knockouts showing about a 10-fold decrease in persister formation. The majority of hits were in global regulators, DksA, DnaKJ, HupAB, and IhfAB. This is an independent indication of redundancy—a global regulator can affect expression of several persister genes simultaneously, resulting in a phenotype (Figure A11-5). The screen also produced two interesting candidate genes that may be more directly involved in persister formation: YgfA, which can inhibit nucleotide synthesis, and YigB, which may block metabolism by depleting the pool of flavin mononucleotide (FMN).
A similar screen of a P. aeruginosa mutant library was recently reported (De Groote et al., 2009). As in E. coli, no persisterless mutant was identified, pointing to the similar redundancy theme.
The main conclusion from the screens is that persister formation does not follow the main design theme of complex cellular functions—a single linear regulatory pathway controlling an execution mechanism. By contrast, persisters are apparently formed through a number of independent parallel mechanisms (Figure A11-5). There is a considerable adaptive advantage in this redundant design—no single compound will disable persister formation.
Screens for persister genes were useful in finding some possible candidate genes and pointing to redundancy of function. It seemed that a method better suited to uncover redundant elements would be transcriptome analysis. For this, persisters had to be isolated.
Persisters form a small and temporary population, making isolation challenging. The simplest approach is to lyse a population of growing cells with a β-lactam antibiotic and collect surviving persisters (Keren et al., 2004). This allows one to isolate enough E. coli cells to perform a transcriptome analysis. A more advanced method aimed at isolating native persisters was developed, based on a guess that these are dormant cells with diminished protein synthesis (Shah et al., 2006). If the strain expressed degradable green fluorescent protein (GFP), then cells that stochastically enter into dormancy will become dim. In a population of E. coli expressing degradable GFP under the control of a ribosomal promoter that is only active in dividing cells, a small number of cells indeed appeared to be dim. The difference in fluorescence allowed for the sorting of the
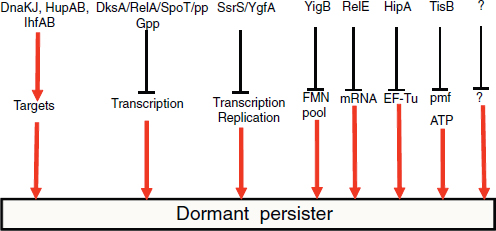
FIGURE A11-5 Candidate persister genes. Persisters are formed through parallel redundant pathways.
two subpopulations. The dim cells were tolerant to ofloxacin, confirming that they are persisters.
Transcriptomes obtained by both methods pointed to downregulation of biosynthesis genes and indicated increased expression of several toxin-antitoxin (TA) modules (RelBE, MazEF, DinJYafQ, YgiU). TA modules are found on plasmids, where they constitute a maintenance mechanism (Gerdes et al., 1986b; Hayes, 2003). Typically, the toxin is a protein that inhibits an important cellular function, such as translation or replication, and forms an inactive complex with the antitoxin. The toxin is stable, while the antitoxin is degradable. If a daughter cell does not receive a plasmid after segregation, the antitoxin level decreases due to proteolysis, leaving a toxin that either kills the cell or inhibits propagation. TA modules are also commonly found on bacterial chromosomes, but their role is largely unknown. In E. coli, MazF and an unrelated toxin RelE induce stasis by cleaving mRNA, which of course inhibits translation, a condition that can be reversed by expression of corresponding antitoxins (Christensen and Gerdes, 2003; Pedersen et al., 2002). This property of toxins makes them excellent candidates for persister genes.
Ectopic expression of RelE (Keren et al., 2004) or MazF (Vazquez-Laslop et al., 2006) strongly increased tolerance to antibiotics. The first gene linked to persisters, hipA (Moyed and Bertrand, 1983), is also a toxin, and its ectopic expression causes multidrug tolerance as well (Correia et al., 2006; Falla and Chopra, 1998; Korch and Hill, 2006; Vazquez-Laslop et al., 2006). Interestingly, a bioinformatics analysis indicates that HipA is a member of the TOR family of kinases, which have been extensively studied in eukaryotes (Schmelzle and Hall, 2000) but have not been previously identified in bacteria. HipA is indeed a kinase; it
autophosphorylates on ser150, and site-directed mutagenesis replaces it, or other conserved amino acids in the catalytic and Mg2+-binding sites abolish its ability to stop cell growth and confer drug tolerance (Correia et al., 2006). The crystal structure of HipA in complex with its antitoxin HipB was recently resolved, and a pull-down experiment showed that the substrate of HipA is elongation factor EF-Tu (Schumacher et al., 2009). Phosphorylated EF-Tu is inactive, which leads to a block in translation and dormancy (Figure A11-6).
The deletion of potential candidates of persister genes noted above does not produce a discernible phenotype affecting persister production, possibly due to the high degree of redundancy of these elements. In E. coli, there are at least 15 TA modules (Alix and Blanc-Potard, 2009; Pandey and Gerdes, 2005; Pedersen and Gerdes, 1999) and there are more than 80 in M. tuberculosis (Ramage et al., 2009).
High redundancy of TA genes would explain the lack of a multidrug-tolerance phenotype in knockout mutants, and therefore it seemed useful to search
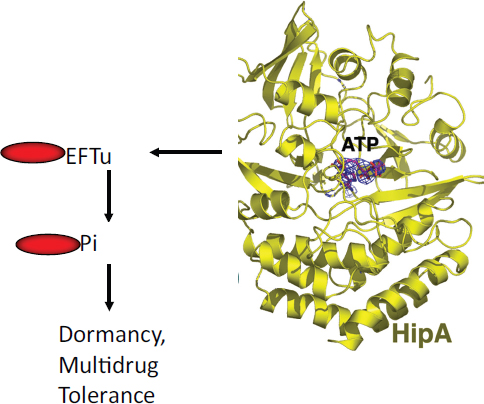
for conditions where a particular toxin would be highly expressed in a wild-type strain and then examine a possible link to persister formation.
Several TA modules contain the Lex box and are induced by the SOS response. These are symER, hokE, yafN/yafO, and tisAB/istr1 (Courcelle et al., 2001; Fernandez De Henestrosa et al., 2000; Kawano et al., 2007; McKenzie et al., 2003; Motiejunaite et al., 2007; Pedersen and Gerdes, 1999; Singletary et al., 2009; Vogel et al., 2004). Fluoroquinolones induce the SOS response (Phillips et al., 1987), and we tested the ability of ciprofloxacin to induce persister formation (Dorr et al., 2009).
Examination of deletion strains showed that the level of persisters dropped dramatically, 10- to 100-fold, in a ΔtisAB mutant. This suggests that TisB was responsible for the formation of the majority of persisters under conditions of SOS induction. The level of persisters was unaffected in strains deleted in the other Lex box containing TA modules. Persister levels observed in time-dependent killing experiments with ampicillin or streptomycin that do not cause DNA damage were unchanged in the ΔtisAB strain. TisB only had a phenotype in the presence of a functional RecA protein, confirming the dependence on the SOS pathway.
Ectopic overexpression of tisB sharply increased the level of persisters. The drop in persisters in a deletion strain and the increase upon overexpression gives reasonable confidence in functionality of a persister gene. The dependence of TisB-induced persisters on a particular regulatory pathway, the SOS response, further strengthens the case for TisB as a specialized persister protein (Figure A11-7). Incidentally, a tisB mutant is not present in the otherwise fairly complete Keio knockout library, and the small open reading frame might have been easily missed by Tn mutagenesis as well, evading detection by the generalized screens for persister genes.
The role of TisB in persister formation is unexpected based on what we know about this type of protein. TisB is a small, 29 amino acid hydrophobic peptide that binds to the membrane and disrupts the proton motive force (pmf), which leads to a drop in ATP levels (Unoson and Wagner, 2008). Bacteria, plants, and animals all produce antimicrobial membrane-acting peptides (Garcia-Olmedo et al., 1998; Sahl and Bierbaum, 1998; Zasloff, 2002). Toxins of many TA loci found on plasmids belong to this type as well. If a daughter cell does not inherit a plasmid, the concentration of a labile antitoxin decreases, and the toxin such as the membrane-acting hok kills the cell (Gerdes et al., 1986a). High-level artificial overexpression of TisB also causes cell death (Unoson and Wagner, 2008). It is remarkable from this perspective that the membrane-acting TisB under conditions of natural (mild) expression has the exact opposite effect of protecting the cell from antibiotics.
Fluoroquinolones such as ciprofloxacin are widely used broad-spectrum antibiotics, and their ability to induce multidrug-tolerant cells is unexpected and a cause of considerable concern. The induction of persister formation by fluoroquinolones may contribute to the ineffectiveness of antibiotics in eradicat-
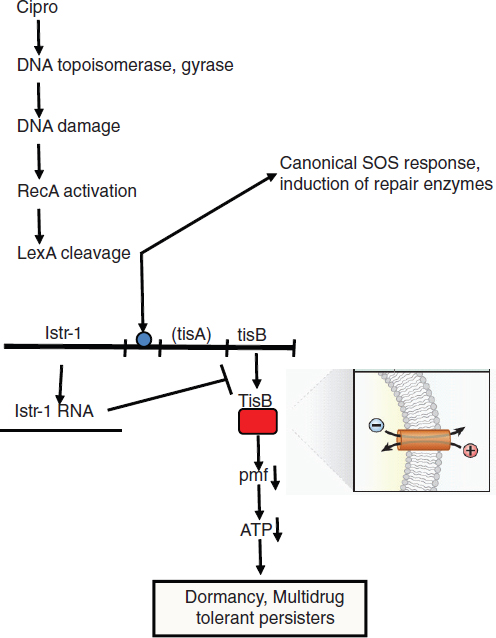
FIGURE A11-7 Persister induction by antibiotic. The common antibiotic ciprofloxacin causes DNA damage by converting its targets, DNA gyrase and topoisomerase, into endonucleases. This activates the canonical SOS response, leading to increased expression of DNA repair enzymes. At the same time, the LexA repressor that regulates expression of all SOS genes also controls transcription of the TisAB toxin-antitoxin module. The TisB toxin is an antimicrobial peptide, which binds to the membrane, causing an increase in pmf and ATP. This produces a systems shutdown, blocking antibiotic targets, which ensures multidrug tolerance.
ing infections. Indeed, preexposure with a low dose of ciprofloxacin drastically increased tolerance to subsequent exposure with a high dose, and TisB persisters are multidrug tolerant.
The finding of the role of TisB in tolerance opens an intriguing possibility of a wider link between other stress responses and persister formation. Pathogens are exposed to many stress factors in the host environment apart from DNA damaging agents: oxidants, high temperature, low pH, and membrane-acting agents. It is possible that all stress responses induce the formation of surviving persisters.
While resistance and tolerance are mechanistically distinct, there is sufficient reason to believe that tolerance may be a major cause for developing resistance. Indeed, the probability of resistance development is proportional to the size of the pathogen population, and a lingering chronic infection that cannot be eradicated due to tolerance will go on to produce resistant mutants and strains acquiring resistant determinants by transmission from other bacteria (Levin and Rozen, 2006). Combating tolerance then becomes a major component in preventing resistance.
The Discovery Challenge
Source Compounds
The discovery of penicillin was an isolated event, but the development of screening for antimicrobial activity from soil actinomycetes by Salman Waxman produced the first, and also the only known, effective platform technology for antibiotic discovery (Schatz et al., 1944). Cultivable actinomycetes, however, are a limited resource; ~99 percent of microbes do not readily grow in the lab and are known as “uncultured” (Lewis et al., 2010). Overmining of actinomycetes by the early 1960s replaced discovery of novel compounds with rediscovery of knowns.
In response to the dwindling returns in natural product antibiotic discovery, the industry responded by focusing on synthetics. Indeed, a number of antimicrobials are synthetic (metronidazole, trimethoprim, isoniazid, ethionamide, pyrazinamide, and ethambutol), and there is one highly effective class of synthetic broad-spectrum antibiotics, the fluoroquinolones. Encouraged by these examples, and by dramatic advances in synthetic and combinatorial chemistry, high-throughput robotics, genomics, and proteomics, a new discovery platform emerged (Figure A11-8). Combinatorial chemistry provided a large number of test compounds, which were screened in high-throughput format against isolated essential target proteins determined by genomics. This platform, however, failed to produce a new class of broad-spectrum antibiotics, leading to the closure of anti-infectives divisions in many of the Big Pharma companies. The main reasons for failure is well understood: high-throughput screening hits were literally running into the penetration barrier of Gram-negative bacteria, which is made of transenvelope multidrug resistance (MDR) pumps that extrude amphipathic
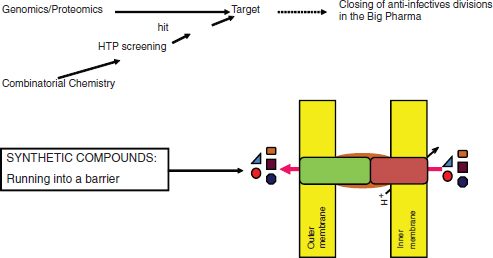
FIGURE A11-8 The high-tech platform. Early leads from screening compound libraries are extruded by transenvelope MDR pumps of Gram-negative species.
compounds across the outer membrane barrier (Lomovskaya et al., 2008). Drugs have to be amphipathic in order to penetrate across the hydrophobic inner membrane, but this is precisely the feature that the outer membrane restricts and the MDRs recognize. There are few compounds that pass this seemingly impenetrable barrier rather effectively: the broad-spectrum aminoglycosides, tetracyclines, fluoroquinolones, some β-lactams, chloramphenicol, and azithromycin. Fluoroquinolones are the only synthetics on this list, and they were discovered 50 years ago.
But what about less challenging narrow-spectrums, with good activity against at least Gram-positive species? Seventy high-throughput screens performed by GlaxoSmithKline, for example, against a large number of targets produced no viable leads (Payne et al., 2007). Glaxo scientists realized that penetration is a serious problem, and therefore also performed in vivo screens against E. coli, but only obtained “nuisance” hits, such as membrane-acting compounds. One obvious conclusion from this negative experience is that the libraries do not carry good starting compounds.
In part this is due to the fact that libraries are constructed based on Lipinski rules (Lipinski, 2003), which are good for predicting druglike properties for compounds acting against mammalian cell targets but do not work well for bacteria because of peculiarities of permeation (O’Shea and Moser, 2008; Silver, 2008). Another important consideration is the probability of resistance development. Pathogen populations produce 109 cells in an infected patient, which means that the probability of resistance development should be <109. This is readily achieved with most of the antibiotic classes currently in use, since they hit more than one
target (fluoroquinolones attack DNA gyrase and topoisomerase, β-lactams inhibit several penicillin-binding proteins, and ribosomal inhibitors bind to rRNA which is coded by multiple genes) (Silver, 2007). This requirement severely limits the number of realistic targets for antimicrobial drug discovery.
The above analysis presents an extremely bleak picture; if we cannot even discover compounds acting against rapidly growing Gram-positive bacteria, what are the prospects of finding broad-spectrum antimicrobials acting against non-growing stationary cells and persisters?
Opportunities
There are many steps in the drug discovery pipeline, but if there are no viable leads, there is no pipeline. Indeed, at the last Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) meeting (2009), there was not a single broad-spectrum lead presented. This means that the number of realistic broad-spectrum leads in the global antimicrobial drug discovery pipeline is zero. This is where the process needs to be restarted, and this is where allocation of resources will make a tangible impact.
A Fresh Look at Potential Sources of Compounds
Natural products There are two largely untapped and potentially enormous new sources of natural products: uncultured microorganisms and silent operons coding for secondary metabolites.
A recent resurgence in cultivation efforts aimed at gaining access to uncultured microorganisms has been sparked by the vast diversity of uncultured bacterial groups revealed by environmental surveys of 16S rRNA (Aoi et al., 2009; Bollmann et al., 2007; Bruns et al., 2002; Connon and Giovannoni, 2002; Davis et al., 2005; Ferrari et al., 2005; Gavrish et al., 2008; Kaeberlein et al., 2002; Nichols et al., 2008; Rappe et al., 2002; Stevenson et al., 2004; Zengler et al., 2002). While some novel bacterial species were successfully cultured by varying media and growth conditions (Joseph et al., 2003), significant departures from conventional techniques were clearly in order, and indeed the new technologies substantially diverged from traditional cultivation methods by adopting single-cell and high-throughput strategies (Connon and Giovannoni, 2002; Nichols et al., 2008; Rappe et al., 2002; Zengler et al., 2002), better mimicking the natural milieu (Aoi et al., 2009; Bruns et al., 2002; Ferrari et al., 2005; Stevenson et al., 2004), increasing the length of incubation, and lowering the concentration of nutrients (Davis et al., 2005). High-throughput extinction culturing is based on the dilution of natural communities of bacteria to 1 to 10 cells per well in low-nutrient, filtered marine water. This strategy resulted in the cultivation of the first member of the ubiquitous, previously uncultured clade, SAR11 (Rappe et al., 2002). Our research group contributed to the effort by developing three
cultivation methodologies (Gavrish et al., 2008; Kaeberlein et al., 2002; Nichols et al., 2008). All three strategies aim to provide microorganisms with their natural growth conditions by incubating them in simulated natural environments.
The diffusion chamber is designed to essentially “trick” cells into thinking they are growing in their natural environment by creating an incubation strategy that very closely mimics their natural habitat (Kaeberlein et al., 2002). The diffusion chamber consists of a stainless steel washer and 0.03-µm pore-size membranes (Figure A11-9). After gluing a membrane to one side of the washer, the inoculum (a mix of environmental cells and warm agar) is introduced, and the second membrane seals the chamber. Nutrients from the environment can diffuse into the chamber; therefore, it is not necessary to add them to the medium. Once inoculated and assembled, the chamber can be returned to the original location of sampling or in a simulated natural environment such as a block of sediment kept in an aquarium in lab. Microcolonies grow in the chamber during such incubation. A recovery rate of 22 percent on average was observed in the diffusion chambers. In this study and follow-up research (Bollmann et al., 2007; Nichols et al., 2008) we isolated numerous species that did not grow in Petri dishes inoculated with environmental samples but were successfully grown in the diffusion chambers.
Reinoculation of material both from marine and soil environments from chamber to chamber produces “domesticated” variants that grow on regular media on a Petri dish and can be exploited for secondary metabolite production (Bollmann et al., 2007; Nichols et al., 2008).
The diffusion chamber typically produces a mixed culture, which requires considerable time to isolate, purify, and reinoculate individual colonies. In order
FIGURE A11-9 A diffusion chamber for growing bacteria in situ. A sample from marine sediment is diluted, mixed with agar, and sandwiched between the two semipermeable membranes of the diffusion chamber, which is returned to the environment.
to streamline this process into a high-throughput system, we developed a variant of the diffusion chamber for massively parallel microbial isolation. The Isolation Chip, or ichip for short (Nichols et al., 2010), consists of hundreds of miniature diffusion chambers that can be loaded with an average of one cell per chamber. The ichip enables microbial growth and isolation in a single step with hundreds of individual cultures incubating on a single chip.
Microorganisms that are particularly important for drug discovery, microscopic fungi and actinomycetes, grow by forming filaments capable of penetrating soft substrates. Since actinomycetes can pass through 0.2-µm pores, we reasoned this could be used to design a trap for the specific capture of these organisms (Gavrish et al., 2008). The trap is similar in design to the diffusion chamber, except the membranes have larger pores and the agar inside the trap is initially sterile when placed in the environment. Any growth observed afterward inside the trap is due to the movement of cells into the trap during incubation. The majority of organisms grown in the traps proved to be actinomycetes, some of which represented rare and unusual species from the genera Dactylosporangium, Catellatospora, Catenulispora, Lentzea, and Streptacidiphilus.
We noticed that some organisms forming colonies in the diffusion chamber can grow on a Petri dish, but only in the presence of other species from the same environment (Kaeberlein et al., 2002; Nichols et al., 2008) and suggested that uncultured bacteria only commit to division in a familiar environment, which they recognize by the presence of growth factors released by their neighbors. In order to assess the commonality of the growth dependence of uncultured organisms on neighboring species and pick good models for study, we chose an environment where bacteria live in a tightly packed community (D’Onofrio et al., 2010). This is a biofilm that envelopes sand particles of a tidal ocean beach (Figure A11-10). There were disproportionately more colonies appearing on densely inoculated plates compared with more dilute plates. This indicated that some of the cells that grew on the densely seeded plates were receiving growth factors from neighboring colonies. To test the possible growth dependence of microorganisms on neighboring species, pairs of colonies growing within a short distance of each other were restreaked in close proximity to each other. Potential uncultured isolates were identified by their diminishing growth with increasing distance from the cultivable “helper” strain on the cross-streak plates. Colonies of the culturable organism Micrococcus luteus KLE1011 (a marine sand sediment isolate 99.5 percent identical to M. luteus DSM 200030T according to 16S rRNA gene sequence) grew larger as their distance from other colonies increased (Figure A11-10). Approximately 100 randomly picked pairs of colonies were restreaked from the high-density plates, and 10 percent of these pairs showed this pattern of growth induction on cross-streaked plates.
In order to isolate growth factors, spent medium from the helper M. luteus KLE1011 was tested and shown to induce growth of the uncultured M. polysiphoniae KLE1104. An assay-guided fractionation led to isolation and structure
FIGURE A11-10 Understanding the mechanism of uncultivability. Marine sand particles are covered by a multispecies biofilm (top). Cells from the biofilm form colonies on a densely seeded plate, and pairing them together reveals that some of them are uncultured bacteria (evenly spread on the plate) that will only grow in the presence of a helper species spotted on the same plate (bottom).
determination of five different siderophores and each of them was able to induce growth of M. polysiphoniae KLE1104. This demonstrated that siderophores represent the growth factors responsible for the helping activity. The siderophores consisted of a central core with alternating N-hydroxycadaverine and succinic acid units and were of the desferrioxamine class (Challis, 2005). Both close relatives of known microorganisms and novel species were isolated by this approach. This study identified the first class of growth factors for uncultured bacteria and suggests that additional ones will come from analyzing organisms growing in co-culture.
Silent operons Whole-genome sequencing of several actinomycetes showed that there are many more potential biosynthetic pathways for production of secondary metabolites than there are known antibiotics made by these organisms (Ikeda et al., 2003). Ecopia used fermentation in 40 different media to entice production of additional compounds and discovered a novel type of enediyne with anticancer activity (Zazopoulos et al., 2003). No novel antimicrobials emerged from this
effort. However, in order to be effective, one needs to develop a high-throughput approach to induce production of such compounds. This is entirely doable.
Synthetics Are existing libraries, both commercially available and proprietary collections in Big Pharma, useless for antibiotic discovery? It does seem so, since they have obviously already been screened for actives, including nonbiased screens for growth inhibition of whole cells, and produced no viable leads. But does it not seem strange that a screen of a collection of 600 dyes by Domagk produced the first viable antibiotic, while a screen of the total global library of ~107 compounds produced nothing at all? As the libraries grew, a number of innovations were introduced aimed at improving the screening outcome; thus we have in vitro screening, targeted screens, Lipinski rules, and specificity validation. My feeling is that each time we tried to improve things, the result was to discard valuable compounds. I think that the existing libraries do harbor useful molecules; the question is how to identify them.
Good Compounds from Bad Libraries
Back to Domagk The first screen was also perfect: Domagk tested compounds against mice infected with streptococci. The result was the discovery of prontosil, a sulfa drug that has no activity in vitro. The compound is cleaved in the intestine by gut bacteria, releasing the active sulfonamide moiety, which inhibits dehydropteroate synthase in the folate pathway. An in vitro test would have missed prontosil. There are obvious advantages to testing compounds in situ—this automatically eliminates the significant burden of toxic molecules and demonstrates efficacy, again automatically eliminating substances with problems of action in an animal, such as serum binding, instability, or poor tissue distribution. In addition, different types of compounds may be uniquely uncovered, such as those requiring activation in situ and those hitting targets that are only important in an infection but not in vitro. While this would theoretically be the perfect way to go, testing in 107 mice is not an option for a variety of reasons, including ethical considerations and the large amounts of required test compound. We therefore considered a useful intermediate between in vitro and a mammal—an animal that, unlike mice, can be dispersed in microtiter wells. Caenorhabditis elegans can be infected with human pathogens by simply ingesting them, and we found that the worm can be cured by common antibiotics such as tetracycline and vancomycin, and at concentrations typically achieved in human plasma (Moy et al., 2006). Worms infected with a pathogen such as Enterococcus feacalis die, stop moving, their shape changes from curved to straight, and they can be detected by typical eukaryotic vital dyes (Figure A11-11). Using these parameters, an automated approach was developed, and a large pilot screen of compound libraries uncovered hits, some of which had no activity in vitro (Moy et al., 2006, 2009). This ap-
FIGURE A11-11 A high-throughput screen for antimicrobials in an animal model. C. elegans are infected with E. faecalis, and cured with ampicillin. This provides for an assay of compounds that cure the worm in situ.
proach shows that C. elegans points us in the right direction—back to Domagk, but with larger libraries.
Better libraries and rules of penetration Of course it would be great to have a better library, constructed based not on Lipinski rules but on “rules of penetration.” We have a small number of broad-spectrum compounds that are able to largely bypass the MDRs and get across the impermeable barrier of Gram-negative membranes—tetracycline, chloramphenicol, aminoglycosides, trimethoprim,
β-lactams (these only need to traverse the outer membrane), fluoroquinolones, and metronidazole. This set is too small to enable us to discern rules for penetration. But testing a large number of unbiased compounds from a library for their ability to enter into the cytoplasm of Gram-negative bacteria should allow us to deduce general rules that favor penetration. Once these are available, this would drive the synthesis and combinatorial chemistry of new compound libraries specifically geared toward antimicrobial discovery.
Prodrugs It is useful to consider the theoretically perfect antibiotic from first principles and then decide whether it is realistic. Approaches we discussed so far do not address the daunting challenge of killing persister cells while at the same time showing broad-spectrum activity. It is useful to start with the end result: a highly reactive compound will kill all cells, including persisters. In order to spare the host, the compound must be delivered as a prodrug, and then a bacteria-specific enzyme will activate it into a generally reactive molecule which will covalently bind to unrelated targets. Importantly, this mechanism creates an irreversible sink, largely resolving the issue of MDR efflux, so the antimicrobial is automatically broad spectrum. Is this realistic? Several existing antimicrobials closely match the properties of this idealized prodrug antibiotic. These are isoniazid, pyrazinamide, ethionamide, and metronidazole. The first three are anti-Mtb drugs, while metronidazole is a broad-spectrum compound acting against anaerobic bacteria. All four compounds convert into active antiseptic-type molecules inside the cell that covalently bind to their targets. It seems to be no accident that prodrug antibiotics make up the core of the anti-Mtb drug arsenal, since an ability to kill the pathogen is critical for treating the disease. Preferred targets have been identified for isoniazid and ethionamide (Vilcheze et al., 2005), suggesting a relatively limited reactivity of these compounds. The existence of preferred targets indicates that the prodrug products are not that reactive, and there is considerable room for developing better sterilizing antibiotics based on the same principle.
References
Al-Dhaheri, R. S., and L. J. Douglas. 2008. Absence of amphotericin B-tolerant persister cells in biofilms of some Candida species. Antimicrobial Agents and Chemotherapy 52:1884-1887.
Alix, E., and A. Blanc-Potard. 2009. Hydrophobic peptides: Novel regulators within bacterial membranes. Molecular Microbiology 72:5-11.
Aoi, Y., T. Kinoshita, T. Hata, H. Ohta, H. Obokata, and S. Tsuneda. 2009. Hollow-fiber membrane chamber as a device for in situ environmental cultivation. Applied Environmental Microbiology 75:3826-3833.
Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Molecular Systems Biology 2:2006.0008.
Barry, C. E., 3rd, H. I. Boshoff, V. Dartois, T. Dick, S. Ehrt, J. Flynn, D. Schnappinger, R. J. Wilkinson, and D. Young. 2009. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nature Reviews 7:845-855.
Bollmann, A., K. Lewis, and S. S. Epstein. 2007. Incubation of environmental samples in a diffusion chamber increases the diversity of recovered isolates. Applied Environmental Microbiology 73:6386-6390.
Bruns, A., H. Cypionka, and J. Overmann. 2002. Cyclic AMP and acyl homoserine lactones increase the cultivation efficiency of heterotrophic bacteria from the central Baltic Sea. Applied Environmental Microbiology 68:3978-3987.
Challis, G. L. 2005. A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. ChemBioChem 6:601-611.
Christensen, S. K., and K. Gerdes. 2003. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Molecular Microbiology 48:1389-1400.
Connon, S. A., and S. J. Giovannoni. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Applied Environmental Microbiology 68:3878-3885.
Correia, F. F., A. D’Onofrio, T. Rejtar, L. Li, B. L. Karger, K. Makarova, E. V. Koonin, and K. Lewis. 2006. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. Journal of Bacteriology 188:8360-8367.
Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: A common cause of persistent infections. Science 284:1318-1322.
Courcelle, J., A. Khodursky, B. Peter, P. Brown, and P. Hanawalt. 2001. Comparative gene expression profiles following UV exposure in wild type and SOS-deficient Escherichia coli. Genetics 158:41-64.
Davis, K. E., S. J. Joseph, and P. H. Janssen. 2005. Effects of growth medium, inoculum size, and incubation time on culturability and isolation of soil bacteria. Applied Environmental Microbiology 71:826-834.
De Groote, V. N., N. Verstraeten, M. Fauvart, C. I. Kint, A. M. Verbeeck, S. Beullens, P. Cornelis, and J. Michiels. 2009. Novel persistence genes in Pseudomonas aeruginosa identified by high-throughput screening. FEMS Microbiology Letters 297:73-79.
Del Pozo, J., and R. Patel. 2007. The challenge of treating biofilm-associated bacterial infections. Clinical Pharmacology & Therapeutics 82:204-209.
D’Onofrio, A., J. M. Crawford, E. J. Stewart, K. Witt, E. Gavrish, S. Epstein, J. Clardy, and K. Lewis. 2010. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chemical Biology 17:254-264.
Dorr, T., K. Lewis, and M. Vulic. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genetics 5:e1000760.
Falla, T. J., and I. Chopra. 1998. Joint tolerance to beta-lactam and fluoroquinolone antibiotics in Escherichia coli results from overexpression of hipA. Antimicrobial Agents and Chemotherapy 42:3282-3284.
Fernandez De Henestrosa, A. R., T. Ogi, S. Aoyagi, D. Chafin, J. J. Hayes, H. Ohmori, and R. Wood-gate. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Molecular Microbiology 35:1560-1572.
Ferrari, B. C., S. J. Binnerup, and M. Gillings. 2005. Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria. Applied Environmental Microbiology 71:8714-8720.
Garcia-Olmedo, F., A. Molina, J. M. Alamillo, and P. Rodriguez-Palenzuela. 1998. Plant defense peptides. Biopolymers 47:479-491.
Gavrish, E., A. Bollmann, S. Epstein, and K. Lewis. 2008. A trap for in situ cultivation of filamentous actinobacteria. Journal of Microbiological Methods 72:257-262.
Gerdes, K., F. W. Bech, S. T. Jorgensen, A. Lobner-Olesen, P. B. Rasmussen, T. Atlung, L. Boe, O. Karlstrom, S. Molin, and K. von Meyenburg. 1986a. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO Journal 5:2023-2029.
Gerdes, K., P. B. Rasmussen, and S. Molin. 1986b. Unique type of plasmid maintenance function: Postsegregational killing of plasmid-free cells. Proceedings of the National Academy of Sciences of the United States of America 83:3116-3120.
Gibson, R. L., J. L. Burns, and B. W. Ramsey. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 168:918-951.
Hansen, S., K. Lewis, and M. Vulić. 2008. The role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrobial Agents and Chemotherapy. 52:2718-2726.
Harrison, J. J., H. Ceri, N. J. Roper, E. A. Badry, K. M. Sproule, and R. J. Turner. 2005a. Persister cells mediate tolerance to metal oxyanions in Escherichia coli. Microbiology 151:3181-3195.
Harrison, J. J., R. J. Turner, and H. Ceri. 2005b. Persister cells, the biofilm matrix and tolerance to metal cations in biofilm and planktonic Pseudomonas aeruginosa. Environmental Microbiology 7:981-994.
Harrison, J. J., R. J. Turner, and H. Ceri. 2007. A subpopulation of Candida albicans and Candida tropicalis biofilm cells are highly tolerant to chelating agents. FEMS Microbiology Letters 272:172-181.
Harrison, J. J., W. D. Wade, S. Akierman, C. Vacchi-Suzzi, C. A. Stremick, R. J. Turner, and H. Ceri. 2009. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrobial Agents and Chemotherapy 53:2253-2258.
Hayes, F. 2003. Toxins-antitoxins: Plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301:1496-1499.
Honda, H., and D. K. Warren. 2009. Central nervous system infections: Meningitis and brain abscess. Infectious Disease Clinics of North America 23:609-623.
Hu, Y., and A. R. Coates. 2005. Transposon mutagenesis identifies genes which control antimicrobial drug tolerance in stationary-phase Escherichia coli. FEMS Microbiology Letters 243:117-124.
Ikeda, H., J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori, and S. Omura. 2003. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nature Biotechnology 21:526-531.
Jesaitis, A. J., M. J. Franklin, D. Berglund, M. Sasaki, C. I. Lord, J. B. Bleazard, J. E. Duffy, H. Beyenal, and Z. Lewandowski. 2003. Compromised host defense on Pseudomonas aeruginosa biofilms: Characterization of neutrophil and biofilm interactions. Journal of Immunology 171:4329-4339.
Joseph, S. J., P. Hugenholtz, P. Sangwan, C. A. Osborne, and P. H. Janssen. 2003. Laboratory cultivation of widespread and previously uncultured soil bacteria. Applied Environmental Microbiology 69:7210-7215.
Kaeberlein, T., K. Lewis, and S. S. Epstein. 2002. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127-1129.
Kawano, M., L. Aravind, and G. Storz. 2007. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Molecular Microbiology 64:738-754.
Keren, I., D. Shah, A. Spoering, N. Kaldalu, and K. Lewis. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. Journal of Bacteriology 186:8172-8180.
Keren, I., S. Minami, E. Rubin, and K. Lewis. 2011. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2:e00100-11.
Korch, S. B., and T. M. Hill. 2006. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: Effects on macromolecular synthesis and persister formation. Journal of Bacteriology 188:3826-3836.
LaFleur, M. D., C. A. Kumamoto, and K. Lewis. 2006. Candida albicans biofilms produce antifungaltolerant persister cells. Antimicrobial Agents and Chemotherapy 50:3839-3846.
LaFleur, M. D., Q. Qi, and K. Lewis. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrobial Agents and Chemotherapy 54:39-44.
Leid, J. G., M. E. Shirtliff, J. W. Costerton, and A. P. Stoodley. 2002. Human leukocytes adhere to, penetrate, and respond to Staphylococcus aureus biofilms. Infection and Immunity 70:6339-6345.
Levin, B. R., and D. E. Rozen. 2006. Non-inherited antibiotic resistance. Nature Reviews 4:556-562. Lewis, K. 2001. Riddle of biofilm resistance. Antimicrobial Agents and Chemotherapy 45:999-1007.
——. 2007. Persister cells, dormancy and infectious disease. Nature Reviews 5:48-56.
——. 2010. Persister cells. Annual Review of Microbiology 64:357-372.
Lewis, K., S. Epstein, A. D’Onofrio, and L. L. Ling. 2010. Uncultured microorganisms as a source of secondary metabolites. Journal of Antibiotics 63:468-476.
Lipinski, C. A. 2003. The Practice of Medicinal Chemistry. Amsterdam: Academic Press. P. 341.
Lomovskaya, O., H. I. Zgurskaya, K. A. Bostian, and K. Lewis. 2008. Multidrug efflux pumps: Structure, mechanism, and inhibition. In Bacterial Resistance to Antimicrobials, edited by R. G. Wax, K. Lewis, A. A. Salyers, and H. Taber. Boca Raton, FL: CRC Press. Pp. 45-70.
McKenzie, M. D., P. L. Lee, and S. M. Rosenberg. 2003. The dinB operon and spontaneous mutation in Escherichia coli. Journal of Bacteriology 185:3972-3977.
Motiejunaite, R., J. Armalyte, A. Markuckas, and E. Suziedeliene. 2007. Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiology Letters 268:112-119.
Moy, T. I., A. R. Ball, Z. Anklesaria, G. Casadei, K. Lewis, and F. M. Ausubel. 2006. Identification of novel antimicrobials using a live-animal infection model. Proceedings of the National Academy of Sciences of the United States of America 103:10414-10419.
Moy, T. I., A. L. Conery, J. Larkins-Ford, G. Wu, R. Mazitschek, G. Casadei, K. Lewis, A. E. Carpenter, and F. M. Ausubel. 2009. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chemical Biology 4:527-533.
Moyed, H. S., and K. P. Bertrand. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. Journal of Bacteriology 155:768-775.
Mulcahy, L. R., J. L. Burns, S. Lory, and K. Lewis. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. Journal of Bacteriology 192:6191-6199.
Nichols, D., K. Lewis, J. Orjala, S. Mo, R. Ortenberg, P. O’Connor, C. Zhao, P. Vouros, T. Kaeberlein, and S. S. Epstein. 2008. Short peptide induces an “uncultivable” microorganism to grow in vitro. Applied Environmental Microbiology 74:4889-4897.
Nichols, D., N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis, and S. S. Epstein. 2010. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Applied Environmental Microbiology 76:2445-2450.
O’Shea, R., and H. E. Moser. 2008. Physicochemical properties of antibacterial compounds: Implications for drug discovery. Journal of Medicinal Chemistry 51:2871-2878.
Pandey, D. P., and K. Gerdes. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Research 33:966-976.
Payne, D. J., M. N. Gwynn, D. J. Holmes, and D. L. Pompliano. 2007. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nature Reviews 6:29-40.
Pedersen, K., and K. Gerdes. 1999. Multiple hok genes on the chromosome of Escherichia coli. Molecular Microbiology 32:1090-1102.
Pedersen, K., S. K. Christensen, and K. Gerdes. 2002. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Molecular Microbiology 45:501-510.
Peterson, W. L., A. M. Fendrick, D. R. Cave, D. A. Peura, S. M. Garabedian-Ruffalo, and L. Laine. 2000. Helicobacter pylori-related disease: Guidelines for testing and treatment. Archives of Internal Medicine 160:1285-1291.
Phillips, I., E. Culebras, F. Moreno, and F. Baquero. 1987. Induction of the SOS response by new 4-quinolones. Journal of Antimicrobial Chemotherapy 20:631-638.
Ramage, H. R., L. E. Connolly, and J. S. Cox. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genetics 5:e1000767.
Rappe, M. S., S. A. Connon, K. L. Vergin, and S. J. Giovannoni. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630-633.
Sahl, H. G., and G. Bierbaum. 1998. Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from Gram-positive bacteria. Annual Review of Microbiology 52:41-49.
Schatz, A., E. Bugie, and S. A. Waxman. 1944. Streptomycin, a substance exhibiting antibiotic activity against Gram positive and Gram negative bacteria. Proceedings of the Society for Experimental Biology and Medicine 55:66.
Schmelzle, T., and M. N. Hall. 2000. TOR, a central controller of cell growth. Cell 103:253-262.
Schumacher, M. A., K. M. Piro, W. Xu, S. Hansen, K. Lewis, and R. G. Brennan. 2009. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323:396-401.
Shah, D., Z. Zhang, A. Khodursky, N. Kaldalu, K. Kurg, and K. Lewis. 2006. Persisters: A distinct physiological state of E. coli. BMC Microbiology 6:53-61.
Silver, L. L. 2007. Multi-targeting by monotherapeutic antibacterials. Nature Reviews Drug Discovery 6:41-55.
——. 2008. Are natural products still the best source for antibacterial discovery? The bacterial entry factor. Expert Opinion on Drug Discovery 3:487-499.
Singletary, L. A., J. L. Gibson, E. J. Tanner, G. J. McKenzie, P. L. Lee, C. Gonzalez, and S. M. Rosenberg. 2009. An SOS-regulated type 2 toxin-antitoxin system. Journal of Bacteriology 191:7456-7465.
Smith, E. E., D. G. Buckley, Z. Wu, C. Saenphimmachak, L. R. Hoffman, D. A. D’Argenio, S. I. Miller, B. W. Ramsey, D. P. Speert, S. M. Moskowitz, J. L. Burns, R. Kaul, and M. V. Olson. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proceedings of the National Academy of Sciences of the United States of America 103:8487-8492.
Spoering, A. 2006. GlpD and PlsB participate in persister cell formation in Escherichia coli. Journal of Bacteriology 188:5136-5144.
Spoering, A. L., and K. Lewis. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. Journal of Bacteriology 183:6746-6751.
Stevenson, B. S., S. A. Eichorst, J. T. Wertz, T. M. Schmidt, and J. A. Breznak. 2004. New strategies for cultivation and detection of previously uncultured microbes. Applied Environmental Microbiology 70:4748-4755.
Stewart, P. S., and J. W. Costerton. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135-138.
Unoson, C., and E. Wagner. 2008. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Molecular Microbiology 70:258-270.
Vazquez-Laslop, N., H. Lee, and A. A. Neyfakh. 2006. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. Journal of Bacteriology 188:3494-3497.
Vilcheze, C., T. R. Weisbrod, B. Chen, L. Kremer, M. H. Hazbon, F. Wang, D. Alland, J. C. Sacchettini, and W. R. Jacobs, Jr. 2005. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrobial Agents and Chemotherapy 49:708-720.
Vogel, J., L. Argaman, E. G. Wagner, and S. Altuvia. 2004. The small RNA Istr inhibits synthesis of an SOS-induced toxic peptide. Current Biology 14:2271-2276.
Vuong, C., J. M. Voyich, E. R. Fischer, K. R. Braughton, A. R. Whitney, F. R. DeLeo, and M. Otto. 2004. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cellular Microbiology 6:269-275.
Walters, M. C., 3rd, F. Roe, A. Bugnicourt, M. J. Franklin, and P. S. Stewart. 2003. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrobial Agents and Chemotherapy 47:317-323.
Wolfson, J. S., D. C. Hooper, G. L. McHugh, M. A. Bozza, and M. N. Swartz. 1990. Mutants of Escherichia coli K-12 exhibiting reduced killing by both quinolone and beta-lactam antimicrobial agents. Antimicrobial Agents and Chemotherapy 34:1938-1943.
Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389-395.
Zazopoulos, E., K. Huang, A. Staffa, W. Liu, B. O. Bachmann, K. Nonaka, J. Ahlert, J. S. Thorson, B. Shen, and C. M. Farnet. 2003. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nature Biotechnology 21:187-190.
Zengler, K., G. Toledo, M. Rappe, J. Elkins, E. J. Mathur, J. M. Short, and M. Keller. 2002. Cultivating the uncultured. Proceedings of the National Academy of Sciences of the United States of America 99:15681-15686.
NEXT-GENERATION SYNTHETIC GENE NETWORKS61
Timothy K. Lu,62,63,64Ahmad S. Khalil,64and James J. Collins64,65
Synthetic biology is focused on the rational construction of biological systems based on engineering principles. During the field’s first decade of development, significant progress has been made in designing biological parts and assembling them into genetic circuits to achieve basic functionalities. These circuits have been used to construct proof-of-principle systems with promising results in industrial and medical applications. However, advances in synthetic biology have been limited by a lack of interoperable parts, techniques for dynamically probing biological systems and frameworks for the reliable construction and operation of complex, higher-order networks. As these challenges are addressed, synthetic biologists will be able to construct useful next-generation synthetic gene networks with real-world applications in medicine, biotechnology, bioremediation and bioenergy.
_______________
61 Reprinted with kind permission from Nature Publishing Group.
62 Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.
63 Harvard-MIT Health Sciences and Technology, Cambridge, Massachusetts, USA.
64 Howard Hughes Medical Institute, Department of Biomedical Engineering, Center for BioDynamics, and Center for Advanced Biotechnology, Boston University, Boston, Massachusetts, USA.
65 Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts, USA. Correspondence should be addressed to T.K.L. (timlu@mit.edu).
Published online 9 December 2009; doi:10.1038/nbt1591
Published online at http://www.nature.com/naturebiotechnology/.
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions/.
Ten years since the introduction of the field’s inaugural devices—the genetic toggle switch (J.J.C. and colleagues) (Gardner et al., 2000) and repressilator (Elowitz and Leibler, 2000)—synthetic biologists have successfully engineered a wide range of functionality into artificial gene circuits, creating switches (Gardner et al., 2000; Kramer et al., 2004; Isaacs et al., 2003; Ham et al., 2006, 2008; Deans et al., 2007; Ajo-Franklin et al., 2007; Friedland et al., 2009), oscillators (Elowitz and Leibler, 2000; Fung et al., 2005; Stricker et al., 2008; Tigges et al., 2009), digital logic evaluators (Rinaudo et al., 2007; Win and Smolke, 2008), filters (Basu et al., 2005; Hooshangi et al., 2005; Sohka et al., 2009), sensors (Bayer and Smolke, 2005; Kobayashi et al., 2004; Win and Smolke, 2007) and cell-cell communicators (Basu et al., 2005; Kobayashi et al., 2004). Some of these engineered gene networks have been applied to perform useful tasks such as population control (You et al., 2004), decision making for whole-cell biosensors (Kobayashi et al., 2004), genetic timing for fermentation processes (J.J.C. and colleagues) (Ellis et al., 2009) and image processing (Levskaya et al., 2005, 2009; Tabor et al., 2009). Synthetic biologists have even begun to address important medical and industrial problems with engineered organisms, such as bacteria that invade cancer cells (Anderson et al., 2006), bacteriophages with enhanced abilities to treat infectious diseases (Lu and Collins, 2007, 2009), and yeast with synthetic microbial pathways that enable the production of antimalarial drug precursors (Ro et al., 2006). However, in most application-driven cases, engineered organisms contain only simple gene circuits that do not fully exploit the potential of synthetic biology. There remains a fundamental disconnect between low-level genetic circuitry and the promise of assembling these circuits into more complex gene networks that exhibit robust, predictable behaviors.
Thus, despite all of its successes, many more challenges remain in advancing synthetic biology to the realm of higher-order networks with programmable functionality and real-world applicability. Here, instead of reviewing the progress that has been made in synthetic biology, we present challenges and goals for next-generation synthetic gene networks, and describe some of the more compelling circuits to be developed and application areas to be considered.
Synthetic Gene Networks: What Have We Learned and What Do We Need?
The engineering of mechanical, electrical and chemical systems is enabled by well-established frameworks for handling complexity, reliable means of probing and manipulating system states and the use of testing platforms—tools that are largely lacking in the engineering of biology. Developing properly functioning biological circuits can involve complicated protocols for DNA construction, rudimentary model-guided and rational design, and repeated rounds of trial and error followed by fine-tuning. Limitations in characterizing kinetic processes and interactions between synthetic components and other unknown constituents in vivo make troubleshooting and modeling frustrating and prohibitively time
consuming. As a result, the design cycle for engineering synthetic gene networks remains slow and error prone.
Fortunately, advances are being made in streamlining the physical construction of artificial biological systems, in the form of resources and methods for building larger engineered DNA systems from smaller defined parts (Ellis et al., 2009; Czar et al., 2009; Guido et al., 2006; Shetty et al., 2008). Additionally, large-scale DNA sequencing and synthesis technologies are gradually enabling researchers to directly program whole genes, genetic circuits and even genomes, as well as to re-encode DNA sequences with optimal codons and minimal restriction sites (see review, Carr and Church [2009]).
Despite these advances in molecular construction, the task of building synthetic gene networks that function as desired remains extremely challenging. Accelerated, large-scale diversification (Wang et al., 2009) and the use of characterized component libraries in conjunction with in silico models for a priori design (Ellis et al., 2009) are proving useful in helping to fine-tune network performance toward desired outputs. Even so, in general, synthetic biologists are often fundamentally limited by a dearth of interoperable and modular biological parts, predictive computational modeling capabilities, reliable means of characterizing information flow through engineered gene networks and test platforms for rapidly designing and constructing synthetic circuits.
In the following subsections, we discuss four important research efforts that will improve and accelerate the design cycle for next-generation synthetic gene networks: first, advancing and expanding the toolkit of available parts and modules; second, modeling and fine-tuning the behavior of synthetic circuits; third, developing probes for reliably quantifying state values for synthetic (and natural) biomolecular systems; and fourth, creating test platforms for characterizing component interactions within engineered gene networks, designing gene circuits with increasing complexity and developing complex circuits for use in higher organisms. These advances will allow synthetic biologists to realize higher-order networks with desired functionalities for satisfying real-world applications.
Interoperable parts and modules for synthetic gene networks
Although there has been no shortage of novel circuit topologies to construct, limitations in the number of interoperable and well-characterized parts have constrained the development of more complex biological systems (Ellis et al., 2009; Guido et al., 2006; Purnick and Weiss, 2009; Lucks et al., 2008). The situation is complicated by the fact that many potential interactions between biological parts, which are derived from a variety of sources within different cellular backgrounds, are not well understood or characterized. As a result, the majority of synthetic circuits are still constructed ad hoc from a small number of commonly used components (e.g., LacI, TetR and lambda repressor proteins and regulated promoters) with a significant amount of trial and error. There is a pressing need
to expand the synthetic biology toolkit of available parts and modules. Because physical interconnections cannot be made in biological systems to the same extent as electrical and mechanical systems, interoperability must be derived from chemical specificity between parts and their desired targets. This limits our ability to construct truly modular parts and highlights the need for rigorous characterization of component interactions so that detrimental interactions can be minimized and factored into computational models.
Engineered zinc fingers constitute a flexible system for targeting specific DNA sequences, one which could significantly expand the available synthetic biology toolkit for performing targeted recombination, controlling transcriptional activity and making circuit interconnections. Zinc-finger technology has primarily been used to design zinc-finger nucleases that generate targeted double-strand breaks for genomic modifications (Maeder et al., 2008). These engineered nucleases may be used to enhance recombination in large-scale genome engineering techniques (Wang et al., 2009). A second and potentially very promising use of engineered zinc fingers is as a source of interoperable transcription factors, which would greatly expand the current and limited repertoire of useful activators and repressors. In fact, zinc fingers have already been harnessed to create artificial transcription factors by fusing zinc-finger proteins with activation or repression domains (Beerli et al., 2000; Park et al., 2003). Libraries of externally controllable transcriptional activators or repressors could be created by engineering protein or RNA ligand-responsive regulators, which control the transcription or translation of zinc finger–based artificial transcription factors themselves (Bayer and Smolke, 2005). These libraries would enable the construction of basic circuits, such as genetic switches (Gardner et al., 2000), as well as more complex gene networks. In fact, several of the higher-order networks we describe below rely on having multiple reliable and interoperable transcriptional activators and repressors for proper functioning.
Even so, these engineered transcription factors have not yet been fully characterized, and if they are to be used as building blocks for complex gene networks, then knowledge of their in vivo kinetics and input-output transfer functions would be beneficial. In addition, much of the rich dynamics associated with small, synthetic gene networks is attributable to the cooperative binding or multimerization of transcription factors, and it is not yet clear what further engineering is required to endow zinc-finger transcription factors with such features.
Nucleic acid–based parts, such as RNAs, are also promising candidates for libraries of interoperable parts because they can be rationally programmed based on sequence specificity (Deans et al., 2007; Isaacs et al., 2004; Win et al., 2009). Novel circuit interconnections could be established using small interfering RNAs (siRNAs) to control the expression of specific components. Recombinases, which target specific DNA recombinase-recognition sites, also represent a fruitful, underutilized source of interoperable parts. Recombinases have been used in the context of synthetic biology to create memory elements and genetic
counters (Friedland et al., 2009). However, more than 100 natural recombinases are known, and these can be engineered by mutagenesis and directed evolution for greater diversity and sequence specificity (Buchholz et al., 1998; Kilby et al., 1993; Santoro and Schultz, 2002; Groth and Calos, 2004).
Libraries of well-characterized, interoperable parts, such as transcription factors and recombinases, would vastly enhance the ability of synthetic biologists to build more complex gene networks with greater reliability and real-world applicability. In addition to libraries of individual parts, it would be of great value to have well-characterized and interoperable modules (e.g., switches, oscillators and interfaces) that could be used in a plug-and-play fashion to create higher-order networks and programmable cells. As the number of parts and modules expands, high-throughput, combinatorial efforts for quantifying the levels of interference and cross-talk between multiple components within cells will be increasingly important as guides for choosing the most appropriate components for network assembly.
Modeling and fine-tuning synthetic gene networks
Integrated efforts for modeling and fine-tuning synthetic gene circuits are useful for ensuring that assembled networks operate as intended. Such approaches will be increasingly important as more complex circuits are constructed along with the expanded development of interoperable parts. Although studies have shown that in some cases, component properties alone are sufficient for predicting network behavior (Ellis et al., 2009; Guido et al., 2006; Kaplan et al., 2008), others have demonstrated the need for modeling and fine-tuning networks after their basic topologies have been established (Gardner et al., 2000; Ellis et al., 2009). A multi-step design cycle that involves creating diverse component libraries, constructing, characterizing and modeling representative network topologies, and assembling and fine-tuning desired circuits, followed by subsequent refinement cycles (Ellis et al., 2009), will be crucial for the successful design and construction of next-generation synthetic gene networks.
The fine-tuning of biomolecular parts and networks can be achieved by developing diverse component libraries through mutagenesis followed by in-depth characterization and modeling (Ellis et al., 2009; Alper et al., 2005; Cox et al., 2007; Hammer et al., 2006; Jensen and Hammer, 1998; Murphy et al., 2007). Significant progress has been made in tuning gene expression by altering transcriptional, translational and degradation activities. For example, promoter libraries with a range of transcriptional activities can be created and characterized, plugged into in silico models and then used to develop synthetic gene networks with defined outputs, without significant post-hoc adjustments (Ellis et al., 2009; Alper et al., 2005; Cox et al., 2007; Hammer et al., 2006; Jensen and Hammer, 1998; Murphy et al., 2007). Alternatively, synthetic ribosome binding site (RBS) sequences can be used to optimize protein expression levels. Recently, Salis et
al. (2009) have developed a thermodynamic model for predicting the relative translational initiation rates for a protein with different upstream RBS sequences, a model that can also be used to rationally forward-engineer RBS sequences to give desired protein expression. In addition, protein degradation can be controlled by tagging proteins with degradation-targeting peptides that impart different degradation dynamics (Andersen et al., 1998).
By automating the construction and characterization of biomolecular components, extensive libraries could be created for the rapid design and construction of complex gene networks. These efforts, coupled with in silico modeling, would serve to fast-track synthetic biology (more detailed discussions of modeling techniques for synthetic biology are found in Ellis et al., 2009; Guido et al., 2006; Nevozhay et al., 2009; Chandran et al., 2009; Kaznessis, 2009; Hasty et al., 2001). However, to build reliable models of biomolecular parts and networks, new methods for probing and acquiring detailed in vitro and in vivo measurements are needed, which we discuss below.
Probes for characterizing synthetic gene networks
Significant advances have been made in the development of new technologies for manipulating biological systems and probing their internal states. At the single-molecule level, for instance, optical tweezers and atomic force microscopes provide new, direct ways to probe the biophysical states of single DNA, RNA and protein molecules as they undergo conformational changes and other dynamical processes (Khalil et al., 2007, 2008; Svoboda and Block, 1994; Bustamante et al., 2003; Neuman and Nagy, 2008). However, we lack similar tools for tracking the in vivo operation of synthetic gene circuits in a high-throughput fashion. Ideally, making dynamical measurements of biological networks would involve placing sensors at multiple internal nodes, akin to how current and voltage are measured in electrical systems. Furthermore, external manipulation of synthetic biomolecular systems is typically accomplished by the addition of chemical inducers, which can suffer from cross-talk (Lee et al., 2007), be difficult to remove and be consumed over time. As a result, inputs are often troublesome to control dynamically.
Microfluidic devices have been coupled to single-cell microscopy and image processing techniques to enable increasingly precise manipulation and measurement of cells, especially since inputs can be modulated over time (Gulati et al., 2009; Bennett and Hasty, 2009). These systems allow the rapid addition and removal of chemical inducers, enabling more sophisticated, time-dependent inputs than conventional step functions, while also enabling researchers to track and quantify single cells for long periods of time. These developments make possible the wider use of well-established engineering approaches for analyzing circuits and other systems in synthetic biology. For example, frequency-domain analysis, a technique used commonly in electrical engineering (Simpson et al., 2003;
Mettetal et al., 2008), can be employed with microfluidics to characterize the transfer functions and noise behaviors of synthetic biological circuits (Simpson et al., 2003; Mettetal et al., 2008; Bennett et al., 2008). Additionally, small-signal linearization of nonlinear gene circuits can be achieved by applying oscillatory perturbations with microfluidics and measuring responses at the single-cell level (Mettetal et al., 2008; Bennett et al., 2008).
Indeed, microfluidics provides a useful platform for perturbing synthetic gene circuits with well-controlled inputs and observing the outputs in high-resolution fashion. Without the proper ‘sensors’ (that is, for quantitatively and simultaneously probing all the internal nodes of a given gene circuit), however, this technology alone is not sufficient to bring full, engineering-like characterization to synthetic gene networks.
Thus far, probes enabling quantitative measurements of synthetic gene circuits have primarily focused on the use of fluorescent proteins for in vivo quantification of promoter activity or protein expression. With the advent of novel mass spectrometry–based methods that provide global, absolute protein concentrations in cells (Malmstrom et al., 2009), quantitative transcriptome data can now be merged with proteome data, improving our ability to characterize and model the dynamics of synthetic gene networks. Global proteomic data may also assist synthetic biologists in understanding the metabolic burden that artificial circuits place on host cells. Further efforts to devise fluorescent-based and other types of reporters for the simultaneous monitoring of transcriptome and proteome dynamics in vivo are needed to close the loop on full-circuit accounting. Some promising tools under development include tracking protein function by incorporating unnatural amino acids that exhibit fluorescence (Summerer et al., 2006; Wang et al., 2006), quantum dots (Michalet et al., 2005) and radiofrequency-controlled nanoparticles (Hamad-Schifferli et al., 2002).
As the field awaits entire-circuit probes, there are, in the meantime, several potentially accessible technologies for increasing the throughput and pace of piecewise gene-circuit characterization. Recent advances in engineering light-inducible biological parts and systems (Levskaya et al., 2005, 2009; Boyden et al., 2005) have unlocked the potential for optical-based circuit characterization, expanding the number and type of tunable knobs available to synthetic biologists. For instance, by coupling a synthetic gene network of interest to a biological light/dark sensor as well as to fluorescent protein outputs, one could potentially measure the network’s input/output transfer function in a high-throughput fashion using spectrophotometric microplate readers, without having to add varying concentrations of chemical inducers. In essence, both control and monitoring of biomolecular systems would be accomplished using reliable and high-speed optics that are typically associated with fluorescence readouts and microscopy. This is an exciting prospect, particularly in the context of microfluidic devices, which would facilitate the focusing of optical inputs and readouts to single cells.
Using electrical signals, in lieu of chemical or optical signals, for control
and monitoring of biological systems would also present high-speed advantages. Recently, advances have been made in integrating silicon electronics with lipid bilayers containing transmembrane pores to perform electronic signal conduction (Misra et al., 2009). This technology may eventually allow direct communication and control between engineered cells and electronic circuits by means of ionic flow. The incorporation of these and other technologies to perturb and monitor the in vivo performance of synthetic gene networks will enable us to achieve desired functionality faster and more reliably.
Test platforms for engineering complex gene circuits
Increasing complexity—whether assembling larger synthetic gene networks from smaller ones or engineering circuits into higher organisms—dramatically increases the number of potential failure modes. In the former case, combining multiple individually functioning genetic circuits into a single cellular background can lead to unintended interactions among the synthetic components or with host factors, and these various failure modes are often difficult to pinpoint and isolate from one another. In the latter case, engineering synthetic networks for mammalian systems poses additional challenges beyond engineering circuits for bacterial and yeast strains, which have comparatively well-characterized genomes, transcriptomes, proteomes and metabolomes. Mammalian systems are much more complex and possess substantially less well-characterized components for engineering (Weber and Fussenegger, 2009), but for these and other reasons, constitute fertile ground for new applications and genetic parts.
The development of test platforms where engineered gene circuits can be designed and validated before being deployed in other or more complex cellular backgrounds would mitigate failure-prone jumps in complexity. These platforms could be used to verify or debug circuit topology and basic functionality in well-controlled environments. For example, cells optimized for testing may be engineered to have minimal genomes to decrease the risk of pleiotropic or uncharacterized interactions between the host and the synthetic networks (Gibson et al., 2008; Glass et al., 2006; Lartigue et al., 2007, 2009; Carrera et al., 2009). The use of orthogonal parts that are decoupled from host cells may enable the dedication of defined cellular resources to engineered functions, which can simplify the construction and troubleshooting of gene circuits. For example, nucleic acid–based parts can be designed to function orthogonally to the wild-type cellular machinery (Rackham and Chin, 2005; Wang et al., 2007; An et al., 2009). Artificial codons and unnatural amino acids, which have enabled new methods for studying existing proteins and the realization of proteins with novel functions, could also be used to produce synthetic circuits that function orthogonally to host cells (Wang et al., 2009). Simplifying backgrounds would additionally enable more accurate computational modeling of complex circuits before they are deployed into their ultimate environments. Furthermore, minimal cells could
themselves contain synthetic circuits that provide useful testing functionalities, such as multiplexed transcriptional and translational controls and output probes.
Lower organisms can also be useful for the construction and characterization of synthetic gene networks before such systems are extended and deployed into higher organisms. In fact, several synthetic circuits, such as clocks and switches, were initially developed in bacteria and later translated into mammalian counterparts using analogous design principles (Kramer et al., 2004; Deans et al., 2007; Tigges et al., 2009). Additionally, lower-organism test platforms could be endowed with certain features of interest from desired higher-organism hosts. For example, RNA interference–based circuits could be built first in Saccharomyces cerevisiae before being used in mammalian cells (Drinnenberg et al., 2009). In one case, mitochondrial DNA was engineered into Escherichia coli before retransplantation into mammalian hosts (Yoon and Koob, 2003). Other biomolecular systems and components that are ripe for engineering in lower organisms include chromatin, ubiquitins and proteosomes.
The introduction of synthetic gene networks into higher organisms also runs the risk of compromising natural networks, which have evolved to maintain cellular robustness. Accordingly, methods for simplifying organisms for designing and testing synthetic circuits could be extended to engineer final deployment hosts, making them more conducive to synthetic gene circuits. Ultimately, in vivo directed evolutionary methods, based on repeated rounds of mutagenesis and selection within final cellular backgrounds, could be used to identify the optimal performance conditions of synthetic gene networks after their basic functionalities have been validated in earlier test platforms (Wang et al., 2009).
Next-Generation Gene Networks
Advancing synthetic gene circuits into the realm of higher-order networks with programmable functionality is one of the ultimate goals of synthetic biology. Useful next-generation gene networks should attempt to satisfy at least one of the following criteria: first, yield insights into the principles that guide the operation of natural biological systems; second, highlight design principles and/or provide modules that can be applied to the construction of other useful synthetic circuits; third, advance the tools available for novel scientific experiments; and fourth, enable real-world applications in medicine, industry and/or agriculture. Below, we describe several next-generation gene circuits and discuss their potential utility in the context of the above criteria.
Tunable filters and noise generators
Fine-tuning the performance of a synthetic gene network typically means reengineering its components, be it by replacing or mutating its parts. Networks whose responses can be tuned without the reengineering of its parts, such as the
biological version of a tunable electronic filter, would enable more sophisticated cellular-based signal processing. Synthetic transcriptional cascades can exhibit low-pass filter characteristics (Hooshangi et al., 2005), and artificial gene circuits with negative autoregulation are capable of pushing the noise spectra of their outputs to higher frequencies, where it can be filtered by the low-pass characteristics of a downstream gene cascade (Austin et al., 2006). Tunable genetic filters with respect to time could be implemented by tuning RNA and/or protein degradation in autoregulated negative-feedback circuits (Simpson et al., 2003; Mar et al., 1999; McGinness et al., 2006; Banaszynski et al., 2006) (Fig. A12-1). Such circuits would be useful in studying and shaping noise spectra to optimize the performance of artificial gene networks.
Recently, an externally tunable, bacterial bandpass-filter has been described (Sohka et al., 2009) that uses low-pass and high-pass filters in series to derive bandpass activity with respect to enzymes and inducer molecules. These types of filters, when coupled to quorum-sensing modules, can be used for spatial patterning applications (Basu et al., 2005; Sohka et al., 2009). They could also be
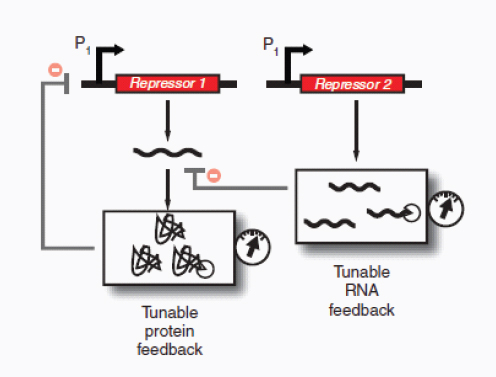
FIGURE A12-1 Tunable genetic filter. Filter characteristics can be adjusted by tuning the degradation of RNA and protein effectors in negative-feedback loops. Examples of RNA effectors include siRNAs, riboregulators and ribozymes. Examples of protein effectors include transcriptional activators and repressors. In this example, the P1 promoter is suppressed by transcriptional repressor proteins expressed from the Repressor 1 gene.
readily extended to complex multicellular pattern formation by engineering a suite of different cells, each carrying filters that respond to different inputs. Synthetic gene circuits based on tunable filters may also make useful platforms for studying cellular differentiation and development, as artificial pattern generation is a model for how natural systems form complex structures (Basu et al., 2005; Sohka et al., 2009).
Along similar lines, recent developments in stem cell biology have unlocked important potential roles for synthetic gene networks (Takahashi et al., 2007). For example, it has been shown that stochastic fluctuations in protein expression in embryonic stem cells are important for determining differentiation fates (Macarthur et al., 2009). Indeed, stochasticity might be harnessed in differentiation to force population-wide heterogeneity and provide system robustness, though it may also be detrimental if it causes uncontrollable differentiation.
The effects of stochasticity in stem cell differentiation could be studied with synthetic gene circuits that act as tunable noise generators. Lu et al., for instance, considered two such designs for modulating the noise profile of an output protein (Lu et al., 2008). This showed that the mean value and variance of the output can be effectively tuned with two external signals, one for regulating transcription and the other for regulating translation, and to a greater extent with three external signals, the third for regulating DNA copy number (Lu et al., 2008). By varying noise levels while keeping mean expression levels constant, the thresholds at which gene expression noise yields beneficial versus detrimental effects on stem cell differentiation can be elucidated (J.J.C. and colleagues) (Blake et al., 2006).
Furthermore, the discovery of induced pluripotent stem cells (iPSCs), based on the controlled expression of four transcription factors (OCT4, SOX2, KLF2 and MYC) in adult fibroblasts, has created a source of patient-specific progenitor cells for engineering (Takahashi et al., 2007). Genetic noise generators and basic control circuits could be used to dissect the mechanism for inducing pluripotency in differentiated adult cells by controlling the expression levels of the four iPSC-dependent transcription factors. Ultimately, these efforts could lead to the development of timing circuits (Ellis et al., 2009) for higher-efficiency stem cell reprogramming.
Lineage commitment to trophectoderm, ectoderm, mesoderm and endoderm pathways are controlled by distinct sets of genes (Macarthur et al., 2009), and many interacting factors, including growth factors, extracellular matrices and mechanical forces, play important roles in cellular differentiation (Discher et al., 2009). As differentiation pathways become better understood, synthetic gene cascades may be used to program cellular commitment with increased fidelity for applications in biotechnology and regenerative medicine.
Analog-to-digital and digital-to-analog converters
Electrical engineers have used digital processing to achieve reliability and flexibility, even though the world in which digital circuits operate is inherently
analog. Although synthetic biological circuits are unlikely to match the computing power of digital electronics, simple circuits inspired by digital and analog electronics may significantly increase the reliability and programmability of biological behaviors.
For example, biological analog-to-digital converters could translate external analog inputs, such as inducer concentrations or exposure times, into internal digital representations for biological processing. Consider, for instance, a bank of genetic switches with adjustable thresholds (Fig. A12-2a). These switches could be made out of libraries of artificial transcription factors, as described above. This design would perform discretization of analog inputs into levels of digital output. Depending on the level of analog inputs, different genetic pathways could be activated. Cells possessing analog-to-digital converters would be useful as biosensors in medical and environmental settings. For example, whole-cell biosensors (Kobayashi et al., 2004), resident in the gut, may be engineered to generate different reporter molecules that could be measured in stool depending on the detected level of gastrointestinal bleeding. Expressing different reporter molecules rather than a continuous gradient of a single reporter molecule would yield more reliable and easily interpretable outputs. Digital-to-analog converters, on the other hand, would translate digital representations back into analog outputs (Fig. A12-2b); such systems could be used to reliably set internal system states. For example, instead of fine-tuning transcriptional activity with varying amounts of chemical inducers, a digital-to-analog converter, composed of a bank of genetic switches, each of which is sensitive to a different inducer, might provide better control. If each activated switch enabled transcription from promoters of varying strengths (Poutput,3 > Poutput,2 > Poutput,1), then digital combinations of inducers could be used to program defined levels of transcriptional activities (Fig. A12-2b). Such a circuit might be useful in biotechnology applications, where reliable expression of different pathways is needed for programming different modes of operation in engineered cells. In addition, digital-to-analog converters may be useful in providing a multiplexed method for probing synthetic circuits. For example, because each analog level is associated with a distinct digital state, a single analog output can allow one to infer the internal digital state of a synthetic gene network (Fig. A12-2b).
Adaptive learning networks
Synthetic gene networks that can learn or adapt to exogenous conditions could provide insight into natural networks and be useful for applications where adaptation to external stimuli may be advantageous, such as autonomous whole-cell biosensors (Fernando et al., 2009; Fritz et al., 2007). Endogenous biomolecular networks in bacteria can exhibit anticipatory behavior for related perturbations in environmental stimuli (Tagkopoulos et al., 2008; Mitchell et al., 2009). This type of behavior and the associated underlying design principles could, in principle, be harnessed to endow transcriptional networks with the ability to learn
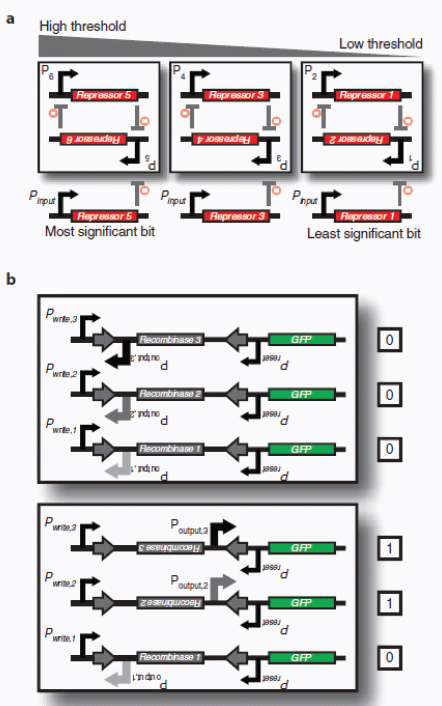
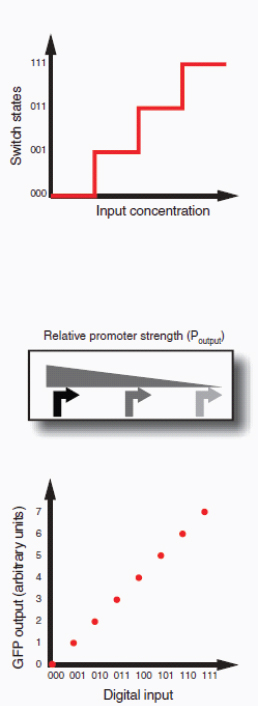
FIGURE A12-2 Genetic signal converters. (a) Analog-to-digital converter circuit that enables the discretization of analog inputs. The circuit is composed of a bank of toggle switches that have increasing response thresholds so that sequential toggling is achieved as input levels increase. The design could enable different natural or synthetic pathways to be activated depending on distinct input ranges, which may be useful in cell-based biosensing applications. Inputs into promoters and logic operations are shown explicitly except when the promoter (P) name is italicized, which represents an inducible promoter. (b) Digital-to-analog converter circuit that enables the programming of defined promoter activity based on combinatorial inputs. The circuit is composed of a bank of recombinase-based switches, known as single-invertase memory modules (SIMMs) (Friedland et al., 2009). Each SIMM is composed of an inverted promoter and a recombinase gene located between its cognate recognition sites, indicated by the arrows. Upon the combinatorial addition of inducers that activate specific Pwrite promoters, different SIMMs will be flipped, enabling promoters of varying strength to drive green fluorescent protein (GFP) expression. This allows combinatorial programming of different levels of promoter activity.
(Fernando et al., 2009), much like synaptic interconnections between neurons. A basic design that would enable this functionality involves two transcriptional activators (Activator A and Activator B), each of which is expressed in the presence of a different stimulus (Fig. A12-3a). Suppose that both transcriptional activators drive the expression of effector proteins (Effector A and Effector B), which control distinct genetic pathways. When both transcriptional factors are active, indicating the simultaneous presence of the two stimuli, a toggle switch is flipped ON. This creates an associative memory. Subsequently, if either of the transcription factors is activated, AND logic between the ON toggle switch and one transcriptional activator produces the effector protein that controls the pathways of the other activators. On the basis of this design, cells could be programmed to associate simultaneous inputs and exhibit anticipatory behavior by activating the pathways of associated stimuli, even in the presence of only one of the stimuli. In another example of a learning network, one could design bacteria that could be taught ‘winner-take-all’ behavior in detecting stimuli, similar to cortical neural processing (Lee et al., 1999). In this example, bacteria could be exposed to different types of chemical stimuli (Inducers A–C; Fig. A12-3b). An exogenously added inducer (Inducer ‘Learn’) acts as a trigger for learning and serves as one input into multiple, independent transcriptional AND gates, which possess secondary inputs for detecting the presence of each of the different chemical stimuli. Each gate drives an individual toggle switch that, when flipped, suppresses the flipping of the other switches. This creates a winner-take-all system in which the presence of the most abundant chemical stimuli is recorded. Furthermore, the toggle switch outputs could be fed as inputs into transcriptional AND gates, which once again possess secondary inputs for detecting the presence of the different stimuli. If these gates drive different fluorescent reporters when activated, then the overall system will associate only a single type of stimuli with the learning trigger and respond with an output only in the presence of the single type of stimuli in the future. This system could potentially be adapted to create chemotactic bacteria that ‘remember’ a particular location or landmark and only respond to the gradient of one chemoattractant.
In more complicated instances of learning networks, it is conceivable that synthetic gene circuits could be designed to adapt on their own, that is, without external mutagenesis or exogenous nucleic acids. For example, transcription-based interconnections could be dynamically reconfigured based on the expression of DNA recombinases (Friedland et al., 2009). Another design could involve error-prone RNA polymerases, which create mutant RNAs that could be reverse-transcribed and joined back into the genome based on double-stranded breaks created by zinc-finger nucleases. Specificity for where the mutations would occur could be achieved by using promoters that are uniquely read by the error-prone RNA polymerases, such as T7 promoters with a T7 error-prone RNA polymerase, and zinc-finger nucleases that define where homologous recombination can occur (Brakmann and Grzeszik, 2001). In this design, enhanced mutagenesis frequencies could be targeted to specific regions of the genome.
Protein-based computational circuits
Beyond DNA- and RNA-based circuits, protein-based synthetic systems have the potential to enable flexible and fast computation through post-translational mechanisms (Yeh et al., 2007; Dueber et al., 2007; Bashor et al., 2008). Protein-based circuits are advantageous in that they can be designed to target synthetic activities to subcellular locations (Levskaya et al., 2009). In this way, different sites within the same cell could have different protein circuit states rather than relying solely on shared cellular promoter states, thereby enabling researchers to explore the functional dynamics and consequences of cellular localization. Protein-based designs can also operate on much shorter time scales than genetic circuits because their operation is independent of the transcription and translation machinery (Goldberg et al., 2009). Accordingly, it would be exciting to develop protein-based circuits that can act as rapidly responding logic gates, smart sensors or memory elements.
With regards to this last application, synthetic amyloids could serve as novel components for epigenetic memory circuits. By fusing a yeast prion determinant from Sup35 to the rat glucocorticoid receptor, a transcription factor regulated by steroid hormone, Li and Lindquist (2000) demonstrated that the state of transcriptional activity from the fused protein could be affected and inherited stably in an epigenetic fashion. Given the increasing number of identified prionogenic proteins (Alberti et al., 2009), there is an opportunity to create amyloid-based memory systems that transmit functionality from one generation to the next (Fig. A12-4). In these systems, aggregation could be induced by the transient expression of the prionogenic domain (PD), whereas disaggregation could be achieved by expressing protein remodeling factors, such as chaperones (heat shock protein 104). Though this system relies on the transcription and translation of prionogenic and disaggregating factors, it may enable the control of protein effectors that can operate on shorter time scales. For example, enzymes fused to a prionogenic domain may exhibit different activity levels depending on whether they are attached to an amyloid core.
Because genetic circuits and proteins function on different time scales, it would also be worthwhile to develop synthetic networks that couple both modalities. For example, the output of protein-based computation could be stored in recombinase-based memory elements (Ham et al., 2006, 2008; Friedland et al., 2009). It would also be conceivable to couple the two types of networks to harness their varied filtering capabilities. For example, the mitogen-activated protein kinase cascade contains both positive-feedback and negative-feedback loops that enable rapid activation followed by deactivation (Bhalla et al., 2002), thus acting like a high-pass filter. On the other hand, transcription- and translation-based gene networks operate on longer time scales rendering them effective low-pass filters. Thus, synthetic kinase/phosphatase circuits that in turn drive gene-based networks could be used to create bandstop filters, which could be coupled with other bandpass filters and used for complex patterning applications.
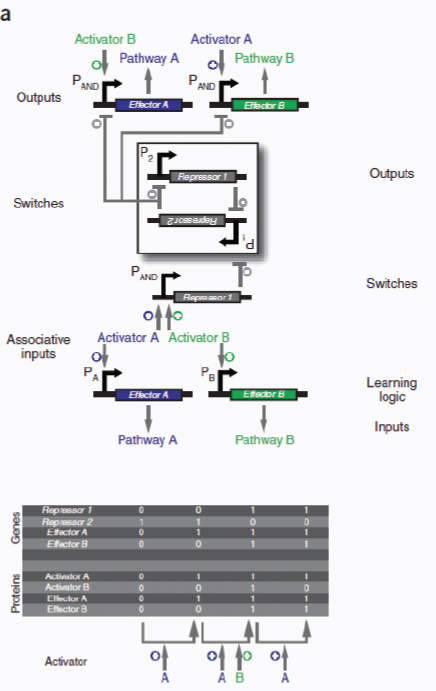
FIGURE A12-3 Adaptive learning networks. (a) Associative memory circuit enables association between two simultaneous inputs (‘Activator A’ and ‘Activator B’) so that the subsequent presence of only a single input can drive its own pathway and the pathway of the other input. Associations between inputs are recorded by a promoter ‘PAND’ that is activated in the presence of Activator A and Activator B to toggle the memory switch. Inputs
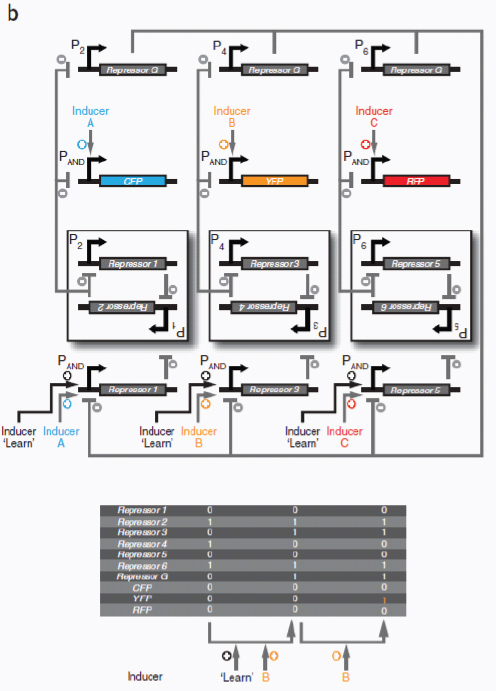
into promoters and logic operations are shown explicitly except when the promoter name is italicized, which represents an inducible promoter. (b) Winner-take-all circuit allows only one input out of many to be recorded. This effect is achieved by a global repressor protein that gates all inputs and prevents them from being recorded if there has already been an input recorded in memory.
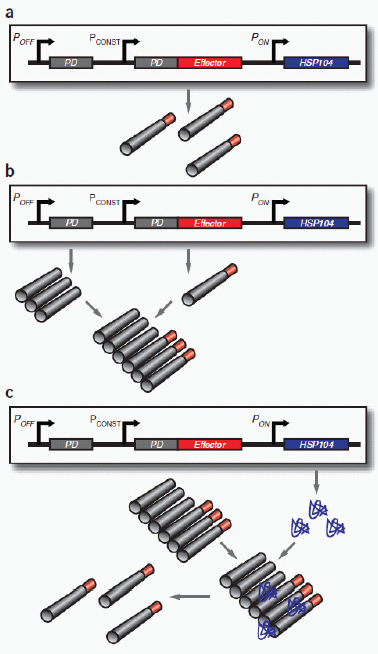
FIGURE A12-4 Amyloid-based memory. (a) Amyloid-based memory can be implemented by fusing a prionogenic domain (PD) to an effector gene, such as a transcriptional activator. (b) Overexpressing the prion-determining region via promoter ‘POFF’ causes aggregation of the fusion protein, rendering the effector inactive. (c) Subsequent overexpression of chaperone proteins (e.g., HSP104), which act to disaggregate amyloids, via promoter ‘PON’ releases the effector from the amyloid state and enables it to fulfill its function. Inputs into promoters and logic operations are shown explicitly except when the promoter name is italicized, which represents an inducible promoter.
Intercell signaling circuits and pulse-based processing for genetic oscillators
Robust genetic oscillators with tunable periods have been developed through a combination of experimental and computational efforts (Stricker et al., 2008; Tigges et al., 2009; Tsai et al., 2008). In addition to shedding light on the design principles guiding the evolution of naturally occurring biological clocks and circadian rhythms, these synthetic oscillators may also have significant utility in biotechnology applications, such as in the synthesis and delivery of biologic drugs. Glucocorticoid secretion, for instance, has a circadian and ultradian pattern of release, resulting in transcriptional pulsing in cells that contain glucocorticoid receptors (Stavreva et al., 2009). Therefore, pulsatile administration of hormones may have therapeutic benefit compared with synthetic hormones applied in a non-ultradian schedule.
An alternative to device-based periodic drug delivery systems could be engineered bacteria that reside in the human gut and synthesize an active drug at fixed time intervals. To realize such an application, one would need to develop and implement intercell signaling circuits for synchronizing and entraining synthetic genetic oscillators (McMillen et al., 2002; Garcia Ojalvo et al., 2004). Such circuits could be based, for example, on modular components from bacterial quorum sensing systems. Along similar lines, one could engineer light-sensitive (Levskaya et al., 2005, 2009) entrainment circuits for synchronizing mammalian synthetic genetic oscillators. This may help in the construction of oscillators that can faithfully follow circadian rhythms. Spike- or pulse-based processing is present in neurons and has been adapted for use in hybrid computation in electrical systems, where interspike times are viewed as analog parameters and spike counts are viewed as digital parameters (Sarpeshkar and O’Halloran, 2002). In synthetic gene circuits, pulse-based processing may open up exciting new methods for encoding information in engineered cells. For example, instead of transmitting information between cells by means of absolute levels of quorum-sensing molecules, the frequency of a robust genetic oscillator could be modulated. This might be useful in delivering information over longer distances, as frequency information may be less susceptible to decay over distance than absolute molecule levels. Representing signals in this fashion is analogous to frequency modulation encoding in electrical engineering.
Engineered circuits for biological containment
Biological containment, which refers to efforts for ensuring that genetically modified organisms do not spread throughout the natural environment, can be achieved by passive or active techniques. In passive containment, cells are engineered to be dependent on exogenous supplementation to compensate for gene defects, whereas in active containment, cells are engineered to directly express toxic compounds when located outside their target environments (Molin et al., 1993). Synthetic genetic counters or timers for programmed cell death could be
used as an active containment tool. Counting circuits could, for example, be designed to trigger cell suicide after a defined number of cell cycles or a sequence of events. Recently, we have developed two designs for synthetic counters—a recombinase-based cascade of memory units and a riboregulated transcriptional cascade—that could be adapted for this purpose (Friedland et al., 2009). In each case, one could incorporate into the counters promoters that are cell cycle–dependent and replace the output reporter proteins with toxic proteins (Fig. A12-5). Circuits of this sort would enable cells to be programmed to have limited, prescribed lifetimes.
Redundant circuits that implement digital logic allowing for the conditional survival of engineered cells only within their desired environments would also potentially reduce the failure rate of biological containment. If a broad set of interoperable parts were developed, multiple layers of control circuits could be built for increased reliability. As in electrical and mechanical engineering, quantitative analysis of failure rates in biological systems would enable improved systems-level design and robustness of synthetic gene networks. This could be accomplished, for example, by subjecting synthetic containment circuits to a variety of stressful conditions that would lead to increased mutation rates and thus improper functioning. Rational and directed evolutionary methods to engineer cells with decreased mutation rates or the application of redundant circuits could then be employed to minimize failure rates.
Whole-cell biosensors and response systems
Programmable cells that act as whole-cell biosensors have been created by interfacing engineered gene networks with the cell’s natural regulatory circuitry (Kobayashi et al., 2004) or with other biological components, such as light-responsive elements (Levskaya et al., 2005, 2009). The development of novel or reengineered sensory modalities and components would expand the range of applications that programmable cells could address. This could involve engineering proteins or RNAs to detect a range of small molecules (Looger et al., 2003; Win et al., 2006), or designing protein-based synthetic signaling cascades by rationally rewiring the protein-protein interactions and output responses of prokaryotic two-component signal transduction systems (Skerker et al., 2008).
The detection of electrical signals or production of biological energy (e.g., mimicking the operation of electrical electrocytes [Xu et al., 2008]) could also be enabled by incorporating natural or synthetic ion channels into engineered cells. In addition, magneto-responsive bacteria could play useful roles in environmental and medical applications (Jogler and Schuler, 2009). Synthetic bacteria, designed to form magnetosomes and seek out cancer cells, could be used to enhance imaging, and magnetic bacteria could be engineered to interact with nanoparticles to enhance the targeting of cancer cells. Moreover, the introduction of mechanosensitive ion channels (e.g., MscL from Mycobacterium tuberculosis and MscS from
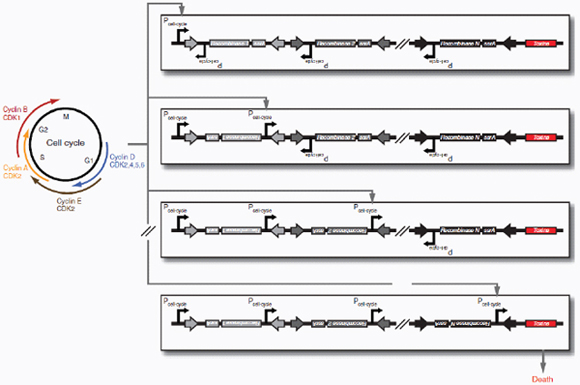
FIGURE A12-5 Cell-cycle counter for biological containment. Cell-cycle counting is accomplished with a cascade of single recombinase-based memory units (e.g., SIMMs [Friedland et al., 2009]), each of which is driven by a cell cycle–dependent promoter. After N cell-cycle events are counted, the gene circuit unlocks the expression of a toxic protein triggering cell death. Protein degradation tags (ssrA) are fused to the recombinase genes to ensure stability of the circuit.
E. coli) could endow designer cells with the ability to detect mechanical forces (Booth et al., 2007). Such cells may be useful in vivo sensors for studying cellular differentiation signals or the effects of external stresses on the body.
Ultimately, programmable cells possessing novel sensory modules could be integrated with mechanical, electrical and chemical systems to detect, process and respond to external stimuli, and exploited for a variety of environmental and biomedical applications. For example, bacteria could be engineered to seek out hazardous chemicals or heavy metals in the environment, perform cleanup and return to their origin to report on the number of hazardous sites encountered via analysis by microfluidic devices. To eventually achieve such complex tasks, an intermediate goal might involve programming chemotactic bacteria to swim from waypoint to waypoint. A dish containing gradients of several chemoattractants would constitute the navigational course (Fig. A12-6a).
At the core of this design could be a synthetic gene network made up of a series of sequential toggle switches that control the expression of receptors needed for bacterial chemotaxis toward chemoattractants (Falke et al., 1997) (Fig. A12-6b). The programmable cells would initially express only a single chemoattractant receptor, and therefore would migrate up only one of the chemoattractant gradients (Falke et al., 1997). To determine that a waypoint has been achieved, a threshold-based toggle switch would be turned ON upon reaching a sufficiently high concentration of the chemoattractant. When the first toggle switch is ON, production of the first chemoattractant receptor would be suppressed and production of a second receptor allowed, resulting in cells swimming up the second chemical gradient. The ON switch would additionally prime the next toggle switch in the series to be switched ON when the second waypoint is reached. When that second toggle flips ON, the previous switch would be flipped OFF to ensure that only one chemoattractant is being followed at a time. The final chemoattractant would lead the bacteria back to its origin so that the engineered cells would complete a multi-stop round trip.
Designer circuits and systems for microbiome engineering
The human microbiome is fertile ground for the application of engineered organisms as scientific tools and therapeutic agents. There are unique bacterial populations residing in distinct locations in the human body that are perturbed in disease states (Gao et al., 2008; Grice et al., 2009). Each represents an exciting opportunity for reengineering the human microbiome and designing targeted therapeutics for a range of conditions, including dermatologic, genitourinary, gastrointestinal, metabolic and immunologic diseases (Ley et al., 2008; Turnbaugh et al., 2006, 2009).
Recently, bacteria have been engineered to infiltrate cellular communities for the purposes of delivering probes, gene circuits or chemicals (Steidler et al., 2000; Braat et al., 2006). In a similar fashion, bacteriophages carrying synthetic
gene circuits could transform existing microbiome bacteria with new functionalities. For instance, given that anaerobic bacteria are known to migrate to hypoxic and necrotic regions of solid tumors (Wei et al., 2008), bacteriophages could be designed to infect cancer-targeting bacteria. These bacteriophages could encode conditional expression of chemotherapeutic agents using synthetic logic gates or switches that are coupled to environmental sensors.
Bhatia and colleagues (von Maltzahn et al., 2007) recently have developed nanoparticles that perform Boolean logic based on proteolytic activity. Viruses that infect tumor cells or bacteria could carry synthetic gene circuits that regulate in a programmable fashion the expression of enzymes that trigger nanoparticle activity. In these ways, one could develop targeted therapies against cancer or infectious diseases that exploit the human microbiome and synthetic gene networks.
Switchboard for dynamically controlling the expression of multiple genes
Engineered cells have long been used to produce recombinant proteins and chemicals for the biotechnology industry, and one of the major applications of synthetic biology to date has been in enhancing microbial production of biofuels (Stephanopoulos, 2007) and biomaterials (Teule et al., 2009; Slotta et al., 2007; Rammensee et al., 2008; Widmaier et al., 2009). Improving production from cells involves numerous engineering decisions related to the entire organism, including codon optimization, choosing whether or not to export recombinant proteins (Choi and Lee, 2004), rational or evolutionary methods for improving metabolic yields (Klein-Marcuschamer and Stephanopoulos, 2008; Dueber et al., 2009), and optimization of growth conditions. Often some or all of the genes required for production are non-optimal for bacterial expression and contain repetitive sequences that are unstable in bacterial hosts. Whole-gene synthesis techniques are increasingly being used to optimize coding sequences for recombinant production (Widmaier et al., 2009).
These innovative approaches, as well as more traditional knockout techniques, introduce hard-wired changes into the genomes of interest. However, for many industrial and bioprocess applications, there is a need to dynamically modulate and control the expression of multiple genes, depending upon the state of the bioreactor. These situations would benefit from the development of a synthetic switchboard, one that could tune the expression of many different genes simultaneously and independently. Such a switchboard could be made up of a series of adjustable threshold genetic switches, riboregulators or riboswitches, and designed to respond to different environmental and intracellular variables, such as pH, light intensity and the metabolic state of the cell. The switchboard design, which would integrate novel sensory modalities with tunable, interoperable genetic circuits, would have broad functionality. It could be programmed, for example, to shift carbon flux between different pathways depending upon cellular
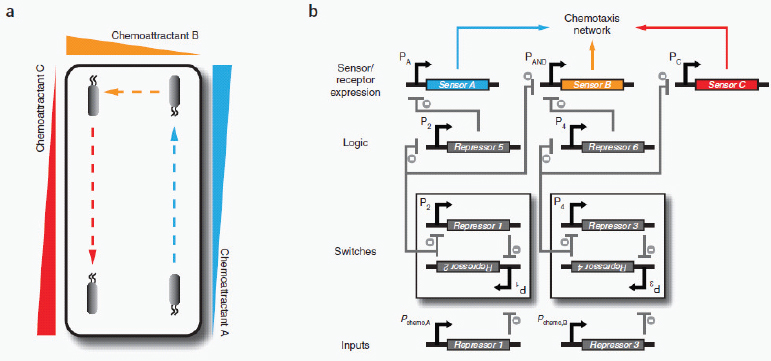
FIGURE A12-6 Autonomous chemotaxis. (a) Chemotactic environment made up of three chemoattractant gradients (A, B, C). (b) The synthetic gene network, whereby toggle switches control the sequential expression of three chemotaxis sensor receptors, for autonomously navigating bacteria down three chemoattractant gradients. Inputs into promoters and logic operations are shown explicitly, except when the promoter name is italicized, which represents an inducible promoter. (c) Boolean ON/OFF values for the network genes illustrate the sequential order of operations.
conditions, thereby optimizing the production of biofuels, specialty chemicals and other materials.
Conclusions
The past decade has witnessed the power of intelligently applying engineering principles to biology in the development of many exciting, artificial gene circuits and biomolecular systems. We are convinced that next-generation synthetic gene networks will advance understanding of natural systems, provide new biological modules and create new tools that will enable the construction of even more complex systems. Most importantly, if the current pace of progress in synthetic biology continues, real-world applications in fields such as medicine, biotechnology, bioremediation and bioenergy will be realized.
Acknowledgments
We would like to thank the Howard Hughes Medical Institute and the National Institutes of Health Director’s Pioneer Award Program for their financial support. We also thank the reviewers for their insights and suggestions.
Competing Interests Statement
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturebiotechnology/.
References
Ajo-Franklin, C.M. et al. Rational design of memory in eukaryotic cells. Genes Dev. 21, 2271–2276 (2007).
Alberti, S., Halfmann, R., King, O., Kapila, A. & Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158 (2009).
Alper, H., Fischer, C., Nevoigt, E. & Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA 102, 12678–12683 (2005).
An, W. & Chin, J.W. Synthesis of orthogonal transcription-translation networks. Proc. Natl. Acad. Sci. USA 106, 8477–8482 (2009).
Andersen, J.B. et al. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl. Environ. Microbiol. 64, 2240–2246 (1998).
Anderson, J.C., Clarke, E.J., Arkin, A.P. & Voigt, C.A. Environmentally controlled invasion of cancer cells by engineered bacteria. J. Mol. Biol. 355, 619–627 (2006).
Austin, D.W. et al. Gene network shaping of inherent noise spectra. Nature 439, 608–611 (2006).
Banaszynski, L.A., Chen, L.C., Maynard-Smith, L.A., Ooi, A.G. & Wandless, T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 (2006).
Bashor, C.J., Helman, N.C., Yan, S. & Lim, W.A. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 319, 1539–1543 (2008).
Basu, S., Gerchman, Y., Collins, C.H., Arnold, F.H. & Weiss, R. A synthetic multicellular system for programmed pattern formation. Nature 434, 1130–1134 (2005).
Bayer, T.S. & Smolke, C.D. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat. Biotechnol. 23, 337–343 (2005).
Beerli, R.R., Dreier, B. & Barbas, C.F. III. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 97, 1495–1500 (2000).
Bennett, M.R. & Hasty, J. Microfluidic devices for measuring gene network dynamics in single cells. Nat. Rev. Genet. 10, 628–638 (2009).
Bennett, M.R. et al. Metabolic gene regulation in a dynamically changing environment. Nature 454, 1119–1122 (2008).
Bhalla, U.S., Ram, P.T. & Iyengar, R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 297, 1018–1023 (2002).
Blake, W.J. et al. Phenotypic consequences of promoter-mediated transcriptional noise. Mol. Cell 24, 853–865 (2006).
Booth, I.R., Edwards, M.D., Black, S., Schumann, U. & Miller, S. Mechanosensitive channels in bacteria: signs of closure? Nat. Rev. Microbiol. 5, 431–440 (2007).
Boyden, E.S., Zhang, F., Bamberg, E., Nagel, G. & Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 (2005).
Braat, H. et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 4, 754–759 (2006).
Brakmann, S. & Grzeszik, S. An error-prone T7 RNA polymerase mutant generated by directed evolution. ChemBioChem 2, 212–219 (2001).
Buchholz, F., Angrand, P.O. & Stewart, A.F. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol. 16, 657–662 (1998).
Bustamante, C., Bryant, Z. & Smith, S.B. Ten years of tension: single-molecule DNA mechanics. Nature 421, 423–427 (2003).
Carr, P.A. & Church, G.M. Genome engineering. Nat. Biotechnol. 27, 1151–1162 (2009).
Carrera, J., Rodrigo, G. & Jaramillo, A. Towards the automated engineering of a synthetic genome. Mol. Biosyst. 5, 733–743 (2009).
Chandran, D., Bergmann, F.T. & Sauro, H.M. TinkerCell: modular CAD tool for synthetic biology. J. Biol. Eng. 3, 19 (2009).
Choi, J.H. & Lee, S.Y. Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl. Microbiol. Biotechnol. 64, 625–635 (2004).
Cox, R.S. III, Surette, M.G. & Elowitz, M.B. Programming gene expression with combinatorial promoters. Mol. Syst. Biol. 3, 145 (2007).
Czar, M.J., Cai, Y. & Peccoud, J. Writing DNA with GenoCAD. Nucleic Acids Res. 37, W40–W47 (2009).
Deans, T.L., Cantor, C.R. & Collins, J.J. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 130, 363–372 (2007).
Discher, D.E., Mooney, D.J. & Zandstra, P.W. Growth factors, matrices, and forces combine and control stem cells. Science 324, 1673–1677 (2009).
Drinnenberg, I.A. et al. RNAi in Budding Yeast. Science 326, 544–550 (2009).
Dueber, J.E., Mirsky, E.A. & Lim, W.A. Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat. Biotechnol. 25, 660–662 (2007).
Dueber, J.E. et al. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 27, 753–759 (2009).
Ellis, T., Wang, X. & Collins, J.J. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol. 27, 465–471 (2009).
Elowitz, M.B. & Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).
Falke, J.J., Bass, R.B., Butler, S.L., Chervitz, S.A. & Danielson, M.A. The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu. Rev. Cell Dev. Biol. 13, 457–512 (1997).
Fernando, C.T. et al. Molecular circuits for associative learning in single-celled organisms. J. R. Soc. Interface 6, 463–469 (2009).
Friedland, A.E. et al. Synthetic gene networks that count. Science 324, 1199–1202 (2009).
Fritz, G., Buchler, N.E., Hwa, T. & Gerland, U. Designing sequential transcription logic: a simple genetic circuit for conditional memory. Syst. Synth. Biol. 1, 89–98 (2007).
Fung, E. et al. A synthetic gene-metabolic oscillator. Nature 435, 118–122 (2005).
Gao, Z., Tseng, C.H., Strober, B.E., Pei, Z. & Blaser, M.J. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE 3, e2719 (2008).
Garcia-Ojalvo, J., Elowitz, M.B. & Strogatz, S.H. Modeling a synthetic multicellular clock: repressilators coupled by quorum sensing. Proc. Natl. Acad. Sci. USA 101, 10955–10960 (2004).
Gardner, T.S., Cantor, C.R. & Collins, J.J. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342 (2000).
Gibson, D.G. et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220 (2008).
Glass, J.I. et al. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. USA 103, 425–430 (2006).
Goldberg, S.D., Derr, P., DeGrado, W.F. & Goulian, M. Engineered single- and multi-cell chemotaxis pathways in E. coli. Mol. Syst. Biol. 5, 283 (2009).
Grice, E.A. et al. Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 (2009).
Groth, A.C. & Calos, M.P. Phage integrases: biology and applications. J. Mol. Biol. 335, 667–678 (2004).
Guido, N.J. et al. A bottom-up approach to gene regulation. Nature 439, 856–860 (2006).
Gulati, S. et al. Opportunities for microfluidic technologies in synthetic biology. J. R. Soc. Interface 6 Suppl 4, S493–S506 (2009).
Ham, T.S., Lee, S.K., Keasling, J.D. & Arkin, A.P. A tightly regulated inducible expression system utilizing the fim inversion recombination switch. Biotechnol. Bioeng. 94, 1–4 (2006).
——. Design and construction of a double inversion recombination switch for heritable sequential genetic memory. PLoS ONE 3, e2815 (2008).
Hamad-Schifferli, K., Schwartz, J.J., Santos, A.T., Zhang, S. & Jacobson, J.M. Remote electronic control of DNA hybridization through inductive coupling to an attached metal nanocrystal antenna. Nature 415, 152–155 (2002).
Hammer, K., Mijakovic, I. & Jensen, P.R. Synthetic promoter libraries–tuning of gene expression. Trends Biotechnol. 24, 53–55 (2006).
Hasty, J., McMillen, D., Isaacs, F. & Collins, J.J. Computational studies of gene regulatory networks: in numero molecular biology. Nat. Rev. Genet. 2, 268–279 (2001).
Hooshangi, S., Thiberge, S. & Weiss, R. Ultrasensitivity and noise propagation in a synthetic transcriptional cascade. Proc. Natl. Acad. Sci. USA 102, 3581–3586 (2005).
Isaacs, F.J., Hasty, J., Cantor, C.R. & Collins, J.J. Prediction and measurement of an autoregulatory genetic module. Proc. Natl. Acad. Sci. USA 100, 7714–7719 (2003).
Isaacs, F.J. et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 22, 841–847 (2004).
Jensen, P.R. & Hammer, K. Artificial promoters for metabolic optimization. Biotechnol. Bioeng. 58, 191–195 (1998).
Jogler, C. & Schuler, D. Genomics, genetics, and cell biology of magnetosome formation. Annu. Rev. Microbiol. 63, 501–521 (2009).
Kaplan, S., Bren, A., Dekel, E. & Alon, U. The incoherent feed-forward loop can generate non-monotonic input functions for genes. Mol. Syst. Biol. 4, 203 (2008).
Kaznessis, Y.N. Computational methods in synthetic biology. Biotechnol. J. 4, 1392–1405 (2009).
Khalil, A.S. et al. Single M13 bacteriophage tethering and stretching. Proc. Natl. Acad. Sci. USA 104, 4892–4897 (2007).
——. Kinesin’s cover-neck bundle folds forward to generate force. Proc. Natl. Acad. Sci. USA 105, 19247–19252 (2008).
Kilby, N.J., Snaith, M.R. & Murray, J.A. Site-specific recombinases: tools for genome engineering. Trends Genet. 9, 413–421 (1993).
Klein-Marcuschamer, D. & Stephanopoulos, G. Assessing the potential of mutational strategies to elicit new phenotypes in industrial strains. Proc. Natl. Acad. Sci. USA 105, 2319–2324 (2008).
Kobayashi, H. et al. Programmable cells: interfacing natural and engineered gene networks. Proc. Natl. Acad. Sci. USA 101, 8414–8419 (2004).
Kramer, B.P. et al. An engineered epigenetic transgene switch in mammalian cells. Nat. Biotechnol. 22, 867–870 (2004).
Lartigue, C. et al. Genome transplantation in bacteria: changing one species to another. Science 317, 632–638 (2007).
Lartigue, C. et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325, 1693–1696 (2009).
Lee, D.K., Itti, L., Koch, C. & Braun, J. Attention activates winner-take-all competition among visual filters. Nat. Neurosci. 2, 375–381 (1999).
Lee, S.K. et al. Directed evolution of AraC for improved compatibility of arabinose- and lactose-inducible promoters. Appl. Environ. Microbiol. 73, 5711–5715 (2007).
Levskaya, A. et al. Synthetic biology: engineering Escherichia coli to see light. Nature 438, 441–442 (2005).
Levskaya, A., Weiner, O.D., Lim, W.A. & Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009).
Ley, R.E., Lozupone, C.A., Hamady, M., Knight, R. & Gordon, J.I. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 (2008).
Li, L. & Lindquist, S. Creating a protein-based element of inheritance. Science 287, 661–664 (2000).
Looger, L.L., Dwyer, M.A., Smith, J.J. & Hellinga, H.W. Computational design of receptor and sensor proteins with novel functions. Nature 423, 185–190 (2003).
Lu, T.K. & Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 104, 11197–11202 (2007).
——. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 106, 4629–4634 (2009).
Lu, T., Ferry, M., Weiss, R. & Hasty, J. A molecular noise generator. Phys. Biol. 5, 036006 (2008).
Lucks, J.B., Qi, L., Whitaker, W.R. & Arkin, A.P. Toward scalable parts families for predictable design of biological circuits. Curr. Opin. Microbiol. 11, 567–573 (2008).
Macarthur, B.D., Ma’ayan, A. & Lemischka, I.R. Systems biology of stem cell fate and cellular reprogramming. Nat. Rev. Mol. Cell Biol. 10, 672–681 (2009).
Maeder, M.L. et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell 31, 294–301 (2008).
Malmstrom, J. et al. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 460, 762–765 (2009).
Mar, D.J., Chow, C.C., Gerstner, W., Adams, R.W. & Collins, J.J. Noise shaping in populations of coupled model neurons. Proc. Natl. Acad. Sci. USA 96, 10450–10455 (1999).
McGinness, K.E., Baker, T.A. & Sauer, R.T. Engineering controllable protein degradation. Mol. Cell 22, 701–707 (2006).
McMillen, D., Kopell, N., Hasty, J. & Collins, J.J. Synchronizing genetic relaxation oscillators by intercell signaling. Proc. Natl. Acad. Sci. USA 99, 679–684 (2002).
Mettetal, J.T., Muzzey, D., Gomez-Uribe, C. & van Oudenaarden, A. The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science 319, 482–484 (2008).
Michalet, X. et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 307, 538–544 (2005).
Misra, N. et al. Bioelectronic silicon nanowire devices using functional membrane proteins. Proc. Natl. Acad. Sci. USA 106, 13780–13784 (2009).
Mitchell, A. et al. Adaptive prediction of environmental changes by microorganisms. Nature 460, 220–224 (2009).
Molin, S. et al. Suicidal genetic elements and their use in biological containment of bacteria. Annu. Rev. Microbiol. 47, 139–166 (1993).
Murphy, K.F., Balazsi, G. & Collins, J.J. Combinatorial promoter design for engineering noisy gene expression. Proc. Natl. Acad. Sci. USA 104, 12726–12731 (2007).
Neuman, K.C. & Nagy, A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods 5, 491–505 (2008).
Nevozhay, D., Adams, R.M., Murphy, K.F., Josic, K. & Balazsi, G. Negative auto-regulation linearizes the dose-response and suppresses the heterogeneity of gene expression. Proc. Natl. Acad. Sci. USA 106, 5123–5128 (2009).
Park, K.S. et al. Phenotypic alteration of eukaryotic cells using randomized libraries of artificial transcription factors. Nat. Biotechnol. 21, 1208–1214 (2003).
Purnick, P.E. & Weiss, R. The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 10, 410–422 (2009).
Rackham, O. & Chin, J.W. A network of orthogonal ribosome x mRNA pairs. Nat. Chem. Biol. 1, 159–166 (2005).
Rammensee, S., Slotta, U., Scheibel, T. & Bausch, A.R. Assembly mechanism of recombinant spider silk proteins. Proc. Natl. Acad. Sci. USA 105, 6590–6595 (2008).
Rinaudo, K. et al. A universal RNAi-based logic evaluator that operates in mammalian cells. Nat. Biotechnol. 25, 795–801 (2007).
Ro, D.K. et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943 (2006).
Salis, H.M., Mirsky, E.A. & Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950 (2009).
Santoro, S.W. & Schultz, P.G. Directed evolution of the site specificity of Cre recombinase. Proc. Natl. Acad. Sci. USA 99, 4185–4190 (2002).
Sarpeshkar, R. & O’Halloran, M. Scalable hybrid computation with spikes. Neural Comput. 14, 2003–2038 (2002).
Shetty, R.P., Endy, D. & Knight, T.F. Jr. Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2, 5 (2008).
Simpson, M.L., Cox, C.D. & Sayler, G.S. Frequency domain analysis of noise in autoregulated gene circuits. Proc. Natl. Acad. Sci. USA 100, 4551–4556 (2003).
Skerker, J.M. et al. Rewiring the specificity of two-component signal transduction systems. Cell 133, 1043–1054 (2008).
Slotta, U. et al. Spider silk and amyloid fibrils: a structural comparison. Macromol. Biosci. 7, 183–188 (2007).
Sohka, T. et al. An externally tunable bacterial band-pass filter. Proc. Natl. Acad. Sci. USA 106, 10135–10140 (2009).
Stavreva, D.A. et al. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat. Cell Biol. 11, 1093–1102 (2009).
Steidler, L. et al. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 289, 1352–1355 (2000).
Stephanopoulos, G. Challenges in engineering microbes for biofuels production. Science 315, 801–804 (2007).
Stricker, J. et al. A fast, robust and tunable synthetic gene oscillator. Nature 456, 516–519 (2008).
Summerer, D. et al. A genetically encoded fluorescent amino acid. Proc. Natl. Acad. Sci. USA 103, 9785–9789 (2006).
Svoboda, K. & Block, S.M. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 23, 247–285 (1994).
Tabor, J.J. et al. A synthetic genetic edge detection program. Cell 137, 1272–1281 (2009).
Tagkopoulos, I., Liu, Y.C. & Tavazoie, S. Predictive behavior within microbial genetic networks. Science 320, 1313–1317 (2008).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Teule, F. et al. A protocol for the production of recombinant spider silk-like proteins for artificial fiber spinning. Nat. Protoc. 4, 341–355 (2009).
Tigges, M., Marquez-Lago, T.T., Stelling, J. & Fussenegger, M. A tunable synthetic mammalian oscillator. Nature 457, 309–312 (2009).
Tsai, T.Y. et al. Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science 321, 126–129 (2008).
Turnbaugh, P.J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Turnbaugh, P.J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
von Maltzahn, G. et al. Nanoparticle self-assembly gated by logical proteolytic triggers. J. Am. Chem. Soc. 129, 6064–6065 (2007).
Wang, H.H. et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898 (2009).
Wang, J., Xie, J. & Schultz, P.G. A genetically encoded fluorescent amino acid. J. Am. Chem. Soc. 128, 8738–8739 (2006).
Wang, K., Neumann, H., Peak-Chew, S.Y. & Chin, J.W. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 25, 770–777 (2007).
Wang, Q., Parrish, A.R. & Wang, L. Expanding the genetic code for biological studies. Chem. Biol. 16, 323–336 (2009).
Weber, W. & Fussenegger, M. Engineering of synthetic mammalian gene networks. Chem. Biol. 16, 287–297 (2009).
Wei, M.Q., Mengesha, A., Good, D. & Anne, J. Bacterial targeted tumour therapy-dawn of a new era. Cancer Lett. 259, 16–27 (2008).
Widmaier, D.M. et al. Engineering the Salmonella type III secretion system to export spider silk monomers. Mol. Syst. Biol. 5, 309 (2009).
Win, M.N. & Smolke, C.D. A modular and extensible rNA-based gene-regulatory platform for engineering cellular function. Proc. Natl. Acad. Sci. USA 104, 14283–14288 (2007).
——. Higher-order cellular information processing with synthetic RNA devices. Science 322, 456–460 (2008).
Win, M.N., Klein, J.S. & Smolke, C.D. Codeine-binding RNA aptamers and rapid determination of their binding constants using a direct coupling surface plasmon resonance assay. Nucleic Acids Res. 34, 5670–5682 (2006).
Win, M.N., Liang, J.C. & Smolke, C.D. Frameworks for programming biological function through RNA parts and devices. Chem. Biol. 16, 298–310 (2009).
Xu, J. & Lavan, D.A. Designing artificial cells to harness the biological ion concentration gradient. Nat. Nanotechnol. 3, 666–670 (2008).
Yeh, B.J., Rutigliano, R.J., Deb, A., Bar-Sagi, D. & Lim, W.A. Rewiring cellular morphology pathways with synthetic guanine nucleotide exchange factors. Nature 447, 596–600 (2007).
Yoon, Y.G. & Koob, M.D. Efficient cloning and engineering of entire mitochondrial genomes in Escherichia coli and transfer into transcriptionally active mitochondria. Nucleic Acids Res. 31, 1407–1415 (2003).
You, L., Cox, R.S. III, Weiss, R. & Arnold, F.H. Programmed population control by cell-cell communication and regulated killing. Nature 428, 868–871 (2004).
ENGINEERING SCALABLE BIOLOGICAL SYSTEMS66
Timothy K. Lu67
Synthetic biology is focused on engineering biological organisms to study natural systems and to provide new solutions for pressing medical, industrial and environmental problems. At the core of engineered organisms are synthetic biological circuits that execute the tasks of sensing inputs, processing logic and performing output functions. In the last decade, significant progress has been made in developing basic designs for a wide range of biological circuits in bacteria, yeast and mammalian systems. However, significant challenges in the construction, probing, modulation and debugging of synthetic biological systems must be addressed in order to achieve scalable higher-complexity biological circuits. Furthermore, concomitant efforts to evaluate the safety and biocontainment of engineered organisms and address public and regulatory concerns will be necessary to ensure that technological advances are translated into real-world solutions.
In the last century, scientists have made giant strides in identifying and studying biological parts such as proteins and nucleic acids (Watson and Crick, 1953; Lander et al., 2001; Venter et al., 2001; Fiers et al., 1976; Fleischmann et al., 1995), understanding regulatory networks (Johnson et al., 1981), and constructing engineered organisms using the ever-advancing tools of genetic engineering (Itakura et al., 1977). In the last decade, synthetic biologists have
_______________
66 Reprinted with permission from Nature Publishing Group.
67 Synthetic Biology Group; Research Lab of Electronics; Department of Electrical Engineering and Computer Science; Massachusetts Institute of Technology; and Broad Institute of MIT and Harvard; Cambridge, MA, USA
Key words: synthetic biology, biological circuits, engineered organisms, modelling, high-throughput design, regulatory issues, biological probes, biological modulators
Abbreviations: PCR, polymerase chain reaction; DNA, deoxyribonucleic acid; RNA, ribonucleic acid; Mbp, mega-basepairs; Kbp, kilo-basepairs; qRT-PCR, quantitative reverse-transcriptase PCR; FRET, fluorescence resonance energy transfer; IPTG, isopropyl β-D-1-thiogalactopyranoside; SELEX, systematic evolution of ligands by exponential enrichment; RNAi, RNA interference; GMOs, genetically modified organisms
Submitted: 06/24/10
Revised: 07/19/10
Accepted: 07/20/10
Previously published online: www.landesbioscience.com/journals/biobugs/article/13086
DOI: 10.4161/bbug.1.6.13086
Correspondence to: Timothy K. Lu; Email: timlu@mit.edu
Commentary to: Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nature Biotechnology 2009; 27:1139–50. PMID: 20010597; DOI: 10.1038/nbt.1591.
leveraged the power of modern molecular biology using frameworks translated from traditional disciplines such as electrical engineering, computer science, mechanical engineering and chemical engineering to create a wide range of synthetic biological circuits, including switches (Gardner et al., 2000; Kramer et al., 2004; Isaacs et al., 2003; Ham et al., 2006, 2008; Deans et al., 2007; Ajo-Franklin et al., 2007; Dueber et al., 2007), oscillators (Elowitz and Leibler, 2000; Stricker et al., 2008; McMillen et al., 2002), digital logic gates (Rinaudo et al., 2007; Win and Smolke, 2008; Anderson et al., 2007; Dueber et al., 2003; Yeh et al., 2007), filters (Hooshangi et al., 2005; Basu et al., 2005; Sohka et al., 2009), modular and interoperable memory devices (Friedland et al., 2009), counters (Friedland et al., 2009), sensors (Win and Smolke, 2007; Bayer and Smolke, 2005), and protein scaffolds (Bashor et al., 2008). Using these circuits, biological engineers have created synthetic organisms that can be used for bioremediation, biosensing, computation, bioenergy and medical therapeutics (reviewed in Lu et al., 2009; Lim, 2010; Khalil and Collins, 2010). Despite these advances, the realization of synthetic-biology-based applications will require future breakthroughs in our ability to create sufficiently complex and reliable biological systems. Here, I will discuss current limitations and potential solutions for the construction, probing, modulation and debugging of scalable biological systems as well as hurdles for the deployment of engineered organisms from bacteria to mammalian cells which adds to the discussion of next-generation synthetic gene networks in Lu et al. (2009) (Fig. A13-1).
Physical Construction of Scalable Biological Systems
Construction of early synthetic circuits largely relied on restriction enzymes and polymerase chain reaction (PCR)-based techniques to assemble existing genetic components. These methods do not scale well with increasing complexity due to a lack of sufficient unique restriction sites and the need to have physical DNA templates from which to amplify genetic parts. Standards for library construction and the assembly of parts libraries (Smolke, 2009) have been integral in circumventing this dependency on templates and restriction sites. However, since these parts must be devoid of restriction sites used in the defined standards and should ideally be optimized for use in one’s organism of choice (Okano et al., 2008), the use of whole-gene DNA synthesis is on the rise (Carlson, 2009). Using direct chemical synthesis, circuits can be designed in silico and implemented in DNA with significantly less effort from researchers. As DNA synthesis becomes increasingly economical and efficient, it will become possible to construct complex systems with less reliance on restriction enzymes. For example, DNA synthesis productivity has exceeded 1 Mbp per person per day while Venter and colleagues recently succeeded in synthesizing a 1.08 Mbp genome (Gibson et al., 2010). However, most synthetic gene circuits to date have not exceeded the 50 Kbp level, indicating that there is a large gap between our ability to read and
FIGURE A13-1 A basic design cycle for synthetic biology includes creating well-characterized parts (e.g., regulatory elements, genes, proteins, RNAs), constructing synthetic devices and modules and designing and assembling higher-order networks. All steps of this cycle are aided by modelling, probes and modulators to analyze circuit performance. Debugging is an iterative process based on parts optimization, fine-tuning regulatory components, modelling and changing circuit architecture.
write DNA and knowing what DNA to write (Fig. A13-2). Just as the decoding of the human genome sequence did not immediately reveal the functions of all human genes, the utility of high-throughput DNA synthesis technology will only gradually become evident as synthetic biologists learn how to create complex systems. For example, future synthesized circuits should be designed with ease of probing, modulating and debugging in mind. These features could be implemented by including validated RNA “handles” that can be easily measured with standard probe sets to determine internal RNA concentrations, gene circuits that allow inducers to modulate synthetic circuit protein levels, and properly situated restriction sites for the rapid cloning of components that need systematic optimization, such as ribosome binding sequences.
Significant advances in well-characterized, interoperable devices are necessary for the construction of higher-order modules that will enable scalable biological systems (Andrianantoandro et al., 2006). The majority of biological
FIGURE A13-2 DNA sequencing and synthesis technologies are advancing at exponential rates, outpacing the ability of synthetic biologists to construct useful and scalable biological circuits (Carlson, 2009). These trends are similar to Moore’s law for integrated circuits (Moore, 1965) and suggest that there is substantial room for growth in the field of synthetic circuits.
circuits have been constructed using a handful of synthetic parts (Lu et al., 2009). Furthermore, it is often the case that when new designs for biological parts are developed, only a few instantiations are created and tested, usually in single cellular backgrounds. As a result, there is a need for the systematic development and characterization of compatible biological parts. Specificity in biological systems largely relies on spatial distribution and chemical interactions. This is in stark contrast to electrical engineering, where specificity is achieved through direct electrical wiring. Thus, strategies for achieving inter-part compatibility include targeting circuits to isolated compartments (Parsons et al., 2010; Tanaka et al., 2010), mutagenesis and directed evolution of existing parts, and using comparative genomics to identify, synthesize, and test homologous proteins or nucleic acids. These efforts may be complicated by unknown global factors (e.g., growth rates, endogenous transcription factors with off-target effects on synthetic circuits, protein-protein interactions, small RNAs) that can confound device testing and render it difficult to use pre-defined parts in a wide range of organisms and environmental conditions without additional alterations and characterization (Klumpp et al., 2009). Therefore, combinatorial methods to test single-component performance, multi-component interactions and biological crosstalk (e.g., cross-activation or cross-repression of transcription, non-specific enzymatic activity, inappropriate triggering of signalling pathways) will be important for parts libraries (Fig. A13-3). These results should be incorporated into mathematical models to aid future model-based design. Indeed, institutions such as BIOFAB are attempting to systematically assemble and characterize libraries of synthetic devices. However, development efforts for certain platforms that are promising for library construction, such as zinc finger proteins and RNA interference, may be slowed by the presence of existing intellectual property (Scott, 2005).
As an example of combinatorial characterization (Fig. A13-3), suppose one would like to construct multiple interoperable NOR (NOT-OR) gates to constitute a universal logic system. NOR functionality can be built by placing pairwise combinations of unique operator sites for transcriptional repressors within synthetic promoters. To identify orthogonal repressors, one can encode individual transcription factors under inducible control on one set of plasmids and individual cognate operator sites driving expression of a reporter gene on another set of plasmids. Then, all possible combinations of transcription factor plasmids and reporter plasmids can be co-transformed into cells and tested for single-component performance (e.g., when a transcription factor is co-transformed with its cognate operator site) and potential crosstalk interactions (e.g., when a transcription factor is co-transformed with non-cognate operator sites). Standard induction curves can be derived by varying the concentration of transcription factors using the inducible promoters and measuring the resulting output (Kelly et al., 2009). Based on these results, an optimal set of non-interacting transcription factors and cognate operators can be selected. To create the NOR gates, all possible pairwise combinations of operators can be constructed in synthetic promoters
FIGURE A13-3 Combinatorial high-throughput methods will be useful in the assembly of well-characterized libraries of synthetic parts and devices. For example, transcriptional regulators and their cognate inducers can be analyzed for (A) single-component performance, (B) interactions between multiple components and (C) inducer crosstalk (e.g., cross-activation and/or cross-inhibition).
and co-transformed into cells with all pairwise combinations of transcription factors under independent inducible control. Proper NOR gate functionality and crosstalk can then be determined in a high-throughput fashion for all potential gates by varying inducer levels and measuring reporter gene output. In addition to enabling interoperable gate selection, large-scale experiments such as these should yield substantial data for models that can predict the orthogonality of transcription factors and operators for future circuits (e.g., using heuristic or
thermodynamically guided algorithms). Moreover, matrices of cross-repression interactions can be constructed and incorporated into transcriptional models when cross-interacting transcription factors must be used in other systems.
Model-guided design is crucial for the construction of complicated electrical and mechanical systems. Time-based simulations for electrical and mechanical systems are possible since mathematical models are established and parameters are well known. In contrast, most parameters in biological circuits are unknown and the computational resources required to accurately simulate noise and multiple component interactions are significant. Recent advances in modelling chemical networks, transcription, translation and biological noise using the inherent physics of solid-state electronic devices should enable the construction of large-scale real-time electronic models of synthetic biological systems (Mandal and Sarpeshkar, 2009). Other techniques from control theory such as small-signal linearization and modularization enable tractable modelling and simulations prior to implementation. Biological systems exhibit nonlinearity (e.g., cooperativity) which can be linearized in different regions of operation (Fig. A13-4A). Frequency-domain analysis in linearized systems allows for block modelling and deeper understanding of system dynamics, such as noise, stability, time constants and performance (Fig. A13-4B) (Cox et al., 2006; Simpson et al., 2003). Small-signal linearization and frequency-domain modelling have not been extensively used for studying and designing synthetic biological circuits even though advances in microfluidics and time-lapse microscopy can now achieve frequency modulation of inputs and long time-scale data collection necessary for frequency-domain analysis (Cox et al., 2006; Mettetal et al., 2008). Furthermore, microfluidics devices can be coupled with electronic controllers to stabilize and alter the dynamics of synthetic biological circuits similar to electronic controllers that are used to control mechanical systems.
The insights that can be gained from linearized block models of complex systems can complement the accuracy of time-domain state-space representation, time-based mathematical simulations and non-linear control theory. To enable successful time-based and frequency-based modelling of biological systems, accurate parameters will need to be derived by high-throughput in vitro and in vivo probes and microarrays, as described below.
Molecular Probes and Modulators for Scalable Biological Systems
High-throughput methods for probing multiple nodes, such as protein and RNA levels, in complex biological circuits are necessary to achieve reliable and scalable performance (Tyagi, 2009). Ideal probe features include high signal-to-noise ratios, specificity, low cost, multiplexability, non-invasiveness and the ability to reveal real-time dynamics. Complementary techniques for the reliable and multiplexed modulation of synthetic circuits are also needed to generate
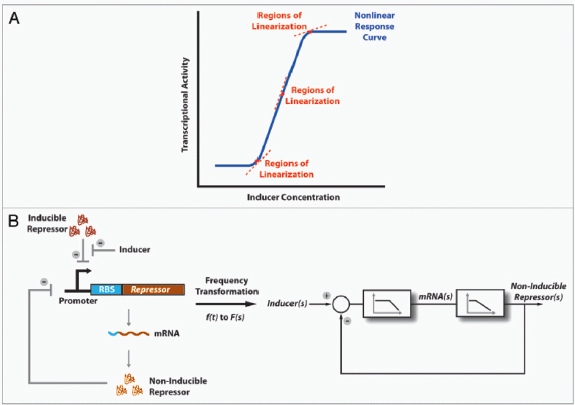
FIGURE A13-4 Control theory techniques for modelling synthetic biological circuits. (A) Small-signal linearization of biological components in different regions of operation enables the development of linear models. (B) Linearization can enable frequency-domain analysis, systems modelling using block diagrams and deeper insights into system dynamics. For example, transcription and translation can be understood as low-pass filters and block diagrams can be drawn for simple negative-feedback loops to yield understanding into system interconnections and responses to different input types (Cox et al., 2006; Simpson et al., 2003). In the block diagram shown, s refers to jω where j is √–1 and ω is angular frequency.
perturbations to obtain system parameters, drive accurate models and monitor system performance.
Analyses of most synthetic circuits rely on fluorescent proteins, which are well-characterized, easy to detect and can be multiplexed, as reporters of combined transcriptional and translational activity. However, fluorescent proteins are large, require folding and maturation and can be too stable to track rapid dynamics without additional modifications (Andersen et al., 1998). To address these issues, methods have recently been developed to create fast-folding fluorescent proteins, degradation tags and in vivo ligation of fluorophores to proteins (Uttamapinant et al., 2010). Alternative methods for monitoring protein levels in synthetic circuits include luminescence or colorimetric assays although these methods can be more difficult to multiplex. Advances in high-throughput proteomic characterization may eventually enable global monitoring of protein levels across time without the need for multiplexed reporter proteins (Malmstrom et al., 2009). Nucleic-acid aptamer beacons may also play a role in protein detection but have not been extensively applied to in vivo settings and require selection protocols since there are no algorithms that can reliably predict the affinity of binding between aptamers and proteins of interest (Hamaguchi et al., 2001). Thus, new techniques for multiplexed and global protein detection are needed for scalable biological system design.
Transcriptional activity can be monitored by quantifying RNA in engineered cells, which is easier to do in a multiplexed and global fashion than quantifying protein levels. Multiplexed RNA levels can be assayed with qRT-PCR or microarrays but these techniques require nucleic acid extraction and cellular destruction. Fusing RNAs of interest to aptamers that bind fluorescent proteins or fluorescent small molecules can be used to monitor in vivo RNA levels but requires modifying RNAs of interest (Tyagi, 2009). Side-by-side fluorescence resonance energy transfer (FRET) probes, quenched autoligation probes and molecular beacons are sequence-specific tools for detecting RNA levels in vivo with multiplexing capabilities but face challenges such as intracellular delivery, compartmentalization and degradation (Tyagi, 2009). Nonetheless, nucleic-acid-based probes are likely to be early enablers for high-throughput in vivo monitoring of transcription in synthetic biological circuits given that they are relatively easy to design for different targets. Thus, methods for achieving efficient delivery of nucleic-acid-based probes are crucially needed to make them widely useful to synthetic biologists for monitoring in vivo RNA levels in real time.
The majority of modulators used in synthetic circuits today are small molecules that induce or repress the activity of existing transcriptional regulators. As increasing number of transcriptional regulators are identified or created, it will be important to identify corresponding inducers that can control in vivo function, characterize crosstalk between different inducers and design interoperable modulators. For example, directed evolution of the AraC transcriptional regulator was necessary to increase compatibility between arabinose-controlled
and IPTG-controlled systems (Lee et al., 2007). Other modulators for synthetic circuits include aptamers that respond to small molecules and trigger translational activities (Bayer and Smolke, 2005). These aptamers can be discovered using techniques such as Systematic Evolution of Ligands by Exponential Enrichment (SELEX). Light-inducible proteins provide rapid and well-controlled signal transduction that could potentially be enhanced by developing variants which preferentially respond to different wavelengths of light (Levskaya et al., 2009; Boyden et al., 2005). An underutilized technology in synthetic biology is RNA interference (RNAi), which allows straightforward design of sequence-specific modulators. However, RNAi is not available in many organisms of interest and must be coupled with efficient delivery mechanisms for nucleic acids (Rinaudo et al., 2007). Fortunately, RNAi has been reconstituted in well-studied organisms such as Saccharomyces cerevisiae and significant advances are being made in RNAi-delivery technology (Whitehead et al., 2009).
Debugging Malfunctioning Biological Systems
The harsh reality of engineering synthetic biological circuits is that most designs fail to perform as expected. Debugging of synthetic circuits is a painstakingly iterative process involving the detailed characterization of all available internal nodes, trial-and-error modification of constituent components and modelling. However, debugging will be greatly facilitated by improved techniques for sequencing, probing and modulating synthetic circuits, as described above. For example, an initial debugging step is usually to fully sequence all circuit components to ensure that the intended genetic program has been constructed. Subsequent debugging can include dynamically probing all possible RNA and protein nodes and interactions using techniques such as qRT-PCR, fluorescent protein fusions and circuit-specific methods (e.g., in vitro gel shift assays, protein phosphorylation assays, single-cell microscopy, and so forth). Based on these results, it can often be determined whether system failure is due to inherent circuit topology or due to poor component performance leading to circuit operation in non-functional parameter regions. In the former case, redesigns are needed and can benefit from detailed modelling using insights and measurements from the failed attempts. In the latter case, systematic and randomized mutagenesis and screening techniques are helpful to optimize system performance. For example, synthetic circuits often do not work properly because of mismatched expression levels resulting in outputs that do not fit into the dynamic range of other inputs. Mismatches in dynamic range can usually be corrected by systematically altering the strengths of promoters and ribosome binding sequences using mutagenesis (Ellis et al., 2009; Salis et al., 2009).
In addition, optimization of synthetic genes can help resolve problems associated with the expression of heterologous genes in foreign hosts including DNA instability and poor translational efficiency (Widmaier et al., 2009). Techniques
for the high-throughput alteration of genetic circuits should also help speed up debugging cycles by enabling testing across larger parameter spaces (Wang et al., 2009). Modelling can generate insights into potential failure points in synthetic circuits but is inadequate at identifying problems with components or parameters that are not included in the models themselves. For example, global effects of host factors can change the performance of synthetic circuits (Klumpp et al., 2009). To model these effects, high-throughput data for global transcriptional responses to defined perturbations should be obtained from large promoter libraries and incorporated into whole-cell simulations (Zaslaver et al., 2006). Moreover, test platforms based on well-characterized host organisms or minimal organisms may be ideal backgrounds for validating system designs prior to deployment in final vehicles (Lu et al., 2009; Forster and Church, 2006). Finally, given the significant time and effort that is spent on fixing biological systems and the wealth of information that can be gleaned from malfunctioning designs, it would be beneficial to the general synthetic biology community to establish repositories where results from debugging cycles and system failures can be disseminated.
Public and Regulatory Considerations for Engineered Biological Systems
In addition to technological advances, efforts to address public and regulatory concerns over synthetic biology are necessary to ensure the successful translation of scalable biological systems to real-world applications. The synthetic biology community has recognized that safety, security and ethical issues must be addressed in an open and earnest fashion (Parens et al., 2008; Bugl et al., 2007). Although there is little data to suggest that genetic engineering or synthetic biology have produced harmful constructs over the last few decades, concerns from the public and the media are often voiced when significant technological milestones in synthetic biology are achieved. Some of these concerns may be rooted in inadequate scientific understanding or communication of the current state of synthetic biology. Thus, enhanced outreach and education efforts between researchers and the public are necessary to ensure the field’s continued progress. Furthermore, global variations in cultures and attitudes towards engineered life should be recognized and addressed by researchers and advocates. For example, differences in opinion between the United States and the European Union regarding the use of Genetically Modified Organisms (GMOs) as food products may translate to synthetic organisms. Thus, it is imperative that synthetic biologists act and communicate their roles as critics of the field to ensure that safety, reliability and the realization of broad social benefits are maintained as the utmost research priorities.
The regulatory issues associated with deploying synthetic biological organisms vary greatly depending on application. The areas associated with the lowest regulatory hurdles include metabolic engineering and bioenergy where biocontainment is straightforward to achieve, direct contact with humans is limited and
ultimate products are chemical compounds produced by engineered cells. For environmental deployment of synthetic organisms in the United States, the Environmental Protection Agency and the Food and Drug Administration can become involved. Regulatory issues for environmental applications are similar to those involved with GMOs, such as the use of recombinant DNA, biocontainment and impact on natural ecosystems. Standardized methods and technologies must be developed with input from regulatory agencies to evaluate these concerns. The most difficult regulatory hurdles pertain to the use of synthetic biological systems for human therapeutics such as adoptive immunotherapy (Varela-Rohena et al., 2008), cancer-seeking bacteria (Anderson et al., 2006), engineered phage targeting bacterial biofilms (Lu and Collins, 2007) and antibiotic-resistant organisms (Lu and Collins, 2009). These hurdles include immunogenicity, biocontainment, recombinant DNA and manufacturing purity. Stem-cell-based therapies face similar hurdles and therefore it will be informative to follow the path of stem cells into the clinic (Kiskinis and Eggan, 2010). Reliable methods to eliminate engineered organisms as needed (Molin et al., 1993), stringent techniques to evaluate safety in animals and humans, techniques for encapsulating and isolating engineered cells (Lim et al., 2010), assays for mutation rates in deployed organisms, utilization of probiotic strains as substrates for engineering microbe-based therapeutics and close communication with regulatory agencies will be necessary to translate synthetic biological organisms into the important frontier of human treatments. Furthermore, synthetic biologists interested in human therapeutics should focus their efforts on areas of significant unmet need to optimize risk-benefit tradeoffs and should be open to proof-of-concept applications in areas with lower regulatory hurdles such as environmental or veterinary use.
Conclusions
The past decade has delivered significant advances in the design and construction of basic synthetic circuits. In the upcoming decade, novel technologies for composing, probing, modulating and debugging scalable biological circuits will enable the robust performance of useful tasks by engineered organisms. Scientific advancements must be accompanied by concomitant efforts to address societal and regulatory concerns over synthetic biology. These endeavors should yield exciting new solutions for real-world problems in critical areas of medical, industrial, environmental and energy applications.
Acknowledgements
I appreciate the insights that the editor and anonymous reviewers have provided. I would also like to thank Michael Koeris and Ahmad Khalil for their critical reading of this manuscript.
References
Ajo-Franklin CM, Drubin DA, Eskin JA, Gee EP, Landgraf D, Phillips I, et al. Rational design of memory in eukaryotic cells. Genes Dev 2007; 21:2271-6.
Andersen JB, Sternberg C, Poulsen LK, Bjorn SP, Givskov M, Molin S. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol 1998; 64:2240-6.
Anderson JC, Clarke EJ, Arkin AP, Voigt CA. Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol 2006; 355:619-27.
Anderson JC, Voigt CA, Arkin AP. Environmental signal integration by a modular AND gate. Mol Syst Biol 2007; 3:133.
Andrianantoandro E, Basu S, Karig DK, Weiss R. Synthetic biology: new engineering rules for an emerging discipline. Mol Syst Biol 2006; 2:2006 0028.
Bashor CJ, Helman NC, Yan S, Lim WA. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 2008; 319:1539-43.
Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R. A synthetic multicellular system for programmed pattern formation. Nature 2005; 434:1130-4.
Bayer TS, Smolke CD. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat Biotechnol 2005; 23:337-43.
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 2005; 8:1263-8.
Bugl H, Danner JP, Molinari RJ, Mulligan JT, Park HO, Reichert B, et al. DNA synthesis and biological security. Nat Biotechnol 2007; 25:627-9.
Carlson R. The changing economics of DNA synthesis. Nat Biotechnol 2009; 27:1091-4.
Cox CD, McCollum JM, Austin DW, Allen MS, Dar RD, Simpson ML. Frequency domain analysis of noise in simple gene circuits. Chaos 2006; 16:026102.
Deans TL, Cantor CR, Collins JJ. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 2007; 130:363-72.
Dueber JE, Yeh BJ, Chak K, Lim WA. Reprogramming control of an allosteric signaling switch through modular recombination. Science 2003; 301:1904-8.
Dueber JE, Mirsky EA, Lim WA. Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat Biotechnol 2007; 25:660-2.
Ellis T, Wang X, Collins JJ. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat Biotechnol 2009; 27:465-71.
Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature 2000; 403:335-8.
Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, et al. Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 1976; 260:500-7.
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995; 269: 496-512.
Forster AC, Church GM. Towards synthesis of a minimal cell. Mol Syst Biol 2006; 2:45.
Friedland AE, Lu TK, Wang X, Shi D, Church G, Collins JJ. Synthetic gene networks that count. Science 2009; 324:1199-202.
Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature 2000; 403:339-42.
Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010; 329:52-6.
Ham TS, Lee SK, Keasling JD, Arkin AP. A tightly regulated inducible expression system utilizing the fim inversion recombination switch. Biotechnol Bioeng 2006; 94:1-4.
Ham TS, Lee SK, Keasling JD, Arkin AP. Design and construction of a double inversion recombination switch for heritable sequential genetic memory. PLoS One 2008; 3:2815.
Hamaguchi N, Ellington A, Stanton M. Aptamer beacons for the direct detection of proteins. Anal Biochem 2001; 294:126-31.
Hooshangi S, Thiberge S, Weiss R. Ultrasensitivity and noise propagation in a synthetic transcriptional cascade. Proc Natl Acad Sci USA 2005; 102:3581-6.
Isaacs FJ, Hasty J, Cantor CR, Collins JJ. Prediction and measurement of an autoregulatory genetic module. Proc Natl Acad Sci USA 2003; 100:7714-9.
Itakura K, Hirose T, Crea R, Riggs AD, Heyneker HL, Bolivar F, et al. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 1977; 198:1056-63.
Johnson AD, Poteete AR, Lauer G, Sauer RT, Ackers GK, Ptashne M. lambda Repressor and cro—components of an efficient molecular switch. Nature 1981; 294:217-23.
Kelly JR, Rubin AJ, Davis JH, Ajo-Franklin CM, Cumbers J, Czar MJ, et al. Measuring the activity of BioBrick promoters using an in vivo reference standard. J Biol Eng 2009; 3:4.
Khalil AS, Collins JJ. Synthetic biology: applications come of age. Nat Rev Genet 2010; 11:367-79.
Kiskinis E, Eggan K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest 2010; 120:51-9.
Klumpp S, Zhang Z, Hwa T. Growth rate-dependent global effects on gene expression in bacteria. Cell 2009; 139:1366-75.
Kramer BP, Viretta AU, Daoud-El-Baba M, Aubel D, Weber W, Fussenegger M. An engineered epigenetic transgene switch in mammalian cells. Nat Biotechnol 2004; 22:867-70.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409:860-921.
Lee SK, Chou HH, Pfleger BF, Newman JD, Yoshikuni Y, Keasling JD. Directed evolution of AraC for improved compatibility of arabinose- and lactose-inducible promoters. Appl Environ Microbiol 2007; 73:5711-5.
Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 2009; 461:997-1001.
Lim GJ, Zare S, Van Dyke M, Atala A. Cell microencapsulation. Adv Exp Med Biol 2010; 670:126-36.
Lim WA. Designing customized cell signalling circuits. Nat Rev Mol Cell Biol 2010; 11:393-403.
Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA 2007; 104:11197-202.
——. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA 2009; 106:4629-34.
Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nat Biotechnol 2009; 27:1139-50.
Malmstrom J, Beck M, Schmidt A, Lange V, Deutsch EW, Aebersold R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009; 460:762-5.
Mandal S, Sarpeshkar R. Log-domain circuit models of chemical reactions. Circuits and Systems, 2009 ISCAS 2009 IEEE International Symposium on 2009; 2697-700.
McMillen D, Kopell N, Hasty J, Collins JJ. Synchronizing genetic relaxation oscillators by intercell signaling. Proc Natl Acad Sci USA 2002; 99:679-84.
Mettetal JT, Muzzey D, Gomez-Uribe C, van Oudenaarden A. The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science 2008; 319:482-4.
Molin S, Boe L, Jensen LB, Kristensen CS, Givskov M, Ramos JL, et al. Suicidal genetic elements and their use in biological containment of bacteria. Annu Rev Microbiol 1993; 47:139-66.
Moore GE. Cramming more components onto integrated circuits. Electronics Magazine 1965; 38.
Okano H, Kobayashi TJ, Tozaki H, Kimura H. Estimation of the source-by-source effect of autorepression on genetic noise. Biophys J 2008; 95:1063-74.
Parens E, Johnston J, Moses J. Ethics. Do we need “synthetic bioethics”? Science 2008; 321:1449.
Parsons JB, Frank S, Bhella D, Liang M, Prentice MB, Mulvihill DP, et al. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation and evidence of filament-associated organelle movement. Mol Cell 2010; 38:305-15.
Rinaudo K, Bleris L, Maddamsetti R, Subramanian S, Weiss R, Benenson Y. A universal RNAi-based logic evaluator that operates in mammalian cells. Nat Biotechnol 2007; 25:795-801.
Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 2009; 27:946-50.
Scott CT. The zinc finger nuclease monopoly. Nat Biotechnol 2005; 23:915-8.
Simpson ML, Cox CD, Sayler GS. Frequency domain analysis of noise in autoregulated gene circuits. Proc Natl Acad Sci USA 2003; 100:4551-6.
Smolke CD. Building outside of the box: iGEM and the BioBricks Foundation. Nat Biotechnol 2009; 27:1099-102.
Sohka T, Heins RA, Phelan RM, Greisler JM, Townsend CA, Ostermeier M. An externally tunable bacterial band-pass filter. Proc Natl Acad Sci USA 2009; 106:10135-40.
Stricker J, Cookson S, Bennett MR, Mather WH, Tsimring LS, Hasty J. A fast, robust and tunable synthetic gene oscillator. Nature 2008; 456:516-9.
Tanaka S, Sawaya MR, Yeates TO. Structure and mechanisms of a protein-based organelle in Escherichia coli. Science 2010; 327:81-4.
Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods 2009; 6:331-8.
Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, et al. A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci USA 2010; 107:10914-9.
Varela-Rohena A, Carpenito C, Perez EE, Richardson M, Parry RV, Milone M, et al. Genetic engineering of T cells for adoptive immunotherapy. Immunol Res 2008; 42:166-81.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science 2001; 291:1304-51.
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009; 460:894-8.
Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953; 171:737-8.
Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov 2009; 8:129-38.
Widmaier DM, Tullman-Ercek D, Mirsky EA, Hill R, Govindarajan S, Minshull J, et al. Engineering the Salmonella type III secretion system to export spider silk monomers. Mol Syst Biol 2009; 5:309.
Win MN, Smolke CD. A modular and extensible RNA-based gene-regulatory platform for engineering cellular function. Proc Natl Acad Sci USA 2007; 104:14283-8.
——. Higher-order cellular information processing with synthetic RNA devices. Science 2008; 322:456-60.
Yeh BJ, Rutigliano RJ, Deb A, Bar-Sagi D, Lim WA. Rewiring cellular morphology pathways with synthetic guanine nucleotide exchange factors. Nature 2007; 447:596-600.
Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, et al. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods 2006; 3:623-8.
Bernhard Palsson69
Abstract
The first full genome sequences were established in the mid-1990s. Shortly thereafter, genome-scale metabolic network reconstructions appeared. Since that time, we have witnessed an exponential growth in their number and uses. Here I discuss, from a personal point of view, four topics: (1) the placement of metabolic systems biology in the context of broader scientific developments, (2) its foundational concepts, (3) some of its current uses, and (4) some of the expected future developments in the field.
Metabolic Systems Biology in the Grand Scheme of Things
Ever since Gregor Mendel discovered discrete quanta of information passed from one generation to the next that determined form and function of an organism, the genotype–phenotype relationship has been of fundamental importance in the life sciences. For monogeneic traits, the genotype–phenotype relationship can be readily understood. However, most phenotypic traits involve multiple gene products. This makes the genotype–phenotype relationship a challenge to reconstruct and understand, given the complex interactions that can form among the gene products.
With the publication of the first full genome sequences in the mid-1990s (Fleischmann et al., 1995) it became possible, in principle, to identify all the gene products involved in complex biological processes in a single organism. The well-studied biochemistry of metabolic transformations made it possible to reconstruct, on a genome-scale, metabolic networks for a target organism in a biochemically detailed fashion (Edwards and Palsson, 1999, 2000). Such
_______________
68 This article was published in FEBS Letters, 583, Palsson, B, Metabolic systems biology, 3900-3904, Copyright Elsevier (2009).
69 Department of Bioengineering, UCSD, La Jolla, CA 92093-0412, USA
Fax: +1 858 822 3120.
E-mail: palsson@ucsd.edu
Article history:
Received 8 July 2009 Revised 5 September 2009 Accepted 15 September 2009
Available online 19 September 2009
Edited by Stefan Hohmann
Keywords: Systems biology, Metabolism
2009 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
metabolic network reconstructions can be converted into a mathematical format yielding mechanistic genotype–phenotype relationships for microbial metabolism (Palsson, 2006). The mathematical format of the underlying biochemical, genetic, and genomic (BiGG) knowledge allows the formulation of genome-scale models (GEMs). GEMs enable the computation of phenotypic traits based on the genetic composition of the target organism (Palsson, 2006; Price et al., 2004).
Since the establishment of the first metabolic genome-scale reconstruction in 1999 and in silico models thereof, many more have followed (Fig. A14-1); perhaps most notably for human metabolism in 2007. The scope and content of network reconstructions continues to grow, for instance to include the entire transcription/translation apparatus of a cell (Thiele et al., 2009) and the structural information about the metabolic enzymes (Zhang et al., in press).
Foundations of Metabolic Systems Biology
The Basic Paradigm
We can now enumerate various cellular components, describe their interactions chemically, formulate a mathematical description of the totality of such interactions, identify the constraints that the resulting network operates under, and apply optimality principles to evaluate likely physiological functions in a
FIGURE A14-1 Growth of genome sequences and genome-scale metabolic reconstructions. The number of network reconstruction has grown exponentially, at a similar pace as genome sequences have appeared (prepared by Adam Feist and Ines Thiele).
given environment. These capabilities provide a consistent framework on which a mechanistic basis for the microbial metabolic genotype–phenotype relationship can be formulated. The underlying process is based on an emerging paradigm to relate the genotype to the phenotype through reconstruction and in silico model building (Fig. A14-2) is comprised of four steps:
- generation of ‘omics’ and collection of literature data on the target organism;
- network reconstruction and the formulation of a BiGG knowledge-base;
- conversion of the reconstruction into a mathematical format and the implementation of in silico query tools; and
- enablement of a variety of basic and applied uses.
This fundamental paradigm allows for the first time the genome-scale computation of phenotypic functions of an organism. The establishment of a mechanistic formulation of the most fundamental relationship in biology – the genotype–phenotype relationship – for a limited number of phenotypic functions, differs from that developed for the basic physical laws about a century ago. This mechanistic description must account for both proximal and distal causation. Proximal (or proximate) causation occurs against a fixed genetic background (i.e., an individual organism), while distal (or ultimate) causation results from (genomic) changes that occur from generation to generation, i.e., evolution.
Some Basic Principles
This basic paradigm has been implemented for a number of organisms and a variety of biological results have been obtained (Feist and Palsson, 2008; Papin et al., accepted for publication). As a result of this successful reduction to practice, one is tempted to try to determine and state the underlying reasons for this success. Below, I attempt to start this process.
Axiom #1: All cellular functions are based on chemistry. A simple but consequential statement, as it implies the fundamental events in a cell can be described by chemical equations. These equations, in turn, come with chemical information and physico-chemical principles.
Axiom #2: Annotated genome sequences along with experimental data enable the reconstruction of genome-scale metabolic networks. The reconstruction process is a grand-scale systematic assembly of information in a quality-controlled/quality assured (QC/QA) setting (Thiele and Palsson, in press) that leads to a BiGG knowledge-base, which is a collection of established biochemical, genetic, and genomic data represented by a network reconstruction. The reconstruction process has been reviewed elsewhere (Feist et al., 2009; Reed et al., 2006b), and a growing number of reconstructions are available (Fig. A14-1).
Axiom #3: Cells function in a context-specific manner. When a cell is placed
FIGURE A14-2 The four-step paradigm for metabolic systems biology. Adapted from Palsson (2006). Prepared by Adam Feist.
in a particular environment, it expresses a subset of its genes in response to environmental cues. The abundance of cellular components can be profiled using ‘omic’ methods (i.e., transcriptomics, proteomics, metabolomics). Such omic data can be mapped onto a network reconstruction to tailor it to the particular condition being considered.
Axiom #4: Cells operate under a series of constraints. Factors constraining cellular functions fall into four principal categories (Palsson, 2006): physico-chemical (see Axiom #5), topological (molecular crowding effects and steric hindrance), environmental (Axiom #3), and regulatory (basically self-imposed constraints, or restraints). These constraints cannot be violated allowing the estimation of all functional (i.e., physiological) states that a genome-scale reconstruction can achieve. Mathematically, such statements are translated into fundamental subspaces associated with the stoichiometric matrix (S), whose properties can be characterized (Palsson, 2006). In this so-called S matrix, where S stands for stoichiometric, the rows correspond to the network metabolites and the columns to the network reactions. The coefficients of the substrates and products of each reaction are entered in the corresponding cell of the matrix.
Axiom #5: Mass (and energy) is conserved. This statement is one of the basic physical laws. Since all proper chemical equations can be described by stoichiometric coefficients, and since a set of chemical equations can be described by the stoichiometric matrix, S, this means that all steady states (normally close to the homeostatic states of interest) of a network can be described by a simple linear equation, S ∙ v = 0, where v is a vector of fluxes through chemical reactions (Palsson, 2006). Thus, the computation of functional states of a network is enabled based on the known underlying chemistry.
Axiom #6: Cells evolve under a selection pressure in a given environment. This Darwinian statement has implicit optimality principles built into it. Consequently, if we know the selection pressure, we can state a so-called objective function and determine optimal states given a network reconstruction and governing constraints.
Each one of these statements by themselves is almost trivial and accepted in various scientific disciplines as being fundamental. Taken together, though, they combine to form the conceptual basis for constraint-based reconstruction and analysis (COBRA), and enable the development of the mechanistic genotype–phenotype relationship for metabolism. The recent emergence of genome-scale reconstructions (Axiom #2) has proven key to this formulation.
Practicing COBRA
The COBRA approach (Palsson, 2006; Price et al., 2004) has been widely used to analyze network reconstructions. It uses stoichiometric information about biochemical transformations taking place in a target organism to construct the model. While a metabolic reconstruction is unique to the target organism, one can derive many different condition-specific models from a single reconstruc-
tion. This conversion of a metabolic reconstruction of an organism into models requires the imposition of physico-chemical and environmental constraints to define systems boundaries. The conversion also includes the transformation of the reaction list into a computable, mathematical matrix format (e.g., using the COBRA toolbox [Becker et al., 2007]).
Formulation of BiGG knowledge-bases: The four-step procedure to reconstruct genome-scale metabolic networks has been detailed elsewhere (Feist et al., 2009; Reed et al., 2006b; Durot et al., 2009). Briefly, all metabolic functions encoded by an organism’s genome are systematically retrieved, curated, and translated into a list of biochemical reactions that comprise the network. The association between the biochemical reactions and the catalyzing gene products is achieved using Boolean logic through the gene-protein-reaction (GPR) associations (Reed et al., 2006b). Further metabolic functions supported by experimental data can be included without gene association. Extensive QA/QC procedures ensure that the BiGG knowledge-base is self-consistent, comprehensive, and exhibits similar physiological properties as the target organism (Thiele and Palsson, in press). A BiGG knowledge-base effectively represents a two-dimensional annotation of a genome (Palsson, 2004) and represents the implementation of Axioms #1 and #2.
From a knowledge-base to a GEM: BiGG knowledge-bases can be converted into GEMs (genome-scale models) by implementing Axioms #3, #4, and #5. While BiGG accounts for genome-scale information, a GEM can represent the capacities of a cell in a particular environmental and genetic state. Subsequently, there are different possible GEMs that can be derived from a given organism’s reconstruction. In this conversion, the BiGG knowledge-base is represented in a mathematical format, S.
System boundaries are defined around the entire reaction network. Exchange reactions are added to all transportable extracellular metabolites and can be constrained in simulations to represent different environmental conditions. Demand reactions are added, including the biomass reaction, that details all precursors and their fractional contributions to a cell’s macromolecular composition, as well as any maintenance energy requirements (Feist et al., 2007).
Computational tools: A broad spectrum of methods has been developed under this umbrella (Palsson, 2006; Price et al., 2004; Durot et al., 2009), collectively called COBRA methods. Constraint-based metabolic models can be imported into Matlab in SBML format. COBRA methods can then be applied (Becker et al., 2007). With a growing number of metabolic reconstructions available (see http://www.systemsbiology.ucsd.edu/In_Silico_Organisms/Other_Organisms) and the accessibility of COBRA tools, the number of practitioners in this field is growing.
In a recent review (Feist and Palsson, 2008), the uses of the Escherichia coli GEM were classified into five categories: (1) metabolic engineering (i.e., genome-scale synthetic biology), (2) gap-filling (i.e., systematic generation of hypothesis), (3) phenotypic screens (data analysis), (4) determining network
properties (in silico systems biology), and (5) evolutionary studies (i.e., COBRA methods can analyze both proximal and distal causation). Similarly, a review of applications to other organisms, (Papin et al., accepted for publication), classifies them as: (1) contextualization of high-throughput data, (2) guidance of metabolic engineering, (3) directing hypothesis-driven discovery, (4) interrogation of multispecies relationships, and (5) network property discovery. Thus, the basic and applied uses of reconstructions and associated GEMs are growing.
Biological Science in the Era of Systems Biology
Genome-scale reconstructions enable biological science to proceed in fundamentally new ways. Here I discuss four new possibilities.
Integration of High-throughput Data
Omics data can be analyzed using a reconstruction as a scaffold. As stated above, a reconstruction is a BiGG knowledge-base, and if ‘omics’ data are mapped onto the reconstruction, it enables the analysis of the ‘omics’ data against the curated knowledge about the target organism as a context.
Several examples of this use of reconstructions have been demonstrated. Tissue-specific expression-profiling data has been mapped against the reconstruction of the global human metabolic map (Shlomi et al., 2008) to yield draft reconstructions of tissue-specific metabolic networks in humans. Proteomic data from the human cardiac mitochondria has been used to form tissue-specific organelle models (Vo et al., 2004) and used for the analysis of SNPs through the use of cosets (Jamshidi and Palsson, 2006). The use of reconstructions to analyze expression-profiling data from E. coli in many studies has been reviewed recently (Lewis et al., 2009). In a similar fashion, metabolomic and fluxomic data can be analyzed, e.g., see Jamshidi and Palsson (2008).
Thus, curated genome-scale reconstructions provide a new way to analyze ‘omics’ data. This ability is likely to help with revealing the information content in an ‘omics’ data set, as purely statistical approaches have proven to be somewhat limited.
Gap-filling
BiGG knowledge-bases are not complete; they have ‘gaps’ in them. These gaps come in at least two fundamental varieties. First, there can be a missing reaction or a path between two metabolites in the reconstruction, and secondly, a metabolite can be detected that has no connections to the network – representing an ‘island’ on the metabolic map. The former can be filled with a gap-filling process (Reed et al., 2006a). Missing links have been discovered this way (Fuhrer et al., 2007) and the methods developed to date for gap-filling have been described
(Breitling et al., 2008); the latter calls for the discovery of a new pathway. Computational tools for suggesting missing pathways have been recently described (Hatzimanikatis et al., 2005; Kumar and Maranas, 2009).
These methods may be viewed as representing interesting computational and algorithmic challenges. However, these developments are more profound and fundamental. They represent the computational generation of hypotheses using genome-scale BiGG knowledge-bases. The reconstruction is used as a context for analyzing data and determining a candidate explanation for discrepancies between experimental data and computational predictions. In fact, mixed-integer linear programming (MILP) algorithms represent the generation of the ‘most parsimonious’ hypothesis as they can be used to grade the complexity of the candidate explanations. In the initial study of this kind, the simplest explanation was always found to be the correct one (Reed et al., 2006a).
Understanding Complex Biological Phenomena
Phenotypic functions rely on the coordinated and simultaneous action of multiple gene products. This makes complex biological processes hard to comprehend. In addition, with changes over generations, such comprehension may be even more challenging. COBRA tools enable the computation of both proximal and distal causation at a genome-scale, and thus possesses the potential to provide a framework for a deep understanding of complex biological phenomena.
This expectation is perhaps being realized through work on bacterial adaptation. The ability of GEMs to predict the outcome of bacterial adaptation to new nutritional environments (Ibarra et al., 2002), even in the face of gene deletions (Fong and Palsson, 2004), opens up new and fascinating avenues to study fundamental biological phenomena. The network level predictions are made using GEMs and phenotypic functions. Fortunately, the third generation sequencing methods enable the full resequencing of microbial genomes following adaptation (Shendure et al., 2005; Herring et al., 2006). With available allelic replacement methods the causality of the mutations can be assessed (Herring et al., 2006; Conrad and Palsson, in press).
The convergence of these developments, COBRA, GEMs, cheap resequencing, and allelic replacement, allows the study of evolution in a laboratory setting. This fascinating prospect is likely to help us understand the plasticity and functions of bacterial genomes better than before. For instance, the RNA polymerase has been shown to be a highly mutable enzyme (Conrad and Palsson, in press) and we are now beginning to determine the objective functions that wild type strains seem to adhere to (Fischer and Sauer, 2005). Taken together, this means that teleology could be studied in an experimental setting.
Thus, metabolic systems biology is enabling a totally new scientific pursuit. Although the application of COBRA methods has been to bacterial adaptation, one might expect similar applications to pathogenic processes. For instance,
carcinogenesis has been conceptualized as the alleviation of a series of constraints (Hanahan and Weinberg, 2000) and metabolism is now seen as an integral part of this process (Vander Heiden et al., 2009). Thus, the analysis of the adjustment of metabolic processes to enable this pathologic state is likely to be possible using COBRA methods.
Metabolic Engineering
Microbial metabolism has been and is being modified to achieve practical ends through willful genetic manipulation of a wild type organism to generate a production strain. This field is called metabolic engineering; with a foundational paper by Bailey appearing in 1991 (Bailey, 1991). A recent review describes three phases in the history of metabolic engineering (Park and Lee, 2008): first is the use of random mutagenesis and screening; second, the use of targeted genetic manipulations to achieve ‘local’ results in the function of a network; and third, the use of GEMs to perform ‘global,’ or genome-scale analysis of genetic manipulations. Algorithms have been developed to perform the computation of genetic changes to achieve such global results (Burgard et al., 2003; Patil et al., 2005; Pharkya et al., 2004).
The engineering dictum, “there is nothing more practical than reliable theory,” is at the foundation of engineering design and practice. Good computational models accelerate design processes, and minimize prototyping, testing, and experimentation. The fact that the third phase in metabolic engineering has been ushered in through the use of GEMs signifies that we are now beginning to contemplate and practice synthetic biology at the genome-scale. Many commercial enterprises that are developing sustainable technologies benefit from this capability.
Beyond Metabolism in Microbes
With the successes of metabolic systems biology in microbes and the statement of some of its underlying axioms, it is natural to wonder what will happen next in this field. The foray into human metabolism seems like an obvious extension with the clear challenges of greater organismic complexity and function. i.e., what are the objectives of various metabolic functions in man?
We have already shown that similar bottom-up reconstructions of the transcription–translation machinery can be achieved (Thiele et al., 2009). Given the chemically detailed representation of this process on a genome-scale, COBRA approaches could be used for the analysis of systems properties. Several chemically detailed reconstructions of signaling systems have appeared (Thiele et al., 2009; Papin and Palsson, 2004) that could be analyzed using COBRA tools. New genome-scale data types (transcription start sites, tiled arrays for expression profiling, ChIP-chip, and proteomic methods) are now yielding the data that is needed to reconstruct transcriptional regulatory networks at a genome-scale (Cho
et al., accepted for publication). Other data types, such as phosphoproteomics (Macek et al., 2009; Soufi et al., 2008) and rapid metabolic regulatory mechanisms (Ralser et al., 2009) will expand the scope of reconstruction of regulatory phenomena.
Closing
The emergences of mechanistic genotype–phenotype relationships that can account for dual causality are likely to have a broad impact on the life sciences. A number of metabolic networks have already been reconstructed for target organisms. New biological science is being performed with these reconstructions. The new avenues that have been opened up by COBRA tools and a growing number of GEMs are probably just at the early stages of their exploration.
This piece is written as a personal account of the development of the field of genome-scale systems biology of metabolism. It is not meant to be a comprehensive view of the field but a discussion of the four topics stated in the summary. Although most of the references are to the author’s own work, many of them are reviews citing the major developments in the field.
Acknowledgements
The author is thankful of the many individuals that have entered the COBRA field and contributed significantly to it. Early adopters include Jens Nielsen, Sang Yup Lee, Costas Maranas, Steve Oliver, Bas Teusink, George Church, and Daniel Segre, entering the field in the early 2000s. Since then, many have followed, making this a vibrant and growing field with an increasing list of accomplishments. The author has been supported by the NIGMS, NIAID, NHRI, and the DOE, and is a co-founder of Genomatica.
References
Bailey, J.E. (1991) Toward a science of metabolic engineering. Science 252, 1668–1675.
Becker, S.A., Feist, A.M., Mo, M.L., Hannum, G., Palsson, B.O. and Herrgard, M.J. (2007) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA toolbox. Nat. Protocol 2, 727–738.
Breitling, R., Vitkup, D. and Barrett, M.P. (2008) New surveyor tools for charting microbial metabolic maps. Nat. Rev. Microbiol. 6, 156–161.
Burgard, A.P., Pharkya, P. and Maranas, C.D. (2003) Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84, 647–657.
Cho, B.K., Zengler, K., Qiu, Y., Knight, E.M., Park, Y.S., Barrett, C.L., Gao, Y. and Palsson, B.O. (accepted for publication.) Elucidation of meta-structure of a bacterial genome. Nat. Biotechnol.
Conrad, T. and Palsson, B.O. (in press) Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations. Genome Biol.
Durot, M., Bourguignon, P.Y. and Schachter, V. (2009) Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol. Rev. 33, 164–190.
Edwards, J.S. and Palsson, B.O. (1999) Systems properties of the Haemophilus influenzae Rd metabolic genotype. J. Biol. Chem. 274, 17410–17416.
——. (2000) The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 97, 5528–5533.
Feist, A.M. and Palsson, B.O. (2008) The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat. Biotech. 26, 659–667.
Feist, A.M. et al. (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3.
Feist, A.M., Herrgard, M.J., Theile, I., Reed, J.L. and Palsson, B.O. (2009) Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 7, 129–143.
Fischer, E. and Sauer, U. (2005) Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat. Genet. 37, 636–640.
Fleischmann, R.D. et al. (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496–498. 507–512.
Fong, S.S. and Palsson, B.O. (2004) Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet. 36, 1056–1058.
Fuhrer, T., Chen, L., Sauer, U. and Vitkup, D. (2007) Computational prediction and experimental verification of the gene encoding the NAD+/NADP+-dependent succinate semialdehyde dehydrogenase in Escherichia coli. J. Bacteriol. 189, 8073–8078.
Hanahan, D. and Weinberg, R.A. (2000) The hallmarks of cancer. Cell 100, 57–70.
Hatzimanikatis, V., Li, C., Ionita, J.A., Henry, C.S., Jankowski, M.D. and Broadbelt, L.J. (2005) Exploring the diversity of complex metabolic networks. Bioinformatics 21, 1603–1609.
Herring, C.D. et al. (2006) Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 38, 1406–1412.
Ibarra, R.U., Edwards, J.S. and Palsson, B.O. (2002) Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420, 186–189.
Jamshidi, N. and Palsson, B.O. (2006) Systems biology of SNPs. Mol. Syst. Biol. 2, 38.
——. (2008) Formulating genome-scale kinetic models in the post-genome era. Mol. Syst. Biol. 4, 171.
Kumar, V.S. and Maranas, C.D. (2009) GrowMatch: an automated method for reconciling in silico/in vivo growth predictions. PLoS Comput. Biol. 5.
Lewis, N.E., Cho, B.K., Knight, E.M. and Palsson, B.O. (2009) Gene expression profiling and the use of genome-scale in silico models of Escherichia coli for analysis: providing context for content. J. Bacteriol. 191, 3437–3444.
Macek, B., Mann, M. and Olsen, J.V. (2009) Global and site-specific quantitative phosphoproteomics: principles and applications. Annu. Rev. Pharmacol. Toxicol. 49, 199–221.
Palsson, B.O. (2004) Two-dimensional annotation of genomes. Nat. Biotechnol. 22, 1218–1219.
——. (2006) Systems Biology: Properties of Reconstructed Networks, Cambridge University Press, New York.
Papin, J.A. and Palsson, B.O. (2004) The JAK-STAT signaling network in the human B-cell: an extreme signaling pathway analysis. Biophys. J. 87, 37–46.
Papin, J.A., Oberhardt, M. and Palsson, B.O. (accepted for publication) Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol.
Park, J.H. and Lee, S.Y. (2008) Towards systems metabolic engineering of microorganisms for amino acid production. Curr. Opin. Biotech. 19, 454–460.
Patil, K.R., Rocha, I., Forster, J. and Nielsen, J. (2005) Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform. 6, 308.
Pharkya, P., Burgard, A.P. and Maranas, C.D. (2004) OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 14, 2367–2376.
Price, N.D., Reed, J.L. and Palsson, B.O. (2004) Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2, 886–897.
Ralser, M., Wamelink, M.M., Latkolik, S., Jansen, E.E., Lehrach, H. and Jakobs, C. (2009) Metabolic reconfiguration precedes transcriptional regulation in the antioxidant response. Nat. Biotechnol. 27, 604–605.
Reed, J.L. et al. (2006a) Systems approach to refining genome annotation. Proc. Natl. Acad. Sci. USA 103, 17480–17484.
Reed, J.L., Famili, I., Thiele, I. and Palsson, B.O. (2006b) Towards multidimensional genome annotation. Nat. Rev. Genet. 7, 130–141.
Shendure, J. et al. (2005) Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732.
Shlomi, T., Cabili, M.N., Herrgard, M.J., Palsson, B.O. and Ruppin, E. (2008) Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 26, 1003–1010.
Soufi, B., Jers, C., Hansen, M.E., Petranovic, D. and Mijakovic, I. (2008) Insights from site-specific phosphoproteomics in bacteria. Biochim. Biophys. Acta 1784, 186–192.
Thiele, I. and Palsson, B.O. (in press) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protocol.
Thiele, I., Jamshidi, N., Fleming, R.M. and Palsson, B.O. (2009) Genome-scale reconstruction of Escherichia coli’s transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS Comput. Biol. 5.
Vander Heiden, M.G., Cantley, L.C. and Thompson, C.B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033.
Vo, T.D., Greenberg, H.J. and Palsson, B.O. (2004) Reconstruction and functional characterization of the human mitochondrial metabolic network based on proteomic and biochemical data. J. Biol. Chem. 279, 39532–39540.
Zhang, Y., Thiele, I., Palsson, B.O., Ostermann, A. and Godzik, A. (in press) Structural genomics provides an atomic level view of the metabolic network of Thermotoga maritima. Science.
Bali Pulendran,71,72,* Shuzhao Li,71and Helder I. Nakaya71
Vaccination is one of the greatest triumphs of modern medicine, yet we remain largely ignorant of the mechanisms by which successful vaccines stimulate protective immunity. Two recent advances are beginning to illuminate such mechanisms: realization of the pivotal role of the innate immune system in sensing microbes and stimulating adaptive immunity, and advances in systems biology. Recent studies have used systems biology approaches to obtain a global picture of the immune responses to vaccination
_______________
70 This article was published in Immunity, 33, Pulendran, B., Li, S., Nakaya, H.I., Systems Vaccinology, 516-529, Copyright Elsevier (2010).
71 Emory Vaccine Center, Yerkes National Primate Research Center, 954 Gatewood Road, Atlanta, GA 30329, USA.
72 Department of Pathology, Emory University School of Medicine, Atlanta, GA, USA.
* Correspondence: bpulend@emory.edu
DOI 10.1016/j.immuni.2010/10.006
in humans. This has enabled the identification of early innate signatures that predict the immunogenicity of vaccines, and identification of potentially novel mechanisms of immune regulation. Here, we review these advances and critically examine the potential opportunities and challenges posed by systems biology in vaccine development.
“We are drowning in a sea of data and thirsting for knowledge. Most biology today is low input, high throughput, no output biology.”
—Sydney Brenner
“We must make this the decade of vaccines.”
—Bill Gates
Introduction
In the epic saga of the evolutionary struggle between microbes and humans, the invention of vaccination is a defining moment, one that represents the victory of our wits over their genes. Ironically, however, despite the common origins of vaccinology and immunology in the pioneering work of giants such as Pasteur and Jenner, the two disciplines have evolved such different trajectories that immunologists remain largely ignorant about the mechanisms of action of successful vaccines (empirically made), and vaccinologists have, until recently, displayed little interest in the intricacies of immune regulation. Understanding the immunological mechanisms of vaccination, however, is of paramount importance in the rational design of future vaccines against pandemics such as HIV, malaria, and tuberculosis and against emerging infections. Recent advances in our understanding of the innate immune system and the use of systems biological approaches are beginning to reveal the fundamental mechanisms by which the innate immune system orchestrates protective immune responses to vaccination (Pulendran and Ahmed, 2006; Steinman, 2008). The innate immune system is capable of sensing viruses, bacteria, parasites, and fungi through the expression of so-called pattern recognition receptors (PRRs), which are expressed by dendritic cells (DCs) and other cells of the innate immune system (Reviewed by Coffman et al. [2010], this issue of Immunity). Toll-like receptors (TLRs) represent the most studied family of PRRs (Iwasaki and Medzhitov, 2010; Kawai and Akira, 2010). However, other non-TLR families of innate receptors, such as C type lectin-like receptors (Geijtenbeek and Gringhuis, 2009), nucleotide-binding oligomerization domain-like receptors (Ting et al., 2010), and retinoic acid-inducible gene I (RIG-I)-like receptors (Wilkins and Gale, 2010), also play critical roles in innate sensing of pathogens and induction of inflammatory responses. Emerging evidence suggests that the nature of the DC subtype, as well as the particular PRR triggered, plays a critical role in modulating the strength, quality, and persistence of adaptive immune responses (Pulendran and Ahmed, 2006; Steinman, 2008). Such insights
about the molecular basis of immune regulation have accrued largely through the traditional scientific method of hypothesis creation, and experimental validation, particularly through the reductionist approaches of molecular biology. However, as powerful as such approaches are, they offer a very limited view of complex biological systems. Thus, there are estimated to be more than 26,000 genes in our genomes, and entry of a vaccine or a pathogen into the body perturbs the expression of a substantial fraction of them. Systems biological tools offer us a solution to this problem. In vaccinology, recent studies have highlighted the use of such approaches in offering a global picture of the biological response to a vaccine. Here we highlight these advances and discuss their potential importance. This review is divided into four parts. In the first part (Biology of the 21st Century), we provide a broad overview of systems biology, its goals and challenges, and highlight the features that distinguish it from reductionistic biology. Next, (in Systems Biology in Vaccinology) we review recent studies that have applied systems biological approaches to vaccinology and suggest key areas where such approaches may impinge on vaccine development. These include identification of potentially novel correlates of immunity, predicting the efficacy of vaccines, accelerating the clinical trial platform of vaccines, and learning new biological insights about immune regulation. In part three (Low-Input, High-Throughput, No Output Biology), we critically examine the challenges and potential pitfalls of systems biological approaches. Finally (in A Framework for Systems Vaccinology), we conclude by offering a conceptual framework of how systems approaches can guide vaccine design and development.
Biology of the 21st Century
Two of the greatest scientific achievements of the 20th century were the discovery of the structure of DNA and the sequencing of the human genome. The grand challenge for biology and medicine at the turn of the 21st century is to understand the biological complexity that emerges from interactions between our genomes and the environment. We are uniquely poised to tackle this challenge of biological complexity by the convergence of a new intellectual framework (a systems rather than a reductionistic view) and new technologies (for measuring and visualizing the behavior of genes, molecules, cells, organs, and organisms), coupled with the innovation of computational and mathematical tools for dealing with complex data sets. The convergence of these disparate threads offers us an unprecedented opportunity to understand the fundamental features of life—from a holistic rather than solely reductionistic; from a predictive rather than descriptive; in short, from a systems biological viewpoint.
Systems biology is an interdisciplinary approach that systematically describes the complex interactions between all the parts in a biological system, with a view to elucidating new biological rules capable of predicting the behavior of the biological system (Kitano, 2002). Although reductionist molecular biology
works by isolating and characterizing each component of the system (e.g., a gene, a protein, or a cell type), systems biology focuses on studying the structure and dynamics of the whole system (Kitano, 2002). Under different types of perturbation, data are collected from all the components of a biological system, analyzed, and integrated in order to generate a mathematical model that describes or predicts the response of the system to individual perturbations (Ideker et al., 2001). A key goal of systems biology is to understand the nature of biological networks, which access, integrate, and communicate information from the genome to the environment, and back (Ideker et al., 2001; Kitano, 2002). These networks represent, in a sense, the lowest functional units of life processes, such as development, disease, immunity, and aging. Therefore, understanding these life processes requires understanding the nature and behavior of these networks, both their robustness and plasticity, in the face of a dynamic environment. What is needed to delineate these networks is the acquisition of high-throughput data on the genes, mRNAs, microRNAs, and proteins that constitute the networks. Systems biology capitalizes on several so-called “omic” technologies that are used to define and monitor all the components of the systems. DNA microarrays and high-throughput sequencing can be applied to identify global differences on gene expression (transcriptomics), genomic rearrangements, and genetic polymorphisms (genomics) as well as to provide a high-resolution global map of protein-DNA interactions (chromatin immunoprecipitation followed by DNA sequencing or hybridization to the array). Other enabling technologies include modern mass spectrometry (powering proteomics, lipidomics, and metabolomics), yeast two-hybrid system (mapping protein interactions), and genome-wide RNA interference screening (identifying genes required for a process). In addition, systems biology features the integration and modeling the huge amount of data generated by high-throughput techniques. An array of computational methods has been developed in the context of systems biology, and data integration and network inference are of special interest (Bansal et al., 2007; Hyduke and Palsson, 2010). Such methods can be closely coupled with experimental studies to generate testable hypotheses and improve the understanding of molecular mechanisms.
Systems biological approaches have changed prognosis and therapy response prediction in oncology (Alizadeh et al., 2000; Sørlie et al., 2001) and are beginning to be applied to understanding mechanisms of innate and adaptive immunity (Aderem and Hood, 2001; Germain, 2001; Gilchrist et al., 2006; Haining et al., 2008; Haining and Wherry, 2010; Kaech et al., 2002; Wherry et al., 2007; Zak and Aderem, 2009), in identifying diagnostic biomarkers of different infections (Chaussabel et al., 2008; Lee et al., 2008; Otaegui et al., 2009; Ramilo et al., 2007), and autoimmunity (Pascual et al., 2010). Systems biological approaches also offer unprecedented opportunities to study immune responses in humans (Aderem and Hood, 2001; Germain, 2001). However, only recently have they started to be applied to vaccinology. There are two broad applications of systems approaches in vaccinology: prediction of immunogenicity and efficacy of vac-
cines and scientific discovery. These two areas use distinct methodologies and have different rationales and output, and they are discussed below.
Systems Biology in Vaccinology
One potential application of systems biology in vaccinology is in predicting vaccine efficacy. The identification of molecular signatures (e.g., patterns of gene expression induced after vaccination), induced rapidly in the blood after vaccination that correlate with and predict the later development of protective immune responses, represents a strategy to prospectively determine vaccine efficacy. In the field of cancer genomics, predictions of cancer outcome have been based on gene expression profiles of the cancer cells themselves (see Box A15-1).
However, in the human immune system, there is no analogous single tissue from which to sample cells for dissecting biology and creating predictors. The immune system spans multiple lineages, is anatomically distributed, and is highly interregulated. Sampling all these cellular components and assaying their gene expression profiles is obviously not feasible. However, two critical features of the
BOX A15-1
Prediction and Classification Based on Gene Expression Signatures
In cancer genomics, gene expression signatures have been used to predict the patient’s clinical outcome and response to therapies. In vaccinology, patterns of gene expression induced after vaccination (i.e., signatures) could be used to predict immunogenicity or efficacy. For example, a particular gene expression signature induced early after vaccination may be able to accurately classify whether the antibody titers are above or below the threshold necessary to confer protective immunity. For many vaccines, such correlates of protection have been established (e.g., Table A15-1). Such a signature can then be used to predict, in an independent trial, whether, for example, a vaccinee would generate an antibody response above the threshold necessary for protection. Toward this end, we have used a machine-learning approach to identify signatures that were capable of predicting the immunogenicity of individuals vaccinated with the yellow fever vaccine 17D (Querec et al., 2009) or the inactivated influenza vaccine.
The identification of signatures that predict the immunogenicity of vaccines could have broad public health utility in several situations: (1) identification of individuals who respond suboptimally to vaccination (e.g., the elderly, infants, the immunocompromised); (2) rapid screening of first responders during emergency outbreaks to identify vaccinees who respond suboptimally; (3) identification of nonresponders in partially effective vaccines (e.g., RTS, S malaria vaccine); (4) accelerated assessment of vaccine immunogenicity and of efficacy (e.g., new meningococcal vaccine); (5) identification of novel correlates of immunity and/or protection.
TABLE A15-1 Methods to Measure Antibody Correlates of Protection.
| Vaccine (Pathogen) | Test | Correlate of Protection |
| Diphtheria (C. diphtheriae) |
Toxin neutralization | 0.01–0.1 IU/ml |
| Hepatitis A | ELISA | 10 mIU/ml |
| Hepatitis B | ELISA | 10 mIU/ml |
| Hib polysaccharide (Hib) | ELISA | 1 µg/ml |
| Hib conjugate (Hib) | ELISA | 0.15 µg/ml |
| Influenza | HAI | 1/40 dilution |
| Lyme disease | ELISA | 1100 EIA U/ml |
| Measles | Microneutralizatoin | 120 mIU/ml |
| Pneumococcus (S. pneumoniae) |
ELISA; opsonophagocytosis | 0.2–0.35 mg/ml (for children); 1/8 dilution |
| Polio | Neutralization | 1/4–1/8 dilution |
| Rabies | Neutralization | 0.5 IU/ml |
| Rubella | Immunoprecipitation | 10–15 mIU/ml |
| Tetanus | Toxin neutralization | 0.1 IU/ml |
| Chickenpox (VZV) | FAMA; gpELISA | ≥1/64 dilution; ≥5 IU/ml |
| Historically, correlates of protection have relied on the measurement of the magnitude of the antigen-specific antibody response stimulated by vaccination. Such measurements typically include the concentration of the binding antibody titers (ELISA) or some measure of the activity of the antibody, such neutralization titers or opsonophagocytic titers. When a given threshold of such a measurement is achieved or exceeded, vaccination is assumed to have reached a signature of protective immunization. These tests have become well standardized and relatively straightforward to perform. The name of the pathogen is included in parenthesis, where its name is different from the commonly used name for the vaccine. The following abbreviations are used: C. diphtheria, Corynebacterium diphtheriae; Hib, Haemophilus influenza type B; S. pneumoniae, Streptococcus pneumonia; HAI, hemagglutination inhibition; EIA, enzyme immunoassay; FAMA, fluorescent antibody to membrane antigens; gpELISA, glycoprotein antibody ELISA; VZV, varicella zoster virus. Adapted from Plotkin (2008). | ||
immune response provide the rationale for applying genomic approaches to study the response to vaccines. First, cells of the immune system are easily accessible in peripheral blood samples. Each blood sample provides a snapshot of many lineages and dozens of differentiation states within the immune system. Moreover, because migration and trafficking is a central and ongoing feature of the immune response, peripheral blood leukocytes represent recent emigrants of peripheral tissues, including vaccine sites. Second, cells of the immune system are uniquely sensitive to perturbation. As discussed below and as we (Querec et al., 2009) and others (Gaucher et al., 2008) have demonstrated, individuals who have been vaccinated manifest marked and characteristic changes in the gene expression profiles of their peripheral blood leukocytes. Thus, the population of immune cells in the peripheral blood provides a sensitive bellwether of localized or systemic immunologic events.
The first examples of studies using systems biological tools to understand vaccine-induced immune responses came from two independent studies that
identified early molecular signatures induced in humans vaccinated with the yellow fever vaccine YF-17D (Gaucher et al., 2008; Querec et al., 2009). YF-17D is a live attenuated vaccine, which was generated after serial passage of a corresponding pathogenic strain (Asibi strain) of the yellow fever virus (Theiler and Smith, 1937) and is one of the most successful vaccines ever developed because it confers protection in nearly 90% of vaccinees. Over 600 million people have received this vaccine and a single immunization results in a broad spectrum of immune responses (neutralizing antibodies, cytototoxic T cells, and T helper 1 (Th1) and Th2 cells) and neutralizing antibody responses that persist for nearly 4 decades. The goal of our study (Querec et al., 2009) was to use YF-17D as a model to determine the feasibility of applying systems biological approaches (1) to identify molecular signatures induced early after vaccination, which could predict the later immunogenicity of the vaccine (i.e., to identify biomarkers of vaccine efficacy) and (2) to obtain biological insights about the mechanism of action of YF-17D. Fifteen individuals who had previously not been vaccinated with YF-17D or infected with yellow fever (and were thus immunologically naive to the vaccine or pathogen) were vaccinated, and blood samples were isolated at baseline and at various time points after vaccination and analyzed with respect to several immunological parameters. There was a striking variation in the magnitude of the antigen-specific CD8+ T cell responses, and the neutralizing antibody titers measured at day 15 or 60, between different individuals (Querec et al., 2009). We then measured cytokine induction in the plasma using a multiplex cytokine assay, and the frequencies and activation status of innate immune cells such as DC and monocyte subsets at days 1, 3, or 7 after vaccination, but these measurements did not correlate with the later T cell or antibody responses. Microarray analyses using the Affymetrix Human Genome U133 Plus 2.0 array of total peripheral blood mononuclear cells (PBMCs) revealed a molecular signature comprised of genes involved in innate sensing of viruses and antiviral immunity in most of the vaccinees. Thus, in addition to enhanced expression of endosomal TLRs, the gene expression of members of the 2′,5′-oligoadenylate synthetase family (e.g., OAS 1,2,3 and L, which are essential proteins involved in the innate immune response to viral infection), DDX58 (RIG-I), and IFIH1 (MDA-5) were all upregulated (Figure A15-1). Two key transcription factors that mediate type I interferon responses, IRF7 and STAT1, were also upregulated. Members of the ISGylation pathway, which preserve essential proteins from being degraded during the IFN-induced cellular antiviral state, were increased, including ISG15, HERC5, and UBE2L6. Another PRR group where both positive and negative regulation is induced by YF-17D is in the complement cascade. The complement signature of YF-17D included the upregulation of genes for C1q and its feedback inhibitor C1IN and the increased expression of the gene-encoding C3a receptor 1 with corresponding increase in the C3a protein in plasma (Figure A15-1). Thus YF-17D activates multiple pathogen surveillance mechanisms in several cellular compartments: extracellular, cell membrane, cytoplasmic, and vesicular
FIGURE A15-1 Using systems biology to predict the immunogenicity of the YF-17D vaccine. Schematic representation of the systems biology approach used to predict the T and B responses of YF-17D vaccinees (Querec et al., 2009). Healthy humans vaccinated with YF-17D are bled at the indicated time points and the innate and adaptive responses studied. Innate signatures obtained with microarrays are found to correlate with the later adaptive immune responses. The predictive power of such signatures is tested in an independent trial (trial 2).
(Figure A15-1). However, these signatures did not correlate with the magnitude of the antigen-specific CD8+ T cell or antibody responses.
We then used additional bioinformatics approaches to identify gene signatures that did correlate with the magnitude of antigen-specific CD8+ T cell responses and antibody titers and that were capable of predicting the magnitude of these responses in an independent trial of YF-17D vaccination in humans. We observed that signatures for CD8+ T cell responses from the first trial were predictive with up to 90% accuracy in the second trial and vice versa. Of the genes present in these predictive signatures, EIF2AK4 is known to be a critical player in the integrated stress response (Wek et al., 2006) and regulates protein synthesis in response to changes in amino acid levels by phosphorylating the elongation initiation factor 2 (eIF2a) (Figure A15-1). This results in a global shutdown of translation of constitutively active proteins by redirection of their mRNAs from polysomes to discrete cytoplasmic foci known as stress granules (SGs), where they are transiently stored (Kedersha and Anderson, 2007). Consistent with this, YF-17D induced the phosphorylation of eIF2a and formation of stress granules (Querec et al., 2009). Moreover, several other genes involved in the stress response pathway, like calreticulin, protein disulfide isomerase, the glucocorticoid receptor, and c-Jun, were observed to correlate with the CD8+ T cell response (Figure A15-1). These observations stimulate the hypothesis that the induction of the integrated stress response in the innate immune system might play a key role in shaping the CD8+ T cell response to YF-17D. Experiments to test the hypothesis are currently underway. In the case of antibody responses, TNFRSF17 (BCMA), a receptor for the B cell growth factor BLyS or BAFF (known to play a key role in B cell differentiation) (Avery et al., 2003), was a key gene in the predictive signatures. Thus, taken together, these studies provide a global description of the innate and adaptive immune responses that are induced after YF-17D vaccination and stimulate the generation of testable hypotheses about the biological mechanisms that regulate the magnitude and nature of the immune response to YF-17D (Figure A15-1).
The utility of such an approach in predicting the immunogenicity and protective efficacy of other vaccines needs to be determined. The question of whether the signatures that predict the T and B cell responses to YF-17D can also predict such responses to other vaccines remains to be determined. In one scenario, it could be envisioned that all vaccines that stimulate antibody responses would induce a common archetypal signature, capable of predicting the magnitude of the antibody response to any vaccine. Similarly, there could be an archetypal signature that predicts the antigen-specific CD8+ T cell responses to any vaccine. However, B and T cell responses come in different flavors, and different vaccines induce different types of B and T cell responses. It seems unlikely, therefore, that a common archetypal signature would be capable of predicting all the different types of B or T cell responses induced by different vaccines. A second scenario is that each vaccine could have a very unique signature, which was capable of predicting the particular type of T or B cell responses only to that vaccine. How-
ever, many vaccines induce similar types of immune responses (e.g., neutralizing antibodies or polyfunctional CD8+ T cells), so it is reasonable to suggest that vaccines that stimulate a similar mechanism of protective immunity will induce similar molecular signatures. For example, vaccine Y that stimulates long-lived plasma cells that produce high-affinity antibody may stimulate a particular signature, whereas vaccine Z that induces polyfunctional CD8+ T cells would stimulate a different signature (Figure A15-2). Vaccine X that induced both types of responses would stimulate a combined signature (Figure A15-2). Other vaccines that relied on opsonophagocytic antibodies for protection may have a different innate signature. Thus, one would have a cluster of signatures that predict various aspects of B cell immunogenicity or T cell immunogenicity. Similarly, there could be a different cluster of signatures that predict protective immunity that is not mediated by T or B cell-dependent mechanisms, but by other mechanisms mediated perhaps by NK cells, DCs, or stress response pathways (Figure A15-2). In this context, our preliminary data with the influenza vaccines suggest that TNFRSF17, which was a key predictor of the neutralizing antibody responses to YF-17D (Querec et al., 2009), is also a predictor of the hemagglutinin antibody titers to vaccination with the inactivated influenza vaccine, suggesting that there are likely to be common predictors of antibody responses to many vaccines (unpublished data). This probably underlies common biological mechanisms by which different vaccines could stimulate antibody responses. The identification of such predictive signatures will facilitate not only the rapid screening of vaccines and the development of a vaccine chip, comprising clusters of a few hundred or fewer genes, each cluster being capable of predicting a facet of immunogenicity (Figure A15-2B). Such a chip would therefore be used to predict the immunogenicity of virtually any vaccine. Indeed, in the field of cancer genomics, after several years of false starts, MammaPrint (http://www.agendia.com), a prognostic chip for breast cancer, was developed by Agendia and approved by the Food and Drug Administration in the United States. Like the story behind this breast cancer prognostic chip, the development of the vaccine chip will probably require the analysis of hundreds of vaccinees over several clinical trials. However, we have already seen how host gene expression profiles induced after vaccination correlate with, and predict, vaccine immunogenicity and also offer mechanistic insights into immune regulation (Querec et al., 2009). This additional layer of knowledge, translated into an array of functional modules on the vaccine chip, gives us extra power that was not utilized in the earlier, brute-force biomarker hunting.
This is likely to have an impact on several public health-related issues in vaccinology. One major issue is that many common vaccines such as the influenza vaccine (Gardner et al., 2006), pneumococcal vaccine (Jackson and Janoff, 2008), and zoster vaccines induce suboptimal immune responses in a substantial proportion of the elderly, in infants, or in immunocompromised populations such as HIV-infected or transplant patients. Therefore, delineation of signatures of immunogenicity would permit such individuals to be identified prospectively.
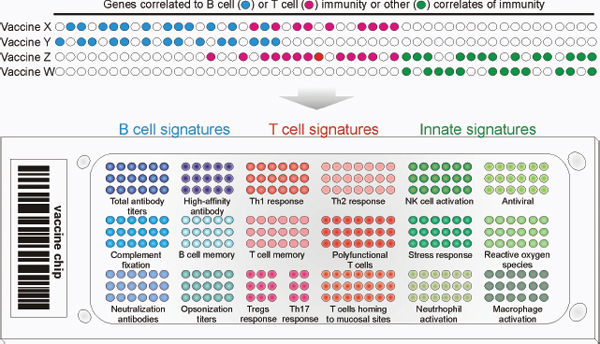
FIGURE A15-2 (Top) Systems biology approaches allow the identification of predictive gene signatures of immunogenicity for many vaccines. Vaccines with similar correlates of protection may or may not share the same gene markers. The identification of predictive signatures of many vaccines would enable the development of a vaccine chip. (Bottom) This chip would consist of perhaps a few hundred genes, subsets of which would predict a particular type of innate or adaptive immune response (e.g., magnitude of effector CD8+ T cell response, frequency of polyfunctional T cells, balance of T helper 1 (Th1), Th2, and Th17 cells, high-affinity antibody titers, and so on). This would allow the rapid evaluation of vaccinees for the strength, type, duration, and quality of protective immune responses stimulated by the vaccine. Thus, the vaccine chip is a device that could be used to predict immunogenicity and protective capacity of virtually any vaccine in the future.
In addition, this strategy will help identify nonresponders when vaccinating first responders during an emerging outbreak or when evaluating the efficacy or immunogenicity of untested vaccines (Table A15-1). Furthermore, the predictive signatures could highlight novel correlates or protective immunity and enable the formulation of new hypotheses about the mechanisms underlying vaccine-induced protective immunity.
Systems biology may also be useful in addressing a major challenge in vaccine development: to determine the correlates of protection against a pathogen. The magnitude of the antigen-specific antibody titers is considered to be the primary correlate of protection against most pathogens (Plotkin, 2008) (Table A15-1). For example, antibodies mediate protection against blood-borne viruses such as hepatitis (Jack et al., 1999; Van Damme and Van Herck, 2007) and yellow fever (Lang et al., 1999; Reinhardt et al., 1998; Wheelock and Sibley, 1965); bacteria that secrete toxins that cause diphtheria (Ipsen, 1946) and tetanus (Looney et al., 1956); viruses that infect via mucosal surfaces such as influenza (Dowdle et al., 1973; Mostow et al., 1973) and rotaviruses (Jiang et al., 2008); rabies virus (Nagarajan et al., 2008), which infect neuronal axons; and pneumococcal and meningococcal bacteria, which are leading causes of pneumonia and meningitis (Andreoni et al., 1993; Romero-Steiner et al., 2006). The antigen-specific antibody responses to such vaccines are measured through standardized assays such as ELISAs (which measure binding antibody titers), hemagglutination inhibition, and functional measures of antibody activity such as neutralization and opsonophagocytosis (Table A15-1). Typically, such assays yield a single value, a threshold, above which antibody responses are considered to be protective.
However, despite the widespread use of such antibody assays to measure the efficacy of current vaccines, in the case of many vaccines humoral immunity may not be the only, or even the best, correlate of protection. Furthermore, protective immunity may not even correlate with the humoral immune response. Varicella virus vaccination efficacy is usually determined by measuring antibody titers with serum neutralization or ELISA. However, persistent varicella-specific T cells have been shown to be indicators of protection from varicella virus infection and have been suggested as possible additional or alternative correlates of protection in children and the elderly (Arvin, 2008; Levin et al., 2008). Furthermore, antibody titers to influenza vaccination may be unreliable for predicting risk of influenza illness in the elderly population (McElhaney et al., 2006). On the contrary, elderly individuals that have strong influenza-specific T cell responses are less likely to develop flu regardless of postvaccination antibody titers (McElhaney et al., 2006). Although antibody titers could not distinguish between elderly subjects that did or did not develop flu, those subjects with high IFN-γ:IL-10 ratios following ex vivo stimulation of PBMCs with live influenza preparations were more likely to be protected from influenza illness (McElhaney et al., 2006). In addition, patients with high frequencies of CMV-specific T cells are less likely to have reactivation of CMV when they are placed on immunosuppressive drugs
to prevent transplant rejection (Bunde et al., 2005; Sester et al., 2001). In fact, many diseases that are a top priority for vaccine development, such as HIV, TB, and malaria, are believed to require strong T cell responses for protection (Hoft, 2008; Pantaleo and Koup, 2004; Reyes-Sandoval et al., 2009). These realizations have led to interest in measuring T cells as correlates of protection.
However, measuring the functional signature of the T cell response as a correlate of protection is more challenging than assessing antibody titers. First, T cell populations are phenotypically and functionally diverse (e.g., CD8+ T cell, CD4+, effector memory, central memory, Th1, Th2, and Th17 cells, etc.). Vaccination can induce the proliferation and differentiation of antigen-specific T cells into effector cells that secrete cytokines such as IFN-γ, IL-4, IL-17, IL-10, IL-9, or effector memory cells and central memory cells, all of which play key roles in mediating short- and/ or long-term protective immunity to the pathogen (Harari et al., 2004; Sallusto et al., 1999, 2010) (this issue of Immunity). Recent studies have monitored activated T cells in humans phenotypically by measuring upregulation of CD38 and HLA-DR or peptide-MHC tetramer-staining cells (Akondy et al., 2009; Appay et al., 2002; Callan et al., 1998; Morgan et al., 2008). Differentiation into effector and memory phenotypes can be assessed by the expression of markers such as CD45RA, CD62L, CD127, and CCR7 (Akondy et al., 2009; Appay et al., 2002; Callan et al., 1998; Morgan et al., 2008). However, the frequencies of differentiated T cell phenotypes may not be adequate correlates of protection, because these may not necessarily correlate with their functional activity. The functions of T cells can be dependent on the cytokines they secrete (e.g., IFN-γ, IL-2, and TNF-α) or production of perforin, as well as other measures of cell proliferation and cell-mediated cytotoxicity. Thus, there are a variety of T cell functional signatures that can be measured as potential correlates of protection in lieu of the traditional antibody response. Importantly, the assessment of a single parameter of T cell function (e.g., IFN-γ secretion) may not be sufficient as a correlate of protection; however, using a functional signature comprised of two or more types of measurements may provide more specific and reliable correlates of protection (Harari et al., 2004). Finally, it may be necessary to abandon the simple linear functional signature model developed for antibody titers where a predetermined threshold is used as a correlate. Instead of using a set threshold of a single variable to determine vaccine efficacy, so called cocorrelates of protection may be more appropriate where it is the balance among multiple variables that indicates efficacy (Qin et al., 2007). For instance, protection against a pathogen may be achieved when two conditions are satisfied: (1) the frequency of Th1 CD4+ effector memory cells meets a given threshold and (2) the magnitude of the neutralizing antibody titers reaches a certain threshold. In individuals in whom the thresholds for each of these conditions are not met, it may be the interaction between various cocorrelates, and not independent levels of each, that provides a functional signature of vaccine efficacy. For instance, in
the control of viruses or intracellular pathogens, the lower the neutralizing antibody titer induced by a vaccine, the higher the cytotoxic T cell response needs to be to enhance the likelihood of protection.
The notion that the innate immune response to vaccination might represent a viable correlate or protection has only recently been considered. Given the pivotal role of the early innate response in regulating the magnitude, quality, and duration of the later adaptive immune responses (Iwasaki and Medzhitov, 2010), specific signatures of innate activation may indicate that the vaccine induced the appropriate quality and sufficient strength of activation to induce protective acquired immunity. As discussed above, it has been shown with yellow fever vaccine 17D that molecular signatures in the blood 3 to 7 days after vaccination, corresponding with vaccine viremia and activation of the innate immune pathways, may be used to predict the peak frequency of activated virus-specific T cells and long-term neutralizing antibody titers (Querec et al., 2009).
How can systems approaches be integrated into the clinical trial framework to identify correlates of protective immunity? At the outset, it is important to clarify a frequent source of confusion that arises regarding correlates of immunogenicity versus correlates of protection. The ultimate goal is of course to determine vaccine-induced signatures a few hours or days after vaccination that can predict whether a given individual will develop long-term protective immunity against the pathogen. The most logical way of addressing this goal is to perform a clinical trial in which vaccinated humans can be challenged with the pathogen and then to identify signatures that would discriminate between those vaccinees who succumbed to the infection versus those who were protected. With very rare exceptions, as in the case of malaria vaccine trials (Vahey et al., 2010), such an approach is clearly untenable ethically and thus alternative approaches must be considered. One alternative approach is to use animal challenge models in which vaccines can be evaluated. Such models, such as the nonhuman primate model for HIV or the ferret model for influenza have greatly accelerated vaccine discovery and offered much insight into the mechanisms of protection (Sui et al., 2010). However, in some cases, opinions vary regarding the relative merits of a given model and how to translate results obtained from such a model into the clinic (Morgan et al., 2008). Therefore, an alternative or even complementary approach is to identify signatures of immunogenicity to the vaccine in humans. This approach relies on the axiom that immune protection against a pathogen is mediated by one or more components of the immune response, which can broadly be divided into the adaptive (antigen-specific B and T cells) and the innate responses. Therefore, if there was a priori knowledge of precisely which component(s) of the immune response (e.g., a combination of persistent neutralizing antibody responses and memory CD8+ T cells that migrate to mucosal tissues), then it becomes relatively straightforward to conduct a phase 0 or 1 clinical trial (similar to the yellow fever vaccine trials) in which early predictive
signatures of such responses can be identified (Figure A15-3). Such signatures can then be applied in the clinic to identify vaccinees who will respond suboptimally to the vaccine. But how do we know what types of immune responses are necessary for protection? In many cases, we can be guided by more than a century of immunological wisdom. For example, few immunologists would deny that the induction of persistent neutralizing antibody responses (Table A15-1) and cytotoxic T cells are beneficial to fight most viral infections. In such cases, early signatures that various aspects of T or B cell immunogenicity can be assessed in a high-throughput manner, using a small number of genes (Vaccine Chip) or an ELISA kit that measured protein expression (Figure A15-3).
But what happens in situations in which the types of immune responses required for protection are not readily apparent or where the full range of responses required for optimally effective protection may be unknown? For example, in HIV infections, although neutralizing antibodies and cytotoxic T cells are thought to be important (Letvin, 2007), there is much interest in ascertaining whether there are additional mechanisms that might confer protection. Here, it is interesting to consider how systems approaches may be integrated into phase II and III clinical trials, with a view to identifying new correlates of protection. Two approaches to integrating systems approaches into phase II and III trials are shown in Figure A15-3. Such trials typically involve thousands of participants, and performing high-throughput analyses on all would be prohibitively expensive. In one approach, signatures of various aspects of T and B cell immunogenicity can be established in a smaller phase I trial, and these signatures can be incorporated into a relatively cheap and high-throughput assay that can be used to predict immunogenicity in phase II and III trials (Figure A15-3). The assumption here is that some aspect of the T or B cell response will be protective. In a different approach, blood samples could be collected at a few strategic time points (e.g., days 0 and 7 after vaccination), put straight into RNA lysis buffer, and stored for future use. Once the trial was completed, a retrospective nested case-control study could be performed using the stored samples in which a detailed analysis of innate and adaptive responses could be performed in, say, 50 vaccinees who acquired the disease and 50 vaccinees who did not. The goal would be to identify signatures induced early on that would discriminate between those who were protected by the vaccine versus those who were not. A caveat with this approach is that one would not know whether those vaccinees who didn’t acquire the disease were actually protected by the vaccine or simply never encountered the pathogen. However, in many endemic areas of infection (e.g., in a rural area where cholera is endemic and access to clean drinking water is absent), it may be assumed that exposure to infection is high.
A potential benefit of using functional signatures of innate immunity as correlates of protection is that they occur quite early after vaccination compared to the development of memory T cells and antibody responses, which can take weeks, months, or years. Being able to determine vaccine efficacy in a short time is useful for many reasons. The current clinical trial format is very lengthy and
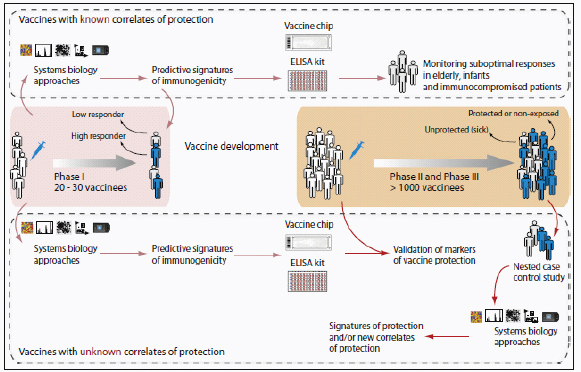
FIGURE A15-3 Integrating systems biology approaches into clinical trials. (Top) For vaccines for which correlates or protection are known (Table A15-1), systems approaches can be used to identify early signatures of protection in a phase 1 trial. The key genes from these signatures can be incorporated into a vaccine chip or ELISA kit, which can then be used to identify nonresponders or suboptimal responders, particularly in special populations such as immunocompromised patients, the elderly, and infants. (Bottom) For new and emerging vaccines, for which correlates of protection are unknown, signatures that predict various aspects of immunogenicity (e.g., CD8+ T cell responses or neutralizing antibody responses) can be assessed in phase I trials. Such signatures can then be incorporated into a vaccine chip or ELISA kit that can then be used in phase II and III trials to determine their capacity to predict protection. Alternatively, a retrospective nested case-control study could be done in a phase II and III trial to identify signatures of protection.
costly and usually offers no insights into why a particular vaccine failed. As such, clinical trials represent a major rate-limiting step in vaccine development. Having a shorter study period increases the probability of retaining all the subjects for the duration of the study, increasing the proportion of subjects that are tracked from vaccination through the final time point. In addition, measuring functional signature of vaccine-induced innate immunity makes high-throughput screening of vaccine candidates more feasible. The short duration of time required to measure innate immune activation relative to the endpoints of acquired immunity means: (1) shorter duration to analyze each batch of vaccine candidates, (2) potentially fewer resources and costs devoted to the early stage analysis of each vaccine candidate, (3) quicker refinement of vaccine formulations and delivery methods, and (4) identification of why a particular vaccine failed (Figure A15-3).
Apart from lack of inducing sufficient protection, another common reason for vaccines to fail is severe side effects. These side effects are often associated with overactivation of certain components of the innate immune system (Gupta et al., 1993; Pulendran et al., 2008). Thus functional signatures of innate immunity may be used to screen adjuvants or as cocorrelates of protection along with parameters of acquired immunity for complete vaccines (antigen + adjuvant). Functional signatures may not only help in the design of protective vaccines but may also help to limit the deleterious side effects.
Finally, systems approaches could also yield biological insights about how vaccines work.
One area that could benefit from systems approaches is delineation of the mechanisms by which adjuvants work. Although the empiric, live attenuated vaccines contain stimuli that activate the innate immune system and, in effect, act as their own adjuvants, recombinant vaccines such as the Hepatitis B vaccine need to be administered with exogenous adjuvants. In the nearly 250 years since the introduction of vaccination, although a great variety of adjuvants have been proposed, until very recently only alum, described by Glenny in 1926, was globally licensed for human use (De Gregorio et al., 2008; Lindblad, 2004). However, alum is a Th2 cell-inducing adjuvant, and does not induce strong Th1 and CTL responses. Thus, there is an urgent need to develop alternative and safe adjuvants that induce different types of immune response that might be optimally effective against different pathogens. Despite its widespread utility, until very recently its mechanism of action has been shrouded in mystery. It has been suggested that alum works by serving as a depot of antigen in the body. It has also been suggested that alum could cause necrosis in the inoculated tissue, which indirectly activates DCs through danger signals in the form of host inflammatory mediators (De Gregorio et al., 2008; Mbow et al., 2010). The details of this mechanism are only now being revealed. Recently it was demonstrated that alum signals via the Nalp3 inflammasome (Eisenbarth et al., 2008; Kool et al., 2008; Li et al., 2008). Thus, DCs or macrophages stimulated in vitro with alum
plus LPS induced IL-1β and IL-18 in a caspase-1- and Nalp3-dependent manner (Eisenbarth et al., 2008; Kool et al., 2008; Li et al., 2008; McKee et al., 2009). Despite the convincing in vitro studies, the question of whether Nalp3 is required for the adjuvanticity of alum remains controversial, with some studies demonstrating abrogation of antibody responses in Nalp3-deficient (Nlrp3–/–) mice (Eisenbarth et al., 2008; Li et al., 2008) and other studies showing partial or no effect (Kool et al., 2008; McKee et al., 2009). Thus, the mechanisms by which alum induces Th2 responses are poorly understood, and a systems biological approach (e.g., microarray analyses of signatures in response to an alum-adjuvanted versus unadjuvanted vaccine) is likely to be useful in providing new insights into the mechanism of action of alum. In this context, Mosca et al. performed an elegant study in mice to assess the molecular and cellular signatures of vaccine adjuvants, including the squalene-based oil-in-water emulsion MF59 (Mosca et al., 2008), which was licensed for human use a decade ago. The molecular mechanism of action and the target cells of alum and MF59 are still unknown. By combining microarray and immunofluorescence analysis, Mosca et al. monitored the effects of the adjuvants MF59, CpG, and alum in the mouse muscle. MF59 induced the expression of 891 genes; in contrast, CpG and alum regulated 387 and 312 genes, respectively. Interestingly, there was a core set of 168 genes that were modulated by all adjuvants. Although all adjuvants promoted the recruitment of antigen-presenting cells, MF59 triggered a more rapid influx of CD11b+ blood cells compared with other adjuvants. Furthermore, MF59 was the most potent inducer of genes encoding cytokines, cytokine receptors, and adhesion molecules involved in leukocyte migration. Intriguingly, two genes identified by microarrays, JunB and Ptx3, suggested skeletal muscle as a direct target of MF59. Taken together, the authors’ interpretation of the data suggests that oil-in-water emulsions are efficient human vaccine adjuvants because they induce an early and strong immunocompetent environment at the injection site by targeting muscle cells. In addition, we have recently applied this approach to identifying a novel mechanism by which adjuvants that induce Th2 responses (e.g., cysteine proteases) program DCs to stimulate Th2 responses (Tang et al., 2010). This involves the induction of reactive oxygen species (ROS) in DCs, which is critical for the induction of Th2 responses (Tang et al., 2010).
These studies demonstrate the utility of systems approaches in understanding the mechanism of action of adjuvants, and in identifying mechanisms that contribute to their toxicity. In addition, emerging work in innate immunity is revealing the mode of action of many adjuvants. Under the brand name AS04, monophosphoryl lipid A (MPL), an LPS derivative and a TLR4 ligand, is used in combination with alum in Cervarix, GlaxoSmithKline’s recently approved human papillomavirus vaccine (Hennessy et al., 2010). With the growing number of adjuvants at our disposal to mimic natural infections, we need a frame of reference as to how to use them for maximum efficacy. Turning to the functional
signatures of innate immunity induced by some of our most successful vaccines is beginning to shed light on this area. For example, YF-17D activates multiple TLRs, including TLR 2, 7, 8, and 9, as well as non-TLR PRRs such as RIG-I and MDA-5 (Querec et al., 2006, 2009), which results in the activation of plasmacytoid DCs and myeloid DCs. Similar approaches are being applied to understand innate responses to other vectors such as the attenuated pox vectors modified vaccinia virus Ankara and New York vaccinia (Guerra et al., 2007) and baculovirusexpressed HIV virus-like particles (Buonaguro et al., 2008).
Systems approaches can also shed light on the mechanisms by which vaccines induce a given type of response. As discussed above, one of the key genes in the predictive signature of YF-17D, EIF2AK4, is known to be a critical player in the integrated stress response (Kedersha and Anderson, 2007) and regulates protein synthesis in response to changes in amino acid amounts by phosphorylating the elongation initiation factor 2 (eIF2a) (Figure A15-1). Our recent data demonstrate that immunization of mice deficient in EIF2AK4 with YF-17D results in substantially diminished CD8+T cell responses (unpublished data). The precise mechanism of this is under investigation, but this result demonstrates that the integrated stress response plays a key role in regulating adaptive immunity to a viral vaccine.
Finally, it is important to remember that the complex behavior of biological systems cannot be understood by studying parts in isolation (Germain, 2001; Ideker et al., 2001; Kitano, 2002; Weng et al., 1999). Therefore, vaccinologists need to move beyond merely understanding each of the parts of the immune system in isolation to instead understand how the different parts of the immune system interact among themselves. Indeed, a unified model of the cellular and molecular mechanisms of vaccine-induced protective immunity is likely to result from studying different hierarchies of organization with the immune system. In such a hierarchy, the cell can be considered to be the ground level, and zooming into the cell to examine innate receptors and signaling networks offers greater conceptual resolution. In contrast, zooming out from the cell, allows more global views of multicellular cooperation (e.g., between DC subsets) and the influence of tissue microenvironments (e.g., intestine versus lung) (Pulendran et al., 2010). In addition, the immune system, as with all biological systems, has redundancies, feedback and feed-forward regulation, and synergism, which all impact how the instruction of the vaccine is processed (Kitano, 2002). For example, combinatorial triggering of specific combinations of TLRs results in a synergistic production of proinflammatory cytokines via a mechanism dependent on TRIF and MyD88 signaling (Napolitani et al., 2005). Consistent with this, vaccination with nanoparticles containing particular combinations of TLR ligands plus antigens induced a synergistic enhancement in the magnitude and persistence of antigen-specific memory B cells and long-lived plasma cells (unpublished data).
Low-Input, High-Throughput, No Output Biology?
Despite the promise of systems approaches in vaccinology, we may do well to heed the advice of Dr. Sydney Brenner: “The idea that we’ll dissect [cellular] complexity by making lots of measurements is bound to fail.…Everyone’s hoping for a magic computer program—experimental data, pharmacogenomics data, the whole lot—and it will come out with the answer. That’s a vague hope. Because I have to tell you, computers are incredibly stupid! It’s better to combine human intelligence with artificial stupidity than the other way around” (Davies, 2008), and “Actually, the orgy of fact extraction in which everybody is currently engaged has, like most consumer economies, accumulated a vast debt. This is a debt of theory, and some of us are soon going to have an exciting time paying it back—with interest, I hope” (Brenner, 1997). The accumulation of a sea of data is but a small stepping stone toward real understanding of biological systems. It is imperative to get beyond colorful heat maps and network maps to an understanding of the functional significance of the molecular signatures of vaccination. This is a daunting challenge because of several intrinsic problems in this approach. These are discussed below.
Conceptual Problems
A major conceptual pitfall lies with the premise that changes in the expression of genes in response to vaccination may necessarily be functionally relevant for generating the immune response to that vaccine. There are many examples where genes that are modulated in response are of no consequence to the biological response to that stimulus because evolution has not had a reason for silencing those genes. Indeed, it is well recognized that gene coexpression only corresponds to causality in very limited cases (Bansal et al., 2007; Schadt et al., 2005). The challenge is to identify true causal relationships among the cooccurring events. One solution is to borrow knowledge from predefined gene modules or pathways. If multiple genes within a module are coordinately regulated by the vaccine, then the likelihood that this module is functionally relevant becomes much higher. Another approach is to combine multiple data types. As Chen et al. (2008) demonstrated, a macrophage-enriched metabolic network, derived by integrating genotyping data and expression data, was causal of obesity traits, whereas each data type alone could not deliver the predictive power. We should be reminded that the current measurements are still a thin slice of immense biological complexity; microarray data, even with a large sample size, may fail to reach any statistical significance (Dixon et al., 2007). The general question is: how much data, what data, at what resolution, at what scale, are needed to explain the immunological phenotypes? This may only be addressed in each individual case through trials and errors. Finally, the results of the analysis have to be validated by functional data via proven techniques, say, gene perturbation or deficient mice. As the study design is closely coupled with computational analysis and
modeling, systems biology is best done in an environment where biologists and computational scientists interact closely.
A second conceptual problem is the premise that we can deduce mechanistic insights about how the vaccine induced immune responses by looking at changes in the expression of genes only in cells isolated from the blood. This is a significant problem because immune response to local vaccinations will be initiated in the draining lymph nodes. However, with many vaccines, such as live viral or bacterial vectors, there is a transient, systemic replication of the vector and, subsequently, a direct activation of blood leukcocytes by it. This is likely to produce the profile of gene expression changes observed in the draining lymph nodes, which serves as a surrogate for immunogenicity. Even in the case of nonreplicating vaccines such as the inactivated influenza vaccine, our results demonstrate that signatures of immunogenicity can be ascertained in the blood (unpublished data). An additional problem is that, for many vaccines that induce mucosal immunity, gene expression signatures in the blood may not predict the strength, quality, and duration of mucosal immunity. Sampling mucosal tissues in human vaccinees is wrought with challenges. Clearly further studies are necessary to ascertain the extent to which immunogenicity of mucosal vaccines can be ascertained from the blood.
Technical Problems
One of the key technical issues is that gene expression signatures are prone to artifacts. Since the early studies of cancer expression microarrays, questions have been raised about how robust the gene signatures are (Ein-Dor et al., 2006). Recently, emphasis has been placed on pathway and network analyses because they incorporate prior knowledge into data analysis and are less prone to spurious errors than analyses of individual genes (Chuang et al., 2007; Dinu et al., 2009). This is particularly relevant to immunological studies where signals are often diluted by cell heterogeneity (Haining and Wherry, 2010). In addition, signatures must be validated with additional techniques and independent samples.
Second, when profiling PBMCs, one is looking at signatures from a mixed bag of cells. Therefore, the extent to which the changes in gene expression reflect alterations in the cellular composition of the blood versus de novo induction of gene expression remains uncertain. One solution to this problem is to FACS sort subpopulations of cells and then to evaluate expression profiles in individual cell types. However, this approach is rather laborious and expensive. An alternative approach is to devise computational strategies for assessing cell type-specific gene expression profiles. Recently, Shen-Orr et al. (2010) have devised such an approach using microarray data and relative cell type frequencies. First they validated their approach using predesigned mixtures of cells, and then they applied it to whole-blood gene expression datasets from stable posttransplant kidney transplant recipients and those experiencing acute rejection.
A third challenge lies in the enormous genetic and environmental heterogeneity in human populations and the impact that such heterogeneity may have on vaccine-induced immunity. Therefore, future studies should strive to conduct such research in populations that are uniform with respect to age, gender, ethnicity, and immune status. Furthermore, studies that aim to compare vaccine-induced immunity between different populations (e.g., frail elderly versus healthy adults) are likely to yield many insights into mechanisms that contribute to impaired immunity in given populations.
Fourth, a major challenge concerns data management and integration of the enormous volume of data generated. The timely sharing of these data is important to the research community. A dedicated database service for vaccine-related data, akin to WormBase (Schwarz et al., 2006) and TB database (Reddy et al., 2009), should be created as soon as possible. Public databases for immunology, including InnateDB (Lynn et al., 2008) and Immgen.org (Heng and Painter, 2008), have been well covered by recent reviews (Gardy et al., 2009; Tong and Ren, 2009). In-house databases often become a necessity for high-throughput projects. Integration of multiple data types is usually driven by the specific modeling approach, for instance by naive Bayesian methods (Huttenhower et al., 2009) or by custom algorithms, or combined by biomolecular concepts (Joyce and Palsson, 2006). For example, transcription factor binding data and gene expression are combined under frameworks of transcriptional regulation; metabolites and enzyme expression are combined in metabolic networks. Broader and more definitive immune parameters are desired (Fauci et al., 2008).
Cultural Problems
Finally, the successful application of systems approaches to vaccinology requires a close transdisciplinary collaboration between biologists and computational scientists. It is critical that such individuals engage in active dialog on a daily basis to combine rigorous bioinformatics analyses of the data with biological insights and intuition. Such intimate collaborations could even take place within a single laboratory where, for example, post-docs trained in bioinformatics and biology interact closely.
A Framework for Systems Vaccinology
At the World Economic Forum’s annual meeting in Davos this year, Bill Gates pledged $10 billion for vaccines over the next decade and said that he hoped that the coming 10 years would be the decade of the vaccine. His words symbolize the unique moment we face today in our millennial war with pathogens. For the first time, we have begun to understand the mechanisms by which highly successful vaccines mediate protective immunity and to begin to harness such insights in designing new vaccines against global pandemics. Systems
biology promises to offer a new paradigm in vaccinology. Recently, the National Institute of Allergy and Infectious Diseases (NIAID) initiated a new nationwide initiative to establish a consortium of human immune profiling research centers (National Institute of Allergy and Infectious Diseases, 2010). The purpose of these centers—which together will receive funding up to $100 million over 5 years—is to characterize the human immune system under normal conditions and to understand how it changes following infection or vaccination to specific viruses and bacteria. Researchers will use the tools of systems biology to follow the global architecture of the immune response to vaccination or infections in humans and integrate information about an individual’s genes, proteins, and metabolic components that are perturbed by vaccination or infection. Such studies will be performed in diverse populations with respect to age (including the elderly and children), immune status (including people with autoimmune diseases such as lupus and transplant patients), gender, and ethnicity. In addition, the initiative will provide support for centralized infrastructure to collect, characterize, and store the human samples; for bioinformatic capacity to analyze the large and complex data sets that will be generated; and for the discovery and development of new immune response-monitoring tools and sample-sparing assays. The results of this initiative are likely to have a major impact on vaccinology and generate an unprecedented volume of data on immune responses in humans. However, we must remember Dr. Brenner’s admonishment and strive to transcend data and discover knowledge and ultimately understanding. The generation of high-throughput data represents but a stepping stone toward understanding. An essential aspect of this is to integrate mechanistic studies involving models, both animal and human, (e.g., knockout mice, transgenic mice, siRNA knock down of genes in humans cells in vitro) that can elegantly validate the functions of genes and proteins picked up in the human immune-profiling studies (Figure A15-4). Therefore, data generated in clinical trials can be mined using bioinformatics tools and used to generate biological hypotheses, which can then be tested with animal models or in vitro systems. The insights gained from experimentation will then guide the design and development of new vaccines (Figure A15-4). Such a framework seeks to bridge the so-called gaps between clinical trials and discovery-based science, between human immunology and mouse immunology, and between translational and basic science and offers a seamless continuum of scientific discovery and vaccine invention. That would be emblematic of 21st century vaccinology!
Acknowledgements
B.P. thanks the National Institutes of Health and the Bill & Melinda Gates Foundation for their generous support, and Rafi Ahmed for review of this manuscript.
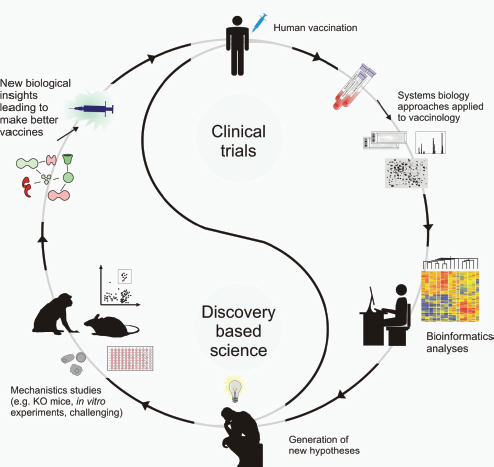
FIGURE A15-4 A framework for systems vaccinology. Systems biology approaches applied to clinical trials can lead to the generation of new hypotheses that can be tested and ultimately lead to developing better vaccines. For example, immune responses to vaccination in clinical trials can be profiled in exquisite depth with technologies such as microarrays, deep sequencing, and proteomics. The high-throughput data generated can be mined using bioinformatics tools and used to create hypotheses about the biological mechanisms underlying vaccine-induced immunity. Such hypotheses can then be tested with animal models or in vitro human systems. The insights gained from experimentation can then guide the design and development of new vaccines. Such a framework seeks to bridge the so-called gaps between clinical trials and discovery-based science, between human immunology and mouse immunology, and between translational and basic science and offers a seamless continuum of scientific discovery and vaccine invention.
References
Aderem, A., and Hood, L. (2001). Immunology in the post-genomic era. Nat. Immunol. 2, 373–375.
Akondy, R.S., Monson, N.D., Miller, J.D., Edupuganti, S., Teuwen, D., Wu, H., Quyyumi, F., Garg, S., Altman, J.D., Del Rio, C., et al. (2009). The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J. Immunol. 183, 7919–7930.
Alizadeh, A.A., Eisen, M.B., Davis, R.E., Ma, C., Lossos, I.S., Rosenwald, A., Boldrick, J.C., Sabet, H., Tran, T., Yu, X., et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511.
Andreoni, J., Käyhty, H., and Densen, P. (1993). Vaccination and the role of capsular polysaccharide antibody in prevention of recurrent meningococcal disease in late complement component-deficient individuals. J. Infect. Dis. 168, 227–231.
Appay, V., Dunbar, P.R., Callan, M., Klenerman, P., Gillespie, G.M., Papagno, L., Ogg, G.S., King, A., Lechner, F., Spina, C.A., et al. (2002). Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8, 379–385.
Arvin, A.M. (2008). Humoral and cellular immunity to varicella-zoster virus: An overview. J. Infect. Dis. 197 (Suppl 2), S58–S60.
Avery, D.T., Kalled, S.L., Ellyard, J.I., Ambrose, C., Bixler, S.A., Thien, M., Brink, R., Mackay, F., Hodgkin, P.D., and Tangye, S.G. (2003). BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 112, 286–297.
Bansal, M., Belcastro, V., Ambesi-Impiombato, A., and di Bernardo, D. (2007). How to infer gene networks from expression profiles. Mol. Syst. Biol. 3, 78.
Brenner, S. (1997). In theory. Curr. Biol. 7, R202.
Bunde, T., Kirchner, A., Hoffmeister, B., Habedank, D., Hetzer, R., Cherepnev, G., Proesch, S., Reinke, P., Volk, H.D., Lehmkuhl, H., and Kern, F. (2005). Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J. Exp. Med. 201, 1031–1036.
Buonaguro, L., Monaco, A., Aricò, E., Wang, E., Tornesello, M.L., Lewis, G.K., Marincola, F.M., and Buonaguro, F.M. (2008). Gene expression profile of peripheral blood mononuclear cells in response to HIV-VLPs stimulation. BMC Bioinformatics 9 (Suppl 2), S5.
Callan, M.F., Tan, L., Annels, N., Ogg, G.S., Wilson, J.D., O’Callaghan, C.A., Steven, N., McMichael, A.J., and Rickinson, A.B. (1998). Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J. Exp. Med. 187, 1395–1402.
Chaussabel, D., Quinn, C., Shen, J., Patel, P., Glaser, C., Baldwin, N., Stichweh, D., Blankenship, D., Li, L., Munagala, I., et al. (2008). A modular analysis framework for blood genomics studies: Application to systemic lupus erythematosus. Immunity 29, 150–164.
Chen, Y., Zhu, J., Lum, P.Y., Yang, X., Pinto, S., MacNeil, D.J., Zhang, C., Lamb, J., Edwards, S., Sieberts, S.K., et al. (2008). Variations in DNA elucidate molecular networks that cause disease. Nature 452, 429–435.
Chuang, H.Y., Lee, E., Liu, Y.T., Lee, D., and Ideker, T. (2007). Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3, 140.
Coffman, R.L., Sher, A., and Seder, R. (2010). Vaccine adjuvants: Putting innate immunity to work. Immunity 33, this issue, 492–503.
Davies, K. (2008) GenBank Celebrates 25th Anniversary. Bio-IT World Magazine, May 2008. http://www.bio-itworld.com/BioIT_Article.aspx?id=75470&LangType=1033.
De Gregorio, E., Tritto, E., and Rappuoli, R. (2008). Alum adjuvanticity: Unraveling a century old mystery. Eur. J. Immunol. 38, 2068–2071.
Dinu, I., Potter, J.D., Mueller, T., Liu, Q., Adewale, A.J., Jhangri, G.S., Einecke, G., Famulski, K.S., Halloran, P., and Yasui, Y. (2009). Gene-set analysis and reduction. Brief. Bioinform. 10, 24–34.
Dixon, A.L., Liang, L., Moffatt, M.F., Chen, W., Heath, S., Wong, K.C., Taylor, J., Burnett, E., Gut, I., Farrall, M., et al. (2007). A genome-wide association study of global gene expression. Nat. Genet. 39, 1202–1207.
Dowdle, W.R., Coleman, M.T., Mostow, S.R., Kaye, H.S., and Schoenbaum, S.C. (1973). Inactivated influenza vaccines. 2. Laboratory indices of protection. Postgrad. Med. J. 49, 159–163.
Ein-Dor, L., Zuk, O., and Domany, E. (2006). Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc. Natl. Acad. Sci. USA 103, 5923–5928.
Eisenbarth, S.C., Colegio, O.R., O’Connor, W., Sutterwala, F.S., and Flavell, R.A. (2008). Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126.
Fauci, A.S., Johnston, M.I., Dieffenbach, C.W., Burton, D.R., Hammer, S.M., Hoxie, J.A., Martin, M., Overbaugh, J., Watkins, D.I., Mahmoud, A., and Greene, W.C. (2008). HIV vaccine research: The way forward. Science 321, 530–532.
Gardner, E.M., Gonzalez, E.W., Nogusa, S., and Murasko, D.M. (2006). Age-related changes in the immune response to influenza vaccination in a racially diverse, healthy elderly population. Vaccine 24, 1609–1614.
Gardy, J.L., Lynn, D.J., Brinkman, F.S., and Hancock, R.E. (2009). Enabling a systems biology approach to immunology: Focus on innate immunity. Trends Immunol. 30, 249–262.
Gaucher, D., Therrien, R., Kettaf, N., Angermann, B.R., Boucher, G., Filali-Mouhim, A., Moser, J.M., Mehta, R.S., Drake, D.R., 3rd, Castro, E., et al. (2008). Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 205, 3119–3131.
Geijtenbeek, T.B., and Gringhuis, S.I. (2009). Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 9, 465–479.
Germain, R.N. (2001). The art of the probable: System control in the adaptive immune system. Science 293, 240–245.
Gilchrist, M., Thorsson, V., Li, B., Rust, A.G., Korb, M., Roach, J.C., Kennedy, K., Hai, T., Bolouri, H., and Aderem, A. (2006). Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178.
Guerra, S., Nájera, J.L., González, J.M., López-Fernández, L.A., Climent, N., Gatell, J.M., Gallart, T., and Esteban, M. (2007). Distinct gene expression profiling after infection of immature human monocyte-derived dendritic cells by the attenuated poxvirus vectors MVA and NYVAC. J. Virol. 81, 8707–8721.
Gupta, R.K., Relyveld, E.H., Lindblad, E.B., Bizzini, B., Ben-Efraim, S., and Gupta, C.K. (1993). Adjuvants—a balance between toxicity and adjuvanticity. Vaccine 11, 293–306.
Haining, W.N., and Wherry, E.J. (2010). Integrating genomic signatures for immunologic discovery. Immunity 32, 152–161.
Haining, W.N., Ebert, B.L., Subrmanian, A., Wherry, E.J., Eichbaum, Q., Evans, J.W., Mak, R., Rivoli, S., Pretz, J., Angelosanto, J., et al. (2008). Identification of an evolutionarily conserved transcriptional signature of CD8 memory differentiation that is shared by T and B cells. J. Immunol. 181, 1859–1868.
Harari, A., Vallelian, F., and Pantaleo, G. (2004). Phenotypic heterogeneity of antigen-specific CD4 T cells under different conditions of antigen persistence and antigen load. Eur. J. Immunol. 34, 3525–3533.
Heng, T.S., and Painter, M.W.; Immunological Genome Project Consortium. (2008). The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094.
Hennessy, E.J., Parker, A.E., and O’Neill, L.A. (2010). Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307.
Hoft, D.F. (2008). Tuberculosis vaccine development: Goals, immunological design, and evaluation. Lancet 372, 164–175.
Huttenhower, C., Haley, E.M., Hibbs, M.A., Dumeaux, V., Barrett, D.R., Coller, H.A., and Troyanskaya, O.G. (2009). Exploring the human genome with functional maps. Genome Res. 19, 1093–1106.
Hyduke, D.R., and Palsson, B.O. (2010). Towards genome-scale signalling-network reconstructions. Nat. Rev. Genet. 11, 297–307.
Ideker, T., Galitski, T., and Hood, L. (2001). A new approach to decoding life: Systems biology. Annu. Rev. Genomics Hum. Genet. 2, 343–372.
Ipsen, J. (1946). Circulating antitoxin at the onset of diphtheria in 425 patients. J. Immunol. 54, 325–347.
Iwasaki, A., and Medzhitov, R. (2010). Regulation of adaptive immunity by the innate immune system. Science 327, 291–295.
Jack, A.D., Hall, A.J., Maine, N., Mendy, M., and Whittle, H.C. (1999). What level of hepatitis B antibody is protective? J. Infect. Dis. 179, 489–492.
Jackson, L.A., and Janoff, E.N. (2008). Pneumococcal vaccination of elderly adults: New paradigms for protection. Clin. Infect. Dis. 47, 1328–1338.
Jiang, B., Gentsch, J.R., and Glass, R.I. (2008). Inactivated rotavirus vaccines: A priority for accelerated vaccine development. Vaccine 26, 6754–6758.
Joyce, A.R., and Palsson, B.O. (2006). The model organism as a system: Integrating ‘omics’ data sets. Nat. Rev. Mol. Cell Biol. 7, 198–210.
Kaech, S.M., Hemby, S., Kersh, E., and Ahmed, R. (2002). Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 11, 373–384.
Kedersha, N., and Anderson, P. (2007). Mammalian stress granules and processing bodies. Methods Enzymol. 431, 61–81.
Kitano, H. (2002). Computational systems biology. Nature 420, 206–210.
Kool, M., Pétrilli, V., De Smedt, T., Rolaz, A., Hammad, H., van Nimwegen, M., Bergen, I.M., Castillo, R., Lambrecht, B.N., and Tschopp, J. (2008). Cutting edge: Alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. 181, 3755–3759.
Lang, J., Zuckerman, J., Clarke, P., Barrett, P., Kirkpatrick, C., and Blondeau, C. (1999). Comparison of the immunogenicity and safety of two 17D yellow fever vaccines. Am. J. Trop. Med. Hyg. 60, 1045–1050.
Lee, E., Chuang, H.Y., Kim, J.W., Ideker, T., and Lee, D. (2008). Inferring pathway activity toward precise disease classification. PLoS Comput. Biol. 4, e1000217.
Letvin, N.L. (2007). Correlates of immune protection and the development of a human immunodeficiency virus vaccine. Immunity 27, 366–369.
Levin, M.J., Oxman, M.N., Zhang, J.H., Johnson, G.R., Stanley, H., Hayward, A.R., Caulfield, M.J., Irwin, M.R., Smith, J.G., Clair, J., et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. (2008). Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J. Infect. Dis. 197, 825–835.
Li, H., Willingham, S.B., Ting, J.P., and Re, F. (2008). Cutting edge: Inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J. Immunol. 181, 17–21.
Lindblad, E.B. (2004). Aluminium compounds for use in vaccines. Immunol. Cell Biol. 82, 497–505.
Looney, J.M., Edsall, G., Ipsen, J., Jr., and Chasen, W.H. (1956). Persistence of antitoxin levels after tetanus-toxoid inoculation in adults, and effect of a booster dose after various intervals. N. Engl. J. Med. 254, 6–12.
Lynn, D.J., Winsor, G.L., Chan, C., Richard, N., Laird, M.R., Barsky, A., Gardy, J.L., Roche, F.M., Chan, T.H., Shah, N., et al. (2008). InnateDB: Facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 4, 218.
Mbow, M.L., De Gregorio, E., Valiante, N.M., and Rappuoli, R. (2010). New adjuvants for human vaccines. Curr. Opin. Immunol. 22, 411–416.
McElhaney, J.E., Xie, D., Hager, W.D., Barry, M.B., Wang, Y., Kleppinger, A., Ewen, C., Kane, K.P., and Bleackley, R.C. (2006). T cell responses are better correlates of vaccine protection in the elderly. J. Immunol. 176, 6333–6339.
McKee, A.S., Munks, M.W., MacLeod, M.K., Fleenor, C.J., Van Rooijen, N., Kappler, J.W., and Marrack, P. (2009). Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J. Immunol. 183, 4403–4414.
Morgan, C., Marthas, M., Miller, C., Duerr, A., Cheng-Mayer, C., Desrosiers, R., Flores, J., Haigwood, N., Hu, S.L., Johnson, R.P., et al. (2008). The use of nonhuman primate models in HIV vaccine development. PLoS Med. 5, e173.
Mosca, F., Tritto, E., Muzzi, A., Monaci, E., Bagnoli, F., Iavarone, C., O’Hagan, D., Rappuoli, R., and De Gregorio, E. (2008). Molecular and cellular signatures of human vaccine adjuvants. Proc. Natl. Acad. Sci. USA 105, 10501–10506.
Mostow, S.R., Schoenbaum, S.C., Dowdle, W.R., Coleman, M.T., and Kaye, H.S. (1973). Inactivated vaccines. 1. Volunteer studies with very high doses of influenza vaccine purified by zonal ultracentrifugation. Postgrad. Med. J. 49, 152–158.
Nagarajan, T., Rupprecht, C.E., Dessain, S.K., Rangarajan, P.N., Thiagarajan, D., and Srinivasan, V.A. (2008). Human monoclonal antibody and vaccine approaches to prevent human rabies. Curr. Top. Microbiol. Immunol. 317, 67–101.
Napolitani, G., Rinaldi, A., Bertoni, F., Sallusto, F., and Lanzavecchia, A. (2005). Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6, 769–776.
National Institute of Allergy and Infectious Diseases. (2010). NIH launches effort to define markers of human immune responses to infection and vaccination: Recovery Act enables research that could help improve vaccines and therapeutics. http://www.niaid.nih.gov/news/newsreleases/2009/pages/hipro.aspx.
Otaegui, D., Baranzini, S.E., Armañanzas, R., Calvo, B., Muñoz-Culla, M., Khankhanian, P., Inza, I., Lozano, J.A., Castillo-Triviño, T., Asensio, A., et al. (2009). Differential micro RNA expression in PBMC from multiple sclerosis patients. PLoS ONE 4, e6309.
Pantaleo, G., and Koup, R.A. (2004). Correlates of immune protection in HIV-1 infection: What we know, what we don’t know, what we should know. Nat. Med. 10, 806–810.
Pascual, V., Chaussabel, D., and Banchereau, J. (2010). A genomic approach to human autoimmune diseases. Annu. Rev. Immunol. 28, 535–571.
Plotkin, S.A. (2008). Vaccines: Correlates of vaccine-induced immunity. Clin. Infect. Dis. 47, 401–409.
Pulendran, B., and Ahmed, R. (2006). Translating innate immunity into immunological memory: Implications for vaccine development. Cell 124, 849–863.
Pulendran, B., Miller, J., Querec, T.D., Akondy, R., Moseley, N., Laur, O., Glidewell, J., Monson, N., Zhu, T., Zhu, H., et al. (2008). Case of yellow fever vaccine–associated viscerotropic disease with prolonged viremia, robust adaptive immune responses, and polymorphisms in CCR5 and RANTES genes. J. Infect. Dis. 198, 500–507.
Pulendran, B., Tang, H., and Manicassamy, S. (2010). Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat. Immunol. 11, 647–655.
Qin, L., Gilbert, P.B., Corey, L., McElrath, M.J., and Self, S.G. (2007). A framework for assessing immunological correlates of protection in vaccine trials. J. Infect. Dis. 196, 1304–1312.
Querec, T., Bennouna, S., Alkan, S., Laouar, Y., Gorden, K., Flavell, R., Akira, S., Ahmed, R., and Pulendran, B. (2006). Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 203, 413–424.
Querec, T.D., Akondy, R.S., Lee, E.K., Cao, W., Nakaya, H.I., Teuwen, D., Pirani, A., Gernert, K., Deng, J., Marzolf, B., et al. (2009). Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 10, 116–125, Published online November 23, 2008.
Ramilo, O., Allman, W., Chung, W., Mejias, A., Ardura, M., Glaser, C., Wittkowski, K.M., Piqueras, B., Banchereau, J., Palucka, A.K., and Chaussabel, D. (2007). Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 109, 2066–2077.
Reddy, T.B., Riley, R., Wymore, F., Montgomery, P., DeCaprio, D., Engels, R., Gellesch, M., Hubble, J., Jen, D., Jin, H., et al. (2009). TB database: An integrated platform for tuberculosis research. Nucleic Acids Res. 37 (Database issue), D499–D508.
Reinhardt, B., Jaspert, R., Niedrig, M., Kostner, C., and L’age-Stehr, J. (1998). Development of viremia and humoral and cellular parameters of immune activation after vaccination with yellow fever virus strain 17D: A model of human flavivirus infection. J. Med. Virol. 56, 159–167.
Reyes-Sandoval, A., Pearson, F.E., Todryk, S., and Ewer, K. (2009). Potency assays for novel T-cell-inducing vaccines against malaria. Curr. Opin. Mol. Ther. 11, 72–80.
Romero-Steiner, S., Frasch, C.E., Carlone, G., Fleck, R.A., Goldblatt, D., and Nahm, M.H. (2006). Use of opsonophagocytosis for serological evaluation of pneumococcal vaccines. Clin. Vaccine Immunol. 13, 165–169.
Sallusto, F., Lenig, D., Förster, R., Lipp, M., and Lanzavecchia, A. (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712.
Sallusto, F., Lanzavecchia, A., Araki, K., and Ahmed, R. (2010). From vaccines to memory and back. Immunity 33, this issue, 451–463.
Schadt, E.E., Lamb, J., Yang, X., Zhu, J., Edwards, S., Guhathakurta, D., Sieberts, S.K., Monks, S., Reitman, M., Zhang, C., et al. (2005). An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 37, 710–717.
Schwarz, E.M., Antoshechkin, I., Bastiani, C., Bieri, T., Blasiar, D., Canaran, P., Chan, J., Chen, N., Chen, W.J., Davis, P., et al. (2006). WormBase: Better software, richer content. Nucleic Acids Res. 34 (Database issue), D475–D478.
Sester, M., Sester, U., Gärtner, B., Heine, G., Girndt, M., Mueller-Lantzsch, N., Meyerhans, A., and Köhler, H. (2001). Levels of virus-specific CD4 T cells correlate with cytomegalovirus control and predict virus-induced disease after renal transplantation. Transplantation 71, 1287–1294.
Shen-Orr, S.S., Tibshirani, R., Khatri, P., Bodian, D.L., Staedtler, F., Perry, N.M., Hastie, T., Sarwal, M.M., Davis, M.M., and Butte, A.J. (2010). Cell type-specific gene expression differences in complex tissues. Nat. Methods 7, 287–289.
Sørlie, T., Perou, C.M., Tibshirani, R., Aas, T., Geisler, S., Johnsen, H., Hastie, T., Eisen, M.B., van de Rijn, M., Jeffrey, S.S., et al. (2001). Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 98, 10869–10874.
Steinman, R.M. (2008). Dendritic cells in vivo: A key target for a new vaccine science. Immunity 29, 319–324.
Sui, Y., Zhu, Q., Gagnon, S., Dzutsev, A., Terabe, M., Vaccari, M., Venzon, D., Klinman, D., Strober, W., Kelsall, B., et al. (2010). Innate and adaptive immune correlates of vaccine and adjuvant-induced control of mucosal transmission of SIV in macaques. Proc. Natl. Acad. Sci. USA 107, 9843–9848.
Tang, H., Cao, W., Kasturi, S.P., Ravindran, R., Nakaya, H.I., Kundu, K., Murthy, N., Kepler, T.B., Malissen, B., and Pulendran, B. (2010). The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat. Immunol. 11, 608–617.
Theiler, M., and Smith, H.H. (1937). The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 65, 787–800.
Ting, J.P., Duncan, J.A., and Lei, Y. (2010). How the noninflammasome NLRs function in the innate immune system. Science 327, 286–290.
Tong, J.C., and Ren, E.C. (2009). Immunoinformatics: Current trends and future directions. Drug Discov. Today 14, 684–689.
Vahey, M.T., Wang, Z., Kester, K.E., Cummings, J., Heppner, D.G., Jr., Nau, M.E., Ofori-Anyinam, O., Cohen, J., Coche, T., Ballou, W.R., and Ockenhouse, C.F. (2010). Expression of genes associated with immunoproteasome processing of major histocompatibility complex peptides is indicative of protection with adjuvanted RTS, S malaria vaccine. J. Infect. Dis. 201, 580–589.
Van Damme, P., and Van Herck, K. (2007). A review of the long-term protection after hepatitis A and B vaccination. Travel Med. Infect. Dis. 5, 79–84.
Wek, R.C., Jiang, H.Y., and Anthony, T.G. (2006). Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11.
Weng, G., Bhalla, U.S., and Iyengar, R. (1999). Complexity in biological signaling systems. Science 284, 92–96.
Wheelock, E.F., and Sibley, W.A. (1965). Circulating virus, interferon and antibody after vaccination with the 17-D strain of yellow-fever virus. N. Engl. J. Med. 273, 194–198.
Wherry, E.J., Ha, S.J., Kaech, S.M., Haining, W.N., Sarkar, S., Kalia, V., Subramaniam, S., Blattman, J.N., Barber, D.L., and Ahmed, R. (2007). Molecular signature of CD8+T cell exhaustion during chronic viral infection. Immunity 27, 670–684.
Wilkins, C., and Gale, M., Jr. (2010). Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22, 41–47.
Zak, D.E., and Aderem, A. (2009). Systems biology of innate immunity. Immunol. Rev. 227, 264–282.
SOLVING VACCINE MYSTERIES: A SYSTEMS BIOLOGY PERSPECTIVE*
Lydie Trautmann and Rafick-Pierre Sekaly73
Systems biology has emerged as a promising research strategy that can be applied to vaccine development. This approach can lead to the identification of new mechanisms and predictors of inactivated vaccine immunogenicity.
The search for vaccines against several incurable diseases, including acquired immunodeficiency syndrome, malaria, tuberculosis, and dengue fever, has largely failed, which highlights the need for new vaccine strategies (Rappuoli and Aderem, 2011). In contrast to the empirical development of most efficacious vaccines, these diseases require a rational approach for directing vaccine responses toward different effector cell subsets of the immune system that vary with the nature or cellular tropism of these pathogens (Pulendran and Ahmed, 2011; Steinman and Banchereau, 2007). The lack of well-defined correlates of protection with most vaccines has been a major impediment to the successful development of new vaccines. The failure to identify such correlates has been mostly a con-sequence of the lack of assays that can measure integrated immune responses and have relied on the evaluation of a limited number of qualitative and quantitative features of effector adaptive immune responses. The efficacy of vaccines is also known to vary considerably in the human population depending on several environmental and genetic parameters, including age, nutrition and preexisting infections. In this issue of Nature Immunology, Nakaya and colleagues use systems biology to identify useful predictors of the efficacy of vaccines against influenza in humans (Nakaya et al., 2011).
Systems biology offers the unique possibility of analyzing the immunological network of complex events and interactions after vaccination (Kitano, 2002). This approach can help accelerate vaccine development by identifying predictors of immunogenicity and previously unknown mechanisms that underlie protective immune responses (Pulendran et al., 2010) (Fig. A16-1). Systems biology has been used to identify early gene signatures that can be used to predict the responses of B cells and CD8+ T cells after vaccination against yellow fever (Querec et al., 2009; Gaucher et al., 2008; Pulendran, 2009). Such studies have highlighted the importance of inducing a potent and diverse innate immune response for the generation of long-term protective adaptive immunity (Querec et al., 2009). Most of these pathways would not have been identified by con-
_______________
* Reprinted with kind permission from Nature Publishing Group.
73 Lydie Trautmann and Rafick-Pierre Sekaly are with the Vaccine and Gene Therapy Institute of Florida, Port Saint Lucie, Florida, USA, e-mail: rpsekaly@vgtifl.org.
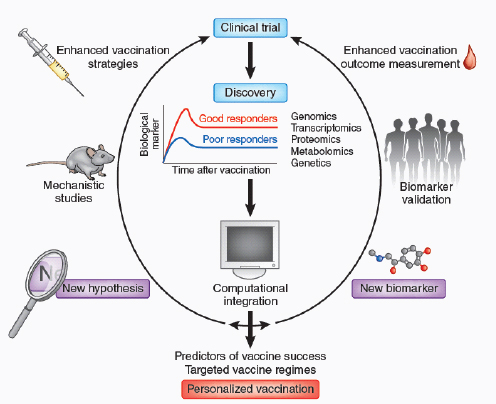
FIGURE A16-1 Systems biology approaches in the vaccine development. Licensed vaccines or new vaccines are tested in clinical trials in humans. Subjects are categorized as good and poor responders to a vaccine on the basis of biological markers. Samples collected are analyzed by one or more ‘-omics’ approaches, and results are integrated by computational methods to generate meaningful data sets. Two outcomes can emerge from these bioinformatic analyses: the generation of new hypotheses that can be tested by mechanistic studies in vitro or in animal models and result in enhanced vaccination strategies, or the determination of new biomarkers that can be validated in other clinical trials and result in enhancement of the vaccination outcome measurement. Both of these arms will ‘feed’ new clinical trials that will follow the same path and, after many iterations of the cycle, lead to the development of better vaccination strategies and define better predictors of immunogenicity. This process might allow the generation of personalized vaccination.
ventional immune response–monitoring strategies such as flow cytometry and analysis of cytokine production.
Despite the demonstrated success of systems approaches in predicting the immunogenicity of the vaccine against yellow fever (Querec et al., 2009; Gaucher et al., 2008) (a live replicating virus), the question of whether such approaches would also be useful in predicting the immunogenicity of inactivated vaccines (which do not replicate) has remained unknown. Furthermore, it has been unclear
whether such approaches could be used to predict the immunogenicity of memory responses. The report from Nakaya et al. addresses these questions directly (Nakaya et al., 2011). Using systems biology to analyze the innate and adaptive responses to seasonal influenza vaccination in humans, the authors define early predictors of a vaccine response and provide new insights about the mechanisms that underlie vaccine immunogenicity. Notably, this work describes a new comprehensive approach with which to tackle the challenge of vaccine development, not only because it defines new predictors of the inactivated influenza vaccine but also because it demonstrates and validates the systems biology approach as a powerful tool for predicting vaccine immunogenicity and discovering additional mechanisms of vaccine efficacy (Fig. A16-1).
This study compares immune responses induced by two highly efficient vaccines against influenza: the trivalent inactivated vaccine (TIV) and the live attenuated influenza vaccine (LAIV). The transcriptional signatures induced by TIV and LAIV are distinct, but both induce common innate immune response pathways, including an inflammatory response characterized by inflammasome and upregulation of the transcription factor NF-κB. In contrast, type I interferons, which are a common feature of the two live attenuated vaccines (LAIV and the YF-17D vaccine against yellow fever) and are potent inducers of innate immunity, are not observed after vaccination with TIV, a whole killed virus. Involvement of innate immunity in modulating the response to TIV is further confirmed by the positive correlation between gene-expression pathways associated with natural killer cell signaling early after vaccination (days 3 or 7) and hemagglutination-inhibition (HAI) titers at day 28. This highlights the fact that the innate immune response is diverse and is critical to the development of a strong adaptive immune response and also that the innate immune response can vary with the type of vaccine platform used. Notably, these different innate immune responses lead to protection. Such diversity of innate immune responses could never have been identified by conventional immune response–monitoring assays. Indeed, counting cells of the innate immune response or assessing cytokine profiles failed to predict the immunogenicity of the YF-17D vaccine (Querec et al., 2009). Comparison of the innate immune responses induced by several other vaccines will provide not only a map of the common patterns induced by all successful vaccines but also the characteristics of particular vaccines that could be classified as either the type of vaccine formulation or the kind or immune response induced (Steinman and Banchereau, 2007; Pulendran et al., 2010; Zak and Aderem, 2009). With this knowledge, the design of new vaccine regimens and adjuvants could focus on recapitulating the common patterns required for vaccine efficiency and selective targeting of the specific type of response to be elicited.
Systems biology approaches can also provide insights into the molecular mechanisms that lead to vaccine efficacy. To reach this objective, however, it is necessary to assess the contributions of the various cellular subsets involved in the immune response that contribute to the overall signature in peripheral blood cells.
Previous attempts aimed at deconvoluting these signatures have relied on the extrapolation of cell subset–specific gene-expression profiles from the frequency of these subsets (Shen-Orr et al., 2010). Nakaya et al. propose an alternative solution for deconvoluting gene-expression profiles (Nakaya et al., 2011). This approach is based on the meta-analysis of cell type–specific gene-expression signatures from publicly available microarray studies and does not require assessment of the frequency of specific immune-response cell subsets. Using this approach, the authors confirm that TIV induces the upregulation of genes mostly in B cells, especially antibody-secreting cells. In contrast, LAIV induces genes in T cells, monocytes and natural killer cells. These findings highlight the need to determine if gene-expression changes are due to changes in the distribution of specific cell subpopulations or to the de novo induction of gene expression in discrete subsets of cells. Hence, as illustrated by this study, such approaches also provide insight into the mechanism of action of vaccines and could guide the development of specific adjuvants.
To identify predictors of TIV vaccine immunogenicity, the authors use the DAMIP (discriminant analysis via mixed integer programming) model to train and test the predictors of two other independent cohorts vaccinated with TIV. By grouping subjects as ‘good responders’ (an increase of fourfold or more in HAI titers) and ‘poor responders’ (an increase of twofold or less in HAI titers), the authors find that gene signatures at days 3–7 that can be used to predict AI titers at day 28 in the first cohort allow the prediction with 90% accuracy the immunogenicity of the inactivated vaccine in the other cohorts. Of note, some of the genes used to predict the magnitude of the TIV response, including TNFRSF17 (a gene expressed during B cell maturation), have also been found to be predictors after vaccination with YF-17D.
This report illustrates all the steps and outcomes of systems biology approaches (Fig. A16-1). Starting from a comparison of two vaccines (TIV and LAIV) or of good and poor immunogenicity as estimated by HAI titers, the authors generate transcriptomics data that they further analyze by bioinformatics analyses. This study confirms the ability of such approaches to tackle complex network interactions that have never been studied before in humans in an unbiased way. For example, five members of the leukocyte immunoglobulin-like receptor family with high expression in monocytes and myeloid dendritic cells at day 3 after vaccination are found by the DAMIP model to be predictors of immunogenicity, which suggests a role for receptors of the innate immune system in regulating adaptive antibody responses. It is still unknown if the best predictors of vaccine immunogenicity would emanate from innate or adaptive immune responses. It is also possible that such predictors would be identified from the analysis of immune responses occurring in tissues or at mucosal sites. The ultimate goal would be to be able to predict if a particular subject would respond to a given vaccine by doing a simple cost-effective assay, such as quantitative
PCR analysis of a drop of blood early after vaccination. The second outcome of this approach is the generation of unexpected hypotheses. This study finds that expression of the gene encoding CaMKIV, an enzyme involved in Ca2+ signaling, is inversely correlated with HAI titers induced by TIV at day 28. The role of CaMKIV in the regulation of antibody response has never been studied before (Racioppi and Means, 2008). The authors demonstrate that TIV induces phosphorylation of CaMKIV and further demonstrate a critical role for CaMKIV in regulating antibody responses, using mice deficient in CaMKIV.
The pioneering approach of Nakaya et al. has led to the generation of several new hypotheses that will pave the way for fundamental research focused on the identification of the mechanisms of protection induced by vaccines and will lead to the improvement of vaccination strategies (Nakaya et al., 2011). Additional studies using similar systems biology approaches to identify determinants of vaccine and adjuvant efficacy will undoubtedly result in the discovery of previously unknown biological mechanisms by which various vaccines trigger and modulate the effector and memory arms of the immune response and thus revitalize immunology. Furthermore, the identification of signatures that can be used to predict vaccine immunogenicity will be of immense value in the early identification of people who respond suboptimally to the vaccine. This in turn will accelerate the pace of clinical trials and allow the design of new vaccine strategies for incurable diseases and personalized vaccine regimens for immunocompromised populations.
Competing Financial Interests
The authors declare no competing financial interests.
References
Gaucher, D. et al. J. Exp. Med. 205, 3119–3131 (2008).
Kitano, H. Nature 420, 206–210 (2002).
Nakaya, H.I. et al. Nat. Immunol. 12, 786–795 (2011).
Pulendran, B. Nat. Rev. Immunol. 9, 741–747 (2009).
Pulendran, B. & Ahmed, R. Nat. Immunol. 131, 509–517 (2011).
Pulendran, B., Li, S. & Nakaya, H.I. Immunity 33, 516–529 (2010).
Querec, T.D. et al. Nat. Immunol. 10, 116–125 (2009).
Racioppi, L. & Means, A.R. Trends Immunol. 29, 600–607 (2008).
Rappuoli, R. & Aderem, A. Nature 473, 463–469 (2011).
Shen-Orr, S.S. et al. Nat. Methods 7, 287–289 (2010).
Steinman, R.M. & Banchereau, J. Nature 449, 419–426 (2007).
Zak, D.E. & Aderem, A. Immunol. Rev. 227, 264–282 (2009).
SYSTEMS BIOLOGY OF VACCINATION FOR SEASONAL INFLUENZA IN HUMANS74
Helder I. Nakaya,75,76Jens Wrammert,75,77Eva K. Lee,78Luigi Racioppi,79,80Stephanie Marie-Kunze,75,76W. Nicholas Haining,81Anthony R. Means,76Sudhir P. Kasturi,75,76Nooruddin Khan,75,76Gui-Mei Li,75,77Megan McCausland,75,77Vibhu Kanchan,75,77Kenneth E. Kokko,82Shuzhao Li,75,76Rivka Elbein,83Aneesh K Mehta,79Alan Aderem,84Kanta Subbarao,85Rafi Ahmed,75,77and Bali Pulendran75,76,86
Here we have used a systems biology approach to study innate and adaptive responses to vaccination against influenza in humans during three consecutive influenza seasons. We studied healthy adults vaccinated with trivalent inactivated influenza vaccine (TIV) or live attenuated influenza vaccine (LAIV). TIV induced higher antibody titers and more plasmablasts than LAIV did. In subjects vaccinated with TIV, early molecular signatures correlated with and could be used to accurately predict later antibody titers in two independent trials. Notably, expression of the kinase CaMKIV at day 3 was inversely correlated with later antibody titers. Vaccination of CaMKIV-deficient mice with TIV induced enhanced antigen-specific antibody titers, which demonstrated an unappreciated role for CaMKIV in the regulation of antibody responses. Thus, systems approaches can be used to predict immunogenicity and provide new mechanistic insights about vaccines.
_______________
74 Reprinted with Permission from Nature Publishing Group.
75 Emory Vaccine Center, Emory University, Atlanta, Georgia, USA.
76 Yerkes National Primate Research Center, Emory University, Atlanta, Georgia, USA.
77 Department of Microbiology and Immunology, Emory University, Atlanta, Georgia, USA.
78 Center for Operations Research in Medicine & Healthcare, School of Industrial & Systems Engineering, Georgia Institute of Technology, Atlanta, Georgia, USA.
79 Department of Pharmacology and Cancer Biology, Duke University, Durham, North Carolina, USA.
80 Department of Cellular and Molecular Biology and Pathology, University of Naples Federico II, Naples, Italy.
81 Dana-Farber Cancer Institute, Boston, Massachusetts, USA.
82 Department of Medicine, Division of Nephrology, Emory University School of Medicine, Atlanta, Georgia, USA.
83 Division of Infectious Diseases, Department of Medicine, School of Medicine, Emory University, Atlanta, Georgia, USA.
84 Institute for Systems Biology, Seattle, Washington, USA.
85 Laboratory of Infectious Diseases, National Institute for Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA.
86 Department of Pathology, Emory University School of Medicine, Atlanta, Georgia, USA. Correspondence should be addressed to B.P. (bpulend@emory.edu).
Received 12 April; accepted 6 June; published online 10 July 2011; doi:10.1038/ni.2067
Annual vaccination is one of the most effective methods for preventing influenza (Sasaki et al., 2007). At present, two types of vaccines for seasonal influenza are licensed for use in the USA: trivalent inactivated influenza vaccine (TIV), given by intramuscular injection; and live attenuated influenza vaccine (LAIV), administered intranasally. These vaccines contain three strains of influenza viruses that are usually changed annually on the basis of the results of global influenza surveillance data (Fiore et al., 2010). The efficacy of a vaccine against influenza, therefore, depends on the match of antigenicity between the vaccine and circulating influenza strains (Sasaki et al., 2008). Additionally, other factors such as the age and immunocompetence of vaccinees, as well as preexisting amounts of antibody derived from prior infection or vaccination, contribute to mechanisms that mediate the efficacy of vaccines against influenza (Sasaki et al., 2007; Fiore et al., 2010; Zeman et al., 2007).
Systems vaccinology has emerged as an interdisciplinary field that combines systems-wide measurements plus network and predictive modeling applied to vaccinology (Pulendran and Nakaya, 2010). A systems biology approach has been used to identify early gene signatures that correlate with and can be used to predict later immune responses in humans vaccinated with the live attenuated vaccine YF-17D against yellow fever (Querec et al., 2009; Gaucher et al., 2008). YF-17D is one of the most successful vaccines ever developed (Pulendran, 2009; Monath, 2005); it stimulates polyvalent innate immune responses (Querec et al., 2006) and adaptive immune responses (Barrett and Teuwen, 2009) that can persist for decades after vaccination (Barrett and Teuwen, 2009). Although systems biology approaches have been used to predict the immunogenicity of YF-17D (Querec et al., 2009; Gaucher et al., 2008), which is a live replicating virus, the extent to which such approaches can be applied to the prediction of the immunogenicity of inactivated vaccines is unknown. Furthermore, it remains unclear whether systems approaches can be used to predict the immunogenicity of recall responses. In the case of influenza, the immune response to vaccination is greatly enhanced by the past history of the vaccine recipient, both by prior infections and vaccinations. Notably, whether such approaches can provide insight into the immunological mechanisms of action of vaccines and help with the discovery of new correlates of protective immunity is untested. To address these issues, we did a series of clinical studies during the annual influenza seasons in 2007, 2008 and 2009, in which we vaccinated healthy young adults with TIV. Our goal was to undertake a detailed characterization of the innate and adaptive responses to vaccination with TIV to identify putative early signatures that correlated with or could be used to predict later immunogenicity and to obtain new insight into the mechanisms that underlie immunogenicity.
The results of our studies demonstrate that systems biology approaches can indeed be used to predict the immunogenicity of an inactivated vaccine such as TIV with up to 90% accuracy. Notably, the expression at day 3 of one of the genes in the predictive signature, encoding the kinase CaMKIV, was inversely correlated with plasma hemagglutination-inhibition (HAI) antibody titers at day 28. Vaccina-
tion of CaMKIV-deficient (Camk4−/−) mice with TIV induced enhanced antigen-specific antibody titers, which demonstrated an unappreciated role for CaMKIV in the regulation of antibody responses. Together our results demonstrate the utility of systems biology not only in the prediction of vaccine immunogenicity but also in offering new insight into the molecular mechanism of influenza vaccines.
Results
Antibody Responses Induced by TIV and LAIV
We evaluated the antibody responses of 56 healthy young adults vaccinated with either LAIV (n = 28) or TIV (n = 28) during the 2008 influenza season. We determined HAI titers for each of the three influenza strains in LAIV and TIV in the plasma of vaccinees at baseline (day 0) and at 28 d after vaccination. We calculated the magnitude of antibody responses to the vaccine (HAI response) as the maximum difference between the HAI titer at day 28 and the baseline titer (day 0) for any of the three influenza strains contained in the vaccine (Fig. A17-1a). The mean HAI response of subjects vaccinated with TIV was sixfold higher than that of those vaccinated with LAIV (Fig. A17-1a), consistent with many published reports (Sasaki et al., 2007; Johnson et al., 1985; Beyer et al., 2002). Furthermore, among the subjects vaccinated with TIV, there was considerable variation in the magnitude of the HAI response (>100-fold; Fig. A17-1a). According to the US Food and Drug Administration Guidance for Industry document for this field (US Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research, 2007), seroconversion can be defined by an HAI titer of 1:40 or more and a minimum fourfold increase in antibody titer after vaccination. Thus, we operationally classified the vaccinees as ‘low HAI responders’ or ‘high HAI responders’ based on whether or not a fourfold increase occurred after vaccination (Fig. A17-1a). Most of the subjects vaccinated with TIV (22 of 28) were classified as high HAI responders; only six were classified as low HAI responders. In contrast, most subjects vaccinated with LAIV (24 of 28) were classified as low HAI responders and only four were classified as high HAI responders (Fig. A17-1a).
Antibodies are produced by antibody-secreting B cells in the blood (plasmablasts) or bone marrow and secondary lymphoid organs (fully differentiated plasma cells). High frequencies of antigen-specific plasmablasts in the blood within a few days of vaccination, reaching a peak at day 7, have been documented (Wrammert et al., 2008). To determine whether the early plasmablast response to influenza vaccination correlated with the later HAI response, we assessed the frequency of influenza-specific plasmablasts at baseline and 7 d after vaccination (Fig. A17-1b,c). As reported before (Wrammert et al., 2008), we observed rapid clonal expansion of influenza-specific plasmablasts 7 d after vaccination with TIV, as measured by enzyme-linked immunospot (ELISPOT) assay (Fig. 1b) and by flow cytometry (Fig. A17-1c). We further found that the population expansion of
circulating plasmablasts secreting immunoglobulin G (IgG) was also greater in subjects vaccinated with TIV than in those vaccinated with LAIV (Fig. A17-1b,c). We obtained similar results for IgA-secreting plasmablasts at day 7 after vaccination (Supplementary Fig. 1a), and a very good correlation was evident between the frequency of plasmablasts as measured by ELISPOT and their frequency as measured by flow cytometry (Fig. A17-1d and Supplementary Fig. 1b).
As we detected a very low HAI response after vaccination with LAIV, we considered only subjects vaccinated with TIV in further correlation analyses. There was a modest positive correlation between the number of IgG-secreting plasmablasts at day 7 and the HAI response at day 28 after vaccination (Fig. A17-1e). Because the frequency of plasmablasts returns to a barely detectable amount by day 14 after vaccination (Wrammert et al., 2008), this correlation suggested that the later antibody response was associated with early circulation of plasmablasts in the blood of vaccinees (Sasaki et al., 2008). However, given the modest correlation (r = 0.43), there was clearly a need for more robust correlates of immunogenicity.
Molecular Signatures of Influenza Vaccines
We first determined whether TIV and LAIV induced molecular signatures that were detectable in the blood. To identify such signatures of immunogenicity, we first measured by multiplex assay the concentrations of key cytokines in the plasma of vaccinees on days 0, 3 and 7 after vaccination (Supplementary Fig. 2a). We selected ten cytokines or chemokines on the basis of their importance as key mediators of host immune responses (CCL5 (RANTES), interleukin 1α, interferon-α2 (IFN-α2), CCL3 (MIP-1α), CCL11 (eotaxin), interleukin 12 subunit p70, IFN-γ, interleukin 1β, CXCL10 (IP-10) and CCL2 (MCP-1)). Among those, only the chemokine CXCL10 (IP-10) was significantly induced by TIV on day 3 relative to its expression on day 0 (P = 0.0189 (t-test); Supplementary Fig. 2b). None of those cytokines were significantly induced or repressed by vaccination with LAIV. The concentration of CXCL10 (IP-10) at day 3 relative to its baseline concentration was negatively correlated to the HAI response at day 28 after vaccination (Supplementary Fig. 2c), which suggested possible involvement of CXCL10 (IP-10) in the antibody response. However, the correlation coefficient was modest (r = −0.48), which again emphasized the need for more robust correlates of immunogenicity.
To determine in an unbiased way the expression changes induced by vaccination against influenza on a genome-wide scale, we did microarray analysis using peripheral blood mononuclear cells (PBMCs) collected from all 56 vaccinees on days 0, 3 and 7 after vaccination. We calculated the change in expression by subtracting the log2 expression value at day 0 from its corresponding value day 3 or 7, and we filtered out genes if we observed no increase or decrease greater than 25% (1.25-fold) in at least 20% of the vaccinees. After that step, we applied three independent statistical tests to the remaining genes and considered only genes identified by all three analyses as being differently expressed.
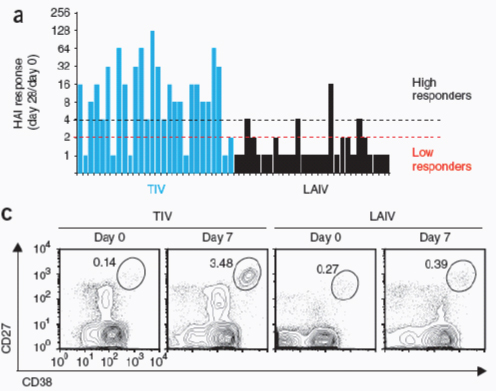
FIGURE A17-1 Analysis of humoral immunity to influenza vaccination. (a) HAI titers in plasma on day 28 after vaccination with TIV or LAIV, relative to baseline (day 0); results are the highest HAI response among all three influenza strains in the vaccine: low responders, no increase above twofold; high responders, fourfold or more above baseline. P < 0.0001, mean HAI response, TIV versus LAIV (t-test). (b) ELISPOT assay of influenza-specific IgG–secreting plasmablasts among PBMCs from all vaccinees at 0 and 7 d after vaccination. Each symbol represents an individual donor; small horizontal lines indicate the median (numbers adjacent median values); dotted lines are the limit of detection. (c) Flow cytometry analysis of plasmablasts in the plasmablast gate (CD3−CD20lo− negCD19+CD27hiCD38hi) in blood from subjects vaccinated with TIV or LAIV. Numbers
Transcriptome analysis of vaccinees showed that LAIV and TIV induced very different gene signatures (Supplementary Fig. 3a). However, the expression of 1,445 probe sets was altered similarly by both vaccines (Supplementary Fig. 3a). Among these common ‘differentially expressed genes’ (DEGs), ingenuity pathway analysis identified a network composed of several genes related to inflammatory and antimicrobial responses (Supplementary Fig. 3b; complete list of DEGs after vaccination with TIV or LAIV, Supplementary Table 1). This indicated that processes related to innate immunity may have influenced the immunogenicity of each vaccine. The expression of several interferon-related genes was altered after vaccination with LAIV but not after vaccination with TIV
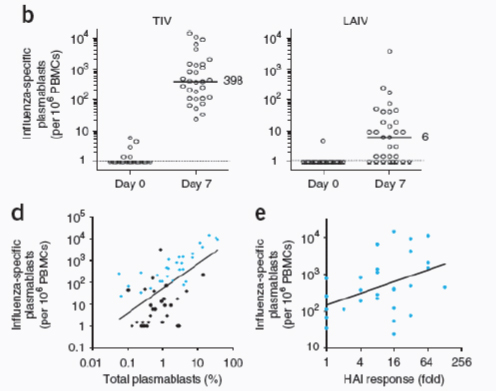
adjacent to outlined areas indicate percent cells in the plasmablast gate. (d) Frequency of plasmablasts, assessed by flow cytometry, versus the number of influenza-specific IgG–secreting plasmablasts, assessed by ELISPOT, at day 7 after vaccination with TIV (blue) or LAIV (black). r = 0.58 (Pearson); P < 0.0001 (for Pearson correlation; two-tailed test). (e) Influenza-specific IgG–secreting plasmablasts at day 7 versus the antibody response at day 28 after vaccination with TIV. r = 0.43 (Pearson); P = 0.02 (for Pearson correlation; two-tailed test). Data are from one experiment with 56 subjects assayed in duplicate (a), 61 subjects assayed in duplicate (b) or 59 subjects assayed once (c) or were generated from data in a–c (d,e).
(Fig. A17-2). Type 1 interferons are central components of the innate immune response to virus16. Therefore, the higher expression of type I interferon–related genes may be attributed to the replication competence of LAIV. Our analysis identified genes encoding molecules closely associated with the interferon signaling pathways, such as STAT1, STAT2, TLR7, IRF3 and IRF7 (Fig. A17-2a). Notably, the difference in expression for many interferon-related genes was greatest at day 3 after immunization with LAIV (Fig. A17-2a).
We also compared the gene signatures of the two influenza vaccines with that of another live attenuated vaccine, the YF-17D vaccine against yellow fever6. For consistency with that publication (Querec et al., 2009), we applied the same
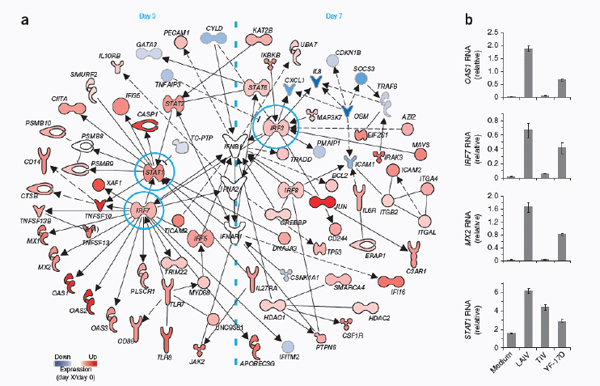
FIGURE A17-2 Molecular signature induced by vaccination with LAIV. (a) Interferon-related genes upregulated (Up) or downregulated (Down) on day 3 or 7 (‘X’ in key) after vaccination with LAIV relative to their expression at day 0 (colors in key): solid lines indicate direct interactions; dashed lines indicate indirect interactions. (b) Quantitative RT-PCR confirmation of the induction of key interferon-related genes (OAS1, IRF7, MX2 and STAT1) in PBMCs obtained from healthy subjects and left unstimulated (Medium) or stimulated for 24 h in vitro with LAIV, TIV or YF-17D; results are normalized to the expression of GAPDH (glyceraldehyde phosphate dehydrogenase) and are presented relative to those of unstimulated PBMCs. Data are representative of one experiment (a) or three independent experiments with one subject each (b; error bars, s.d.).
stringency and criteria to identify genes with differences in expression in subjects vaccinated with YF-17D, as follows: we filtered out genes if we found no increase or decrease in expression (on day 3 or 7 relative to baseline) greater than 1.41-fold in at least 60% of the vaccinees; we used one-way analysis of variance with the Benjamini and Hochberg false-discovery-rate method with a cutoff of 0.05; and genes had to have a difference in expression in both YF-17D trials (Querec et al., 2009). However, this time we did the analysis at the level of the probe set instead of defining genes based on the UniGene database (National Center for Biotechnology Information). Although subjects vaccinated with YF-17D had a gene-expression profile distinct from that of those vaccinated against influenza, many interferon-related genes were commonly induced by YF-17D and LAIV (data not shown). RT-PCR analysis of RNA from PBMCs stimulated in vitro with LAIV, TIV or YF-17D confirmed that interferon-related genes were upregulated 24 h after treatment with LAIV or YF-17D but not after stimulation with TIV (Fig. A17-2b). Together these data demonstrated that vaccination with TIV or LAIV induced distinct molecular signatures in the blood.
Molecular Signatures of Sorted Cell Subsets
We did microarray analyses of the gene-expression profiles of PBMCs isolated from the blood of vaccinees at baseline and at days 3 and 7 after vaccination. One confounding variable here was that the observed transcriptional changes may have resulted from new induction of gene expression or may have simply reflected the changing cellular composition of the PBMC compartment. To overcome this issue, we used the approach of isolating and identifying the genomic signatures of each subset in the PBMC pool. We did microarray experiments with the following four different cell types, obtained from subjects vaccinated with LAIV (n = 6) or TIV (n = 6) and sorted by flow cytometry: CD19+ B cells, CD14+ monocytes, CD-11chiCD123lo myeloid dendritic cells (DCs) and CD123hi CD11clo plasmacytoid DCs. We extracted, amplified and labeled total RNA from 96 sorted cell samples at baseline and day 7 and hybridized the RNA on microarray chips (Supplementary Fig. 4a). We did significance analysis of microarrays (Tusher et al., 2001) for each subset, separately comparing the values at day 7 with the corresponding baseline values. This approach identified hundreds to thousands of probe sets with differences in expression after vaccination with TIV or LAIV (Supplementary Fig. 4b and Supplementary Table 2), which demonstrated that vaccines against influenza produced global expression changes for each cell type.
In subjects vaccinated with TIV, myeloid DCs and B cells had the most DEGs (Supplementary Fig. 4b). Notably, there was an enrichment for DEGs associated with plasmablasts (Supplementary Fig. 4c). However, because a substantial proportion of plasmablasts die after being frozen and thawed (data not shown), the DEGs observed in the B cell compartment were probably an underestimation of the DEGs associated with plasmablasts (Fig. A17-3). Nevertheless,
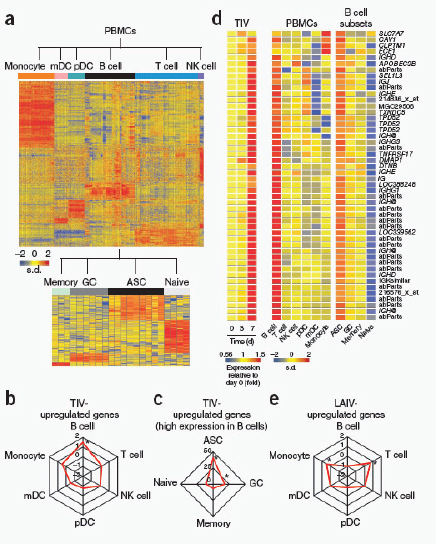
FIGURE A17-3 Molecular signatures induced by vaccination with TIV. (a) Heat map of gene signatures of cells of the immune response, identified by meta-analysis. Expression of each gene (rows) is presented as s.d. above (red) or below (blue) the average value for that gene for all samples (columns). mDC, myeloid DC; pDC, plasmacytoid DC; NK, natural killer. (b) Enrichment for genes upregulated by TIV among genes with high expression in any PBMC subset (numbers in plot indicate enrichment (fold)). (c) Enrichment for genes upregulated by TIV among genes with high expression in B cells and also in a specific B cell subset. (d) Heat map of genes upregulated after vaccination with TIV and also with high expression in B cells (PBMCs) and ASCs (B cell subsets); ‘abParts’ indicates probe sets mapping to antibody variable regions, and Affymetrix probe identifiers are provided for probe sets not annotated. (e) Enrichment for genes upregulated by LAIV among genes with high expression in any PBMC subset. *P < 10−10 (two-tailed Fisher’s exact test). Data are representative of 28 experiments with 281 samples.
we were still able to identify DEGs related to antibody-secreting cells (ASCs) and the unfolded protein response in sorted B cells after immunization with TIV (Supplementary Fig. 4c,d). To cope with the large amount of immunoglobulin proteins that are produced, ASCs must greatly increase the function of their secretion machinery, which may lead to the accumulation of misfolded proteins in the endoplasmic reticulum (Iwakoshi et al., 2003a,b). In response to such stress, the cells activate intracellular signal-transduction pathways and the unfolded protein response, which protects the cells by enhancing the capacity of the secretory apparatus and by diminishing the endoplasmic reticulum load (Ron and Walter, 2007). After vaccination with TIV, upregulation of genes encoding two transcription factors, XBP-1 and ATF6B, which are central orchestrators of the unfolded protein response, was detectable in sorted B cells but not in PBMCs (Supplementary Fig. 4d).
In subjects vaccinated with LAIV, in contrast to results obtained with those vaccinated with TIV, the plasmacytoid DC subset generated the most DEGs (Supplementary Fig. 4b). Of the many interferon-related genes induced by LAIV (Fig. A17-2a), we found that 37 were induced in at least one subset of the sorted cells. Of those, 17 and 14 were upregulated in monocytes and plasmacytoid DCs, respectively (Supplementary Fig. 4e). In addition, there were 44 interferon-related genes that were induced or repressed in at least one subset of the sorted cells but not in the PBMCs (Supplementary Fig. 4e). Most were upregulated in myeloid and plasmacytoid DCs (Supplementary Fig. 4e). These data suggest that antigen-presenting cells may be important in the innate response to vaccination with LAIV. The large number of interferon-related genes ‘missing’ from the PBMC analysis may have been due to the fact that myeloid DCs and plasmacytoid DCs together represent <1% of total PBMCs (Ueda et al., 2003).
The observations reported here indicated the type of information that can be obtained by examination of the gene-expression profiles of sorted cell types. However, evaluating the gene-expression signatures of individual subsets of cells isolated by flow cytometry presents a considerable challenge. The practical use of such an approach is very limited, both logistically (that is, the need to use freshly isolated samples to prevent the ‘preferential’ loss of certain cell types, such as plasmablasts and effector T cells) and financially (that is, the need for large numbers of gene chips). Therefore, as described below, we devised an alternative strategy.
Meta-Analysis of Cell Type–Specific Signatures
Human PBMCs consist of many different cell types, each with a distinct transcriptome. A published study has demonstrated the use of a deconvolution method to analyze cell type–specific gene expression differences in complex tissues (Shen-Orr et al., 2010). We devised an independent strategy to discern cell type–specific transcriptional signatures with the results of the PBMC microarray analyses. We did a meta-analysis of publicly available microarray studies in
which the gene-expression profiles of isolated individual cell types of PBMCs (such as T cells, B cells, monocytes, natural killer cells and so on) or B cell subsets (such as naive, memory and germinal center B cells and ASCs from blood or tonsils) had been analyzed (Supplementary Fig. 5a,b). To avoid issues of cross-platform normalization and probe selection, we used only samples hybridized to Affymetrix Human Genome U133 Plus 2.0 Arrays or Affymetrix Human Genome U133A Arrays in our meta-analysis. Additionally, for each study, we manually removed samples based on the severity of the disease or treatment and/ or the method of cell purification (samples and studies, Supplementary Table 3). We included in our meta-analysis microarray data of flow cytometry–sorted plasmacytoid and myeloid DCs obtained from PBMCs of subjects before and after vaccination with TIV or LAIV (Supplementary Fig. 4a). We compared the expression profile of a given cell subset with the expression profile of all other subsets by t-test (P < 0.05; mean change, over twofold). We designated a gene as having high expression in a particular cell type by determining the number of times the gene was upregulated in the cell type by all possible pairwise comparisons with its expression in other cell types (Supplementary Fig. 5b and Supplementary Methods). We then compared the genomic signatures of cells of the immune response obtained by this approach (Fig. A17-3a and Supplementary Table 4) with the genomic signatures of subjects vaccinated against influenza.
Our meta-analysis confirmed that the group of genes upregulated by TIV was enriched for genes with high expression in B cells (Fig. 3b) and, among those, genes with high expression in ASCs (Fig. A17-3c). We prepared a heat map of the genes upregulated in ASCs after vaccination with TIV (Fig. A17-3d). Among the genes upregulated were those encoding ‘antibody parts’ (rearranged variable-diversity-joining immunoglobulin gene segments) and several other genes encoding parts of immunoglobulins (IGH@, IGHE, IGHG3, IGHG1 and IGHD), as well as TNFRSF17 (which encodes BCMA, a member of the BAFF-BLyS family of receptors (Avery et al., 2003), and whose expression has been shown before to be a key feature of the best predictive signatures of neutralizing antibody responses to YF-17D6). These results confirmed the results obtained by flow cytometry and ELISPOT, with which we observed a greater frequency of IgG+ and IgA+ ASCs in the blood of vaccinees at day 7 after vaccination with TIV (Fig. A17-1 and Supplementary Fig. 1).
In addition to the ASC signature, we observed a signature composed of several genes encoding molecules that orchestrate the unfolded protein response (Iwakoshi et al., 2003b; Ron and Walter, 2007) (data not shown). The large number of XBP-1 target genes with differences in expression after vaccination was consistent with a role for XBP-1 in orchestrating the differentiation of plasma cells (Iwakoshi et al., 2003b). Among those, genes such as ATF6, MANF, CREB3, PDIA4, DNAJB11, HSP90B1, HERPUD1 and DNAJB9 encode molecules that are already known to be involved in the unfolded protein response (Park et al., 2010; Liu and Li, 2008; Apostolou et al., 2008).
In contrast to results obtained for TIV, analysis of the transcriptional signature induced by LAIV by meta-analysis showed considerable enrichment for genes with high expression in T cells and monocytes (Fig. A17-3e). We also found many genes with high expression in natural killer cells, although these results did not reach statistical significance (data not shown). Among the interferon-related genes upregulated after vaccination with LAIV (Fig. A17-2a), most had high expression in monocytes and natural killer cells (data not shown). That result was similar to our microarray analysis of flow cytometry–sorted cells obtained from subjects vaccinated with LAIV, in which most interferon-related genes with differences in expression in PBMCs and at least one cell subset had high expression in monocytes (Supplementary Fig. 4e). These results indicate that the innate immune responses can have an important role in the mechanism of action of this live attenuated virus vaccine.
Signatures That Correlate With the Antibody Response
Vaccination with TIV induced considerable variation in the magnitude of the HAI response (Fig. A17-1a). To gain insight into the potential mechanisms underlying that variation and to identify gene signatures with which we could predict the magnitude of the HAI response, we searched for early gene signatures that correlated with the B cell responses at days 7 and 28 after vaccination with TIV (complete list, Supplementary Table 5). Pearson correlation analysis identified 600–1,100 probe sets that correlated, either directly or inversely, with the magnitude of the HAI response (Fig. A17-4a). Among those were several genes known to be regulated by XBP-1 and to be involved in the differentiation of plasma cells and the unfolded protein response (Fig. A17-4b).
Ingenuity pathway analysis of the genes that were either positively or negatively correlated with HAI titers showed enrichment for genes related to the cell-mediated immune response and to the infection mechanism and inflammatory response, respectively (Supplementary Fig. 6a,b). The identification of genes such as TLR5, CASP1, PYCARD, NOD2 and NAIP suggested previously unknown mechanistic links between host innate immunity and humoral responses to influenza vaccination. In fact, research has shown that a candidate vaccine against influenza composed of a recombinant fusion protein linking influenza antigens to the Toll-like receptor 5 ligand flagellin may induce potent immunogenicity in mice (Huleatt et al., 2008) and humans (Treanor et al., 2010). In addition, canonical pathways, such as T cell receptor antigen receptor signaling and CTLA-4 signaling in cytotoxic T lymphocytes, included many of the genes present in the cell-mediated immune response network and were among those with the highest enrichment score by ingenuity pathway analysis (Supplementary Fig. 7a,b). Although further experimentation is needed, these data indicated a possible association between cellular responses and humoral responses to vaccination with TIV (He et al., 2006). Among the top canonical pathways enriched
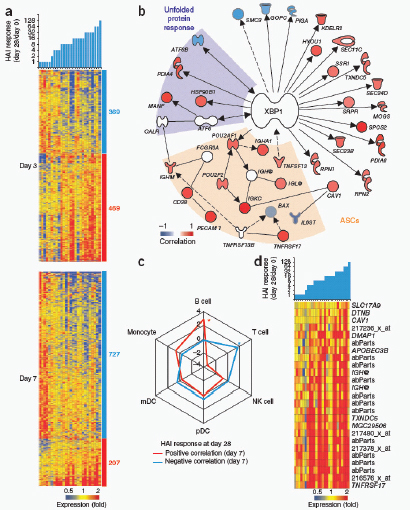
FIGURE A17-4 Molecular signatures that correlate with titers of antibody to TIV. (a) Heat map of probe sets (rows) and subjects (columns) whose baseline-normalized expression at day 3 (top) or day 7 (bottom) correlated with baseline-normalized antibody response at day 28 after vaccination with TIV (colors in map indicate gene expression at day 3 or 7 relative to expression at day 0). Right margin, number of probe sets with negative correlation (blue) or positive correlation (red). Probe sets that correlated with the HAI response on both day 3 and day 7 were considered ‘day 7’. P < 0.05 (Pearson). (b) HAI response–correlated genes associated with the unfolded protein response (purple shading) or ASC differentiation (tan shading) and/or regulated by XBP-1 (solid and dashed lines as in Fig. A17-2a). P < 0.05 (Pearson). (c) Enrichment for genes (among those with high expression in any PBMC subset) whose expression on day 3 or 7 after vaccination with TIV was positively or negatively correlated with HAI titers (cutoff, P < 0.05 (Pearson)). *P < 10−10 (two-tailed Fisher’s exact test). (d) Heat map of probe sets with high expression in B cells and ASCs whose baseline-normalized expression correlated with the baseline-normalized HAI response. P < 0.05 (Pearson). Data are representative of one experiment with 28 subjects.
for genes positively correlated to HAI response (Supplementary Fig. 7c), we found networks associated with innate immunity, such as the natural killer cell signaling network, and network for the production of nitric oxide and reactive oxygen species in macrophages (Supplementary Fig. 7d). Our analysis also showed that the expression of interferon-related genes (including those encoding the receptors for interferon-α and interferon-γ) on day 3 after vaccination was correlated to the HAI response (Supplementary Fig. 8), which suggested a link between the interferon response and the antibody response (Le Bon et al., 2006).
Next we compared the genes whose expression correlated with the HAI response at day 28 after vaccination of subjects with TIV with the genomic signatures of the cells of the immune response defined by our meta-analysis. This approach showed that the set of genes positively correlated to HAI response was enriched for genes with high expression in B cells (Fig. A17-4c) and, more specifically, in the ASC subset (Fig. A17-4d). The genes with negative correlation to the HAI response were substantially enriched among the genes with high expression in T cells (Fig. A17-4c), which supported the identification of the T cell pathways by ingenuity pathway analysis (Supplementary Fig. 7a,b). Together these data demonstrated the identification of early signatures that correlated with later HAI titers induced by TIV.
Molecular Signatures to Predict Antibody Responses
Once we had delineated signatures that correlated with the magnitude of HAI response, our next step was to identify the minimum sets of genes we could use to predict such a response. Ideally, such sets of genes must be able to be used to accurately classify high responders versus low responders in additional and independent TIV trials. For this, we used DAMIP (discriminant analysis via mixed integer programming [Lee, 2007; Brooks and Lee, 2010]), which is a very powerful supervised-learning classification method for predicting various biomedical and ‘biobehavioral’ phenomena (Lee, 2007; Brooks and Lee, 2010).
In initial analyses, we classified the subjects vaccinated with TIV into two ‘extreme’ groups: very low HAI responders, and very high HAI responders. The former group consisted of subjects with an increase of twofold or less in HAI titers against any of the three influenza strains of the vaccine (Fig. A17-1a). The latter group consisted of subjects with an increase of eightfold or more in the HAI response for at least one of the three influenza strains of the vaccine. We did not analyze subjects with intermediate HAI response (between twofold and eightfold) and subjects for whom microarray data were not available at either day 3 or day 7 after vaccination (n = 7). We used that trial (the 2008–2009 trial) to train the DAMIP model to establish an unbiased estimate of correct classification. We used a second, independent trial to evaluate the predictive accuracy of the classification rules identified in the first trial (Fig. A17-5a). The second trial (the 2007–2008 trial) consisted of the microarray gene-expression profiles of subjects
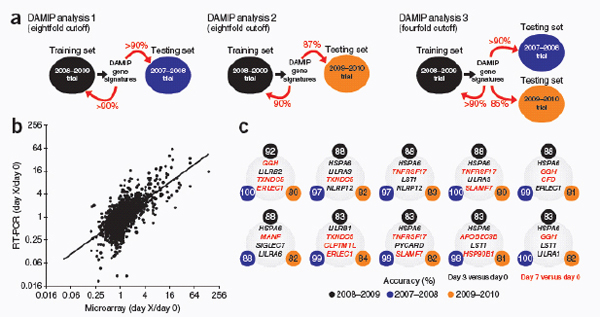
FIGURE A17-5 Signatures that can be used to predict the antibody response induced by TIV. (a) Experimental design used to identify the early gene signatures that can be used to predict antibody responses to vaccination with TIV: the 2008–2009 trial was used as a training set to identify predictive signatures with the DAMIP model; those signatures were then tested on the data from the 2007–2008 trial (the testing set). The expression of a subset of genes in the DAMIP predictive signatures of the 2007–2008 and 2008–2009 trials was then quantified by RT-PCR in a third independent trial (2009–2010 trial); the DAMIP model was again used to confirm the predictive signatures. (b) RT-PCR confirmation of the expression of a subset of genes in the predictive signatures generated by the DAMIP model. Each symbol represents a single gene at a given time point. P < 10−11, microarray versus RT-PCR (Pearson); r = 0.68; n = 2,897 XY pairs. Data are representative of one experiment with 44 genes from 28 subjects at two time points. (c) DAMIP gene signatures identified with the 2008–2009 trial as the training set and the 2007–2008 and 2009–2010 trials as the validation sets (DAMIP model 3); the accuracy represents the number of subjects correctly classified as ‘low responders’ or ‘high responders’ (Fig. A17-1a). Data are representative of three independent experiments.
(n = 9) vaccinated with TIV in the previous year. With this approach, DAMIP model identified 12 sets of genes containing two to four genes each (each set associates with one predictive rule) from 2008–2009 trial with a tenfold cross-validation accuracy over 90%. The resulting ‘blind prediction’ accuracy of the 2007–2008 trial (predicting low or high responders) was over 90%. Furthermore, some of the 271 sets of discriminatory genes offered an accuracy of over 90% in both tenfold cross-validation in the training trial and ‘blind prediction’ accuracy (Fig. A17-5a and Supplementary Table 6).
We then used real-time RT-PCR to confirm that 44 genes from the DAMIP gene signatures encoded molecules with potential biological relevance and/or utility as a predictor of influenza vaccine immunogenicity. We found a significant positive correlation (r = 0.679; P = 3.25 × 10−12) for changes in expression on day 3 or 7 relative to baseline expression as detected by microarray and RT-PCR (Fig. A17-5b and Supplementary Table 7), which confirmed the correctness of the microarray data. More notably, that result gave us confidence to test some of the candidate predictors of immunogenicity in a third and independent influenza vaccine trial (Fig. A17-5a). We collected RNA from PBMCs of subjects (n = 30) vaccinated with TIV during the 2009–2010 influenza season and analyzed this RNA by real-time RT-PCR. We then used the expression of the 44 genes selected from the initial DAMIP gene signatures to confirm their utility in predicting the magnitude of antibody response in this third TIV trial (Fig. A17-5a). To avoid the identification of ‘over-trained’ rules, we re-ran the DAMIP analysis using the 2008–2009 trial as the training set and the 2007–2008 and 2009–2010 trials as the blind predictive sets. This approach identified 47 sets of genes; some of these we used to correctly classify >85% of the vaccines as being very low HAI responders or very high HAI responders in any of the three trials (Supplementary Table 8).
Because seroconversion after vaccination is widely defined as a fourfold increase in HAI titers (Sullivan et al., 2010), we ran an additional DAMIP analysis using a cutoff of fourfold to classify the vaccinees (Fig. A17-5a). Thus, we classified subjects with an increase of fourfold or greater in the HAI titers after vaccination as ‘high responders’ and those with an increase of twofold or less as ‘low responders’. With the 2008–2009 trial as a training set and 2007–2008 and 2009-2010 trials as blind predictive sets, the DAMIP model generated 42 sets of gene signatures (Fig. A17-5c), each composed of three to four discriminatory genes, some of which had an unbiased estimate of correct classification above 85%, as determined by tenfold cross-validation and blind prediction (Supplementary Table 9).
One of the genes present in the DAMIP gene signatures, TNFRSF17, was also identified in DAMIP models used to predict antibody responses to vaccination with YF-17D6.
Among the genes in the TIV DAMIP models, we found five members of the leukocyte immunoglobulin-like receptor family (Supplementary Table 9). These genes are expressed by immune-response cells of both myeloid and lymphoid lineages and the molecules they encode are thought to have an immunomodulatory
role in the innate and adaptive immune systems by regulating T cells and autoimmunity (Anderson and Allen, 2009; Thomas et al., 2010; Brown et al., 2004). Our meta-analysis showed that these genes had high expression in monocytes and myeloid DCs at day 3 after vaccination (data not shown). These results and the presence of five members of this family among markers of antibody responses to influenza vaccination raised the possibility of previously unknown roles for these innate immune receptors in regulating antibody responses.
CaMKIV Regulates the Antibody Response
To demonstrate that the gene signatures identified in our study could be used to generate new hypotheses, we selected one gene in the predictive signature, CAMK4, for functional confirmation experiments. CaMKIV is involved in several processes of the immune system, such as T cell development (Krebs et al., 1997; Wang et al., 2001; Anderson and Means, 2002), inflammatory responses (Illario et al., 2008; Sato et al., 2006) and the maintenance of hematopoietic stem cells (Kitsos et al., 2005). However, nothing is known about the possible role of CaMKIV in B cell responses.
The change in CAMK4 expression on day 3 after vaccination with TIV was negatively correlated with the antibody response on day 28 after vaccination in two independent trials (Fig. A17-6a). Additionally, the change in CAMK4 expression was negatively correlated with the population expansion of IgG-secreting plasmablasts at day 7 (Fig. A17-6b), which suggested a possible role for CaMKIV in the regulation of antibody responses to vaccination against influenza.
In vitro stimulation of mouse splenocytes with TIV resulted in phosphorylation of CaMKIV (Fig. A17-6c), which suggested that this vaccine may trigger activation of CaMKIV. That finding was further demonstrated in human PBMCs, in which in vitro stimulation with influenza vaccine resulted in phosphorylation of CaMKIV as early as 2 h after stimulation (Fig. A17-6d). The mechanism by which this occurs remains to be identified.
To check if CaMKIV regulates the antibody response to influenza vaccine, we immunized wild-type and Camk4−/− mice with TIV and measured serum concentrations of IgG1 and IgG2c on days 7, 14 and 28 after vaccination (Fig. A17-6e). After immunization, Camk4−/− mice had a significantly greater antibody response than that of wild-type mice (Fig. A17-6e). The biggest difference was on day 7, with 3- to 6.5-fold higher antibody titers in Camk4−/− mice than in wild-type mice (Fig. A17-6e). These results supported our prediction based on the microarray results and suggested that CaMKIV is important in the regulation of B cell response.
Discussion
Despite the great success of vaccines, little is understood about the mechanisms by which effective vaccines stimulate protective immune responses. Two
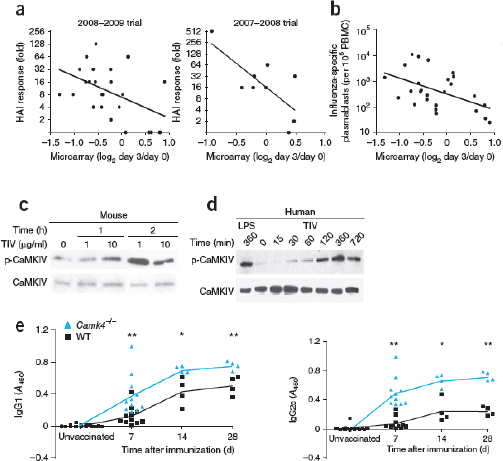
FIGURE A17-6 CaMKIV regulates the antibody response to vaccines against influenza. (a) HAI response at day 28 versus microarray analysis of CAMK4 mRNA in PBMCs at day 3 after vaccination with TIV in the 2008–2009 trial (left; r = −0.47 (Pearson); P = 0.016 (for Pearson correlation; two-tailed test) or the 2007–2008 trial (right; r = −0.73 (Pearson); P = 0.024 (for Pearson correlation; two-tailed test). (b) ELISPOT analysis of influenza-specific IgG–secreting plasmablasts at day 7 versus microarray analysis of CAMK4 mRNA on PBMCs at day 3 after vaccination with TIV. (c) Immunoblot analysis of the phosphorylation (p-) of mouse CaMKIV after in vitro stimulation of splenocytes for 1 or 2 h with various doses of TIV (above lanes). (d) Immunoblot analysis of the phosphorylation of CaMKIV after in vitro stimulation of human PBMCs for 0–720 min with lipopolysaccharide (LPS) or TIV. (e) Serum antigen-specific IgG1 (top) and IgG2c (bottom) responses of wild-type and Camk4−/− mice at days 7, 14 and 28 after immunization with TIV (symbols represent individual mice), presented as absorption at 450 nm (A450). *P < 0.05 and **P < 0.01 (Student’s t-test). Data are representative of one trial each with 26 subjects (2008–2009) or 9 subjects (2007–2008); (a), one experiment with 26 subjects (b), three experiments (c,d) or at least four independent experiments (e).
developments are beginning to offer such understanding: increasing appreciation of the key roles of the innate immune system in sensing vaccines and tuning immune responses, and emerging advances in systems biology (Pulendran and Ahmed, 2011). A systems biology approach has been used to obtain a global picture of the immune responses in humans to vaccine YF-17D against yellow fever, one of the most successful vaccines ever developed. This approach has identified unique biomarkers (molecular signatures) used to predict the magnitude of the antigen-specific CD8+ T cell and antibody responses induced by YF-17D6,7 and has resulted in the formulation of new hypotheses about the mechanism of action of this vaccine. However, whether such an approach could have broad utility in the identification of signatures of immunogenicity of other kinds of vaccines, particularly inactivated vaccines, and whether such signatures would be informative about the underlying mechanisms of immunity remain unknown. To address these issues, we did a series of studies over three consecutive influenza seasons. The goal of these studies was to analyze in detail the innate and adaptive immune responses to vaccination with two vaccines against influenza, TIV and LAIV, to identify early molecular signatures that can be used to predict later immune responses and to obtain insight into the mechanisms that underlie immunogenicity. According to guidelines established by the US Food and Drug Administration (US Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research, 2007), seroconversion can be defined as an HAI titer of 1:40 or more and a minimum increase of fourfold in antibody titer after vaccination. However, it often takes several weeks after vaccination to achieve this titer; therefore, the ability to predict seroconversion just a few days after vaccination and identify nonresponders would be of great value from a public health perspective. We thus used systems biology approaches to identify early signatures that we used to predict HAI titers 4 weeks after vaccination. To accomplish this goal, we used an interdisciplinary approach, including gene-expression profiling by microarray, RT-PCR and computational methods, combined with cellular and molecular biological approaches, as well as experiments involving genetically deficient mice. Our data have demonstrated that such a systems biology approach can indeed be used not only to identify predictive signatures but also to obtain new insights about the immunological mechanisms involved.
Although the clinical effectiveness of both vaccines is similar, LAIV induces lower serum antibody response in adults than does TIV (Sasaki et al., 2007, 2008; Moldoveanu et al., 1995). This probably reflects the lower ‘take’ of LAIV because of preexisting mucosal IgA that can neutralize the virus (Beyer et al., 2002). Nevertheless, our microarray analysis identified a large number of genes with differences in expression, most related to the type I interferon response, in the PBMCs of subjects vaccinated with LAIV. Future studies should focus on analyzing changes in the transcriptome of the nasal mucosa after vaccination with LAIV and how that correlates with or can be used to predict local antibody responses.
Among the genes induced by vaccination with TIV, we found enrichment
for genes with high expression in ASCs. This result may have reflected the rapid proliferation of plasmablasts at day 7 after vaccination (Wrammert et al., 2008); however, our microarray analysis of B cells sorted from subjects vaccinated against influenza indicated that the changes in expression observed in PBMCs could also have been derived from real transcriptional changes in B cells. The transcription factor XBP-1, which is essential for the differentiation of ASCs and the unfolded protein response (Iwakoshi et al., 2003a), and its target genes were upregulated after vaccination with TIV and correlated with IgG and HAI responses. The genes identified by our study may offer new opportunities for studying the complex mechanisms involved in the unfolded protein response and its link to ASC differentiation (Iwakoshi et al., 2003a).
A key question was whether the signatures that can be used to predict the T cell and B cell response to one vaccine can also be used to predict such responses to another vaccine. Notably, of the 133 genes present in the 271 DAMIP gene signatures that we used to predict the antibody response to vaccination with TIV, 7 were also predictors of the antibody response to vaccination with the YF-17D vaccine against yellow fever6. Key genes in the predictive signatures were TNFRSF17, which encodes BCMA, a receptor for the B cell growth factor BLyS (known to have a key role in B cell differentiation; Avery et al., 2003), and CD38, which encodes a surface protein important in lymphocyte development (Shubinsky and Schlesinger, 1997; Deaglio et al., 2001). BCMA belongs to a family of molecules (BAFF, APRIL, BAFF-R and TACI) that regulate the differentiation of plasma cells and antibody production (Avery et al., 2003). Notably, there were strong correlations between the expression of genes encoding APRIL, BAFF-R and TACI and the magnitude of the HAI titers in response to vaccines against influenza and the magnitude of neutralizing antibody response to YF-17D (data not shown), which suggested that this network may be critically involved in regulating antibody responses to different vaccines. The functional relevance of this network in mouse models remains to be determined. It also remains to be seen whether this network represents a common predictor of antibody responses induced by many vaccines.
A second issue was whether the data generated from such studies would be useful in providing new biological insights into the regulatory mechanisms that underlie vaccine immunogenicity. Our experiments with Camk4−/− mice demonstrated that such data can indeed identify unexpected biological targets, which can be mechanistically confirmed by mouse models. Although the data demonstrated a potent role for CaMK4 in regulating antibody responses to vaccines against influenza, further work is needed to delineate the cellular mechanisms involved.
Third, whether signatures that can be used to predict immunogenicity can also be used to predict efficacy must be considered. Several studies have shown that serum HAI antibody concentrations correlate with protection against influenza (Clements et al., 1986; Potter and Oxford, 1979; Hirota et al., 1997). Seroconversion after vaccination, commonly defined as an increase of fourfold in HAI titers (Sullivan et al., 2010), represents a useful surrogate for vaccine ef-
ficacy when applied to a population. However, this parameter may not provide the optimal prediction of protection in an individual vaccinee or a group of vaccinees. In addition, protective concentrations of antibody may vary according to the prevalent virus subtype and laboratory doing the assay (Belshe, 2004). Therefore, we used a more stringent parameter (an increase of eightfold or more in HAI response) to classify subjects with very high antibody responses. Using this cutoff in our analyses, the DAMIP method was able to identify gene signatures that we could use to predict the antibody response induced by vaccination with TIV. We confirmed the validity of these gene signatures in three independent trials, which demonstrated the robustness of our approach. To meet the definition of seroconversion in the US Food and Drug Administration Guidance for Industry document for this field (an HAI titer of 1:40 or more and a minimum increase of fourfold in antibody titer after vaccination) (US Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research, 2007), we reran the DAMIP analysis using an increase of fourfold as a cutoff for defining high HAI responders. Again, the DAMIP method was able to identify sets of three to four discriminatory genes with an unbiased estimate of correct classification up to 90% for the three influenza trials. However, the generality of our findings in terms of using gene signatures in PBMCs to predict the immunogenicity and/or efficacy of other vaccines such as mucosal vaccines must be tested. It is likely that different signatures could be generated by analysis of mucosal tissues.
Finally, although the main goal of our study was a proof-of-concept demonstration of the feasibility of this approach in predicting vaccine immunogenicity, (rather than a demonstration of cost effectiveness), in ascertaining the predictive value of our signature in the 2009–2010 trial, we used a PCR-based assay (instead of an assay with gene-expression chips) of only a handful of genes. This demonstrated the feasibility of designing a cost-effective, PCR-based ‘vaccine chip’ that can be used to predict the immunogenicity of vaccines. Thus, we have shown how systems biology approaches can be applied to elucidate the molecular mechanisms of influenza vaccines. We envision that the predictive signatures of influenza vaccine–induced antibody responses may have implications in vaccine development, in the monitoring of suboptimal immune responses (in the elderly, infants or immunocompromised populations) or perhaps in identifying new correlates of protection.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/natureimmunology/.
Accession codes. GEO: microarray data, GSE29619.
Note: Supplementary information is available on the Nature Immunology website.
Acknowledgments
We thank B.T. Rouse and R. Compans for discussion and comments on the manuscript, and H. Oluoch for technical assistance. Supported by the US National Institutes of Health (U19AI090023, HHSN266200700006C, U54AI057157, R37AI48638, R01DK057665, U19AI057266 and N01 AI50025 for the B.P. laboratory; AI30048 and AI057266 for the R.A. laboratory; DK074701 for the A.R.M. laboratory; Intramural Research Program of the National Institute of Allergy and Infectious Diseases for the K.S. laboratory; and UL1 RR025008 from the Clinical and Translational Science Award program, National Center for Research Resources for clinical work), the Bill & Melinda Gates Foundation (Collaboration for AIDS Vaccine Discovery 38645 to the R.A. and B.P. laboratories), the National Science Foundation (E.K.L. laboratory) and the Centers for Disease Control (E.K.L. laboratory).
Author Contributions
H.I.N. did all the experiments and analyses in Figures A17-2–6 and Supplementary Figures 2–8; J.W., G.-M.L., M.M. and V.K. did the analyses in Figure A17-1 and Supplementary Figure 1; E.K.L. did the DAMIP model analyses in Figure A17-5; L.R., A.R.M., S.P.K. and N.K. did the mouse experiments in Figure A17-6; W.N.H. helped with the microarray analyses in Supplementary Figure 4; S.L. assisted with the bioinformatics analyses of the data in Figure A17-3; A.A. did the microarray analysis of samples from the 2007 influenza annual season; S.M.-K., K.E.K., R.E. and A.K.M. assisted with the collection and processing of samples; K.S. measured HAI titers; R.A. helped conceive of and design the study and supervised the studies in Figure A17-1 and Supplementary Figure 1; B.P. conceived of the study and designed and supervised the experiments and analyses in Figures A17-1–6 and Supplementary Figures 1–8; and B.P. and H.I.N. wrote the paper.
Competing Financial Interests
The authors declare no competing financial interests.
Published online at http://www.nature.com/natureimmunology/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
Anderson, K.A. & Means, A.R. Defective signaling in a subpopulation of CD4+ T cells in the absence of Ca2+/calmodulin-dependent protein kinase IV. Mol. Cell. Biol. 22, 23–29 (2002).
Anderson, K.J. & Allen, R.L. Regulation of T-cell immunity by leucocyte immunoglobulin-like receptors: innate immune receptors for self on antigen-presenting cells. Immunology 127, 8–17 (2009).
Apostolou, A., Shen, Y., Liang, Y., Luo, J. & Fang, S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp. Cell Res. 314, 2454–2467 (2008).
Avery, D.T. et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 112, 286–297 (2003).
Barrett, A.D. & Teuwen, D.E. Yellow fever vaccine–how does it work and why do rare cases of serious adverse events take place? Curr. Opin. Immunol. 21, 308–313 (2009).
Belshe, R.B. Current status of live attenuated influenza virus vaccine in the US. Virus Res. 103, 177–185 (2004).
Beyer, W.E., Palache, A.M., de Jong, J.C. & Osterhaus, A.D. Cold-adapted live influenza vaccine versus inactivated vaccine: systemic vaccine reactions, local and systemic antibody response, and vaccine efficacy. A meta-analysis. Vaccine 20, 1340–1353 (2002).
Brooks, J.P. & Lee, E.K. Analysis of the consistency of a mixed integer programming-based multicategory constrained discriminant model. Ann. Oper. Res. 174, 147–168 (2010).
Brown, D., Trowsdale, J. & Allen, R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens 64, 215–225 (2004).
Clements, M.L., Betts, R.F., Tierney, E.L. & Murphy, B.R. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J. Clin. Microbiol. 24, 157–160 (1986).
Deaglio, S., Mehta, K. & Malavasi, F. Human CD38: a (r)evolutionary story of enzymes and receptors. Leuk. Res. 25, 1–12 (2001).
Fiore, A.E. et al. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm. Rep. 59, 1–62 (2010).
Gaucher, D. et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 205, 3119–3131 (2008).
He, X.S. et al. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J. Virol. 80, 11756–11766 (2006).
Hirota, Y. et al. Antibody efficacy as a keen index to evaluate influenza vaccine effectiveness. Vaccine 15, 962–967 (1997).
Huleatt, J.W. et al. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine 26, 201–214 (2008).
Illario, M. et al. Calmodulin-dependent kinase IV links Toll-like receptor 4 signaling with survival pathway of activated dendritic cells. Blood 111, 723–731 (2008).
Iwakoshi, N.N. et al. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4, 321–329 (2003a).
Iwakoshi, N.N., Lee, A.H. & Glimcher, L.H. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 194, 29–38 (2003b).
Johnson, P.R. Jr., Feldman, S., Thompson, J.M., Mahoney, J.D. & Wright, P.F. Comparison of long-term systemic and secretory antibody responses in children given live, attenuated, or inactivated influenza A vaccine. J. Med. Virol. 17, 325–335 (1985).
Kitsos, C.M. et al. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J. Biol. Chem. 280, 33101–33108 (2005).
Krebs, J., Wilson, A. & Kisielow, P. Calmodulin-dependent protein kinase IV during T-cell development. Biochem. Biophys. Res. Commun. 241, 383–389 (1997).
Le Bon, A. et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 176, 2074–2078 (2006).
Lee, E.K. Large-scale optimization-based classification models in medicine and biology. Ann. Biomed. Eng. 35, 1095–1109 (2007).
Liu, B. & Li, Z. Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood 112, 1223–1230 (2008).
Moldoveanu, Z., Clements, M.L., Prince, S.J., Murphy, B.R. & Mestecky, J. Human immune responses to influenza virus vaccines administered by systemic or mucosal routes. Vaccine 13, 1006–1012 (1995).
Monath, T.P. Yellow fever vaccine. Expert Rev. Vaccines 4, 553–574 (2005).
Park, S.W. et al. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16, 429–437 (2010).
Potter, C.W. & Oxford, J.S. Determinants of immunity to influenza infection in man. Br. Med. Bull. 35, 69–75 (1979).
Pulendran, B. Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat. Rev. Immunol. 9, 741–747 (2009).
Pulendran, B. & Ahmed, R. Immunological mechanisms of vaccination. Nat. Immunol. 131, 509–517 (2011).
Pulendran, B., Li, S. & Nakaya, H.I. Systems vaccinology. Immunity 33, 516–529 (2010).
Querec, T. et al. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 203, 413–424 (2006).
Querec, T.D. et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 10, 116–125 (2009).
Ron, D. & Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 (2007).
Sasaki, S. et al. Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J. Virol. 81, 215–228 (2007).
——. Influence of prior influenza vaccination on antibody and B-cell responses. PLoS ONE 3, e2975 (2008).
Sato, K. et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat. Med. 12, 1410–1416 (2006).
Shen-Orr, S.S. et al. Cell type-specific gene expression differences in complex tissues. Nat. Methods 7, 287–289 (2010).
Shubinsky, G. & Schlesinger, M. The CD38 lymphocyte differentiation marker: new insight into its ectoenzymatic activity and its role as a signal transducer. Immunity 7, 315–324 (1997).
Sullivan, S.J., Jacobson, R. & Poland, G.A. Advances in the vaccination of the elderly against influenza: role of a high-dose vaccine. Expert Rev. Vaccines 9, 1127–1133 (2010).
Takaoka, A. & Yanai, H. Interferon signalling network in innate defence. Cell. Microbiol. 8, 907–922 (2006).
Talbot, H.K. et al. Immunopotentiation of trivalent influenza vaccine when given with VAX102, a recombinant influenza M2e vaccine fused to the TLR5 ligand flagellin. PLoS ONE 5, e14442 (2010).
Thomas, R., Matthias, T. & Witte, T. Leukocyte immunoglobulin-like receptors as new players in autoimmunity. Clin. Rev. Allergy Immunol. 38, 159–162 (2010).
Treanor, J.J. et al. Safety and immunogenicity of a recombinant hemagglutinin influenza-flagellin fusion vaccine (VAX125) in healthy young adults. Vaccine 28, 8268–8274 (2010).
Tusher, V.G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98, 5116–5121 (2001).
Ueda, Y. et al. Frequencies of dendritic cells (myeloid DC and plasmacytoid DC) and their ratio reduced in pregnant women: comparison with umbilical cord blood and normal healthy adults. Hum. Immunol. 64, 1144–1151 (2003).
US Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research. Guidance for Industry: Clinical Data Needed to Support the Licensure of Pandemic Influenza Vaccines (Office of Communication, Training and Manufacturers Assistance, Rockville, Maryland, 2007).
Wang, S.L., Ribar, T.J. & Means, A.R. Expression of Ca2+/calmodulin-dependent protein kinase IV (caMKIV) messenger RNA during murine embryogenesis. Cell Growth Differ. 12, 351–361 (2001).
Wrammert, J. et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008).
Zeman, A.M. et al. Humoral and cellular immune responses in children given annual immunization with trivalent inactivated influenza vaccine. Pediatr. Infect. Dis. J. 26, 107–115 (2007).
Sean C. Sleight,87Bryan A. Bartley, and Herbert M. Sauro88
Abstract
One problem with engineered genetic circuits and metabolic pathways in synthetic microbes is their stability over evolutionary time in the absence of selective pressure. We recently measured the evolutionary stability dynamics and determined the loss-of-function mutations for a wide variety of BioBrick-assembled genetic circuits in order to develop simple design principles for engineering mutationally robust genetic circuits (Sleight et al., 2010b). In this report, we focus on experiments performed with the LuxR receiver device, T9002, and reengineered versions of this circuit. T9002 loses function in fewer than 20 generations and the mutation that repeatedly causes its loss of function is a deletion between two homologous transcriptional terminators. To measure the effect between transcriptional terminator homology levels and evolutionary stability, we reengineered six versions of T9002 with a different transcriptional terminator at the end of the circuit. When there is no homology between terminators, the evolutionary half-life of this circuit is significantly improved by more than twofold and is independent of the expression level. Removing all homology between terminators and decreasing expression level fourfold increases the evolutionary half-life over 17-fold. We also investigated the predictability of loss-of-function mutations between nine replicate evolved populations. T9002 circuits reengineered to have no sequence similarity between terminators effectively removes a certain class of mutations from occurring, where the most common loss-of-function mutation is a deletion between 8-base pair (bp) scar sequences to remove the luxR promoter. Finally, we evolved T9002 and a reengineered version in media with different levels of inducer to measure the relationship between expression level and evolu-
_______________
87 Corresponding author: Sean C. Sleight, e-mail: sleight@u.washington.edu.
88 Department of Bioengineering, University of Washington, Seattle, WA 98195, USA.
tionary stability. The results show that on average, like other circuits we studied, evolutionary half-life exponentially decreases with increasing expression levels.
Introduction
Experimental evolution is a powerful system for studying evolution in the laboratory over short or long timescales (Elena and Lenski, 2003). For these experiments, normally bacteria or other microbes are propagated over multiple generations in certain environmental conditions to understand the phenotypic and genetic differences between evolved strains and their progenitors (Herring et al., 2006; Riehle et al., 2005; Sleight and Lenski, 2007). Experimental evolution experiments also allow for the study of parallel genetic or phenotypic changes between replicate evolved populations. When multiple evolved populations converge on a similar phenotype, it is often a strong indicator of evolutionary adaptation, as opposed to the product of genetic drift (Bull et al., 1997; Colosimo et al., 2005; Sleight et al., 2008). Understanding convergent evolution is of interest because it increases our ability to predict the evolution of unknown strains in different environments.
Synthetic biologists use engineering principles to design and construct new organisms that do not exist in nature (Endy, 2005). Often these synthetic organisms are programmed with genetic circuits that are constructed from biological parts such as promoters, ribosome binding sites, coding sequences, and transcriptional terminators called BioBricks (Endy, 2005; Shetty et al., 2008). The MIT Registry of Standard Biological Parts (called “the Registry” from here on) maintains over 3,000 BioBricks encoded on plasmids that are available to researchers with a wide variety of functions, from bacterial photography, to quorum sensing, to odor production and sensing. BioBricks are widely available for the design of more complex systems but in general are not well characterized (Canton et al., 2008; Endy, 2005). The most well characterized part to date is a cell-to-cell communication receiver device (Canton et al., 2008), which was provided with a published prototype “biological part data sheet” containing information engineers would need to use it in their own designs. One of the figures in this data sheet describes the reliability of this circuit over evolutionary time. Connecting the receiver device to a green fluorescent protein (GFP)-reporter device causes this circuit to repeatedly lose function in fewer than 100 generations due to a deletion mutation between transcriptional terminators that are repeated in both the receiver and reporter devices.
From a synthetic biologist’s perspective, ideally synthetic organisms will be robust to environmental conditions and mutations to allow for optimal function over evolutionary time. However, the genetic circuits encoded on plasmids in synthetic organisms are often unstable over evolutionary time if there is no selective pressure to maintain function of the circuit. This loss of function occurs because a cell in the population that has a mutation in one of its plasmids may have a slight growth advantage. When this cell divides, the daughter cell may acquire
more copies of the mutant plasmid, giving it a greater growth advantage. Over multiple generations, the functional cells in the population may be outcompeted by nonfunctional cells, unless these evolutionary events are controlled.
From the public health perspective, if a synthetic microbe escaped the laboratory into the environment, how long would it survive and how many generations would it take before its synthetic function was lost over evolutionary time? Could a synthetic pathogenic strain evolve to become more virulent over time? To our knowledge, these questions are largely unexplored, but from this perspective we hope that synthetic cells will not be robust to environmental conditions and mutations. As a first step toward understanding the evolutionary stability dynamics and predictability of loss-of-function mutations in genetic circuits, we evolved replicate populations of Escherichia coli carrying several different genetic circuits for multiple generations (Sleight et al., 2010b).
In this study, we found that a wide variety of loss-of-function mutations are observed in BioBrick-assembled genetic circuits including point mutations, small insertions and deletions, large deletions, and insertion sequence (IS) element insertions that often occur in the scar sequence between parts. Promoter mutations are selected more than any other biological part. We also found that, on average, evolutionary half-life exponentially decreases with increasing expression levels. Surprisingly, one particular circuit that was expressed at a low level due to having a weak promoter was able to maintain function for 500 generations, but most circuits lost function within 100 generations. We were able to reengineer genetic circuits to be more mutationally robust using a few simple design principles: high expression of genetic circuits comes with the cost of low evolutionary stability, avoid repeated sequences, and the use of inducible promoters increases stability. Interestingly, inclusion of an antibiotic resistance gene within the circuit does not ensure evolutionary stability, but a reengineered version of this circuit may improve stability when certain environmental conditions are used.
In this report, we focus on the results of this study from experiments performed with the LuxR receiver device, T9002, and six reengineered versions of this circuit. We then discuss the relevance of these results for emerging microbial pathogens and future directions.
Results
Loss-of-Function Mutations and Evolutionary Stability Dynamics in the T9002 Circuit
We first measured the evolutionary stability dynamics of the T9002 genetic circuit (Canton et al., 2008) propagated in Escherichia coli MG1655 in order to determine the loss-of-function mutations that cause its instability. High-copy plasmids were used instead of low- or medium-copy plasmids to maximize selective pressure so that evolution would occur more rapidly since replication and expression of genetic circuits encoded on high-copy plasmids will increase
metabolic load and lower fitness. Cells with a low metabolic load (e.g., cells with mutant plasmids) have greater fitness than cells with a higher metabolic load (e.g., cells with functional plasmids) (unpublished results). Therefore, we expect that mutants will be able to rapidly outcompete functional cells that have a high expression level. However, other factors besides expression level will play a role in this evolutionary process such as mutation rate and the metabolic load associated with plasmid replication.
T9002 is a Lux receiver circuit that expresses luxR that activates GFP expression when the inducer AHL is added to the media (Figure A18-1). The evolutionary stability dynamics were measured by serial propagation with a dilution factor that allows for about 10 generations per day. Figure A18-2 shows the evolutionary stability dynamics of the T9002 circuit propagated in high-input (with AHL) and low-input (without AHL) conditions. From different time points in the experiment, the low- and high-input populations were induced with AHL to measure their normalized expression (here measured by fluorescence divided by cell density) over time. The low-input evolved populations slowly lose their function to about 50 percent of the maximum after 300 generations. The evolved populations in high-input conditions rapidly lose their function in fewer than 30 generations. The dynamics of this evolutionary stability are described below in Figure A18-5. No functional clones were observed after 30 generations as determined by measurement of individual colonies. The mutation that repeatedly causes loss of function in the high-input evolved populations is a deletion between two homologous transcriptional terminators (Figure A18-3), the same mutation described by Canton et al. (2008). This mutation evidently occurs at such a high rate that mutants quickly take over the population. In fact, Canton et al. (2008) were unable to isolate a population derived from a single isolate that did not already carry the deletion. The mutant plasmid was transformed back into the progenitor and was shown not to fluoresce after induction with AHL.
Evolution Experiment with Reengineered Circuits
Based on the results of the previous experiments, we reengineered the T9002 circuit to test various predictions of evolutionary stability and mutational predictability. The loss-of-function mutation was a deletion that repeatedly occurred between two homologous transcriptional terminators. Mutations and genetic rearrangements can occur due to misalignment of homologous sequences during replication (termed “replication slippage”) (Lovett, 2004). Deletion mutations between repeated sequences are known to be dependent on repeat length, proximity, and homology level (Bzymek and Lovett, 2001). These deletions are recAindependent if the repeat length is less than 200 bp (Bi and Liu, 1994; Lovett, 2004), as is the case with the repeated terminators in the T9002 circuit. Thus, we reengineered the last terminator of T9002 with various terminators available in the Registry to measure the effect of terminator homology level and orientation with evolutionary stability (Figure A18-4). We predicted that we could increase
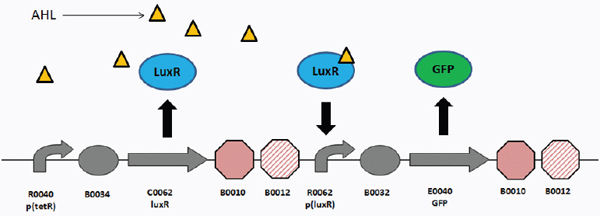
FIGURE A18-1 The T9002 genetic circuit. Symbols depict promoters (bent arrows), ribosome binding sites (ovals), coding sequences (arrows), and transcriptional terminators (octagons). T9002 consists of two devices, a LuxR receiver device and a GFP-expressing device. The first device is composed of the tetR-regulated promoter R0040 (which is constitutively expressed in the MG1655 strain since it does not produce TetR), B0034 RBS, C0062 luxR coding sequence, and B0010-B0012 (B0015) transcriptional terminator. The second device is composed of the R0062 luxR promoter, B0032 RBS, E0040 gfp coding sequence, and B0015 transcriptional terminator. LuxR is constitutively expressed from the tetR promoter. When the inducer 3OC6HSL (AHL) is added to the media, it binds with LuxR to activate transcription of GFP from the luxR promoter. If no AHL is in the media, the circuit is off.
SOURCE: This figure is taken from the Journal of Biological Engineering 4(1), 12. doi: 10.1186/1754-1611-4-12.
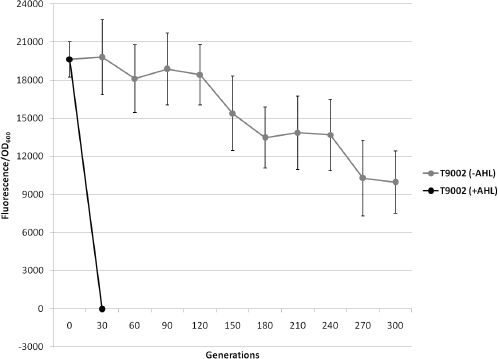
FIGURE A18-2 Evolutionary stability dynamics of T9002 evolved under low-input (−AHL) and high-input (+AHL) conditions. Low- and high-input evolved populations are shown with solid gray circles and solid black circles, respectively. Evolved populations at different time points were grown with AHL to measure relative GFP levels. Relative fluorescence normalized by OD is plotted versus generations. Error bars represent one standard deviation from the mean of nine independently evolved populations.
SOURCE: This figure is taken from the Journal of Biological Engineering, 4(1), 12. doi: 10.1186/1754-1611-4-12.
evolutionary robustness by decreasing the mutation rate of this deletion. Furthermore, although there have been several studies on recombination between repeated sequences, this phenomenon has not been studied in the context of synthetic biology using genetic circuits constructed from BioBricks. For instance, we do not know the effect of using various BioBrick terminators with different homology levels in the same circuit. The use of different terminators will become increasingly important when more complex circuits are constructed and BioBricks become even more widespread in the community. Also, many of the studies on recombination between repeated sequences use antibiotic resistance genes to measure recombination rates and may not relate to actual functioning circuits.
Since we learned from previous experiments that evolutionary stability dynamics of genetic circuits have high variability between replicate populations, we evolved nine independent populations of each reengineered circuit for at least 300 generations. Three experimental replicate populations of three independent
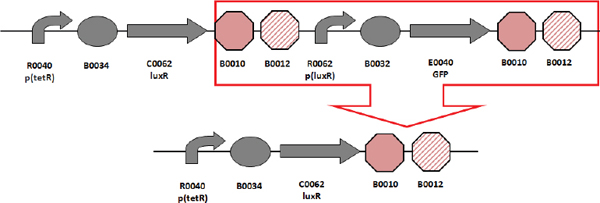
FIGURE A18-3 T9002 loss-of-function mutation. This circuit repeatedly has a deletion between homologous repeated terminators after 30 generations in the high-input evolved populations. This is the same mutation found by Canton et al. (2008).
SOURCE: This figure is taken from the Journal of Biological Engineering, 4(1), 12. doi: 10.1186/1754-1611-4-12.
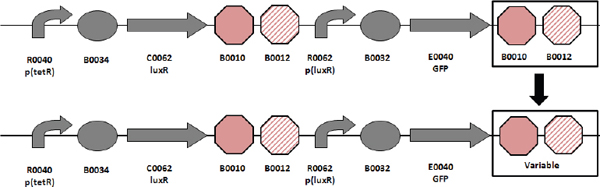
FIGURE A18-4 T9002 reengineering. T9002 reengineering involves changing the second double transcriptional terminator with varying degrees of homology and orientation to the first double transcriptional terminator.
SOURCE: This figure is taken from the Journal of Biological Engineering 4(1), 12. doi: 10.1186/1754-1611-4-12.
transformants were used to test for independent mutational events. A single transformant may have a mutation at a low level that will eventually sweep through the population, so if only one transformant was used, the same mutation may show up in all replication populations. For each of the nine populations in every circuit, the evolutionary half-life was measured to quantitate the number of generations until the expression level had decreased to half of its initial level (Table A18-1). Plasmids from a single clone from each evolved population were then sequenced after the population level had decreased to less than 10 percent of the original expression level, or after 500 generations, whichever came first.
Reengineered T9002 Circuits with Different Transcriptional Terminators: Loss-of-Function Mutations and Evolutionary Stability Dynamics
Figure A18-4 shows the schematic for reengineering the last transcriptional terminator in the T9002 circuit. The evolutionary stability dynamics for six reengineered T9002 circuits and the original T9002 circuit are shown in Figure A18-5. Figure A18-6 shows the types of mutations that occurred in each of the nine replicate evolved populations. Finally, Figure A18-7 shows the most common mutations for each reengineered circuit. The six reengineered circuits are labeled T9002-A through T9002-F in Figure A18-5 and are color coded to correspond to the same circuit mutations shown in Figure A18-7. These circuits were all propagated with the inducer AHL to allow for GFP expression.
In the following paragraphs, the loss-of-function mutations and evolutionary stability dynamics for the original T9002 circuit and for each reengineered circuit are described in detail.
T9002 The original T9002 circuit decreases rapidly to about 30 percent of the original level after only 10 generations and all function is lost by 20 generations (Figure A18-5). The same deletion between homologous terminators as was observed in previous experiments (Figure A18-3) was found in all nine replicate populations (Figure A18-6). The evolutionary half-life of this circuit was found to be about 7.1 generations on average (Table A18-1).
TABLE A18-1 Evolutionary Half-Life of T9002 and Reengineered T9002 Genetic Circuits
| Circuit | Evolutionary Half-life | SD |
| T9002 | 7.056 | 3.657 |
| T9002-A | 5.563 | 1.687 |
| T9002-C | 6.650 | 3.250 |
| T9002-D | 56.986 | 6.188 |
| T9002-E | 16.701 | 4.966 |
| T9002-F | 125.443 | 47.664 |
| NOTE: The mean evolutionary half-life for nine independently evolved populations is shown for each genetic circuit with the standard deviation (SD). See the Methods section on page 414 for details. | ||
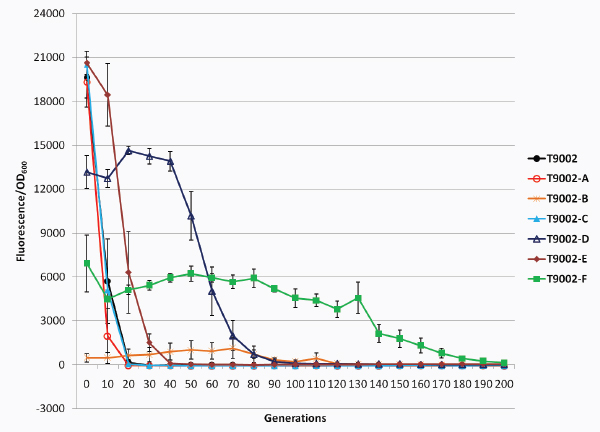
FIGURE A18-5 Evolutionary stability dynamics of T9002 and reengineered T9002 circuits. The fluorescence/OD600 is plotted versus the generations for T9002 (solid black circles) and T9002 reengineered circuits (various shapes and colors) under high-input (+AHL) conditions. Error bars represent one standard deviation from the mean of nine independently evolved populations.
SOURCE: This figure is taken from the Journal of Biological Engineering 4(1), 12. doi: 10.1186/1754-1611-4-12.
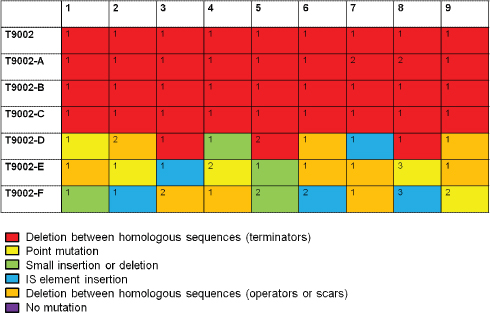
FIGURE A18-6 Loss of mutations in nine independently evolved populations. For nine independently evolved populations, colored boxes correspond to the mutation legend below the table. The most common mutation for a particular type of mutation is labeled with “1” in the boxes above and less common mutations are labeled with increasing numbers.
SOURCE: This figure is taken from the Journal of Biological Engineering 4(1), 12. doi: 10.1186/1754-1611-4-12.
T9002-A The final double terminator in T9002 was reengineered in the reverse complementary orientation (called B0025 in the Registry) to make T9002-A. The stability of this circuit has approximately the same expression level and stability dynamics as T9002, but it has an evolutionary half-life of about 5.6 generations (Figure A18-5, Table A18-1). This decreased stability may be because the terminator in the reverse orientation is more likely to cause replication slippage. Since the expression level is similar to T9002 and therefore the metabolic load should be roughly equivalent, the difference in stability is primarily due to an increased mutation rate. Seven of nine replication populations have a deletion between the first B0010 terminator and the reverse complement of B0010 (Figures A18-6 and A18-7). This effect likely occurs because B0010 has a long hairpin structure, so one-half of B0010 can interact with the other half of the reverse complementary B0010 sequence during DNA replication since they have perfect homology. Two of the nine populations had a deletion that formed a triple terminator of B0010-B0012-B0010.
T9002-B The T9002-B circuit was reengineered to rearrange the B0010 and B0012 terminators to have B0012 first and then B0010. The rearrangement de-
creases the expression level to almost zero initially and this expression drifts up over time and then decreases to zero (Figure A18-5). For this circuit, evolutionary half-life measurements are essentially meaningless due to the randomness of low expression. Notice that other reengineered T9002 circuits also have decreased expression levels relative to T9002, presumably due to weaker terminator hairpin structures having increased mRNA degradation (Smolke and Keasling, 2002) or transcriptional readthrough that can decrease plasmid copy number (Stueber and Bujard, 1982). Others have observed that removal of transcriptional terminators can decrease expression levels in general (Kaur et al., 2007). All nine populations have the same deletion between B0010 terminators (Figures A18-6 and A18-7). Because the B0010 terminator is an inexact hairpin (there are some mismatches), one-half of the first B0010 interacts with the other half of the second B0010 terminator, causing a hybrid B0010 terminator (Figure A18-7).
T9002-C The T9002-C circuit was reengineered to have B0012 and B0011 as the final double terminator instead of B0010 and B0012. This circuit has nearly identical stability dynamics as T9002, with an evolutionary half-life of about 6.7 generations on average (Figure A18-5, Table A18-1). All nine populations have the same deletion between B0012 terminators that make a triple terminator of B0010-B0012-B0011 (Figures A18-6 and A18-7). Since the expression level and stability dynamics are roughly equivalent to that of T9002, the mutation rate between repeated B0010-B0012 terminators (129 bp) is probably about the same as that between repeated B0012 terminators (41 bp). Interestingly, no significant stability difference was observed between T9002-C (41-bp homology) and T9002 or T9002-A (both 129-bp homology), despite having similar expression levels. This result suggests that shortening the repeated regions of homologous terminators did not increase evolutionary robustness, contrary to what we expected.
T9002-D The T9002-D circuit has the same final B0012-B0011 terminator but is the reverse complement of this sequence. The inclusion of this terminator decreases the initial expression level to about 65 percent of T9002 (Figure A18-5). The evolutionary half-life of this circuit is about 57 generations (Figure A18-5, Table A18-1). Also, the slope of the stability plot is decreased relative to other circuits with higher expression (T9002, T9002-A, T9002-C, and T9002-E) and the stability lag (time for expression to decrease to zero along the x axis) is increased (Figure A18-5). In contrast to other circuits with repeated terminators, only three of nine have deletions between homologous terminators, forming a hybrid B0012 terminator (Figures A18-6 and A18-7). This result is probably because, unlike T9002-C, the second B0012 is the reverse complement, and therefore the only homology in this circuit is the 8-bp hairpin structure having complementary sequences; the rest of the terminator has a loop structure of noncomplementary sequences. In other words, B0010 has sufficient homology in either the forward or reverse orientation to cause replication slippage, but in B0012 replication slippage is more likely to occur only in the forward orientation. The other mutations
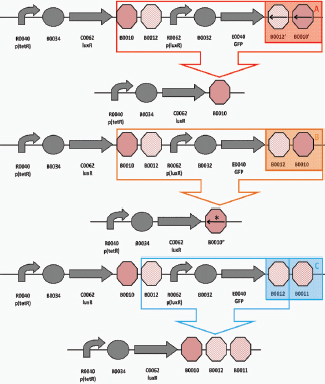
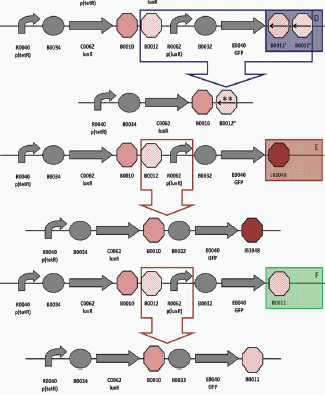
FIGURE A18-7 Most common loss-of-function mutations in reengineered T9002 circuits. The original circuit is shown above the loss-of-function mutation circuit for each of the six reengineered T9002 circuits.
SOURCE: This figure is taken from the Journal of Biological Engineering 4(1), 12. doi: 10.1186/1754-1611-4-12.
in this circuit are composed of point mutations, short insertions or deletions, deletions between scar sequences, or IS mutations (Figure A18-6).
T9002-E The T9002-E circuit, like the T9002-F circuit, was reengineered to have no homology between terminator sequences. This circuit has the highest initial expression level on average probably because J61048 is a very strong terminator, but has similar expression relative to the T9002, T9002-A, and T9002C circuits (Figure A18-5). Its evolutionary half-life is about 16.7 generations (Figure A18-5, Table A18-1). Thus, relative to other circuits with similar expression levels, it is the most evolutionary robust circuit, having over twofold higher stability than T9002. When the evolutionary half-life is measured for the nine replicate populations, this evolutionary half-life difference compared to T9002 is highly significant (one-tailed t-test, p = 0.0003). Notice that the stability slope is similar to that for T9002, T9002-A, and T9002-C circuits, but the stability lag is increased by about 10 generations. This difference in lag is likely due to a decreased mutation rate since mutations are more random compared to the other similar expression-level circuits (Figure A18-6). The most common mutation is a deletion between repeated scar sequences that removes the luxR promoter and effectively inactivates the circuit function (Figure A18-7).
T9002-F The T9002-F circuit was reengineered with the B0011 terminator, so it also has no homology between terminator sequences. The B0011 is evidently a weak terminator since its initial expression level is about fourfold lower than that of T9002. Its stability dynamics show that it is the most evolutionary robust of the reengineered T9002 circuits, with an evolutionary half-life of about 125 generations (Figure A18-5, Table A18-1). This result indicates that decreasing homology levels and expression through terminator reengineering increased the evolutionary half-life of this circuit over 17-fold relative to T9002. Like T9002-E, the mutations in each of the nine populations are mostly random (Figure A18-6). Also like T9002-E, the most common mutation is a deletion between repeated scar sequences that removes the luxR promoter driving GFP expression (Figure A18-7). Since T9002-E and T9002-F likely have similar mutation rates with zero terminator homology, the large stability difference between these circuits can be explained by expression levels alone.
Overall, excluding T9002-B, three of the five reengineered T9002 circuits are more evolutionary robust than the original circuit. The order of evolutionary robust genetic circuits is as follows: T9002-F > D > E > A = C = T9002. This increase in evolutionary robustness can be attributed to decreased expression levels (due to the terminator reengineering) and to decreased mutation rate between homologous transcriptional terminators. The reengineered circuits with homologous transcriptional terminators almost always have deletions between homologous regions, whereas circuits without homology have mutations in other locations in the circuit. Reengineering this circuit to remove all homology effectively removes a certain class of mutations from occurring. The T9002-E circuit is more
evolutionarily robust than other circuits with similar expression levels, which is likely due to decreased mutation rate alone. Thus, evolutionary robustness can be increased by removing long repeated sequences from genetic circuits, but even short 8-bp scar sequences have the potential for replication slippage.
Evolutionary Half-Life Versus Initial Expression Level in T9002 and T9002-E Circuits Evolved with Different Inducer Concentrations
From previous results (Figure A18-5, Table A18-1), we noticed that circuits with a high initial expression level tended to have low evolutionary stability. Also, particular circuits with high mutation rates had lower stability compared to circuits with lower mutation rates. To test the relationship between initial expression level and evolutionary half-life directly, we evolved the T9002 (high mutation rate) and T9002-E (lower mutation rate) circuits propagated with different AHL concentrations.
Figure A18-8 shows the mean initial expression level versus mean evolutionary half-life for eight replicate populations from three different AHL concentrations in T9002 (black) and T9002-E (red). An exponential fit of these mean data points gives a much higher r2 value than a linear fit (>0.1) in both cases. T9002 has an r2 value of 0.954 compared to the r2 value of 0.955 in T9002-E. The curve for T9002 is shifted to the left from T9002-E due to its higher mutation rate (expression alone cannot account for the shift), but as expression is decreased the evolutionary half-life difference between these two circuits also decreases. This decrease may be because, at high expression levels, the fitness difference between the progenitor and mutant cells is the highest, and therefore mutants outcompete functional cells in the population more quickly.
Discussion
Genetic circuits lose function over evolutionary time and are found to have a wide variety of mutations that cause their loss of function. Circuits with repeated sequences almost always have deletions between these sequences, but the effect of repeat length is not well understood. In one reengineered T9002 circuit, shortening the length of homology from 129 to 41 bp did not significantly increase evolutionary stability. Stability was only increased when there was no homology whatsoever between transcriptional terminators. Mutations between repeated sequences without perfect homology in the case of some reengineered T9002 circuits are usually, but not always, predictable.
In circuits without repeated sequences, mutations are more random between evolved replicate populations. In other circuits we studied (not shown here), mutations that remove promoter function are most often selected for among all the genetic circuits tested (Sleight et al., 2010b). This result is likely because promoter mutations remove the metabolic load at both the transcriptional and translational levels. Mutations within RBSs are not found and mutations in
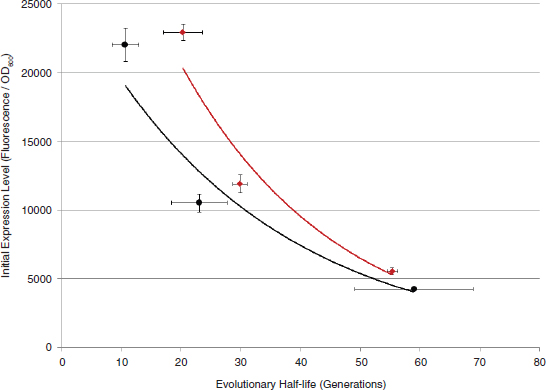
FIGURE A18-8 Evolutionary half-life versus initial expression level in T9002 and T9002-E circuits evolved with different inducer concentrations. Evolutionary half-life versus initial expression level is plotted for T9002 (solid black circles) and T9002-E (solid dark red diamonds) circuits. An exponential fit for the mean of each evolutionary half-life versus initial expression data point is shown by the black (T9002) or red (T9002-E) line. Error bars for both the x and y axes represent one standard deviation from the mean of eight independently evolved populations.
SOURCE: This figure is taken from the Journal of Biological Engineering, 4(1), 12. doi: 10.1186/1754-1611-4-12.
coding sequences are rare except when that coding sequence is an activator of transcription downstream (as in the case of the luxR coding sequence in T9002). In the case of T9002, removing homology between transcriptional terminators only shifts the mutation to one that often removes function of the luxR promoter or luxR coding sequence instead.
What is needed is a method to predict the evolutionary stability of circuits from the properties of their parts, but the emergent behaviors of circuits will likely make prediction difficult. At the very least, publishing the evolutionary stability properties of simple circuits in future data sheets may allow engineers to calculate the expected evolutionary stability of more complex circuits. This calculation would likely require software (Chandran et al., 2009) and mathematical modeling (Chandran et al., 2008) that analyzes each part individually and the entire DNA sequence as a whole to determine the expected evolutionary stability. This calculation would also require standardization for methods to measure evolutionary stability, and the methods described here are not necessarily the best way. On the other hand, more general methods may be developed that focus less on design of the circuit and more on design of the environment to impose a selective pressure for function of the circuit (Bayer et al., 2009). Design of a selective environment is ideal, but it is difficult to do when the output of most circuits (e.g., GFP) is not linked to survival or growth rate. A cell-sorter device that sorts between functional and nonfunctional cells may help with this issue, but it may not be practical for performing routine experiments.
From our results of what types of mutations are selected for in genetic circuits and the evolutionary stability dynamics, a few simple design principles can be proposed when engineering circuits. The first principle is that high expression of genetic circuits comes with the cost of low evolutionary stability. Although exceptions to this rule certainly occur, a genetic circuit with high expression correlates with a large metabolic load and therefore is predicted to have decreased cellular fitness. When the fitness difference between the functional and nonfunctional cells in the population is large, evolutionary stability will decrease quickly. Therefore, the initial expression level of the circuit is likely to be a good predictor of evolutionary stability when a circuit with high mutational robustness is desired. Using a low- or medium-copy plasmid will help with stability as long as the expression level does not need to be high. For more complex circuits where a high expression level is needed for proper functioning of the circuit, decreasing expression level then comes at the cost of changing the function of the circuit.
The second design principle is to avoid repeated sequences. This principle may be obvious, but nearly every circuit in the Registry with more than one coding sequence has repeated B0015 terminators. When a circuit has a high metabolic load (higher than T9002) and repeated sequences on a high-copy-number plasmid, when using MG1655 as the host strain, the circuit will almost always lose function during overnight growth (unpublished results). Reengineering the T9002 circuit to have two different transcriptional terminators (T9002-E) does significantly increase evolutionary half-life by more than twofold and is inde-
pendent of expression levels. However, since this circuit has high expression, this improvement only results in an increase of about 10 generations. Decreasing the expression level along with the mutation rate will increase the evolutionary half-life about 17-fold, as seen in the T9002-F circuit. This result suggests that simple ways to increase evolutionary stability can be used without changing the function of the circuit. For more complex circuits, the community will need to identify many more terminators than those that currently exist in the Registry to design circuits without repeated sequences.
The third design principle is that use of inducible promoters generally increases evolutionary stability (not shown here) (Sleight et al., 2010b). This principle may or may not be significant depending on the circuit used. Inducible circuits are likely more stable due to decreased metabolic load and are preferred since expression can be controlled and fine-tuned, though in some circumstances a constitutive promoter may be desired. Therefore, the use of inducible promoters can be thought of as one extra precaution to maximize evolutionary stability, but expression levels and repeated sequences should first be considered.
We emphasize that the design principles proposed may not be general since only relatively simple circuits were tested in this study. Evolutionary stability should be measured in larger and more complex circuits to understand if these design principles apply. Furthermore, these simple design principles should not necessarily be all used simultaneously. A researcher may not want only to design circuits that have low expression, have no repeated regions, and use a promoter that is inducible. For instance, if recombination sites are needed in the circuit, then repeated or inverted sequences may be impossible to avoid. In addition to the design for the proper function of the circuit, the design for evolutionary robustness should be carefully considered. For this, we need to think about the probability of mutations occurring when the expression level, and therefore metabolic load, is high. In this study, removing repeated regions often shifts mutations to the promoter, and putting a selection on the promoter often shifts the mutation to the chromosome (not shown here) (Sleight et al., 2010b).
Thus, mutations are unavoidable without a selective pressure, but evolutionary stability can likely be improved in the future by better design of selective environments where the circuit is linked to survival and/or growth rate, understanding of mutation rates in genetic circuits, fitness differences between functional and nonfunctional cells, and improvements to the host strain that decrease mutation rates or buffer metabolic loads more efficiently. Another way to improve evolutionary stability is to engineer an error detection and correction circuit that will correct mutations, but it will need careful design since this circuit itself will be prone to mutation. Recently, a technique called ClChE was developed to insert and amplify (increase copy number of) genetic circuits on the chromosome to avoid the problem of maintaining evolutionary stability of function on plasmids (Tyo et al., 2009). This is a powerful method that should be used, but it is also somewhat tedious to insert and amplify circuits on the chromosome. There are also examples of chromosomal genes that are evolutionarily unstable when not
under selection (Cooper and Lenski, 2000; Cooper et al., 2001) and therefore chromosomally integrating synthetic circuits will only delay this problem. Designing mutationally robust genetic circuits, therefore, is somewhat of an art form at the moment besides a few simple design rules, but it should be seen as something the engineer can eventually control.
From the opposite perspective, if a synthetic microbe is released into the environment, we need to hope it will not be mutationally robust. For this, we need to be able to predict how it will evolve if it is able to survive and compete with microbes in their natural environment. One circuit with a weak constitutive promoter was stable for 500 generations in the presence of ampicillin (Sleight et al., 2010b), so it is theoretically possible for circuits to maintain their function for many generations in the right environmental conditions. For the T9002 circuits described here, if the cells carrying these plasmids escaped into the environment, there would likely not be any ampicillin to maintain the plasmid itself, so plasmid stability may follow similar dynamics, as shown by Lenski and Bouma (1987). As far as circuit stability goes, since it is unlikely there will be AHL in the environment to keep circuit function on, function of the circuit will likely decay with similar dynamics as that shown in Figure A18-2 in the T9002 populations evolved without AHL. A circuit with a lower expression level and lower mutation rate (such as T9002-F) will decay more slowly, but it really depends on the interaction between circuit function and the selective pressure in a particular environment.
When thinking about the evolutionary trajectory of a synthetic microbe, if the circuit function imparts a selective advantage over natural microbes, it should be of concern. For example, it is unlikely that GFP will provide a fitness boost in a particular environment, but a different circuit that allows for the degradation of a carbon source may allow cells with this function to have a growth advantage and permanently change an ecosystem. More worrisome is the release of a synthetic microbe that evolves to become pathogenic by acquiring genes from a virus, for instance. Here, one could imagine cells with T9002 mutating out the GFP part of the circuit and inserting a virulence factor instead. Then, the new cells would express the virulence factor only if AHL was present—a remote possibility if there are other microbes that use this molecule for quorum sensing, but still possible. The results shown in this report illustrate the evolution of genetic circuits when no selective pressure is imposed to maintain function of the circuit, but it is unknown what will happen in a natural environment where real selective pressures exist. The study of natural pathogen evolution in E. coli and other pathogens in combination with evolutionary studies in synthetic biology will allow us to understand the risks involved.
Acknowledgments
We thank members of the Sauro and Klavins labs for materials and useful discussions. A special thanks goes to Lucian Smith, Wilbert Copeland, and Kyung Kim for valuable suggestions. We also thank three anonymous reviewers
for valuable feedback. This research was funded by the National Science Foundation (NSF) Grant in Theoretical Biology (0827592), and BEACON: An NSF Center for the Study of Evolution in Action (0939454). Part of this research was previously published in the Journal of Biological Engineering. This journal is an open-access journal where the authors retain copyright and hold an Open Access license (http://www.biomedcentral.com/info/authors/license).
Methods
Circuit Engineering and Use of Strains
All circuits were either obtained from the Registry of Standard Biological Parts or engineered using the Clontech In-Fusion PCR Cloning Kit with the specific methods described by Sleight et al. (2010a). All circuits are encoded on the pSB1A2 plasmid, a high-copy-number plasmid (100-300 plasmids/cell) with an ampicillin resistance gene. Plasmids were transformed into strains via chemical transformation. Escherichia coli MG1655 was the strain used for all circuits described.
Evolution Experiment
For each engineered circuit, plasmid DNA that had been fully sequenced was transformed into either MG1655 competent cells. Three individual transformant colonies were grown overnight at 37°C, shaking at 250 rpm in +100 µg/mL ampicillin. Freezer stocks were saved of these cultures in 15 percent glycerol and stored at −80°C. These freezer stocks were streaked out on LB +100 µg/mL ampicillin plates with appropriate inducer (1 × 10−7 M AHL for T9002 circuits) and grown overnight at 37°C. Three colonies were chosen from each transformant (nine total colonies) and inoculated into 1.5 mL LB + 100 µg/mL ampicillin media in Eppendorf deep-well plates sealed with a Thermo Scientific gas-permeable membrane to allow for maximum oxygen diffusion. T9002 circuit cultures were supplemented with the inducer 1 × 10−7 M 3OC6HSL (AHL). Cultures were propagated with a serial dilution scheme using a 1:1000 dilution to achieve about 10 generations per day (log21000 = 9.97). Evolved populations were grown for 24 hours at 37°C, shaking at 250 rpm. Freezer stocks (with 15 percent glycerol) of each population were saved at −80°C every day for future study. All replicate populations were evolved in parallel to minimize experimental variability.
Evolutionary Stability Measurements
Every 24 hours, cell density (OD600) and fluorescence (excitation wavelength, 485/15; emission wavelength, 516/20) of evolved populations were measured in a Biotek Synergy HT plate reader. Twenty-four hours was used as the measurement time point because the rate of change of GFP was closest to zero
(i.e., closest to steady state). Evolved populations thus spent about 8-12 hours in lag or exponential phase and the remaining time in stationary phase. For each time point, all populations were thoroughly mixed and 200 µL was transferred into black, clear-bottom 96-well plates (Costar). OD was subtracted from blank media to measure the OD without background. Fluorescence was subtracted from the R0011 + E0240 circuit with a mutation in the promoter to measure background fluorescence. Fluorescence was then divided by OD to measure the normalized expression (fluorescence/OD600).
Plasmid Sequencing
At appropriate evolutionary time points, usually when circuit function had decreased to less than 10 percent of the original expression level, or 500 generations, the evolved populations were streaked out on LB + 100 µg/mL ampicillin plates, supplemented with 1 × 10−7 M AHL. Colonies were visualized for fluorescence on a Clare Chemical Dark Reader Transilluminator. Nonfluorescing colonies, or weakly fluorescing colonies if no nonfluorescing colonies were present, were grown overnight in 5 mL of LB + 100 µg/mL ampicillin. Plasmids were extracted using the Qiagen Miniprep Kit or glycerol stocks were sent to the University of Washington High-Throughput Genomics Unit facility (http://www.htseq.org). Purified plasmid DNA was sequenced using the VF2 (5′-TGC-CACCTGACGTCTAAGAA-3′) and VR (5′-ATTACCGCCTTTGAGTGAGC-3′) primers specific to the pSB1A2 vector (about 100 bp on either side of the circuit) or primers specific to the circuit.
Quantitative Analysis of Evolutionary Half-Life
Evolutionary half-life was calculated for each independently evolved population. First, the slope and y-intercept were calculated using the two normalized expression (fluorescence/OD600) data points on either side of the half-maximum expression value on the evolutionary stability plot. A linear regression on those two data points was performed using the equation y = ax + b, where y is the half-maximum initial expression, a is the slope of the two data points, b is the y-intercept of the two data points, and solving for x gives the evolutionary half-life. This method gave a very accurate half-life estimate in terms of generations and was a better estimate than using third-order polynomial equations, which we also calculated.
Experiment to Measure Evolutionary Half-Life Versus Initial Expression Level in T9002 and T9002-E Circuits Evolved with Different Inducer Concentrations
This experiment was performed as described earlier in the Evolution Experiment section, except that eight replicate populations were propagated with different inducer concentrations. The results of this experiment are shown in Figure
A18-8. The AHL concentrations used were 1 × 10−7 M (high-expression-level data point on the far left side of Figure A18-7a), 2 × 10−9 M (medium expression level), and 1 × 10−9 M (low expression level). The evolutionary half-life for individual evolved populations was determined as described earlier in the Quantitative Analysis of Evolutionary Half-Life section. For each inducer concentration, the mean evolutionary half-life versus initial-expression-level data point was plotted. These data points were fit to an exponential curve since this relationship always had the highest r2 value.
References
Bayer, T. S., K. G. Hoff, C. L. Beisel, J. J. Lee, and C. D. Smolke. 2009. Synthetic control of a fitness tradeoff in yeast nitrogen metabolism. Journal of Biological Enginerring 3:1.
Bi, X., and L. F. Liu. 1994. recA-independent and recA-dependent intramolecular plasmid recombination. Differential homology requirement and distance effect. Journal of Molecular Biology 235(2):414-423.
Bull, J. J., M. R. Badgett, H. A. Wichman, J. P. Huelsenbeck, D. M. Hillis, A. Gulati, C. Ho, and I. J. Molineux. 1997. Exceptional convergent evolution in a virus. Genetics 147(4):1497-1507.
Bzymek, M., and S. T. Lovett. 2001. Instability of repetitive DNA sequences: The role of replication in multiple mechanisms. Proceedings of the National Academy of Sciences of the United States of America 98(15):8319-8325.
Canton, B., A. Labno, and D. Endy. 2008. Refinement and standardization of synthetic biological parts and devices. Nature Biotechnology 26(7):787-793.
Chandran, D., W. B. Copeland, S. C. Sleight, and H. M. Sauro. 2008. Mathematical modeling and synthetic biology. Drug Discovery Today: Disease Models 5(4):299-309.
Chandran, D., F. T. Bergmann, and H. M. Sauro. 2009. TinkerCell: Modular CAD tool for synthetic biology. Journal of Biological Engineering 3:19.
Colosimo, P. F., K. E. Hosemann, S. Balabhadra, G. Villarreal, M. Dickson, J. Grimwood, J. Schmutz, R. M. Myers, D. Schluter, and D.M. Kingsley. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307(5717):1928-1933.
Cooper, V. S., and R. E. Lenski. 2000. The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407(6805):736-739.
Cooper, V. S., D. Schneider, M. Blot, and R. E. Lenski. 2001. Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. Journal of Bacteriology 183(9):2834-2841.
Elena, S. F., and R. E. Lenski. 2003. Evolution experiments with microorganisms: The dynamics and genetic bases of adaptation. Nature Reviews Genetics 4(6):457-469.
Endy, D. 2005. Foundations for engineering biology. Nature 438(7067):449-453.
Herring, C. D., A. Raghunathan, C. Honisch, T. Patel, M. K. Applebee, A. R. Joyce, T. J. Albert, F. R. Blattner, D. van den Boom, C. R. Cantor, and B. Palsson. 2006. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nature Genetics 38(12):1406-1412.
Kaur, J., G. Rajamohan, and K. L. Dikshit. 2007. Cloning and characterization of promoter-active DNA sequences from Streptococcus equisimilis. Current Microbiology 54(1):48-53.
Lenski, R. E., and J. E. Bouma. 1987. Effects of segregation and selection on instability of plasmid pACYC184 in Escherichia coli B. Journal of Bacteriology 169(11):5314-5316.
Lovett, S. T. 2004. Encoded errors: Mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Molecular Microbiology 52(5):1243-1253.
Riehle, M., A. F. Bennett, and A. D. Long. 2005. Changes in gene expression following high-temperature adaptation in experimentally evolved populations of E. coli. Physiological and Biochemical Zoology 78(3):299-315.
Shetty, R. P., D. Endy, and T. F. Knight. 2008. Engineering BioBrick vectors from BioBrick parts. Journal of Biological Engineering 2:5.
Sleight, S. C., and R. E. Lenski. 2007. Evolutionary adaptation to freeze-thaw-growth cycles in Escherichia coli. Physiological and Biochemical Zoology PBZ 80(4):370-385.
Sleight, S. C., C. Orlic, D. Schneider, and R. E. Lenski. 2008. Genetic basis of evolutionary adaptation by Escherichia coli to stressful cycles of freezing, thawing and growth. Genetics 180(1):431-443.
Sleight, S. C., B. A. Bartley, J. A. Lieviant, and H. M. Sauro. 2010a. In-Fusion BioBrick assembly and re-engineering. Nucleic Acids Research 38(8):2624-2636.
——. 2010b. Designing and engineering evolutionary robust genetic circuits. Journal of Biological Engineering 4(1):12.
Smolke, C. D., and J. D. Keasling. 2002. Effect of gene location, mRNA secondary structures, and RNase sites on expression of two genes in an engineered operon. Biotechnology and Bioenginerring 80(7):762-776.
Stueber, D., and H. Bujard. 1982. Transcription from efficient promoters can interfere with plasmid replication and diminish expression of plasmid specified genes. EMBO Journal 1(11):1399-1404.
Tyo, K. E. J., P. K. Ajikumar, and G. Stephanopoulos. 2009. Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nature Biotechnology 27(8):760-765.
ISOPRENOID PATHWAY OPTIMIZATION FOR TAXOL PRECURSOR OVERPRODUCTION IN ESCHERICHIA COLI89
Parayil Kumaran Ajikumar,90,91Wen-Hai Xiao,90Keith E. J. Tyo,90Yong Wang,92Fritz Simeon,90Effendi Leonard,90Oliver Mucha,90Too Heng Phon,91Blaine Pfeifer,92,* and Gregory Stephanopoulos90,91,*
Taxol (paclitaxel) is a potent anticancer drug first isolated from the Taxus brevifolia Pacific yew tree. Currently, cost-efficient production of Taxol and its analogs remains limited. Here, we report a multivariate-modular approach to metabolic-pathway engineering that succeeded in increasing titers of taxadiene—the first committed Taxol intermediate—approximately 1 gram per liter (~15,000-fold) in an engineered Escherichia coli strain. Our approach partitioned the taxadiene metabolic pathway into two modules: a native upstream methylerythritol-phosphate (MEP) pathway forming isopentenyl pyrophosphate and a heterologous downstream terpenoid–forming pathway.
_______________
89 From Ajikumar, P.K., et al. 2010. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 330: 70-74. Reprinted with permission from AAAS.
90 Department of Chemical Engineering, Massachusetts Institute of Technology (MIT), Cambridge, MA 02139, USA.
91 Chemical and Pharmaceutical Engineering Program, Singapore-MIT Alliance, 117546 Singapore.
92 Department of Chemical and Biological Engineering, Tufts University, 4 Colby Street, Medford, MA 02155, USA.
* To whom correspondence should be addressed. E-mail: gregstep@mit.edu (G.S.); blaine.pfeifer@tufts.edu (B.P.).
Systematic multivariate search identified conditions that optimally balance the two pathway modules so as to maximize the taxadiene production with minimal accumulation of indole, which is an inhibitory compound found here. We also engineered the next step in Taxol biosynthesis, a P450-mediated 5α-oxidation of taxadiene to taxadien-5α-ol. More broadly, the modular pathway engineering approach helped to unlock the potential of the MEP pathway for the engineered production of terpenoid natural products.
Taxol (paclitaxel) and its structural analogs are among the most potent and commercially successful anticancer drugs (Kingston, 2007). Taxol was first isolated from the bark of the Pacific yew tree (Wani et al., 1971), and early-stage production methods required sacrificing two to four fully grown trees to secure sufficient dosage for one patient (Suffness and Wall, 1995). Taxol’s structural complexity limited its chemical synthesis to elaborate routes that required 35 to 51 steps, with a highest yield of 0.4% (Nicolaou et al., 1994; Holton et al., 1994; Walji and MacMillan, 2007). A semisynthetic route was later devised in which the biosynthetic intermediate baccatin III, isolated from plant sources, was chemically converted to Taxol (Holton et al., 1995). Although this approach and subsequent plant cell culture–based production efforts have decreased the need for harvesting the yew tree, production still depends on plant-based processes (Frense, 2007), with accompanying limitations on productivity and scalability. These methods of production also constrain the number of Taxol derivatives that can be synthesized in the search for more efficacious drugs (Roberts, 2007; Goodman and Walsh, 2001).
Recent developments in metabolic engineering and synthetic biology offer new possibilities for the overproduction of complex natural products by optimizing more technically amenable microbial hosts (Tyo et al., 2007; Ajikumar et al, 2008). The metabolic pathway for Taxol consists of an upstream isoprenoid pathway that is native to Escherichia coli and a heterologous downstream terpenoid pathway (fig. S1). The upstream methylerythritol-phosphate (MEP) or heterologous mevalonic acid (MVA) pathways can produce the two common building blocks, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), from which Taxol and other isoprenoid compounds are formed (Ajikumar et al., 2008). Recent studies have highlighted the engineering of the above upstream pathways to support the biosynthesis of heterologous isoprenoids such as lycopene (Farmer and Liao, 2000; Alper et al., 2005), artemisinic acid (Chang and Keasling, 2006; Martin et al., 2003), and abietadiene (Morrone et al., 2010; Leonard et al., 2010). The downstream taxadiene pathway has been reconstructed in E. coli and Saccharomyce cerevisiae together with the overexpression of upstream pathway enzymes, but to date titers have been limited to less than 10 mg/liter (Huang et al., 2001; Engels et al., 2008).
The above rational metabolic engineering approaches examined separately either the upstream or the downstream terpenoid pathway, implicitly assuming that modifications are additive (a linear behavior) (Farmer and Liao, 2000;
Morrone et al., 2010; Yuan et al., 2006). Although this approach can yield moderate increases in flux, it generally ignores nonspecific effects, such as toxicity of intermediate metabolites, adverse cellular effects of the vectors used for expression, and hidden pathways and metabolites that may compete with the main pathway and inhibit the production of the desired molecule. Combinatorial approaches can overcome such problems because they offer the opportunity to broadly sample the parameter space and bypass these complex nonlinear interactions (Yuan et al., 2006; Jin and Stephanopoulos, 2007; Wang et al., 2009). However, combinatorial approaches require high-throughput screens, which are often not available for many desirable natural products (Klein-Marcuschamer, 2007).
Considering the lack of a high-throughput screen for taxadiene (or other Taxol pathway intermediate), we resorted to a focused combinatorial approach, which we term “multivariate-modular pathway engineering.” In this approach, the overall pathway is partitioned into smaller modules, and the modules’ expression are varied simultaneously—a multivariate search. This approach can identify an optimally balanced pathway while searching a small combinatorial space. Specifically, we partition the taxadiene-forming pathway into two modules separated at IPP, which is the key intermediate in terpenoid biosynthesis. The first module comprises an eight-gene, upstream, native (MEP) pathway of which the expression of only four genes deemed to be rate-limiting was modulated, and the second module comprises a two-gene, downstream, heterologous pathway to taxadiene (Figure A19-1). This modular approach allowed us to efficiently sample the main parameters affecting pathway flux without the need for a high-throughput screen and to unveil the role of the metabolite indole as inhibitor of isoprenoid pathway activity. Additionally, the multivariate search revealed a highly nonlinear taxadiene flux landscape with a global maximum exhibiting a 15,000-fold increase in taxadiene production over the control, yielding 1.02 ± 0.08 g/liter (SD) taxadiene in fed-batch bioreactor fermentations.
We have further engineered the P450-based oxidation chemistry in Taxol biosynthesis in E. coli to convert taxadiene to taxadien-5α-ol and provide the basis for the synthesis of subsequent metabolites in the pathway by means of similar cytochrome P450 (CYP450) oxidation chemistry. Our engineered strain improved taxadiene-5α-ol production by 2400-fold over the state of the art with yeast (Dejong et al., 2006). These advances unlock the potential of microbial processes for the large-scale production of Taxol or its derivatives and thousands of other valuable terpenoids.
The multivariate-modular approach in which various promoters and gene copy-numbers are combined to modulate diverse expression levels of upstream and downstream pathways of taxadiene synthesis is schematically described in fig. S2. A total of 16 strains were constructed in order to widen the bottleneck of the MEP pathway as well as optimally balance it with the downstream taxadiene pathway. (Materials and methods are available as supporting material on Science Online.) The dependence of taxadiene accumulation on the upstream pathway for constant values of the downstream pathway is shown in Figure A19-2A, and the
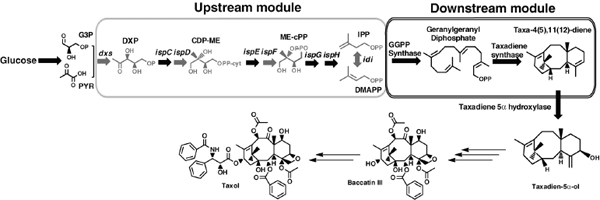
FIGURE A19-1 Multivariate-modular approach for isoprenoid pathway optimization. To increase the flux through the upstream MEP pathway, we targeted reported enzymatic bottlenecks (dxs, idi, ispD, and ispF) (gray) for overexpression by an operon (dxs-idi-ispDF) (Yuan et al., 2006). To channel the overflow flux from the universal isoprenoid precursors, IPP and DMAPP, toward Taxol biosynthesis, we constructed a synthetic operon of downstream genes GGPP synthase (G) and taxadiene synthase (T) (Walker and Croteau, 2001). Both pathways were placed under the control of inducible promoters in order to control their relative gene expression. In the E. coli metabolic network, the MEP isoprenoid pathway is initiated by the condensation of the precursors glyceraldehyde-3 phosphate (G3P) and pyruvate (PYR) from glycolysis. The Taxol pathway bifurcation starts from the universal isoprenoid precursors IPP and DMAPP to form geranylgeranyl diphosphate, and then the taxadiene. The cyclic olefin taxadiene undergoes multiple rounds of stereospecific oxidations, acylations, and benzoylation to form the late intermediate Baccatin III and side chain assembly to, ultimately, form Taxol.
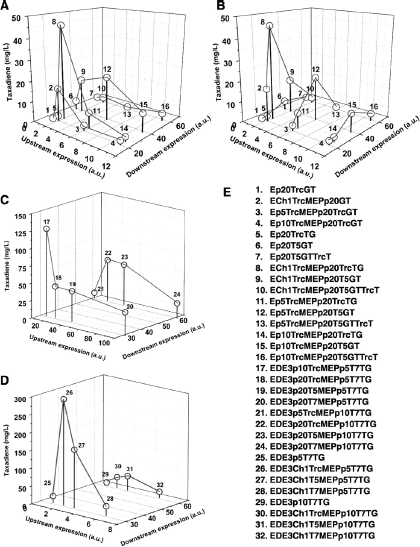
FIGURE A19-2 Optimization of taxadiene production through regulating the expression of the upstream and downstream modular pathways. (A) Response in taxadiene accumulation to changes in upstream pathway strengths for constant values of the downstream pathway. (B) Dependence of taxadiene on the downstream pathway for constant levels of upstream pathway strength. (C) Taxadiene response from strains (17 to 24) engineered with high upstream pathway overexpressions (6 to 100 a.u.) at two different downstream expressions (31 a.u. and 61 a.u.). (D) Modulation of a chromosomally integrated upstream pathway by using increasing promoter strength at two different downstream expressions (31 a.u. and 61 a.u.). (E) Genotypes of the 32 strain constructs whose taxadiene phenotype is shown in Figure A19-2, A to D. E, E. coli K12MG1655 ΔrecAΔendA; EDE3, E. coli K12MG1655 ΔrecAΔendA with DE3 T7 RNA polymerase gene in the chromosome; MEP, dxs-idi-ispDF operon; GT, GPPS-TS operon; TG, TS-GPPS operon; Ch1, 1 copy in chromosome; Trc, Trc promoter; T5, T5 promoter; T7, T7 promoter; p5, pSC101 plasmid; p10, p15A plasmid; and p20, pBR322 plasmid.
dependence on the downstream pathway for constant upstream pathway strength is shown in Figure A19-2B (table S1, calculation of the upstream and downstream pathway strength from gene copy number and promoter strength). As the upstream pathway expression increases in Figure A19-2A from very low levels, taxadiene production also rises initially because of increased supply of precursors to the overall pathway. However, after an intermediate value further upstream pathway increases cannot be accommodated by the capacity of the downstream pathway. For constant upstream pathway expression (Figure A19-2B), a maximum in downstream expression was similarly observed owing to the rising edge to initial limiting of taxadiene production by low expression levels of the downstream pathway. At high (after peak) levels of downstream pathway expression, we were probably observing the negative effect on cell physiology of the high copy number.
These results demonstrate that dramatic changes in taxadiene accumulation can be obtained from changes within a narrow window of expression levels for the upstream and downstream pathways. For example, a strain containing an additional copy of the upstream pathway on its chromosome under Trc promoter control (strain 8) (Figure A19-2A) produced 2000-fold more taxadiene than one expressing only the native MEP pathway (strain 1) (Figure A19-2A). Furthermore, changing the order of the genes in the downstream synthetic operon from GT (GGPS-TS) to TG (TS-GGPS) resulted in a two- to threefold increase (strains 1 to 4 as compared with strains 5, 8, 11, and 14). Altogether, the engineered strains established that the MEP pathway flux can be substantial if an appropriate range of expression levels for the endogenous upstream and synthetic downstream pathway are searched simultaneously.
To provide ample downstream pathway strength while minimizing the plasmid-born metabolic burden (Jones et al., 2000), two new sets of four strains each were engineered (strains 17 to 20 and 21 to 24), in which the downstream pathway was placed under the control of a strong promoter (T7) while keeping a relatively low number of five and 10 plasmid copies, respectively. The taxadiene maximum was maintained at high downstream strength (strains 21 to 24), whereas a monotonic response was obtained at the low downstream pathway strength (strains 17 to 20) (Figure A19-2C). This observation prompted the construction of two additional sets of four strains each that maintained the same level of downstream pathway strength as before but expressed very low levels of the upstream pathway (strains 25 to 28 and 29 to 32) (Figure A19-2D). Additionally, the operon of the upstream pathway of the latter strain set was chromosomally integrated (fig S3). Not only was the taxadiene maximum recovered in these strains, albeit at very low upstream pathway levels, but a much greater taxadiene maximum was attained (~300 mg/liter). We believe that this significant increase can be attributed to a decrease in the cell’s metabolic burden.
We next quantified the mRNA levels of 1-deoxy-d-xylulose-5-phosphate synthase (dxs) and taxadiene synthase (TS) (representing the upstream and downstream pathways, respectively) for the high-taxadiene-producing strains (25 to
32 and 17 and 22) that exhibited varying upstream and downstream pathway strengths (fig. S4, A and B) to verify our predicted expression strengths were consistent with the actual pathway levels. We found that dxs expression level correlates well with the upstream pathway strength. Similar correlations were found for the other genes of the upstream pathway: idi, ispD, and ispF (fig. S4, C and D). In downstream TS gene expression, an approximately twofold improvement was quantified as the downstream pathway strength increased from 31 to 61 arbitrary units (a.u.) (fig. S4B).
Metabolomic analysis of the previous strains led to the identification of a distinct metabolite by-product that inversely correlated with taxadiene accumulation (figs. S5 and S6). The corresponding peak in the gas chromatography–mass spectrometry (GC-MS) chromatogram was identified as indole through GC-MS, 1H, and 13C nuclear magnetic resonance (NMR) spectroscopy studies (fig. S7). We found that taxadiene synthesis by strain 26 is severely inhibited by exogenous indole at indole levels higher than ~100 mg/liter (fig. S5B). Further increasing the indole concentration also inhibited cell growth, with the level of inhibition being very strain-dependent (fig. S5C). Although the biochemical mechanism of indole interaction with the isoprenoid pathway is presently unclear, the results in fig. S5 suggest a possible synergistic effect between indole and terpenoid compounds of the isoprenoid pathway in inhibiting cell growth. Without knowing the specific mechanism, it appears that strain 26 has mitigated the indole’s effect, which we carried forward for further study.
In order to explore the taxadiene-producing potential under controlled conditions for the engineered strains, fed-batch cultivations of the three highest taxadiene accumulating strains (~60 mg/ liter from strain 22; ~125 mg/liter from strain 17; and ~300 mg/liter from strain 26) were carried out in 1-liter bioreactors (Figure A19-3). The fed-batch cultivation studies were carried out as liquid-liquid two-phase fermentation using a 20% (v/v) dodecane overlay. The organic solvent was introduced to prevent air stripping of secreted taxadiene from the fermentation medium, as indicated by preliminary findings (fig. S8). In defined media with controlled glycerol feeding, taxadiene productivity increased to 174 ± 5 mg/liter (SD), 210 ± 7 mg/liter (SD), and 1020 ± 80 mg/liter (SD) for strains 22, 17, and 26, respectively (Figure A19-3A). Additionally, taxadiene production significantly affected the growth phenotype, acetate accumulation, and glycerol consumption [Figure A19-3, B and D, and supporting online material (SOM) text]. Clearly, the high productivity and more robust growth of strain 26 allowed very high taxadiene accumulation. Further improvements should be possible through optimizing conditions in the bioreactor, balancing nutrients in the growth medium and optimizing carbon delivery.
Having succeeded in engineering the biosynthesis of the “cyclase phase” of Taxol for high taxadiene production, we turned next to engineering the oxidation-chemistry of Taxol biosynthesis. In this phase, hydroxyl groups are incorporated by oxygenation at seven positions on the taxane core structure, mediated by CYP450-dependent monooxygenases (Kaspera and Croteau, 2006). The first
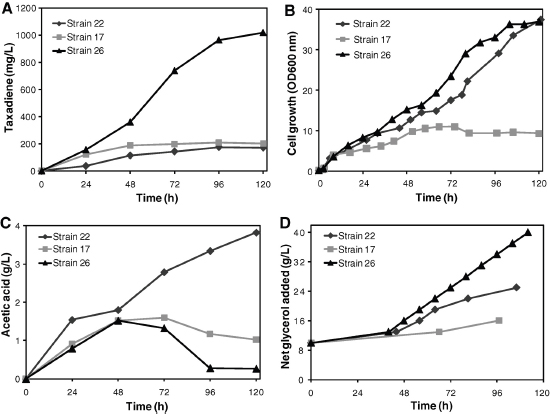
FIGURE A19-3 Fed-batch cultivation of engineered strains in a 1-liter bioreactor. Time courses of (A) taxadiene accumulation, (B) cell growth, (C) acetic acid accumulation, and (D) total substrate (glycerol) addition for strains 22, 17, and 26 during 5 days of fed-batch bioreactor cultivation in 1-liter bioreactor vessels under controlled pH and oxygen conditions with minimal media and 0.5% yeast extract. After glycerol depletes to ~0.5 to 1 g/liter in the fermentor, 3 g/liter of glycerol was introduced into the bioreactor during the fermentation. Data are mean of two replicate bioreactors.
oxygenation is the hydroxylation of the C5 position, followed by seven similar reactions en route to Taxol (fig. S1) (Jennewein et al., 2004a). Thus, a key step toward engineering Taxol-producing microbes is the development of CYP450based oxidation chemistry in vivo. The first oxygenation step is catalyzed by a CYP450, taxadiene 5α-hydroxylase, which is an unusual monooxygenase that catalyzes the hydroxylation reaction along with double-bond migration in the diterpene precursor taxadiene (Figure A19-1).
In general, functional expression of plant CYP450 in E. coli is challenging (Schuler and Werck-Reichhart, 2003) because of the inherent limitations of bacterial platforms, such as the absence of electron transfer machinery and CYP450-reductases (CPRs) and translational incompatibility of the membrane signal modules of CYP450 enzymes because of the lack of an endoplasmic reticulum. Recently, through transmembrane (TM) engineering and the generation of chimera enzymes of CYP450 and CPR, some plant CYP450s have been expressed in E. coli for the biosynthesis of functional molecules (Chang and Keasling, 2006; Leonard and Koffas, 2007). Still, every plant CYP450 has distinct TM signal sequences and electron transfer characteristics from its reductase counterpart (Nelson, 1999). Our initial studies were focused on optimizing the expression of codon-optimized synthetic taxadiene 5α-hydroxylase by N-terminal TM engineering and generating chimera enzymes through translational fusion with the CPR redox partner from the Taxus species, Taxus CYP450 reductase (TCPR) (Figure A19-4A) (Jennewein et al., 2004a, 2005; Leonard and Koffas, 2007). One of the chimera enzymes generated, At24T5αOH-tTCPR, was highly efficient in carrying out the first oxidation step, resulting in more than 98% taxadiene conversion to taxadien-5α-ol and the by-product 5(12)-Oxa-3(11)-cyclotaxane (OCT) (fig. S9A).
Compared with the other chimeric CYP450s, At24T5αOH-tTCPR yielded twofold higher (21 mg/liter) production of taxadien-5α-ol (Figure A19-4B). Because of the functional plasticity of taxadiene 5α-hydroxylase with its chimeric CYP450’s enzymes (At8T5αOH-tTCPR, At24T5αOH-tTCPR, and At42T5αOHtTCPR), the reaction also yields a complex structural rearrangement of taxadiene into the cyclic ether OCT (fig. S9) (Rontein et al., 2008). The by-product accumulated in approximately equal amounts (~24 mg/liter from At24T5αOH-tTCPR) to the desired product taxadien-5α-ol.
The productivity of strain 26-At24T5αOH-tTCPR was significantly reduced relative to that of taxadiene production by the parent strain 26 (~300 mg/liter), with a concomitant increase in indole accumulation. No taxadiene accumulation was observed. Apparently, the introduction of an additional medium copy plasmid (10-copy, p10T7) bearing the At24T5αOH-tTCPR construct disturbed the carefully engineered balance in the upstream and downstream pathway of strain 26 (fig S10). Small-scale fermentations were carried out in bioreactors so as to quantify the alcohol production by strain 26-At24T5αOH-tTCPR. The time course profile of taxadien-5α-ol accumulation (Figure A19-4C) indicates alcohol production of up to 58 ± 3 mg/liter (SD) with an equal amount of the
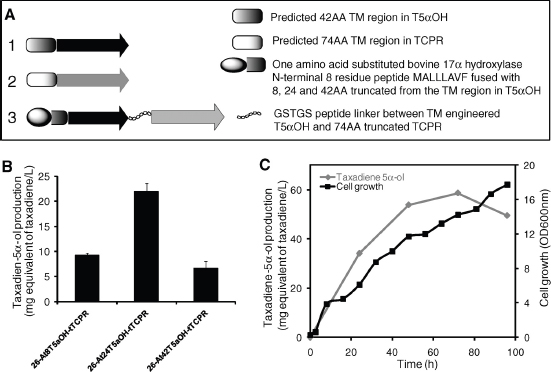
FIGURE A19-4 Engineering Taxol P450 oxidation chemistry in E. coli. (A) TM engineering and construction of chimera protein from taxadien5α-ol hydroxylase (T5αOH) and Taxus cytochrome P450 reductase (TCPR). The labels 1 and 2 represent the full-length proteins of T5αOH and TCPR identified with 42 and 74 amino acid TM regions, respectively, and 3 represents chimera enzymes generated from three different TM engineered T5αOH constructs [At8T5αOH, At24T5αOH, and At42T5αOH constructed by fusing an 8-residue synthetic peptide MALLLAVF (A) to 8, 24, and 42AA truncated T5αOH] through a translational fusion with 74AA truncated TCPR (tTCPR) by use of linker peptide GSTGS. (B) Functional activity of At8T5αOH-tTCPR, At24T5αOH-tTCPR, and At42T5αOH-tTCPR constructs transformed into taxadiene producing strain 26. Data are mean ± SD for three replicates. (C) Time course profile of taxadien-5α-ol accumulation and growth profile of the strain 26-At24T5αOH-tTCPR fermented in a 1-liter bioreactor. Data are mean of two replicate bioreactors.
OCT by-product produced. The observed alcohol production was approximately 2,400-fold higher than previous production in S. cerevisiae (Dejong et al., 2006).
The MEP pathway is energetically balanced and thus overall more efficient in converting either glucose or glycerol to isoprenoids (fig. S11). Yet, during the past 10 years many attempts at engineering the MEP pathway in E. coli in order to increase the supply of the key precursors IPP and DMAPP for carotenoid (Yuan et al., 2006; Farmer and Liao, 2001), sesquiterpenoid (Martin et al., 2003), and diterpenoid (Morrone et al., 2010) overproduction met with limited success. This inefficiency was attributed to unknown regulatory effects associated specifically with the expression of the MEP pathway in E. coli (Martin et al., 2003). Here, we provide evidence that such limitations are correlated with the accumulation of the metabolite indole, owing to the non-optimal expression of the pathway, which inhibits the isoprenoid pathway activity. Taxadiene overproduction (under conditions of indole-formation suppression), establishes the MEP pathway as a very efficient route for biosynthesis of pharmaceutical and chemical products of the isoprenoid family (fig. S11). One simply needs to carefully balance the modular pathways, as suggested by our multivariate-modular pathway–engineering approach.
For successful microbial production of Taxol, demonstration of the chemical decoration of the taxadiene core by means of CYP450-based oxidation chemistry is essential (Kaspera and Croteau, 2006). Previous efforts to reconstitute partial Taxol pathways in yeast found CYP450 activity limiting (Dejong et al., 2006), making the At24T5αOH-tTCPR activity levels an important step to debottleneck the late Taxol pathway. Additionally, the strategies used to create At24T5αOHtTCPR are probably applicable for the remaining monooxygenases that will require expression in E. coli. CYP450 monooxygenases constitute about one half of the 19 distinct enzymatic steps in the Taxol biosynthetic pathway. These genes show unusually high sequence similarity with each other (>70%) but low similarity (<30%) with other plant CYP450s (Jennewein et al., 2004b), implying that these monooxygenases are amenable to similar engineering.
To complete the synthesis of a suitable Taxol precursor, baccatin III, six more hydroxylation reactions and other steps (including some that have not been identified) need to be effectively engineered. Although this is certainly a daunting task, the current study shows potential by providing the basis for the functional expression of two key steps, cyclization and oxygenation, in Taxol biosynthesis. Most importantly, by unlocking the potential of the MEP pathway a new more efficient route to terpenoid biosynthesis is capable of providing potential commercial production of microbially derived terpenoids for use as chemicals and fuels from renewable resources.
References and Notes
P. K. Ajikumar et al., Mol. Pharm. 5, 167 (2008).
H. Alper, K. Miyaoku, G. Stephanopoulos, Nat. Biotechnol. 23, 612 (2005).
M. C. Chang, J. D. Keasling, Nat. Chem. Biol. 2, 674 (2006).
J. M. Dejong et al., Biotechnol. Bioeng. 93, 212 (2006).
B. Engels, P. Dahm, S. Jennewein, Metab. Eng. 10, 201 (2008).
W. R. Farmer, J. C. Liao, Nat. Biotechnol. 18, 533 (2000).
——, Biotechnol. Prog. 17, 57 (2001).
D. Frense, Appl. Microbiol. Biotechnol. 73, 1233 (2007).
J. Goodman, V. Walsh, The Story of Taxol: Nature and Politics in the Pursuit of an Anti-Cancer Drug. (Cambridge Univ. Press, Cambridge, 2001).
R. A. Holton et al., J. Am. Chem. Soc. 116, 1597 (1994).
R. A. Holton, R. J. Biediger, P. D. Boatman, in Taxol: Science and Applications, M. Suffness, Ed. (CRC, Boca Raton, FL, 1995), pp. 97–119.
Q. Huang, C. A. Roessner, R. Croteau, A. I. Scott, Bioorg. Med. Chem. 9, 2237 (2001).
S. Jennewein, R. M. Long, R. M. Williams, R. Croteau, Chem. Biol. 11, 379 (2004a).
S. Jennewein, M. R. Wildung, M. Chau, K. Walker, R. Croteau, Proc. Natl. Acad. Sci. U.S.A. 101, 9149 (2004b).
S. Jennewein et al., Biotechnol. Bioeng. 89, 588 (2005).
Y. S. Jin, G. Stephanopoulos, Metab. Eng. 9, 337 (2007).
K. L. Jones, S. W. Kim, J. D. Keasling, Metab. Eng. 2, 328 (2000).
R. Kaspera, R. Croteau, Phytochem. Rev. 5, 433 (2006).
D. G. Kingston, Phytochemistry 68, 1844 (2007).
D. Klein-Marcuschamer, P. K. Ajikumar, G. Stephanopoulos, Trends Biotechnol. 25, 417 (2007).
E. Leonard, M. A. Koffas, Appl. Environ. Microbiol. 73, 7246 (2007).
E. Leonard et al., Proc. Natl. Acad. Sci. U.S.A. 107, 13654 (2010).
V. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman, J. D. Keasling, Nat. Biotechnol. 21, 796 (2003).
D. Morrone et al., Appl. Microbiol. Biotechnol. 85, 1893 (2010).
D. R. Nelson, Arch. Biochem. Biophys. 369, 1 (1999).
K. C. Nicolaou et al., Nature 367, 630 (1994).
S. C. Roberts, Nat. Chem. Biol. 3, 387 (2007).
D. Rontein et al., J. Biol. Chem. 283, 6067 (2008).
M. A. Schuler, D. Werck-Reichhart, Annu. Rev. Plant Biol. 54, 629 (2003).
M. Suffness, M. E. Wall, in Taxol: Science and Applications, M. Suffness, Ed. (CRC, Boca Raton, FL, 1995), pp. 3–26.
K. E. Tyo, H. S. Alper, G. N. Stephanopoulos, Trends Biotechnol. 25, 132 (2007).
A. M. Walji, D. W. C. MacMillan, Synlett 18, 1477 (2007).
K. Walker, R. Croteau, Phytochemistry 58, 1 (2001).
H. H. Wang et al., Nature 460, 894 (2009).
M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon, A. T. McPhail, J. Am. Chem. Soc. 93, 2325 (1971).
L. Z. Yuan, P. E. Rouvière, R. A. Larossa, W. Suh, Metab. Eng. 8, 79 (2006).
We thank R. Renu for extraction, purification, and characterization of metabolite Indole; C. Santos for providing the pACYCmelA plasmid, constructive suggestions during the experiments, and preparation of the manuscript; D. Dugar, H. Zhou, and X. Huang for helping with experiments and suggestions; and K. Hiller for data analysis and comments on the manuscript. We gratefully acknowledge support by the Singapore-MIT Alliance (SMA-2) and NIH, grant 1-R01-GM085323-01A1.
B.P. acknowledges the Milheim Foundation Grant for Cancer Research 2006-17. A patent application that is based on the results presented here has been filed by MIT. P.K.A. designed the experiments and performed the engineering and screening of the strains; W-H.X. performed screening of the strains, bioreactor
experiments, and GC-MS analysis; F.S. carried out the quantitative PCR measurements; O.M. performed the extraction and characterization of taxadiene standard; E.L., Y.W., and B.P. supported with cloning experiments; P.K.A., K.E.J.T., T.H.P., B.P., and G.S. analyzed the data; P.K.A., K.E.J.T., and G.S. wrote the manuscript; G.S. supervised the research; and all of the authors contributed to discussion of the research and edited and commented on the manuscript.
Supporting Online Material
www.sciencemag.org/cgi/content/full/330/6000/70/DC1
Materials and Methods
SOM Text
Figs. S1 to S11
Tables S1 to S4
References
29 April 2010; accepted 9 August 2010
10.1126/science.1191652
PROGRAMMING CELLS: TOWARDS AN AUTOMATED ‘GENETIC COMPILER’93
Kevin Clancy94and Christopher A Voigt95
One of the visions of synthetic biology is to be able to program cells using a language that is similar to that used to program computers or robotics. For large genetic programs, keeping track of the DNA on the level of nucleotides becomes tedious and error prone, requiring a new generation of computer-aided design (CAD) software. To push the size of projects, it is important to abstract the designer from the process of part selection and optimization.
_______________
93 This article was published in Current Opinion in Biotechnology, Vol 21, K. Clancy and C.A. Voigt, Programming cells: towards an automated ‘Genetic Compiler’, 572-581, Copyright Elsevier (2010).
94 Life Technologies, 5791 Van Allen Way, Carlsbad, CA 90028, United States.
95 Department of Pharmaceutical Chemistry, University of California-San Francisco, MC 2540, Room 408C, 1700 4th Street, San Francisco, CA 94158, United States.
Corresponding author: Voigt, Christopher A (cavoigt@gmail.com)
Current Opinion in Biotechnology 2010, 21:1–10
This review comes from a themed issue on Systems biology
Edited by Vitor Martins dos Santos and Jiri Damborsky
0958-1669/$ – see front matter Published by Elsevier Ltd.
DOI 10.1016/j.copbio.2010.07.005
The vision is to specify genetic programs in a higher-level language, which a genetic compiler could automatically convert into a DNA sequence. Steps towards this goal include: defining the semantics of the higher-level language, algorithms to select and assemble parts, and biophysical methods to link DNA sequence to function. These will be coupled to graphic design interfaces and simulation packages to aid in the prediction of program dynamics, optimize genes, and scan projects for errors.
Introduction
Numerous genetic circuits have been built that encode functions that are analogous to electronic circuits (Stricker et al., 2008; Ham et al., 2006; Voigt, 2006). When multiple circuits are connected to sensors and actuators, this forms a genetic program. For example, we constructed an ‘edge detector’ program that combines a ANDN gate, light sensor, and cell–cell communication that give bacteria the ability to draw the edge between the light and dark regions of an image projected onto a plate (Salis and Kaznessis, 2006). Other genetic programs have been built that combine circuits to produce a push-on/push-off circuit (Lou et al., 2009+), implement a counter (Friedland et al., 2009++), and reproduce predator–prey dynamics (Balagadde et al., 2008). These represent toy systems, but the implementation of such programs in applications for industrial biotechnology is inevitable.
Automated DNA synthesis gives genetic engineers an unprecedented design capacity (Czar et al., 2008). This technology enables the specification of every basepair for long sequences, without having to be concerned about the path to construction. Together with methods to rapidly combine genetic parts (Gibson et al., 2009++; Shetty et al., 2008) and assembly methods that scale to whole genomes (Gibson et al., 2008, 2010; Wang et al., 2009), the problem of DNA construction has far outpaced our capacity for design (Purnick and Weiss, 2009+). A good example of this is the 2006 UCSF iGEM team to build a ‘remote-controlled bacterium.’ DNA synthesis was used to build the first construct (requiring a few weeks), but after four years of additional tinkering, the paper will be submitted in 2010.
Our ability to design programs has been hampered by three problems. First, there is a lack of good, robust genetic circuits that can be easily connected. Second, there are few design rules that are sufficiently quantitative to be carried out algorithmically. Modeling can be helpful before the experiments to determine the topologies and parameter regimes required to obtain a particular function. However, simulations cannot be used to ‘reach down’ to the DNA and suggest a specific mutation or select a part. Third, the frequency of mistakes in the DNA sequence increases quickly with size. Currently, to scan for potential errors (e.g. transposon insertion sites or putative internal promoters), it requires the running
of multiple (usually web-based) programs. There is no unified software package to date that addresses all of these issues.
The creation of a simulation environment for genetic engineering is complicated by the diversity of cellular functions. When studying natural networks, there is a feeling of ‘peeling an onion,’ where there are seemingly endless redundancies and classes of biochemical interactions. Even within the Registry of Standard Biological Parts (www.partsregistry.org), there are a wide variety of cellular functions: from enzymes and transcription factors to multi-gene gas vesicles and secretion chaperones. Each specific problem requires its own style of simulation; a dynamic program may be well satisfied by sets of differential equations, pattern formation by cellular automata, and enzymes by metabolic flux analysis. It would be daunting to create a simulation package that could encompass all of this diversity.
To reduce the problem complexity and to frame recent computational work, we introduce the concept of a ‘Genetic Compiler,’ whose inputs are high-level instructions (equivalent to VHDL or Verilog) and whose output is a DNA sequence. The sequence can be sent to a company for DNA synthesis or a robot for automated assembly. The problem is constrained by focusing on genetic programs that encode a desired logical or dynamical function, which can be integrated into many applications in biotechnology (Figure A20-1). This avoids the application-specific portions of the problem; for example, building a butanol sensor a particular metabolic pathway. It is distinct from tools for protein or metabolic engineering (Cho et al., 2007).
The scope of this review is on the underlying algorithms and biophysical methods that would power such a compiler (Figure A20-2). Realizing this goal will require: 1. Libraries of reliable genetic circuits designed specifically to be part of a CAD program, 2. the definition of a higher-level language, 3. algorithms to assemble circuits according to a specified program, 3. biophysical methods to connect and optimize circuits, 4. simulation programs to debug the program dynamics, 5. algorithms for DNA assembly and experimental design. The scope has been limited to exclude several topics that are crucial to synthetic biology, but have been well-reviewed elsewhere, notably codon optimization and tools from systems biology and metabolic engineering (Cho et al., 2007; Richardson et al., 2010; Villalobos et al., 2006).
Main Text
Robust combinatorial logic
Combinatorial logic is implemented by Boolean circuits and is the basis for digital computing. It is used to build circuits that apply Boolean algebra on a set of inputs to transform them into a set of desired outputs. Simple circuits can be layered in different configurations in order to achieve a computational operation.
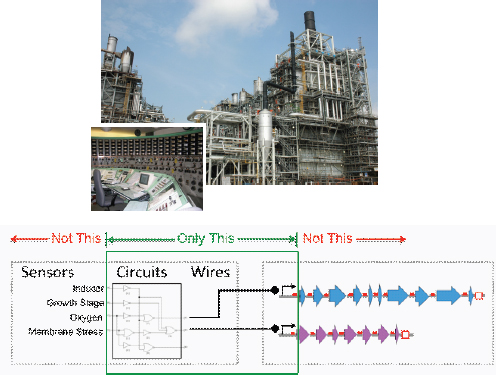
FIGURE A20-1 The compiler is focused on assembling the circuitry that links the inputs and outputs of a larger project. The inputs include genetic sensors that can respond to diverse signals, such as temperature, light, stress, or metabolites. The circuitry encodes the logic and dynamics. The output of the circuits control actuators, such as a metabolic pathway, the activation of cell–cell communication, or a stress response. The inputs and outputs tend to be problem-specific and the diversity of biological applications makes this difficult to encompass in a single simulation package. By contrast, the circuitry can be reconfigured to build programs relevant to diverse problems in biotechnology.
This has enabled the automated design that underlies VLSI. The ability for digital circuits to be flexibly used and easily captured by CAD comes at a cost of speed, design size, and power (Sarpeshkar, 2010++). There are ongoing efforts to design the core biochemistry that would act as genetic logic gates (Salis and Kaznessis, 2006; Ramalingam et al., 2009; Kinkhabwala and Guet, 2008; Cox et al., 2007; Rackham and Chin, 2006; Kramer et al., 2005; Benenson, 2009; Sharma et al., 2008+; Dueber et al., 2003; Anderson et al., 2007; Rinaudo et al., 2007; Buchler et al., 2003). There are several important considerations in designing genetic logic gates to be used in CAD:
- Scalability. Each circuit in a project needs to be orthogonal because all of the circuits operate based on biochemical interactions within the cell. A circuit is scalable when the underlying genetic parts can be swapped
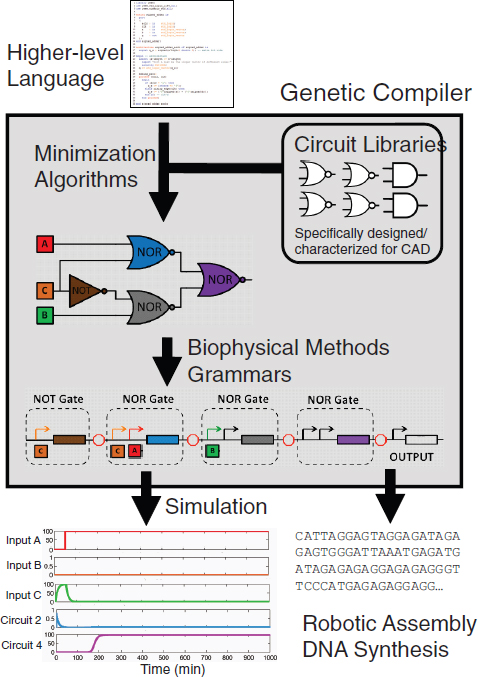
FIGURE A20-2 A genetic compiler. The compiler automatically converts a higher-level language to a DNA sequence. Ideally, the designer would be completely blind to these steps (gray box). Simulations aid the debugging of the program, but the debugging would occur within the higher-level language.
-
to create orthogonal circuits. For example, orthogonal NOT gates could be built based on new repressor-operator pairs (Yokobayashi et al., 2002). By contrast, an AND gate that we constructed is not scalable because its underlying parts do not lend themselves to making multiple gates (Anderson et al., 2007).
- Extensibility. In order to layer genetic circuits, the inputs and outputs of the circuit have to have the same signal carrier (Canton et al., 2008). Put simply, for circuits to be connected, the inputs and the outputs need to be the same biochemical form (phosphorylation, transcription, etc.). For transcriptional circuits, it has been proposed that the flux of RNAPs on DNA could serve this purpose (Canton et al., 2008). In practice, this can be implemented by making the circuit inputs and the outputs be promoters. Several examples of this are a NOT gate (Yokobayashi et al., 2002) and an AND gate (Anderson et al., 2007). This is in contrast to circuits where, for example, the inputs are small molecules and the output is the activation of a riboswitch (Sharma et al., 2008+).
- Modularity. The inputs and outputs of the circuits need to be able to be easily changed. Considering CAD, the ease has to be sufficient for a computer to be able to reliably perform this function. Promoters, protein–protein interaction domains, and RNA are sufficiently modular for this purpose (Sharma et al., 2008+). Some biological systems that are considered modular—such as the protein domains of bacterial two-component systems—are not sufficiently modular for CAD. This is because small libraries are required to identify a functional chimera, as opposed to design rules that can be implemented by a machine.
- Robustness. The circuits must remain functional over large regions of parameter space. This is particularly important when connecting multiple circuits in series, where robust circuits will allow for more uncertainty while still producing a functional connection (Salis and Kaznessis, 2006; Salis et al., 2009). The circuits also have to be non-toxic and have minimal impact on host processes (Tan et al., 2009++).
- Speed. The circuits also need to be reasonably fast. For example, a three-layer cascade of NOT gates required 20 min for each layer to compute (Hooshangi et al., 2005), but a DNA inversion switch requires 4 h (Ham et al., 2006). Even 20 min may be too slow for some applications, thus requiring logic to be implemented by phosphorelays or protein–protein interactions, which can occur in milliseconds (Groban et al., 2009).
Higher-level languages and their deconstruction
A higher-level language abstracts the designer from the details of the implementation. In electronics, it enables the CPU operations to be hidden. In synthetic biology, it would hide the details of the molecular biology and DNA sequence, and possibly even the choice of circuits. Impeding the development of a higher-
level language in synthetic biology is the diversity of cellular functions. This can be partially overcome by limiting the language to describe problems that are common to many applications, while leaving the application-specific aspects unabstracted (Figure A20-1). There are two areas that have shown promise in moving towards such a language. First, vocabularies and grammars are being developed that implement design rules and can be used to describe genetic parts and their combination. This will form the backbone of the higher-level language. Second, there are a number of algorithms that can be borrowed from electrical engineering, notably logic minimization, which can convert the language into integrated circuits.
The semantics of genetic programs
A higher-level language in synthetic biology needs a vocabulary and grammar to represent genetic parts and the rules underlying how they are combined to build circuits and programs (Cai et al., 2007). A vocabulary captures how genetic functions are described and how the data underlying a part is stored and accessed. A rule is an enforced relationship between parts. For example, an expression cassette is defined as needing a promoter, cistron, and terminator and a cistron requires a RBS sequence and gene (Cai et al., 2009+).
An example of a program that describes a well-known genetic oscillator (the ‘repressilator’) is shown in Figure A20-3. At first glance, this appears unnecessarily complex compared to the more familiar plasmid visualization. However, it becomes advantageous as the designs get larger and more complex. Plasmid construction programs (like Vector NTI or ApE) toggle between the plasmid map for visualization and the DNA sequence for design. Both are difficult for debugging large programs, whereas formal grammars give an intermediate level of abstraction for writing programs and the rules enable automated troubleshooting (Cai et al., 2009+). Rules also allow for the permutation of parts in order to create multiple designs (Densmore et al., 2010b). The high-level language is also very modular and creates and separates the rules from the construction of parts and devices. This allows the complexity of a genetic program to be in-lined by a compiler/interpreter and never seen by the user after the parts are specified.
An emerging group of tools (GenoCAD, Eugene, and GEC) propose formalisms for vocabularies and provide a means to embed domain expertise in software allowing less experienced users to benefit from it (Czar et al., 2009; Cai et al., 2010; Bilitchenko et al., 2010+). Domain experts can express problem-specific design strategies as formal languages using a controlled vocabulary to represent the types of genetic parts usable in a particular organism or even for a particular application (Cai et al., 2010). Formal languages provide a means to develop a knowledge base on a collection of defined terms organized with respect to their structural relationships to each other in the DNA sequence (Cai et al., 2007). The advantage of this approach is that the relationship of the individual parts to each other are defined in the software and can provide a means of indexing and re-
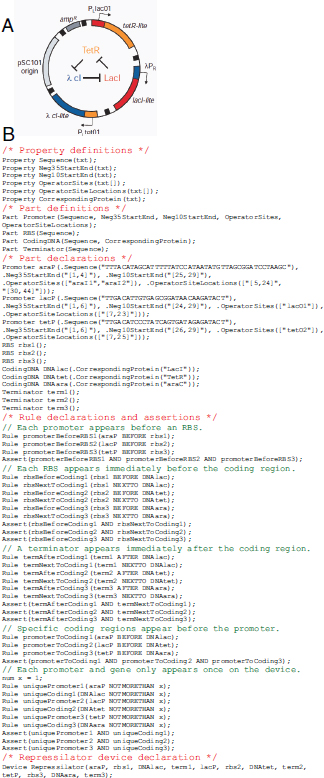
FIGURE A20-3 Semantics of genetic programs. The plasmid map (a) and Eugene code (b) for a genetic oscillator (Elowitz and Leibler, 2000) is shown (b). The author of the Eugene code is Adam Liu (Bilitchenko et al., 2010+).
trieving knowledge about parts and designs associated with defined terms. When expressing design strategies with formal languages it is possible to use parsers to verify that parts used in a design are properly positioned with respect to each other (Goler et al., 2008). Thus, users have a level of in silico validation through the use of such systems. Beyond this syntactic or structural layer, languages can be augmented by adding a semantic layer to derive dynamics encoded in complex DNA sequences composed of interacting parts using algorithms originally developed to compile the source code of computer programs (Cai et al., 2009+).
Instead of simulating the function of a DNA sequence, it is possible to perform a semantic analysis of DNA sequences using expert systems capable of reasoning using the relationships between defined terms to infer many aspects of their use, assembly based on data present in the part or associated with the biology from which the part was derived. An expert system would scan in silico designs for logical errors in construction, identify constraints that would preclude use of particular combinations of parts. Such an expert system can be constructed using ontologies—a rigorous organization of knowledge concerning a particular domain of knowledge (Arp and Smith, 2008). Ontologies are particularly good examples of expert systems in that they permit the development of a model of the entities and their relationships within the domain (Shah et al., 2009). Such definitions are logically consistent and can be used by reasoner softwares to verify known information on entities tagged by the ontology and to infer new information based upon these definitions. Softwares developed to use ontologies have the advantage that as data models change in the ontology, the software simply needs to adapt to the ontology, simplifying many aspects of software data model development and design (Shah et al., 2009). The synthetic biology community has started the Synthetic Biology Ontology Language to represent concepts tied to synthetic biology (Jiang and Hui, 2008).
A larger model that the synthetic biology community can take advantage of is the National Center for Biomedical Ontologies (Clevers, 2006). The NCBO supports researchers by providing resources to access, review, and integrate many biomedical ontologies. The Ontology for Biomedical Research (OBI) is especially interesting as it has been developed to describe the disparate elements and their relations that are necessary and sufficient to describe an experiment (Smith et al., 2007). Many of the concepts from OBI are very applicable to the description of synthetic biology workflows, protocols, parts, and device usage. NCBO also promotes standards for sharing data across ontologies, most notably the Minimum Information to Reference an External Ontology (Xiang et al., 2010). Use of MIREOT guidelines during the development of an ontology will support the consistent inclusion of terms from ontologies as diverse as Gene, Chemical Information and Pathway Information and Phenotypic Information.
Logic minimization
Several programs developed in electrical engineering have the ability to take a truth table as an input and then output a wiring diagram from simpler circuits (Figure A20-4). The ESPRESSO program, developed in the early 1980s, has been used extensively for logic minimization in VLSI design (Rudell, 1986) and it is embedded along with other tools in the abc program that is currently maintained by UC-Berkeley (Mishchenko, 2010). The output of the logic minimization tools feeds into programs, such as Logic Friday (Wu et al., 2009+), which both act as a visualization tool and enable constraints to be applied to the construction of a circuit diagram. The minimum identified by these programs is not guaranteed to be the global minimum. The logic minimization programs will have to be modi-
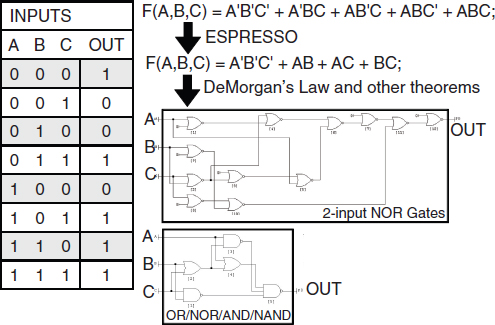
FIGURE A20-4 Automated program design using logic minimization algorithms. An example of a multi-input single-output truth table is shown. The truth table is converted to an equation F, which is a function of the four inputs (a,b,c,d). Each term corresponds to each row of the truth table where the output is 1 and the prime (′) is shorthand for the NOT function. Logic minimization algorithms, such as ESPRESSO (Rudell, 1986), can be used to simplify the full equation to its minimal form (simplest sum of products). This equation is then converted to circuit diagrams using programs such as Logic Friday (Wu et al., 2009+), which can also implement constraints. For example, the large wiring diagram consists of only 2-input NOR gates (top), whereas the smaller wiring diagram was built allowing for multiple-input OR/NOR/AND/NAND gates. Biological constraints, such as gate availability and DNA size can then be applied to search for the optimal diagram.
fied to include biological constraints, including the size of the DNA, and availability and orthogonality of the gates. Two additional considerations in choosing a wiring diagram are:
- Fault tolerance with respect to asynchronous computing. Genetic circuits are asynchronous because there is no clock controlling when each layer of the computation is performed (Seitz, 1979). Delays in the progression of the signal can lead to faults in the calculation. One approach to this problem is to build a genetic clock, and progress towards a robust oscillator (Stricker et al., 2008; Danino et al., 2010++) and counter (Friedland et al., 2009++) indicate that this may be possible. The design of asynchronous logic blocks that are robust to faults is also an active area of research, especially with respect to minimizing power requirements (Cortadella et al., 2006; Manohar, 2006; Rahbaran and Steininger, 2009). The principles from this work could be applied to the construction of integrated genetic logic, although unlike simple logic minimization, there has been historically a lack of CAD tools for asynchronous circuit design (Gibson et al., 2009++; Josephs and Furey, 2002). Recently, a system for asynchronous system design (RALA) has been developed and this framework is particularly applicable for biological systems (Gershenfeld et al., 2010+).
- Robustness to perturbations. The wiring diagrams need to be robust with respect to perturbations due to fluctuations in molecules and environmental conditions (Rodrigo and Jaramillo, 2008). In the design of electronic circuits, it has been noted that asynchronous circuits are also more robust (Gibson et al., 2009++; Rahbaran and Steininger, 2009). Macia and Sole enumerated digital systems composed of layered NAND gates and found that fault tolerance resulted from designs that contained a high degree of degeneracy, defined as occurring when multiple subsets of a circuit diagram that are structurally different perform the same function (Macia and Sole, 2009+).
Biophysical models of part function
The automated selection of genetic parts is one of the most difficult aspects of synthetic biology CAD (Kazenesis, 2007). Biophysical models can map the DNA sequence of a genetic part to its function. This enables the impact of an individual mutation to calculate for a part and then coupled to a dynamical model to determine the effect on circuit function. In other words, they facilitate linking a simulation of reaction kinetics to physical DNA. Biophysical models are also useful in screening parts selected from a registry for context effects and can scan the DNA sequence of programs to identify unintended functional sequences (e.g. internal promoters or terminators) and correct potential errors.
Cellular regulation is highly redundant and there are often many ways to control the same parameter. For example, there are many options in controlling the concentration of a protein, from plasmid copy number to protein stability. This gives flexibility to the designer to take whatever route is convenient. In terms of design automation, it will be important to choose those routes that are the most reliable when modeled on the computer. For example, the interactions between RNA basepairs is relatively straightforward to calculate and efficient algorithms have been developed to calculate the folding of RNA secondary structure (Dirks et al., 2007; Markham and Zuker, 2008). This has formed the core of predictive methods to capture how changes in the sequence affect the strength of ribosome binding sites (Salis et al., 2009; De Smit and van Duin, 1990; Na et al., 2010), the efficiency of terminators (Carafa et al., 1990; Yager and von Hippel, 1991; Kingsford et al., 2007+), the shape of the transfer function of an shRNA switch controlled by a small molecule (Beisel et al., 2008), and to create orthogonal rRNA–mRNA binding sequences (Chubiz and Rao, 2008). In each of these models, the effect of arbitrary mutations on part function can be quantitatively calculated.
The activity of promoters has been successfully modeled using position weight matrixes (PWMs) (Rhodius and Mutalik, 2010++; Li et al., 2002). These models predict the free energy by which a transcription factor binds to its operator sequence in a promoter. They make an additive assumption, where each base in the operator is assumed to be independent and a relatively small data set is used to parameterize a model. PWMs have suffered from high false positive rates, which has been a challenge in using them to scan genomes for promoters. However, they have been very successful at modeling how mutations affect the strength of a given promoter (Rhodius and Mutalik, 2010++). In this sense, they are ideal for applications where it is necessary to link how mutations will affect circuit dynamics by impacting promoter function.
Biophysical models are particularly applicable to a common problem that occurs when connecting genetic circuits in series (Figure A20-5). If the output of the first circuit is not in the correct dynamic range to trigger the second circuit, then the combined circuits will not function properly. One way to fix this is to vary the ribosome binding site (RBS) linking the circuits, typically by substituting different sites from a part library (Tabor et al., 2009; Temme et al., 2008) or through random mutagenesis (Anderson et al., 2006, 2007). More recently, a biophysical model of RBS function was developed and shown to be able to identify de novo RBS sequences that can connect two previously characterized circuits (Salis et al., 2009).
This approach could accelerate the automated connection and optimization of many genetic circuits.
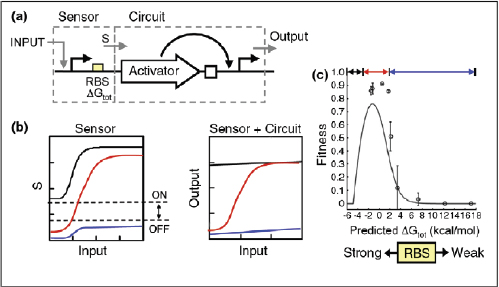
FIGURE A20-5 Connecting genetic circuits. (a) A simple program is shown consisting of a sensor and circuit. The sensor turns on a promoter in response to an INPUT. A RBS controls a translation initiation rate S that then serves as an input to the circuit. The circuit consists of an activator that turns on a promoter that serves as the output. (b) If the transfer function of the sensor is too low (blue) or the basal level is too high (black), then it will not cross the dynamic range to turn on the circuit (dotted lines). When the sensor output crosses that which is required to trigger the circuit, then the program is functional (red). (c) Experimental data is shown where the RBS is modified in the connection of a sensor to an AND gate. The solid line is the predicted fitness curve derived from the transfer functions of the sensor and circuit measured in isolation of the program.
Simulation and design environments
Kinetic simulations are commonly used to study the dynamics of molecules in the cells and there are a number of packages from systems biology that aid the modeling of signaling and regulatory networks (Andrews et al., 2010; Tomita et al., 1999; Casanova et al., 2004; Ander et al., 2004). A limitation in applying these tools to synthetic biology is the need to connect the output of a simulation to a specific design choice that is on the level of the DNA sequence, whether it be a mutation or the choice of a part. In the context of a compiler, the role of simulations would be to predict the dynamics of the assembled genetic circuits and serve as a debugging tool that would aid the user in writing the higher-level language (Figure A20-2).
A number of software tools have been developed to facilitate the selection and combination of parts gleaned from the Registry of Standard Biological Parts. For example, BioJade, SynBioSS, and TinkerCell enable the import of BioBricks
along with descriptive parameters sets that can form the basis for simulations or algorithms for part selection (Hill et al., 2008; Goler, 2004; Chandran and Bergmann, 2009; Cooling et al., 2010). Several algorithms enable the selection of parts to match an objective function that describes a desired circuit, such as an oscillator, toggle switch or logic gate (Rodrigo and Jaramillo, 2008; Dasika and Maranas, 2008). All of these simulation packages suffer from a lack of parts and circuits whose kinetics are well-documented and easily imported.
An underlying assumption of this approach is that parts will identical kinetic parameters when measured individually or in the context of different genetic programs. There are several elegant examples where this assumption has been shown to hold, including in the construction of AND gates (Salis and Kaznessis, 2006), toggle switches, and feedforward loops (Ellis et al., 2009++) through the assembly of different permutations of underlying parts. In our work with the edge detector, we found that the behavior of the program could be explained based on the component circuits (Salis and Kaznessis, 2006). However, it is not clear how often this is true and there are many published (as well as unpublished!) reports where the final behavior could not be predicted based on the components (Tan et al., 2009; Guet et al., 2002; Murphy et al., 2007). How to quantitatively measure and report this information is still an open question.
Genetic circuits usually consist of small numbers of molecules, and stochastic effects can dominate. There has been much work performed in the area of stochastic modeling and this has been applied to understanding natural and synthetic genetic networks (Salis and Kaznessis, 2006). A challenge is to convert the results of a stochastic simulation to a design choice; for example, the selection of a promoter. Noise can be controlled through the position of an operator in a promoter (Murphy et al., 2007), and it is influenced by the number of layers in a genetic program (Pedraza and van Oudenaarden, 2005). In a theoretical study, Hasty and co-workers demonstrated that the variability of the circuit response can be tuned by independently controlling the transcription and translation rates and DNA copy number (Lu et al., 2008). Cytometry data is the most common source of data in the characterization of genetic circuits and this contains significant and often unutilized information about the variability in the circuits. Munsky et al. have developed a method that enables the full cytometry distributions to be used to parameterize a genetic circuit, including its stochastic behavior (Munsky et al., 2009++).
Optimizing DNA assembly and experimental design
Once the DNA sequence is debugged on the computer, it needs to be constructed. This can either be by automated DNA synthesis or by the robotic assembly of parts. Despite the declining cost of DNA synthesis, the ability to automatically assemble parts using BioBricks and related cloning strategies will remain advantageous when it is desired to make many combinatorial variants of
a genetic system. This is useful when there is potential degeneracy in the design, allowing multiple constructs to be tested for function. Often, only a subset of the permutations function as expected.
Grammars have been modified to include information for assembly (Cai et al., 2010). The inclusion of rules can significantly reduce the construction cost by reducing the number of variants that need to be assembled (Densmore et al., 2010b). This can be further coupled to kinetic simulations to reduce the permutations to those that are likely to perform a given circuit function. For example, Peccoud and co-workers have applied grammars to permutations of the toggle switch in order to identify those that are likely to perform the correct function (Cai et al., 2009+). After rules and simulations constrain the set of permutations, a path to fabrication needs to be determined. Densmore and co-workers have developed algorithmic methods to reduce the path length in the number of assembly steps when constructing a genetic system (Densmore et al., 2010a+). The same approach can be applied to reducing the cloning steps when using the same set of underlying parts to assemble many systems.
Methods from the statistical design of experiments (DOE) could also be integrated into a CAD program (Bailey, 2008; Atkinson et al., 2007). Of particular interest is the design of sequential experiments, where the design of the next round of DNA constructs is influenced by the results from the previous round. This has been applied to highly dimensional problems, such as the optimization of fermentation, where there are many variables [pH, feed rate, oxygen, media, etc.] that need to be optimized. In the context of genetic programs, the rigorous application of DOE methods would randomize an initial set of constructs. After the constructs were measured for the desired function, this would be fed back to the algorithm to identify the next set of constructs. The process is iterated until the function is optimized. This is a particularly powerful approach when coupled with optimization algorithm, such as Nelder-Mead or ant colonization (Dorigo et al., 2006).
Conclusions
Genetic engineering is moving towards becoming an information science. The model of storing and distributing genetic material is slowly losing relevance. It is routine to outsource the task of constructing DNA from the designer to synthesis facilities. This has created a strong need for computer-aided design programs that are able to facilitate the organization and construction of large projects. Once the parts are experimentally characterized, it is unnecessary to distribute the DNA. Rather, the CAD program can access the sequence and function information to design a genetic program. The sequence information corresponding to the desired program is sent to a synthesis/assembly facility for construction.
Existing genetic circuits have not been designed to be sufficiently robust to be automatically connected using CAD. These circuits and the genetic parts on
which they are based need to be constructed specifically with the intent of coupling with a CAD program. One of the key problems is that there simply are not enough robust circuits. There are significant efforts underway to develop computational methodologies specifically to create the orthogonal parts that would underlie such programs (Chubiz and Rao, 2008; Mandell and Barbas, 2006; Desai et al., 2009++). What is particularly needed are sets of orthogonal transcription factors that bind to different operator sequences. It is the co-development of fundamental circuits with the computational algorithms to assemble them that will allow us to move towards the vision of being able to program living cells.
Acknowledgements
The authors thank Ron Weiss (MIT), Rahul Sarpeshkar (MIT), Alan Mishchenko (UC-Berkeley), Jean Peccoud (VPI), Costas Maranas (Penn State), and Douglas Densmore (BU) for helpful discussions. CAV is supported by Life Technologies, ONR, Packard Foundation, NIH, NSF (synBERC: Synthetic Biology Engineering Research Center, www.synberg.org) and a Sandpit on Synthetic Biology hosted by EPSRC/ NSF.
References and Recommended Reading
Papers of particular interest, published within the annual period of review, have been highlighted as: + of special interest
++ of outstanding interest
Ander M, Beltrao P, Di Ventura B, Ferkinghoff-Borg J, Foglierini M, Kaplan A, Lemerle C, Tomas-Oliveira I, Serrano L: SmartCell, a framework to simulate cellular processes that combines stochastic approximation with diffusion and localization: analysis of simple networks. Syst Biol 2004, 1:129-138.
Anderson JC, Clarke EJ, Arkin AP, Voigt CA: Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol 2006, 355:619-627.
Anderson JC, Voigt CA, Arkin AP: Environmental signal integration by a modular AND gate. Mol Syst Biol 2007, 3:1-8.
Andrews SS, Addy NJ, Brent R, Arkin AP: Detailed simulations of cell biology with Smoldyn 2.1. PLoS Comput Biol 2010, 6:1-10.
Arp R, Smith B: Function, role, and disposition in basic formal ontology. Nat Preceed 2008:1-4. hdl:10101/npre.2008.1941.1.
Atkinson A, Doney A, Tobias R: In Optimum Experimental Designs, with SAS. Oxford Statistical Science Series. Edited byAtkinson RJCA, Hand DJ, Pierce DA, Schervish MJ, Titterington DM. Oxford: Oxford University Press; 2007.
Bailey RA: In Design of Comparative Experiments. Cambridge Series in Statistical and Probabilistic Mathematics. Edited by Gill BDRR, Ross S, Silverman BW, Stein M. Cambridge: Cambridge University Press; 2008.
Balagadde FK, Song H, Ozaki J, Collins CH, Barnet M, Arnold FH, Quake SR, You L: A synthetic Escherichia coli predator–prey system. Mol Syst Biol 2008, 4:1-8.
Beisel CL, Bayer TS, Hoff KG, Smolke CD: Model-guided design of ligand-regulated RNAi for programmable control of gene expression. Mol Syst Biol 2008, 4:1-12.
+Bilitchenko L, Liu A, Chung S, Weeding E, Xia B, Leguia M, Anderson JC, Densmore D: Eugene—a domain specific language for specifying and constraining synthetic biological parts, devices, and systems. PLoS ONE 2010.
Eugene provides a vocabulary and grammar for the description of genetic devices. An example of which is shown in Figure A20-3. This can be compiled into a machine language that controls a part assembly robot.
Benenson Y: RNA-based computation in live cells. Curr Opin Biotechnol 2009, 20:471-478.
Buchler NE, Gerland U, Hwa T: On schemes of combinatorial transcription logic. Proc Natl Acad Sci USA 2003, 100:5136-5141.
Cai Y, Hartnett B, Gustafsson C, Peccoud J: A syntactic model to design and verify synthetic genetic constructs derived from standard biological parts. Bioinformatics 2007, 23:2760-2767.
+Cai Y, Lux MW, Adam L, Peccoud J: Modeling structure–function relationships in synthetic DNA sequences using attribute grammars. PLoS Comput Biol 2009, 5:1-10.
A description of a grammar that captures synthetic systems. The grammar was used to constrain the computational enumeration of toggle switches by varying the underlying genetic parts.
Cai Y, Wilson ML, Peccoud J: GenoCAD for iGEM: a grammatical approach to the design of standard-compliant constructs. Nucleic Acids Res 2010, 38:2637-2644.
Canton B, Labno A, Endy D: Refinement and standardization of synthetic biological parts and devices. Nat Biotechnol 2008, 26:787-793.
Carafa Yd, Brody E, Thermes C: Prediction of rho-independent Escherichia coli transcription terminators. J Mol Biol 1990, 216:835-858.
Casanova H, Berman F, Bartol T, Gokcay E, Sejnowski T, Brinbaum A, Dongarra J, Miller M, Ellisman M, Faerman M et al.: The virtual instrument: support for grid-enabled MCell simulations. Int J High Perform Comput Appl 2004, 18:3-17.
Chandran D, Bergmann FT, Sauro HM: TinkerCell: modular CAD tool for synthetic biology. Journal of Biological Engineering 2009, 3:19.
Cho B-K, Charusanti P, Herrgard MJ, Palsson BO: Microbial regulatory and metabolic networks. Curr Opin Biotechnol 2007, 18:360-364.
Chubiz LM, Rao CV: Computational design of orthogonal ribosomes. Nucleic Acids Res 2008, 36:4038-4046.
Clevers H: Wnt/beta-catenin signaling in development and disease. Cell 2006, 127:469-480.
Cooling MT, Rouilly V, Misirli G, Lawson J, Yu T, Hallinan J, Wipat N: Standard virtual biological parts: A repository of modular modeling components for synthetic biology. Bioinformatics 2010. in press.
Cortadella J, Kondratyev A, Lavago L, Sotiriou CP: Desynchronization: synthesis of asynchronous circuits from synchronous specification. IEEE Trans Comput Aided Des Integr Circuits Syst 2006, 25:1904-1921.
Cox RS, Surette M, Elowitz MB: Reprogramming gene expression with combinatorial promoters. Mol Syst Biol 2007, 3:1-11.
Czar MJ, Anderson JC, Bader JS, Peccoud J: Gene synthesis demystified. Trends Biotechnol 2008, 27:64-72.
Czar MJ, Cai Y, Peccoud J: Writing DNA with GenoCAD. Nucleic Acids Res 2009, 37:W40-W47.
++Danino T, Mondragon-Palomino O, Tsimring L, Hasty J: A synchronized quorum of genetic clocks. Nature 2010, 463:326-329.
A robust genetic oscillator is connected to cell–cell communication to synchronize cells within a population.
Dasika MS, Maranas CD: OptCircuit: an optimization based method for computational design of genetic circuits. BMC Syst Biol 2008, 2:2.
De Smit MH, van Duin J: Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc Natl Acad Sci USA 1990, 87:7668-7672.
+Densmore D, Hsiau THC, Kittleson JT, DeLoache W, Batten C, Anderson JC: Algorithms for automated DNA assembly. Nucleic Acids Res 2010a, 38:2607-2616.
Graph searching algorithms are applied to identify the optimal path in assembling constructs using BioBrick and related cloning strategies. The method is particularly suited for robotic automation.
Densmore D, Kittleson JT, Bilitchenko L, Liu A, Anderson JC, Rule based constraints for the construction of genetic devices 2010b. IEEE International Symposium on Circuits and Systems.
++Desai TA, Rodionov DA, Gelfand MS, Alm EJ, Rao CV: Engineering transcription factors with novel DNA-binding specificity using comparative genomics. Nucleic Acids Res 2009, 37:2493-2503.
An approach using comparative genomics is used to create an orthogonal set of CRP mutants that bind to different DNA sequences. This approach could be used to create many classes of orthogonal transcription factors.
Dirks RM, Bois JS, Schaeffer JM, Winfree E, Pierce NA: Thermodynamics analysis of interacting nucleic acid strands. SIAM Rev 2007, 49:65-68.
Dorigo M, Birattari M, Stutzle T: Ant colony optimization. IEEE Comput Intell Mag 2006:28-49.
Dueber JE, Yeh BJ, Chak K, Lim WA: Reprogramming control of an allosteric signaling switch through modular recombination. Science 2003, 301:1904-1908.
++Ellis T, Wang X, Collins JJ: Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat Biotechnol 2009, 27:465-471.
A parameterized set of parts was used to construct a large library of feedforward and toggle switch circuits that implement logic and time delay functions. Mathematical models of the combinations of parts accurately predicted the circuit dynamics.
Elowitz MB, Leibler S: A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403:335-338.
++Friedland AE, Lu TK, Wang X, Shi D, Church G, Collins JJ: Synthetic gene networks that count. Science 2009, 324:1199-1202.
Genetic programs based on DNA recombinases and riboswitches are implemented that respond after counting a series of pulses.
++Gershenfeld N, Dalrymple D, Chen K, Knaian A, Green F, Demaine ED, Greenwald S, SchmidtNielson P: Reconfigurable Asynchronous Logic Automata (RALA). POPL; Madrid, Spain: 2010.
A cell-based CAD program is developed for the automated design of asynchronous circuits. The operations and framework are simple and should be directly applicable to genetic circuits.
Gibson DG, Benders GA, Axelrod KC, Zaveri J, Algire MA, Moodle M, Montague MG, Venter JC, Smith HO, Hutchison CA: One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc Natl Acad Sci USA 2008, 105:20404-20409.
++Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchinson CA, Smith HO: Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009, 6:343-345.
A method is presented for the highly efficient single reaction assembly of many genetic parts. This eliminates the need for restriction enzymes in combining parts.
Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM et al.: Creation of a bacterial cell controlled by a chemically synthesized genome. Science (1-8)2010.
Goler JA: Masters Thesis. Massachusettes Institute of Technology: Cambridge, MA; 2004.
Goler JA, Bramlett BW, Peccoud J: Genetic design: rising above the sequence. Trends Biotechnol 2008, 26:538-544.
Groban ES, Clarke EJ, Salis HM, Miller SM, Voigt CA: Kinetic buffering cross talk between bacterial two-component systems. J Mol Biol 2009, 390:380-393.
Guet CC, Elowitz MB, Hsing W, Leibler S: Combinatorial synthesis of genetic networks. Science 2002, 296:1466-1470.
Ham TS, Lee SK, Keasling JD, Arkin AP: A tightly regulated inducible expression system utilizing the fim inversion recombination switch. Biotechnol Bioeng 2006, 94:1-4.
Hill AD, Tomshine JR, Weeding EMB, Sotiropoulos V, Kaznessis YN: SynBioSS: the synthetic biology modeling suite. Bioinformatics 2008, 24:2551-2553.
Hooshangi S, Thiberge S, Weiss R: Ultrasensitivity and noise propegation in a synthetic transcriptional cascade. Proc Natl Acad Sci USA 2005, 102:3581-3586.
Jiang J, Hui C: Hedgehog signaling in development and cancer. Dev Cell 2008, 15:801-812.
Josephs MB, Furey DP: A programming approach to the design of asynchronous logic blocks. In Concurrency and Hardware Design. Edited by al. JCe. Berlin: Springer-Verlag; 2002:33-60.
Kazenesis YN: Models for synthetic biology. BMC Syst Biol 2007, 1:1-4.
+Kingsford CL, Ayanbule K, Salzberg SL: Rapid, accurate, computational discovery of rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 2007, 8:R22.
The authors describe a computational method to scan genomes for terminators (TransTermHP). This is also useful to scan synthetic designs for inadvertent terminator sequences.
Kinkhabwala A, Guet CC: Uncovering cis regulatory codes using synthetic promoter shuffling. PLoS ONE 2008, 3:1-10.
Kramer BP, Fischer M, Fussenegger M: Semi-synthetic mammalian gene regulatory networks. Metabol Eng 2005, 7:241-250.
Li H, Rhodius V, Gross C, Siggia ED: Identification of the binding sites of regulatory proteins in bacterial genomes. Proc Natl Acad Sci USA 2002, 99:11772-11777.
Lou C, Liu, Xili, Ni M, Huang Y, Huang Q, Huang L, Jiang L, Lu D, Wang M et al.: Synthesizing a novel genetic sequential logic circuit: a push-on push-off switch. Mol Syst Biol 2009, 6:1-11.
Lu T, Ferry M, Weiss R, Hasty J: A molecular noise generator. Phys Biol 2008, 8:1-8.
+Macia J, Sole RV: Distributed robustness in cellular networks: insights from synthetic evolved circuits. J R Soc Interface 2009, 6:393-400.
Various wiring diagrams consisting of NAND gates are analyzed for fault tolerance. The measures of robustness applied here could be broadly applied to designing integrated genetic circuits.
Mandell JG, Barbas CF: Zinc finger tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res 2006, 34:W516-W523.
Manohar R: Reconfigurable asynchronous logic. IEEE Custom Integr Circuits Conf 2006.
Markham NR, Zuker M: UNAFold—software for nucleic acid folding and hybridization. Methods in Molecular Biology. Volume Structure, Function, and Applications. 2008: pp. 3–31.
Mishchenko, A. ABC A system for sequential synthesis and verification. 2010; Available from: http://www.eecs.berkeley.edu/ http://www.eecs.berkeley.edu/~alanmi/abc/~alanmi/abc/.
++Munsky B, Trinh B, Khammash M: Listening to the noise: random fluctuations reveal network parameters. Mol Syst Biol 2009, 5:1-7.
A method is presented to fit cytometry data and use it to estimate the parameters for a genetic circuit.
Murphy KF, Balazsi G, Collins JJ: Combinatorial promoter design for engineering noisy gene expression. Proc Natl Acad Sci USA 2007, 104:12726-12731.
Na D, Lee S, Lee D: Mathematical modeling of translocation initiation for the estimation of its efficiency to computationally design mRNA sequences with desired expression levels in prokaryotes. BMC Syst Biol 2010, 4:p71.
Pedraza JM, van Oudenaarden A: Noise propegation in gene networks. Science 2005, 307:1965-1969.
+Purnick PE, Weiss R: The second wave of synthetic biology: from modules to systems. Nat Rev 2009, 10:410-421.
A very interesting review that outlines the difficulties that underlie increasing the complexity of genetic programs.
Rackham O, Chin JW: Synthesizing cellular networks from evolved ribosome-mRNA pairs. Biochem Soc Trans 2006, 34:328-329.
Rahbaran B, Steininger A: Is asynchronous logic more robust than synchronous logic? IEEE Trans Depend Secure Comput 2009, 6:282-294.
Ramalingam KI, Tomshine JR, Maynard JA, Kaznessis YN: Forward engineering of synthetic biological AND gates. Biochem Eng J 2009, 47:38-47.
++Rhodius VA, Mutalik VK: Predicting strength and function for promoters of the Escherichia coli alternative sigma factor, σE. Proc Natl Acad Sci USA 2010, 107:2854-2859.
A predictive model of promoter strength is developed using position weighted matrixes and data from a comprehensive set of promoters activated by the σEsigma factor. They also demonstrate equivalence between in vitro and in vivo measurement methods. This approach could be broadly applied to modeling of the impact of basepair changes on promoter strength.
Richardson SM, Nunley PW, Yarrington RM, Boeke JD, Bader JS: GeneDesign 3.0 is an updated synthetic biology toolkit. Nucleic Acids Res 2010, 38:2603-2606.
Rinaudo K, Bleris L, Maddamsetti R, Subramanian S, Weiss R, Benenson Y: A universal RNAi-based logic evaluator that operates in mammalian cells. Nat Biotechnol 2007, 25:795-801.
Rodrigo G, Jaramillo A: Computational design of digital and memory biological devices. Syst Synth Biol 2008.
Rudell RL: Multiple-Valued Logic Minimization for PLA Synthesis. Berkeley, CA: UC-Berkeley; 1986.
Salis H, Kaznessis YN: Computer-aided design of modular protein devices: Boolean AND gene activation. Phys Biol 2006, 3:310-395.
Salis HM, Mirsky EA, Voigt CA: Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 2009, 27:946-951.
++Sarpeshkar R: Ultra Low Power Bioelectronics. Cambridge, MA: Cambridge University Press; 2010.
Some outstanding discussions comparing digital and analog circuits and the interpretation of genetic regulation from the perspective of electrical engineering. Read Chapters 1, 2, 7, 9, and 22–24.
Seitz CL: In Introduction to VLSI Systems. Edited by Conway CMaL. 1979.
Shah NH, Jonquet C, Chiang AP, Butte AJ, Chen R, Musen MA: Ontology-driven indexing of public datasets for translational bioinformatics. BMC Bioinformatics 2009, 10:pS1.
+Sharma V, Nomura Y, Yokobayashi Y: Engineering complex riboswitch regulation by dual genetic selection. J Am Chem Soc 2008, 48:16310-16315.
A selection method is developed that optimizes genetic AND/NAND gates composed of riboswitches that respond to small molecules. This approach is broadly useful in optimizing circuit function and creating orthogonal gates.
Shetty RP, Endy D, Knight TF: Engineering BioBrick vectors from BioBrick parts. J Biol Eng 2008, 2:1-12.
Smith B, Ashburner M, Rosse C, Bard J, Bug W, Ceusters W, Goldberg LJ, Eilbeck K, Ireland A, Mungall CJ et al.: The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nat Biotechnol 2007, 25:1251-1255.
Stricker J, Cookson S, Bennett MR, Mather WH, Tsimring LS, Hasty J: A fast, robust and tunable synthetic gene oscillator. Nature 2008, 456:516-519.
Tabor JJ, Salis HM, Simpson ZB, Chevalier AA, Levskaya A, Marcotte EM, Voigt CA, Ellington AD: A synthetic genetic edge detector program. Cell 2009, 137:1272-1281.
++Tan C, Marguet P, You LC: Emergent bistability by a growth-modulating positive feedback circuit. Nat Chem Biol 2009, 5:842-848.
A positive feedback loop constructed using T7 is shown to impact host processes, which then changes the dynamics of the circuit. A rare quantitative look at the impact of off target effects.
Temme K, Salis H, Tullman-Ercek D, Levskaya A, Hong SH, Voigt CA: Induction and relaxation dynamics of the regulatory network controlling the type III secretion system encoded within Salmonella pathogenicity island 1. J Mol Biol 2008, 377:47-61.
Tomita M, Hashimoto K, Takahashi K, Shimizu TS, Matsuzaki Y, Miyoshi F, Saito K, Tanida S, Yugi K, Venter JC, Hutchison CA: E-Cell: software environment for whole-cell simulation. Bioinformatics 1999, 15:72-84.
Villalobos A, Ness JE, Gustafsson C, Minshull J, Govindarajan S: Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC Bioinformatics 2006, 7:285.
Voigt CA: Genetic parts to program bacteria. Curr Opin Biotechnol 2006, 17:548-557.
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM: Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460:894-898.
+Wu Y, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM: A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 2009, 461:104-108.
A simple graphic interface to minimize logic operations, apply constraints, and display the circuit diagram. The minimization procedure is based on the ESPRESSO algorithm.
Xiang Z, Courtot M, Brinkman RR, Ruttenberg A, He Y: OntoFox: web-based support for ontology reuse. BMC Res Notes 2010, 3:175.
Yager TD, von Hippel PH: A thermodynamic analysis of RNA transcript elongation and termination in Escherichia coli. Biochemistry 1991, 30:1097-1118.
Yokobayashi Y, Weiss R, Arnold FH: Directed evolution of a genetic circuit. Proc Natl Acad Sci USA 2002, 99:16587-16591.
PROKARYOTIC GENE CLUSTERS: A RICH TOOLBOX FOR SYNTHETIC BIOLOGY96
Michael Fischbach97and Christopher A. Voigt98
Bacteria construct elaborate nanostructures, obtain nutrients and energy from diverse sources, synthesize complex molecules, and implement signal processing to react to their environment. These complex phenotypes require the coordinated action of multiple genes, which are often encoded in a contiguous region of the genome, referred to as a gene cluster. Gene clusters
_______________
96 Reprinted with permission from Fischbach, M., and Voigt, C. A: Prokaryotic gene clusters: A rich toolbox for synthetic biology. Biotechnology Journal 2010. 5. 1277-1296. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
97 Department of Bioengineering and Therapeutic Sciences, University of California–San Francisco, San Francisco, CA, USA.
98 Department of Pharmaceutical Chemistry, University of California–San Francisco, San Francisco, CA, USA.
Correspondence: Professor Christopher A. Voigt, Department of Pharmaceutical Chemistry, University of California – San Francisco, MC 2540, Room 408C, 1700 4th Street, San Francisco, CA 94158, USA.
E-mail: cavoigt@gmail.com Fax: +1-415-502-4690
Abbreviations: PCB, polychlorinated biphenyls; PHA, poly(hydroxyalkanoate); PHB, poly(3-hydroxybutyrate)
Keywords: Biotechnology ∙ Devices ∙ Genetic parts ∙ Refactoring ∙ Systems biology
This work was funded by NIH grant R01 AI067699. The authors have declared no conflict of interest.
sometimes contain all of the genes necessary and sufficient for a particular function. As an evolutionary mechanism, gene clusters facilitate the horizontal transfer of the complete function between species. Here, we review recent work on a number of clusters whose functions are relevant to biotechnology. Engineering these clusters has been hindered by their regulatory complexity, the need to balance the expression of many genes, and a lack of tools to design and manipulate DNA at this scale. Advances in synthetic biology will enable the large-scale bottom-up engineering of the clusters to optimize their functions, wake up cryptic clusters, or to transfer them between organisms. Understanding and manipulating gene clusters will move towards an era of genome engineering, where multiple functions can be “mixed-and-matched” to create a designer organism.
1. Introduction
Gene clusters are the genetic building blocks of bacteria and archaea. Prokaryotic genomes are highly organized and the genes associated with a particular function often occur near each other (Fischbach et al., 2008). Occasionally, all of the genes that are necessary for a discrete function form a cluster in the genome. These clusters encode functions that affect all aspects of the life style of bacteria, including nutrient scavenging, energy production, chemical synthesis, and environmental sensing. Large protein scaffolds, nanomachines, and cytoplasmic organelles are also encoded within clusters. These functions could play a central role for many applications in biotechnology; however, their complexity makes them difficult to engineer. Here, we survey the wide range of cellular functions that are known to be encoded in these genetically compact units with an eye on their potential ability to be transferable modules in multiple host species for engineering applications.
The organization of genes into clusters may facilitate the transfer of complete functions during evolution (Fischbach et al., 2008; Lawrence and Roth, 1996). All of the gene clusters presented in this review have some evidence for horizontal transfer, including phylogenetic trees disparate from ribosomal RNA, differing G+C content, and the presence of flanking transposon/integron genes (Ochman et al., 2000; Karlin, 2001). Phage genomes and conjugative plasmids also contain bacterial gene clusters, implying that a mobile element can confer a fitness advantage on its host by adding a novel function. For example, the photosynthetic apparatus (Lindell et al., 2004) and type IV pili (Karaolis et al., 1999) have been observed in phage genomes. Because gene clusters appeared and were shaped by interspecies transfer, it is intriguing that they could be fodder for genome building, where they provide a convenient unit of DNA that could be utilized to introduce a novel function into a synthetic organism. To date, such transfers are sometimes successful and sometimes fail for unknown reasons (Dixon and Postgate, 1972; Hansen-Wester et al., 2004). Potential problems include that the
cluster may rely on regulatory interactions that are not present in the new host, the genes do not express or express at the wrong ratios, or there are auxiliary interactions with, or dependencies on, the host (Fischbach et al., 2008).
Within gene clusters, there can also be sub-gene clusters that evolve separately. This modular organization of clusters within clusters enables rapid diversification and can replicate a useful function in multiple contexts. Two examples of such sub-clusters are microcompartments that can sequester toxic metabolic intermediates (Section 2.2) and the stressosome that can integrate signals and control different signaling mechanisms (Section 6.1). Sub-clusters also occur within metabolic pathways, where particular conversion (e.g., the modification of a sugar moiety) can occur in different contexts. Examples of such sub-clusters are present in the erythromycin pathway (Section 4.3). The useful functions encoded by these sub-clusters have propagated into different metabolic and signaling pathways.
As the number of complete sequenced genomes grows, it has become clear that many gene clusters are “cryptic;” in other words, there are no known conditions under which the genes are expressed (Challis, 2008). Homology analysis can be useful (albeit inexact) in predicting the general classes of molecules produces. For example, there may be many novel antibiotics and other pharmaceuticals that are encoded by such clusters. Sometimes, it is possible to “wake up” a cluster by engineering its regulatory circuitry (Scherlach and Hertweck, 2009). This could either be through the deletion of a repressor or the addition of an inducible system. However, many clusters remain intransigent to these approaches. As the sequence databases grow, it is going to be increasingly tempting to access the functions encoded therein.
Genetic engineering is moving towards the era of the genome. Automated DNA synthesis has continued to advance, with the size of routine orders increasing to >50 000 bp, and declining cost and turnaround time (Czar et al., 2008). Recently, the entire wild-type genome of Mycoplasma was synthesized and transferred into a new cell, producing a living organism (Gibson et al., 2010). However, no design was implemented in this tour-de-force project; essentially, a natural genome was replicated. In an attempt to improve our design capacity, synthetic biology has been contributing a growing toolbox of genetic parts (e.g., ribosome binding sites, promoters, terminators) and devices (e.g., genetic circuits, sensors) that enable programmable control over transcription and translation (Voigt, 2006). In addition, methods have been developed to rapidly assemble these parts into intermediate 10-kb fragments (Gibson et al., 2009), which can then be further assembled to multi-100-kb pieces (Benders et al., 2010).
As synthetic biology moves towards genome design, gene clusters are an appropriate intermediate stepping stone. On one hand, they are themselves composed of genetic parts and devices. On the other, they could be hierarchically combined to add functions to a genome. Indeed, this type of construction occurs frequently in nature, where large plasmids have been discovered that contain
multiple gene clusters (e.g., the pRSB107 plasmid combines nine antibiotic resistance gene clusters and one that scavenges iron) (Szczepanowski et al., 2005).
This review is organized to describe and compare clusters that encode a variety of functions from different species. The clusters are loosely organized into five classes according to the type of function they encode: structural, scavenging, synthesis, energy, and sensing. Each example focuses on a well-studied instance of that cluster. Many variations of each cluster exist. We also describe the current and potential applications in biotechnology for each of the gene clusters.
2. Nano-Machines, Organelles, and Large Protein Structures
2.1 Molecular Hypodermic Needle: Type III Secretion System Salmonella typhimurium (sprB to invH)
The type III secretion system (T3SS) is a molecular machine that exports proteins from the cytoplasm to the extracellular environment (Fig. A21-1) (Cornelis, 2006). Many Gram-negative pathogens have such systems, where it forms a syringe-like structure (Schraidt et al., 2010) that injects proteins into animal or plant cells to hijack a variety of host processes. The length of the needle varies depending on the function, from 60 nm in Yersinia to 2 µm in Pseudomonas syringae, where it needs to penetrate a thick plant cell wall. Effector proteins that are exported have an N-terminal secretion tag and a chaperone-binding domain that direct it to the needle. The proteins are actively unfolded before being transported through a 2-nm pore. All of the genes that are required to form the needle, as well as chaperones and effectors, are encoded within a single gene cluster (Fig. A21-2). Additional effector proteins can be scattered throughout the genome. A complex regulatory network encoded within the cluster integrates environmental signals and controls the dynamics of gene expression (Temme et al., 2008). Pathogens often have multiple clusters encoding needles that are responsible for different phases of an infection. The flagellum, involved in propulsion when bacteria swim, is a distantly related T3SS, but its genes are usually not organized into a single cluster.
Salmonella and other pathogenic bacteria have been explored for therapeutic uses as a mechanism to deliver vaccines and to block the growth of tumors. As a live vaccine, Salmonella has been used to deliver heterologous antigens from diverse sources to the immune system, including through the integration of peptides into effectors that are secreted from the T3SS encoded in Salmonella pathogenicity island 1 (SPI-1) (Galen et al., 2009). Salmonella naturally localizes to tumors after infection and it has been explored as an anticancer therapeutic, and has gone through clinical trials. The growth suppression of tumors has been shown to be dependent on the T3SS encoded within Salmonella pathogenicity island 2 (SPI-2) (Pawelek et al., 2002).
FIGURE A21-1 Gene clusters encode organelles and molecular machines. A schematic (left) and image (right) is shown for each system that appears in this review. Clockwise from top left: A reconstruction of the cryo-EM structure of the Salmonella type III secretion system is shown along with an image of multiple needle complexes spanning the inner and outer membranes (Kubori et al., 1998, 2000). A schematic of the C. thermocellum cellulosome is shown with their surface localization (Fontes and Gilbert, 2010). The crystal structure of the R. sphaerodes Reaction center and LH1 (center) and LH2 (outer eight rings) are shown next to a high-resolution AFM of the photosynthetic membrane of Rsp. photometricum (inset) (Scheuring et al., 2004). A cartoon of the type I pili from G. sulfurreducens (Fe3+ oxide is shown localized at the tip) (Lovley, 2006) is compared to a SEM of the pilus-like appendages from S. oneidensis MR-1 (Ntarlagiannis et al., 2007). The cryo-EM structure of the B. subtilis stressosome and their localization using RsbR-specific antibodies are shown (Marles-Wright et al., 2008). An idealized {100}+{111} Fe3O4 crystal is shown with an image of a magnetosome chain (Jogler and Schuler, 2009). An electron micrograph of a gas vesicle from Hfx. Mediterranei (Pfeifer, 2006) and the packing of multiple vesicles in Microcystis sp. (Walsby, 1994) are shown. Carboxysomes are shown from Synechocystis sp. PCC 6803 along with the pathway for carbon dioxide fixation (Yeats et al., 2010). All images reproduced with permission.
FIGURE A21-2 Gene clusters described in this review are compared. The colors of the genes loosely classify their functions. Many genes contain multiple functions. A classification of being “structural” includes genes that associate with a large complex and are necessary for function; for example, ATPases in type I pili and type III secretion.
2.2 Chemical Reactors Within Bacteria: Microcompartments Salmonella typhimurium (pduA to pduX)
Many bacteria build geometrically regular polyhedral organelles with a diameter of 80–200 nm (Fig. A21-1) (Yeats et al., 2010; Kerfeld et al., 2010). These act as microcompartments to encapsulate enzymes that participate in metabolic pathways where an intermediate is toxic or requires concentration. The canonical example is the carboxysome, which co-localizes carbonic anydrase, which increases the concentration of CO2 with the CO2-fixing enzyme RuBisCo. There are several examples where a pathway produces an intermediate molecule that causes toxicity when it is produced in the cytoplasm. Examples include pathways for the utilization of propanediol and ethanolamine, whose toxic intermediates are aldehyde and acetaldehyde, respectively. An example of the former is the pdu gene cluster in Salmonella typhimurium. The proteins that form the microcompartments produce a sixfold symmetry with a 2–3-Å pore that can potentially expand to 11–13 Å. The pore controls the transport of molecules in and out of the microcompartment and has a variable amino acid sequence. The core structural components of the microcompartment are conserved and appear in gene clusters with the enzymes that make up the metabolic pathway (Fig. A21-2). For example the ethanolamine utilization gene cluster has 17 genes, 4 of which are the compartment shell proteins.
Microcompartments exist in natural pathways to solve many of the problems that emerge in metabolic engineering. Frequently, an intermediate metabolite is toxic and this is detrimental to the cell if the flux in and out are unbalanced, causing the intermediate to accumulate (Mukhopadhyay et al., 2008). Further, some enzymes have an intrinsically high Km, requiring that the substrate be concentrated. Other functions require an anaerobic environment, and it has been shown that microcompartments can exclude oxygen molecules. Effectively harnessing microcompartments will require that targeting sequences be available to direct enzymes to the organelle (Fan et al., 2010) and protein engineering methods to alter the pores to allow desired substrates to enter (Yeats et al., 2010). Several microcompartments have been predicted to be involved in pathways of direct relevance to biotechnology, including in the production of ethanol in Vibrio (Wackett et al., 2007) and butyrate in Clostridium (Seedorf et al., 2008).
2.3 Balloons in Bacteria: Gas Vesicles Halobacterium salinarum (gvpA to gvpM)
A variety of species of archaea and bacteria control their buoyancy by forming gas-filled balloons in their cytoplasm (Fig. A21-1) (Pfeifer, 2006; Walsby, 1994). They function to maintain a desired depth in an aquatic environment, and their inflation and deflation are regulated by environmental signals that are used to identify the correct depth, including UV, light intensity, salinity, and oxygen. To have an effect on buoyancy, at least 10% of the cell volume needs to be composed of gas vesicles; thus, some bacteria have >10 000 per cell depend-
ing on their size. These vesicles are large, ranging in size from 33 to 250 nm in diameter, but they have very thin walls—about 2 nm—made entirely of protein. The protein that is the major component of the wall is GpvA, which forms a hydrophobic inner surface that blocks the formation of water droplets via surface tension. Gas enters the vesicles via diffusion and it is not stored there, rather it is in equilibrium with the surrounding concentration of dissolved gas (Walsby, 1994). The filling time is 0.4 µs. The vesicles can be filled by small and large gas molecules, including O2, N2, H2, CO2, CO, CH4, Ar, and C4F8. All of the genes necessary for gas vesicle formation occur together in a gene cluster, including the core structural proteins, accessory proteins that control the size and shape, chaperones, seeding proteins, and regulators (Fig. A21-2). Vesicles are produced when the gene cluster from Bacillus megaterium is transferred into Escherichia coli (Li and Cannon, 1998).
Recombinant proteins can be inserted to an accessory protein (GvpC) that participates in the formation of the shell of the gas vesicle. Antibodies have been produced and purified in this way, where the buoyancy of the vesicle aids purification and delivery (Sremac and Stuart, 2008). Gas vesicles isolated from cyanobacteria have been shown to improve oxygen transport in a mammalian cell fermentation (Sundararajan and Ju, 2000). Very interestingly, vesicles have been discovered in terrestrial bacteria that have been studied for their diverse secondary metabolisms, including antibiotic production (such as Streptomyces avermitilis), although their role in these organisms has yet to be determined (van Keulen et al., 2005). Perhaps the most interesting aspect of blowing up balloons in bacteria is that it is fun (Endy et al., 2005).
2.4 Metal Nanoparticles: Magnetosomes Magnetospirillum magneticum AMB-1 (mamC to mamF)
Biomineralization is a process by which bacteria build intricate 3-D nanostructures from dissolved metal. One of the most dramatic structures is the magnetosome, which is used by bacteria that can orient their swimming to align with the geomagnetic field (magnetotaxis) (Fig. A21-1) (Jogler and Schuler, 2009). Spherical nanocrystals (diameter ~30–50 nm) of iron oxide magnetite (Fe3O4) are contained within lipid organelles (Komeili, 2007). A string of these crystals are held in a chain orientation by proteins that resemble those involved in the cytoskeleton. All of the genes that encode the proteins involved in the biosynthesis, organization, and regulation of the magnetosome are encoded in a conserved Magnetosome Island (MAI) region of the genome that spans 80–150 kb (Fig. A21-2). This region has been noted to be unstable, and is frequently lost in lab culture. MAIs are phylogenetically widespread and there is diversity in the size, shape, and mineral composition of the crystals (Komeili, 2007).
The process by which magnetosome crystals are grown produces remarkably uniform and highly ordered structures (Jogler and Schuler, 2009). The bacteria also have the ability to concentrate low environmental abundances of metals and
form crystals under mild conditions compared to chemical routes (Schuler and Frankel, 1999). Further, their synthesis in a lipid membrane makes them easily dispersed in an aqueous environment. Thus, magnetosome-producing cells have the potential to be harnessed for the industrial production of magnetic nanoparticles, with diverse applications in medicine, imaging, and commercial uses in magnetic tape and ink (Schuler and Frankel, 1999; Matsunaga et al., 2007). Beyond Fe-based magnetosomes, many bacteria are able to build structures of diverse size (1–6000 nm), complexity (spherical, triangular, octahedral, decahedral, cubic, plates), and out of a variety of materials (gold, silver, cadmium, palladium, selenium, titanium, lanthanum, zinc, uranium, lead) (Korbekandi et al., 2009).
3. Scavenging for Nutrients
3.1 Molecular Chainsaws: The Cellulosome Clostridium cellulolyticum (cipC to cel5N)
Cellulose is a polymer of sugar molecules and is an abundant component of plant cell walls. It is the most abundant polymer in biomass and many organisms have the ability to degrade this material to obtain carbon and energy. They do this by secreting cellulases and other enzymes that are able to break down complex cell walls to release simpler sugars that can diffuse into the cell. In clostridia and rumen organisms, these enzymes frequently cluster to form a large cellulosome that protrudes from the cell surface (Fig. A21-1) (Fontes and Gilbert, 2010).
The cellulosome consists of a scaffolding protein (scaffoldin) that contains a series of cohesin domains. These domains bind to dockarin domains at the N termini of the cellulases that are involved in the assembly of the cellulosome. The scaffoldin is tethered to the cell surface (often to the S layer) and it and many of the cellulases contain cellulose-binding domains so that they bind to the cellulose in the plant cell wall (Desvaux, 2005). The advantage of having a cellulosome has been postulated to be that there is a higher likelihood that the released sugars will be consumed by the organism (Fontes and Gilbert, 2010). There is much diversity in the size and composition of cellulosomes and additional enzyme activities are commonly present, including hemicellulases and pectins to aid the decomposition of the plant cell wall. The cellulosomes can be very large, with up to 200 enzymes, and can be up to 16 MDa (Desvaux, 2005; Ding et al., 2001). Within species, the cohesin-dockarin interactions are not specific and individual enzymes are not discriminated at each position in the scaffoldin. Remarkably, the enzyme composition of the cellulosome reflects the substrate on which the cells were grown; for example, on grass clippings, pectins are expressed, and on sewage soils consisting of insect biomass, chitinases are expressed (Desvaux, 2005). The genetic regulation that controls this adaptation is unknown.
The scaffolding protein and many of the enzymes are often organized into a gene cluster (Fig. A21-2).The cluster in C. cellolyticum is a model system with 12 genes encoded in a 26-kb region, and the pattern of phylogenetic distribution
implies horizontal transfer (Desvaux, 2005). CipC is the scaffoldin and there are 8 cellulases, 1 hemicellulase (Man5K), and 1 pectinase (Cc-Rgl11Y) present in the cluster. The breakdown of crystalline cellulose is complex and requires synergistic activities between multiple enzymes. Some enzymes cut the polymer at random locations (exoglucanase), whereas others start from either free reducing or non-reducing ends and progressively cleave the polymer (endoglucanase). The most prevalent enzymes of this cellulosome represent one from each category: Cell9E and Cell48F (Desvaux, 2005). Cell9E randomly cuts the cellulose strands. In contrast, Cell48F forms a long hydrophobic tunnel, through which cellulose strands are threaded as it progressively cuts and releases simpler sugars. C. cellulolyticum contains 62 enzymes that contain dockarin domains and the enzyme composition has been shown to vary based on the substrate on which bacteria grow (Blouzard et al., 2010).
Biofuels and renewable chemicals require a source of carbon. Currently, this is frequently obtained in the form of sugar from crops such as corn and sugarcane. Particularly for fuels, this poses sustainability problems as agricultural crops would be diverted from food. In contrast, the amount of carbon that could be extracted from biomass is on the same scale as the fuels industry (and microbes have been estimated to naturally release the equivalent of 640 billion barrels of crude per year!) (Fontes and Gilbert, 2010). The problem is in the efficient liberation of carbon from cellulosic feedstocks, and this is currently a very active area of research. Significant effort has been put into the engineering of the cellulosome for this purpose (Bayer et al., 1994). One approach is to metabolically engineer natural cellulosic microbes to produce valuable products. A problem with this approach is that organisms containing cellulosomes are often not adapted for high carbon fluxes (Desvaux, 2005). A variety of metabolic engineering approaches have been used to increase the catabolism of these organisms. Another approach is to move the cellulosome into a noncellulolytic organism. This has been achieved for moving a minimal cellulosome to C. acetobutylicum (Sabathe and Soucaille, 2003) and the yeast S. cerevisiae (Lilly et al., 2009). Finally, the adhesin-dockarin domains have been harnessed as modules that control protein-protein interactions for a variety of applications outside of bioenergy, including protein purification and display (Nordon et al., 2009).
3.2 Eating Oil: Alkane Degradation Pathways Pseudomonas putida GPO1 (alkB to alkS)
Numerous marine and terrestrial bacteria have the ability to utilize hydrocarbons as a carbon and energy source (van Hamme et al., 2003). When oil leaks into seawater, this leads to the growth of a bloom of bacteria that are obligate consumers of alkanes (Yakimov et al., 2007). Many of the genes involved in the utilization of hydrocarbons occur together in a gene cluster (van Beilen et al., 2001). Petroleum is a chemically diverse substance and there are a range of en-
zymes and related pathways that break down different classes of molecules (van Beilen et al., 2001; van Beilen and Funhoff, 2007). The gene cluster in P. putida is one of the most well-studied systems and is able to degrade medium-length alkanes (Fig. A21-2) (van Beilen et al., 2001). The metabolic pathway begins with an alkane hydroxylase (AlkB—a membrane-associated non-heme diiron monooxygenase), which converts the alkane to an alcohol (Fig. A21-3) (van Beilen and Funhoff, 2007). Often, strains contain multiple alkane hydroxylases to broaden the range of substrates that can be consumed (van Hamme et al., 2003). Electrons are delivered to AlkB by two rubredoxins (AlkF and AlkG).The alcohol is converted to acyl-CoA in three steps (AlkHJK), at which point it can enter metabolism. Two additional proteins, AlkL and AlkN, putatively encode an importer and chemotaxis sensory protein, respectively. AlkS acts as an alkane
FIGURE A21-3 Utilization and breakdown pathways encoded in gene clusters are shown. The alkane degradation pathway from P. putida is adapted from Witholt and co-workers (van Beilen et al., 2001). Nitrogenase is shown along with the pathway for the production of FeMoCo (Rubio and Ludden, 2008). All images reproduced with permission.
sensor and up-regulates gene expression. The alk gene cluster occurs in many phylogenetically distinct bacteria (van Hamme et al., 2003). It has a lower G+C content than the genome and is flanked by transposon genes, which indicate frequent horizontal transfer.
Petroleum-degrading organisms have been proposed to be used in a wide variety of industrial applications. This includes a variety of potential roles in environmental cleanup, from biosensing and site evaluation to environmental dispersal, fermenter-based waste treatment, refinery waste treatment, and tanker ballast cleaning (van Hamme et al., 2003). Organisms and related pathways have been identified that can break down nearly all of the components of petroleum, including benzene, ethylbenzene, trimethylbenzene, toluene, ethyltoluene, xylene, naphthalene, methylnapthalene, phenanthrene, C6–C8 alkanes, C14–C20 alkanes, branched alkanes, and cymene (van Hamme et al., 2003). In addition, alkane-degrading organisms could be used as biocatalysts to add value to petroleum products (van Hamme et al., 2003). For example, Alcanivorax has been engineered to direct the carbon flux from alkanes to the production and export of the bioplastic precursor poly(hydroxyalkanoate) (PHA) (Sabirova et al., 2006). A particularly interesting use is for microbial enhanced oil recovery (MEOR), where bacteria are introduced into oil wells to facilitate secondary recovery (van Hamme et al., 2003). The injection of oil-degrading organisms can increase recovery by reducing viscosity or secreting surfactants. MEOR has been tested worldwide, including in the USA, and has led to increases of 15–23% for oil wells in Japan and China (van Hamme et al., 2003). Finally, the alkane-sensing transcription factor (AlkS) and the AlkB promoter have been transferred into E. coli to construct a genetic biosensor (Sticher et al., 1997).
3.3 Fertilizer Factories: Nitrogen Fixation Klebsiella pneumoniae (nifJ to nifQ)
The availability of nitrogen limits the growth of many organisms (Igarashi and Seefeldt, 2003). In agriculture, fixed nitrogen is a critical component of fertilizer and its availability has been linked to the growth of the human population. The primary source of nitrogen is from the atmosphere in the form of N2. Converting this into a form that can enter metabolism—such as ammonia (NH3)—is a difficult chemical reaction. The Haber-Bosch process can chemically convert N2 to NH3 using high temperatures and pressures using an iron catalyst. In contrast, biological nitrogen fixation uses a complex enzyme (nitrogenase) to perform this reaction (Fig. A21-3). Remarkably, the current flux of fixed nitrogen from synthetic chemical and natural biological processes is about equal (Igarashi and Seefeldt, 2003).
Only prokaryotes and some archaea have the ability to fix nitrogen (Dixon and Kahn, 2004). Often, all of the genes necessary for nitrogen fixation are encoded in a gene cluster. One of the simplest and most well-studied clusters is from K. pneumoniae, which consists of 20 genes encoded in 23 kb (Fig. A21-2) (Rubio and Ludden, 2008). These genes encode all of the necessary components for nitrogen fixation, including the nitrogenase, a metabolic pathway for the synthesis
of metal co-factors, e– transport, and a regulatory network. Nitrogenase consists of two core proteins (NifH and the NifDK complex) that participate in a reaction cycle (Igarashi and Seefeldt, 2003). The reaction itself is very energy and redox intensive with the balance
N2 + 8e– + 16ATP + 8H+ → 2NH3 + 16ADP + 16Pi + H2.
Each reaction cycle consists of the transfer of 1 e– and the consumption of 2 ATP (the energy of which is used to greatly accelerate e– transfer). It is marked by a transient interaction between NifH, which receives an e– from a variety of sources, and NifDK, which contains the reaction center where N2 binds and fixation occurs. The cycle of binding, electron transfer, and dissociation needs to be repeated eight times to fix a single N2 molecule. Nitrogenase is slow (kcat = 5 s–1) and is thought to be limited by the dissociation step (Igarashi and Seefeldt, 2003). Three co-factors form the core of the e– transfer and catalysis: [Fe4-S4] in NifH, the P cluster [Fe8-S7] in NifDK, and FeMo-co [Mo-Fe7-S9-X] (Fig. A21-4) where the reaction occurs (Rubio and Ludden, 2008). The enzymes involved in the synthesis of these co-factors and chaperones for their incorporation to form mature nitrogenase make up the majority of the cluster (Fig. A21-3). It has been proposed that these proteins all form a macromolecular “biosynthetic factory” centered on the NifEN proteins (Rubio and Ludden, 2005). NifF and NifJ are a flavodoxins that feed electrons to NifH, with pyruvate as one source (Rubio and Ludden, 2005). Nitrogenase is extremely oxygen sensitive and expensive for the cells to make and run (Fischer, 1994). A simple regulatory cascade is formed by the activator NifA and the anti-activator NifL, which integrate signals to ensure that the genes are only expressed in the absence of oxygen and fixed nitrogen (Dixon and Kahn, 2004). Since the earliest tools in genetic engineering were developed, it has been a dream of biotechnology to create cereal crops that can fix their own nitrogen. The complexity of the nitrogen fixation pathway and a lack of efficient tools for modifying non-model plants have hindered progress in this area (Dixon et al., 1997). In contrast, the complete gene cluster was functionally transferred from Klebsiella to E. coli relatively early in 1972 (Dixon and Postgate, 1972). The chloroplast may be a potentially good target for the maturation and function of nitrogenase because: (1) it is where ammonia assimilation occurs, (2) ATP is generated there, (3) there is evidence that the ancillary proteins for Fe-S formation exist, and (4) the genetic context is similar to a prokaryote, including the ability to transcribe operons (Cheng, 2008). Individual genes from the pathway have been transferred to the Tobacco genome with a chloroplast-targeting peptide and to the plastid in the algae Chlamydomonas reinhardtii (Dixon et al., 1997). Neither of these efforts yielded appreciable expression. Besides the difficulty of expressing Klebsiella-encoded genes in these contexts, there are regulatory issues around the oxygen sensitivity of nitrogenase and, thus, its inconsistency with the photosynthetic processes in the chloroplast. One way to overcome this would be to place the nitrogenase under the control of light or oxygen sensitive
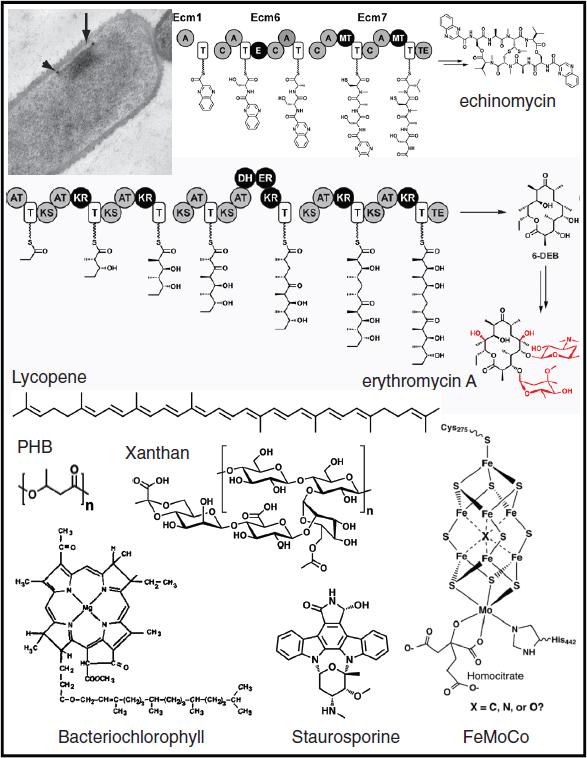
FIGURE A21-4 Chemical production pathways are often encoded within gene clusters. The image is of an organelle containing 10–100 associated 2.5 MDa NRPS-PKS mega-complexes from B. subtilis (Straight et al., 2007). The erythromycin pathway is shown from Saccharopolyspora erythraea NRRL 2338 and echinomycin pathway from Streptomyces lasaliensis. For erythromycin, chemical groups added by post-assembly-line tailoring enzymes (two P450s and two glycosyltransferases) are shown in red. A, adenylation; T, thiolation; C, condensation; E, epimerization; MT, methyltransferase; TE, thioesterase; AT, acyltransferase; KS, ketosynthase; KR, ketoreductase; DH, dehydratase; ER, enoylreductase. Bacteriochlorophyll (bottom left) is incorporated into light harvesting complexes. FeMoCo (bottom right) is produced by a metabolic pathway and incorporated into nitrogenase (image from Noodleman et al. [2004]). All images reproduced with permission.
transcription factors (Dixon et al., 1997), or to express oxygen-protective factors, including the “Shethna” protein from Azotobacter vinelandii (Moshiri et al., 1994) and some nitrogenases are intrinsically less sensitive to oxygen (Cheng, 2008). Several applications of nitrogenase have been proposed that do not involve moving the system into a plant, including for the use of N2 as a cheap source of nitrogen during fermentation (Chen et al., 2001), cyanide detoxification (Gupta et al., 2010), and the use of bacteria as biofertilizer (Bhattacharjee et al., 2008), and for the industrial production of ammonia (Brouers and Hall, 1986).
3.4 Bioremediation: Polychlorinated Biphenyl Degradation Burkholderia xenovorans LB400 (orf0 to bphD)
Some bacteria can use harmful organic pollutants as their sole source of carbon and energy (Pieper, 2005). For example, Burkholderia xenovorans LB400 can subsist on polychlorinated biphenyls (PCBs), which are used industrially as, among others, fire retardants and plasticizers (Fig. A21-4) (Pieper and Seeger, 2008). This capability has made B. xenovorans and other PCB-metabolizing bacteria key elements of bioremediation strategies for chemical spills. Highly chlorinated PCBs are reductively dehalogenated by organisms such as Dehalococcoides, which can use PCBs as a terminal electron acceptor for anaerobic respiration (Pieper and Seeger, 2008). These lower chlorinated PCBs are the substrate for the B. xenovorans degradation pathway, which consists of a series of enzyme-mediated oxidations culminating in the cleavage of one of the linked aromatic rings by the ring-opening dioxygenase BphC. The cleaved ring is converted to two equivalents of acetate in a three-step pathway, while the uncleaved ring is released as benzoic acid and then further processed to catechol by the protein products of the benABCD gene cluster (Fig. A21-2) (Pieper and Seeger, 2008).
Several strategies are being employed to increase the number of PCBs that can be degraded microbially, including directed evolution of a ring-cleaving dioxygenase (Fortin et al., 2005) and functional screening of metagenomic libraries from activated sludge (Suenaga et al., 2007). Future efforts may attempt to introduce PCB degradation gene clusters into bacterial strains that synthesize compounds of industrial value, which would allow these strains to consume feedstocks that would otherwise require expensive and environmentally unfriendly disposal.
4. Biosynthesis of Chemicals
4.1 Bioplastic Biosynthesis: Poly(3-hydroxybutryrate) Ralstonia eutropha H16 (phbA to phbC)
Many bacteria synthesize poly(3-hydroxybutyrate) (PHB) and other PHAs as a means of storing carbon and energy intracellularly (Fig. A21-4). The biosynthetic pathway for PHB, exemplified by the phb gene cluster in Ralstonia eutropha (Fig. A21-2) (Pohlmann et al., 2006), consists of three steps: PhbA catalyzes a
Claisen condensation to convert two molecules of acetyl-CoA to acetoacetyl-CoA, PhbB reduces acetoacetyl-CoA to 3-hydroxybutryl-CoA, and PhbC polymerizes 3-hydroxybutryl-CoA with release of CoA to form PHB (Madison and Huisman, 1999). PHB is hydrophobic and accumulates in cytoplasmic granules.
PHB and other PHAs are versatile bioplastics; biodegradable forms of a diverse set of products ranging from plastic bottles to golf tees are produced commercially from bacterially synthesized PHAs (Madison and Huisman, 1999). Efforts to metabolically engineer the synthesis of bioplastics are proceeding along two tracks. First, the genes for the production of PHB and other PHAs have been introduced into plants to realize the benefits of using CO2 as a carbon source rather than fermentation feedstocks (Slater et al., 1999). However, these efforts have been only modestly successful; to date, the best PHA production titer seen in plants is only ~10% of dry weight. Second, a variety of engineering efforts including genetic engineering and the provision of unnatural substrate derivatives in the fermentation broth have led to the optimization of PHA yields in native and engineered hosts and the production of novel PHA derivatives (Aldor and Keasling, 2003).
4.2 Nonribosomal Peptide Biosynthesis: Echinomycin Streptomyces lasaliensis (ecm1 to ecm18)
Nonribosomal peptides (NRPs) are a class of peptidic small molecules that includes the antibiotic vancomycin and the immunosuppressant cyclosporine (Fischbach and Walsh, 2006). The gene cluster for echinomycin (Fig. A21-4), a DNA-damaging NRP from the quinoxaline class, is typical in encoding four categories of gene products (Fig. A21-2): (1) Genes for miniature, self-contained metabolic pathways that provide unusual monomers. Eight ecm-encoded enzymes convert tryptophan into quinoxaline-2-carboxylic acid (QC), an unusual monomer that enables echinomycin to intercalate between DNA base pairs; (2) Genes for an assembly-line-like enzyme known as an NRP synthetase (NRPs) that link monomers (typically amino acids) into a peptide and then release it from covalent linkage to the assembly line, often with concomitant macrocyclization. The ecm gene cluster encodes two NRPS enzymes, Ecm6 (2608 amino acids) and Ecm7 (3135 amino acids), that convert QC, serine, alanine, cysteine, and valine into a cyclic, dimeric decapeptidolactone; (3) Genes for chemical ‘tailoring’ after release from the NRPS. Two ecm-encoded enzymes oxidatively fuse the two cysteine side chains into a thioacetal; and (4) Genes that encode regulatory and resistance functions. Transporters are also commonly found in NRP gene clusters (Gorby et al., 2006).
There are two ways in which synthetic biology is being used in the area of NRPS engineering. First, efforts are being made to express NRPS gene clusters in heterologous hosts, either in their native form (Penn et al., 2006) or re-engineered for E. coli (Gorby et al., 2006). Expression in a heterologous host can serve three purposes: making the encoded NRP accessible for structure elucidation or
biological characterization, particularly useful if the native host is unknown or unculturable; making the genes easier to manipulate, which is useful if the native host is not amenable to genetics; and improving the production titer of its small molecule product, which is helpful if the gene cluster is repressed by an external regulatory system in the native host. Second, the production of NRP derivatives has been engineered by replacing portions of NRPs genes with variants from other gene clusters that lead to the incorporation of alternative amino acid building blocks. This technique has been used most extensively to generate derivatives of the NRP antibiotic daptomycin (Baltz, 2009).
4.3 Polyketide Biosynthesis: Erythromycin Saccharopolyspora erythraea NRRL 2338 (SACE_0712 to eryCI)
Polyketides (PKs) are a class of acetate- and propionate-derived small molecules that includes the immunosuppressant FK506, the antibiotic tetracycline, the cholesterol-lowering agent lovastatin, and a number of rapamycin analogues made by genetic engineering are in clinical trials (Fischbach and Walsh, 2006). The biosynthetic pathways for PKs and fatty acids are similar in their chemical logic and use related enzymes: both involve the polymerization of acetate- or propionate-derived monomers by a series of Claisen condensations followed by reduction of the resulting β-ketothioester (Fischbach and Walsh 2006). The gene cluster for erythromycin (Fig. A21-4), an antibacterial PK from the macrolide class, encodes the following classes of gene products (Fig. A21-2):
- (1) 3 large PK synthase (PKS) enzymes—DEBS 1 (3545 amino acids), DEBS 2 (3567 amino acids), and DEBS 3 (3171 amino acids)—that convert seven equivalents of the propionate-derived monomer methylmalonyl-CoA into the intermediate 6-deoxyerythronolide B (6-DEB);
- (2) 2 P450s that hydroxylate the nascent scaffold;
- (3) 12 enzymes that synthesize the unusual sugars desosamine and mycarose from glucose and attach them to the nascent scaffold. Without these sugars, erythromycin does not have appreciable antibiotic activity; and
- (4) an erythromycin resistance gene that modifies the 50S subunit of the ribosome to prevent erythromycin from binding (Staunton and Weissman, 2001).
Many PKSs have been expressed in heterologous hosts such as E. coli, including the PKSs for erythromycin and the anticancer agent epothilone (Fujii, 2009). Another notable heterologous host is a variant of the Streptomyces fradiae strain used for the industrial production of the antibiotic tylosin; having gone through many rounds of classical strain improvement, the metabolism of this strain is well suited to the production of PKs. A variant of the strain was created in which the tylosin gene cluster was replaced by the erythromycin PKS yielding
a high titer of the non-native product (Rodriguez et al., 2003). The PKS genes have been mutated or replaced with variants from other gene clusters to generate PK derivatives (McDaniel et al., 1999), or to create custom PKSs that synthesize small PK fragments by assembling portions of several PKS genes (Menzella et al., 2005).
4.4 Terpenoid Biosynthesis: Lycopene Rhodobacter capsulatus (crtE to crtY)
Terpenoids are a class of molecules that include the anticancer agent taxol, the antibiotic pleuromutilin, and the carotenoid pigments. While terpenoids are more common among plants than bacteria (Walsh and Fischbach, 2010), carotenoids are produced by a range of bacteria. Lycopene and other carotenoids are generally used in one of two ways: to harvest light (either for energy or photo-protection) or as antioxidants (Fig. A21-4). As with other terpenoids, the first step in the biosynthetic pathway for lycopene is the CrtE-catalyzed polymerization of the C5 monomer isopentenyl pyrophosphate (IPP) or its Δ2 isomer dimethallyl pyrophosphate (DMAPP), in this case to the C20 polymer geranylgeranyl diphosphate (GGDP). CrtB then dimerizes two equivalents of GGDP in a tail-to-tail fashion, resulting in the formation of the linear C40 polymer phytoene. CrtI catalyzes four successive desaturations to yield lycopene. Alternative products such as beta-carotene are formed by the action of CrtY, which cyclizes the termini of the linear polymer (Umeno et al., 2005). All of the genes in this pathway occur together in a cluster (Fig. A21-2).
The colored nature of carotenoids has enabled their pathways to be engineered by genetic screens with colony color phenotypes. For example, a library of shuffled phytoene desaturases was screened in an E. coli strain harboring the crt gene cluster, resulting in the identification of desaturase clones that enabled the production of two lycopene variants, 3,4,3′,4′-tetradehydrolycopene and torulene (Schmidt-Dannert et al., 2000). Much synthetic biological work has been done by Keasling and coworkers on the production of plant terpenes (e.g., artemisinin) in the microbial hosts S. cerevisiae (Cogdell et al., 2006) and E. coli (Martin et al., 2003). This effort has involved two key challenges. First, since biosynthetic genes are not physically clustered in plant genomes, identifying the genes involved in terpenoid biosynthesis has been difficult, although the ongoing projects to sequence the genomes of hundreds of plants should enable bioinformatic efforts to identify biosynthetic genes. Second, the metabolism of S. cerevisiae and E. coli has been optimized for the production of terpenoids by increasing the flux of carbon toward IPP and DMAPP; in E. coli this was accomplished by supplementing the endogenous IPP biosynthetic pathway with the one from S. cerevisiae.
4.5 Oligosaccharide Biosynthesis: Xanthan Xanthomonas campestris pv. campestris (gumB to gumM)
Every year, 10 000–20 000 tons of xanthan are produced for use in foods (e.g., to control the crystallization of ice cream and to emulsify salad dress-
ings) and in industry (e.g., to modulate the viscosity of explosives and laundry detergents) (Becker et al., 1998). Xanthan, an oligosaccharide produced by the plant pathogen Xanthomonas campestris, is composed of a cellulose backbone, on alternating sugars of which a mannose-β-1,4-glucuronate-β-1,2-mannose trisaccharide is appended (Fig. A21-4). A portion of the terminal mannoses have pyruvate linked as a ketal to the 4′- and 6′-hydroxyls, and some of the internal mannoses are acetylated on the 6′-hydroxyl. Owing to the glucuronate units and pyruvoyl substituents, xanthan is an acidic polymer. Xanthan biosynthesis involves the action of five glycosyltransferases (GumDMHKI), and the growing chain is anchored on undecaprenyl pyrophosphate, similarly to peptidoglycan biosynthesis (Fig. A21-2). Three tailoring enzymes (GumFGL) add the aforementioned pyruvoyl and acetyl substitutents, and GumBCE are required for xanthan export (Becker et al., 1998).
Future efforts are likely to proceed along two tracks. First, while substrate to xanthan conversion rates of 60–70% have been achieved (Becker et al., 1998), X. campestris could be engineered to grow on cheaper feedstocks or to make the separation of the cells from the xanthan less costly; alternatively, the gum gene cluster could be moved to an alternative host. Second, changes to the structure of xanthan have important effects on its rheological properties. Efforts to use genetic engineering to alter the structure (and therefore the rheological properties) of xanthan—or of other microbial exopolysaccharides such as alginate or gellan (Sa-Correia et al., 2002)—have the potential to create new polymers, e.g., with altered viscosity and shear stability.
4.6 Indolocarbazole Biosynthesis: Staurosporine Streptomyces sp. TP-A0274 (staR to staMB)
Indolocarbazoles are natural products formed by the oxidative fusion of primary metabolic monomers (Walsh and Fischbach 2010). Staurosporine, an indolocarbazole, is a promiscuous, nanomolar inhibitor of serine/threonine protein kinases that binds in an ATP-competitive manner to these enzymes (Fig. A21-4) (Sanchez et al., 2006; Nakano and Omura, 2009). The staurosporine gene cluster encodes three categories of gene products (Fig. A21-2): (1) Four oxidoreductases (two P450s and two flavoenzymes) that catalyze a net 10-electron oxidation to fuse two molecules of tryptophan into the indolocarbazole aglycone (Howard-Jones and Walsh, 2006); (2) nine enzymes to synthesize and attach an unusual hexose to the indolocarbazole scaffold at the indole nitrogens; and (3) a transcriptional activator that regulates the expression of the gene cluster. Other naturally occurring indolocarbazoles differ in the oxidation state of the indolocarbazole scaffold, the derivatization of the indole ring by chlorination, and the sugar substituent appended to the indolocarbazole aglycone.
More than 50 unnatural indolocarbazole derivatives have been made by assembling artificial gene clusters in a non-native host (Salas and Mendez, 2009). These molecules harbor chemical modifications that would be difficult to introduce by semisynthetic derivatization of naturally occurring indolocarbazoles
or by total synthesis. The majority of these efforts have used genes from the gene clusters for indolocarbazoles as the building blocks for the artificial gene clusters. Future efforts to explore the activities of completely unrelated enzymes (e.g., ring-opening dioxygenases) may enable the modification of portions of the indolocarbazole scaffold—such as the external six-membered rings—that would be difficult to access using synthetic organic chemistry or enzymes from indolocarbazole gene clusters.
5. Energy Generation and Transfer
5.1 Solar Powerpacks: Photosynthetic Light Harvesting Rhodobacter sphaeoroides (pufH to pufX—and puc genes)
Sunlight is converted into power by the light harvesting system of anaerobic photosynthetic bacteria (Fig. A21-1) (Jones, 2009). Light energy is captured by two light harvesting complexes (LH1 and LH2) and is funneled to a reaction center (RC) (Cogdell et al., 2006). The RC uses the energy to produce a transmembrane charge separation mediated by the reduction of a quinone. This ultimately causes a proton to move from the cytoplasm to periplasm, which powers the production of ATP via the protonmotive force. The R. sphaeroides membrane has spherical invaginations that increase the surface area and number of RCs. Each RC occupies a hole in a ring formed by LH1. LH2 also forms rings that surround the RC:LH1 complex in the membrane. The LH complexes use carotenoids and bacteriochlorophyll (Fig. A21-4) to absorb green and near-infrared light, respectively (Cogdell et al., 2006). The photosynthetic genes are frequently found in a single cluster in purple bacteria (Naylor et al., 1999; Alberti et al., 1995). In R. sphaeroides, the photosynthesis gene cluster is 40.7 kb long and contains all of the necessary genes for the formation of the RC/LH1 (puf genes), and LH2 (puc genes) (Fig. A21-2). Two biosynthetic pathways make up the bulk of the cluster, where bacteriochlorophyll is produced from heme in a 16-gene pathway (bch genes) and the carotenoid sphaeroidine is produced from isopentenyl pyrophosphate in a 7-gene pathway (crt genes). The gene cluster is regulated by oxygen concentration, as well as the light intensity and color (Cogdell et al., 2006).
The Rhodobacter light harvesting system has been a model system for studying photosynthesis. It is relatively simple, there is only one photosystem, and the organism is genetically tractable. This has enabled detailed quantum mechanical measurements to be made on light absorption and electron transfer (Cogdell et al., 2006), which may enable the design of next-generation “biologically inspired” photovoltaic cells. Going one step further, the light harvesting complex can be functionally reconstituted in vitro and this has led to the construction of various hybrid systems, where the electrons are shuttled to inorganic materials (Lu et al., 2007). A particularly interesting approach is the development of a self-assembled monolayer, where the RC is tethered to a metal (Pt, Hg or Au) surface by an organic molecule that ends with a quinone. Multi-layer films have been constructed
and shown to efficiently capture electrons. Rhodobacter has also been harnessed for producing H2 from light for use in fuel cells or microbial fuel cells (Logan and Regan, 2006).
5.2 Nanowires: Conductive Surface Pili Geobacter sulfurreducens (pilB to pilA)
Metal-reducing bacteria are able to discharge electrons to solid surfaces through “wires” formed by pili that extrude from the cell surface (Fig. A21-1) (Reguera et al., 2005). This gives these bacteria the required terminal electron acceptor for oxidative phosphorylation in the absence of other dissolved acceptors (oxygen, nitrate, sulfate, etc) (El-Naggar et al., 2008). Geobacter is able to form pili that attach to Fe(III) oxide surfaces. The genes that form the pilus are encoded in a gene cluster, with PilA being the pilin subunit that is homologous with the Type IV pili from other organisms (Fig. A21-2) (Reguera et al., 2005).The pili have a diameter of 50 nm and can extend up to 20 µm from the cell surface (Revil et al., 2010). High electron transfer rates of 1011/s through the pili have been observed (El-Naggar et al., 2008). The nanowires can also connect multiple bacteria, implying that a community of cells can be wired for rapid electron transfer (Fig. A21-1) (Gorby et al., 2006). Beyond Geobacter, many other species have been shown to produce conductive pili in response to electron-acceptor limitation (Gorby et al., 2006).
Microbial fuel cells have emerged as a potential source of alternative energy (Gorby et al., 2006). A microbial fuel cell involves bacteria that are sequestered such that the only mechanism of electron transfer during respiration is to a graphite or gold anodes (Richter et al., 2008). The closest applications for microbial fuel cells are for long-term sensors deployed in the ocean and in wastewater treatment. Ocean sensors make use of the natural generation of electrical currents in the sea floor for power (Revil et al., 2010). Electricity can also be recovered from wastewater treatment as a byproduct of the breakdown of biomass. To deliver electricity to the anode, either a dissolved electron carrier or pili nanowires are required. Geobacter is one of the most efficient electron donors where it forms 50 µm thick biofilms on the surface (Revil et al., 2010). Pili are critical for electron transfer through the biofilm (Richter et al., 2008).
6. Environmental Sensing and Signal Processing
6.1 Physical Integrated Circuits: The Stressosome Bacillus subtilis (rsbR to rsbX)
One goal of synthetic biology is to build genetic circuits that can integrate information from environmental sensors or produce a dynamic response. To date, connecting circuits to form a program involves building a cascade at the level of transcription or translation. Each layer of the cascade requires about 20 min to complete (Hooshangi et al., 2005). More complex operations that require multiple layers can be particularly slow in the propagation of the signal. It would be much
faster if the circuitry could be built as a molecular machine, where signals are directly received and signal integration occurs due to conformational changes or signal propagation (e.g., via a phosphorelay).
Bacteria have such a machine, known as the stressosome (Marles-Wright et al., 2008). In B. subtilis, it is in the gene cluster that contains many regulatory factors that converge on the anti-sigma factor σB, which controls the general stress response. This pathway integrates energy and environmental stresses through a complex partner-switching mechanism (Fig. A21-5) involving anti-sigma factors, anti-anti-sigma factors, kinases, and phosphatases, most of which occur together in a gene cluster (Fig. A21-2). Three of these proteins (RsbRST) form
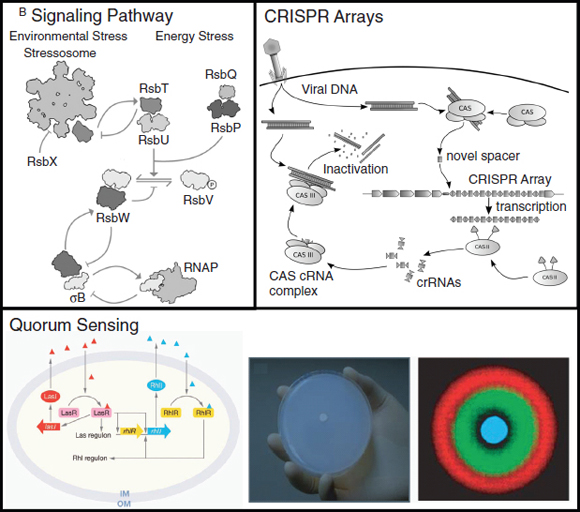
FIGURE A21-5 Complex regulatory pathways can be encoded by gene clusters. The signaling network formed by the σB gene cluster is shown (Marles-Wright et al., 2008). Environmental stress is received by the stressosome, whereas energy stress is sensed by a different branch of the pathway. Quorum sensing pathways from Pseudomonas aeruginosa (Waters and Bassler, 2005) are shown with their synthetic use to build pattern-forming programs in E. coli (Basu et al., 2005). All images reproduced with permission. The CRISPR image adapted from one drawn by James Atmos.
the structure of the stressosome, which is as large as a ribosome (1.8 MDa, 300 Å diameter) and appears as a spiked ball, with a core and protrusions (Fig. A21-1) (Marles-Wright and Lewis, 2010). The protrusions are composed of RsbR as well as four RsbR paralogs (Marles-Wright et al., 2008). The N-terminal domains of these proteins are variable, leading to the hypothesis that they act like sensors to receive diverse signals, including small molecules, protein-protein interactions, and even light (Marles-Wright and Lewis, 2010; Hecker et al., 2007). These signals are integrated by the core of the stressosome through conformational changes or other biochemical mechanisms. The RbsT protein interacts with the stressosome and transmits the signal to the σB pathway. The stressosome is induced by environmental stress and the release of RsbT is highly cooperative (a Hill factor of n=8). About 20 stressosomes are present in a single B. subtilis cell and they are closely associated with nucleoids and exhibit little diffusion. The σB gene cluster contains a number of other regulatory proteins that participate in partner-swapping and kinase/phosphatase interactions and there are internal transcriptional positive and negative feedback loops (Fig. A21-5). Together, this regulation ensures that the response is: (1) transient with a 30-min pulse of activity, (2) fast, (3) graded, and (4) the magnitude matches the degree of stress (Hecker et al., 2007; Igoshin et al., 2007).
The stressosome and the σB stress response pathway have several applications in biotechnology. The core proteins of the stressosome (RsbRST) are present in many species, including Gram negatives, and are associated with a variety of regulatory mechanisms, including aerotaxis, two-component sensors, and the biosynthesis of signaling molecules (Marles-Wright et al., 2008; Hecker et al., 2007). It may be that this structure is a common mechanism by which signals are integrated and understanding how to “reprogram” this structure would potentially enable much faster signal integration than transcriptional circuitry. In B. subtilis, there is evidence that five signals are integrated, but given the size of the structure, many more may be possible. The complete σB gene cluster is much less distributed than the stressosome. Several industrially relevant strains contain σB and related gene clusters, including Bacillus and Streptomyces (Hecker et al., 2007). The general stress response mediated by σB is involved with a number of stresses that are relevant to biotechnology, including response to shifts in salt concentration, pH, ethanol, ATP, cell wall stress, and UV light (Marles-Wright and Lewis, 2010). These types of stresses are common as the result of product accumulation, shifts in growth phase, and occur in different microenvironments in a bioreactor. Understanding how to rapidly integrate these signals would enable the construction of cell controllers that could regulate metabolic flux based on the changing environmental conditions of a fermentation.
6.2 An Immune System Against Phage: CRISPR Arrays Escherichia coli (cas3 to CRISPR4)
Many bacteria and archea contain an “immune system” that recognizes and intercepts foreign DNA based on previous exposure (Horvath and Barrangou,
2010; Marraffini and Sontheimer, 2010). This improves resistance against phage and the conjugative transfer of plasmid (Fig. A21-5). This function is encoded by a clustered, regularly spaced short palindromic repeat (CRISPR) region that occurs next to a gene cluster (Fig. A21-2). Each repeat spacer in the CRISPR region represents a DNA sequence of a phage or plasmid to which the bacteria have been exposed. The region is actively reprogrammed to respond to new challenges, which lead to the extension of the CRISPR region. Each repeat consists of a ~31-bp region of the targeted DNA and up to 374 repeats in a sinster (Marraffini and Sontheimer, 2010). Several genes are encoded in the cluster that form the Cas complex, which performs the tasks for the insertion of new spacer repeats and for the destruction of foreign DNA. The whole Cas-CRISPR cluster has undergone frequent horizontal transfer and some organisms have multiple clusters. An extreme example is Methanocaldoccus jannaschii, which contains 18 complete clusters with a total of 1188 repeat elements (Marraffini and Sontheimer, 2010).
CRISPR operates as an immune system by incorporating foreign DNA as a new repeat spacer and then recognizing this sequence in foreign DNA and destroying it (Fig. A21-5) (Horvath and Barrangou, 2010). The Cas complex cleaves foreign DNA and integrates it into the CRISPR region. The spacers are then transcribed together and processed into individual CRISPR RNAs (crRNAs), which associate with the Cas complex. The Cas-crRNA complex then recognizes the sequences in foreign DNA based on the crRNA sequence and the DNA is directed for degradation.
Bacteriophages are relevant in biotechnology as they are notorious for disrupting fermentations involving bacteria (Jones et al., 2000; Los et al., 2004). Traditionally, this is done through a process of “phage immunization” where resistant bacteria are identified through serial dilution of surviving cells (Jones et al., 2000). In one industrial example, dairy starter cultures of S. thermophilus were isolated and are now in use where phage resistance is conferred by CRISPR (Marraffini and Sontheimer, 2010). Bacteriophages have also been harnessed in biotechnology as agents for self-organization in the construction of materials (Flynn et al., 2003) and to be used as antibiotics (Sulakvelidze et al., 2001; Lu and Collins, 2007). For the latter, one of the issues that arises is rapid resistance that arises in the bacteria. In a study where bacteriophages were used to treat tooth decay, resistance in Streptococcus mutans occurred due to CRISPR immunity (van der Ploeg, 2009). The use of CRISPR elements has also been proposed as a mechanism to block the transfer of plasmids that confer antibiotic resistance and the horizontal transfer of pathogenicity islands that confer virulence (Horvath and Barrangou, 2010; Marraffini and Sontheimer, 2010).
6.3 Smelling Bacteria: Quorum Sensing Receiver Clusters Photorhabdus luminescens (yhfS to rafZ)
A nemotode (Heterorhabditis bacteriophora) implements biological warfare on its insect targets (Clarke and Photorhabdus, 2008). It has developed a symbiotic relationship with the bacterium P. luminescens to attack and digest a
wide range of insects. The genome of the bacterium is filled with more toxins and virulence factors than any known organism (Duchaud et al., 2003). After the nemotode invades, the bacteria are released into the bloodstream where they kill and breakdown the host. P. luminescens is filled with gene clusters that encode multiple type I pili for adhesion, a type III secretion system, many toxin and virulence factor pathways, and 22 clusters that encode PKSs and NRPSs. These produce a variety of small molecules, including antibiotics that kill other bacteria that may compete for the dead insect’s nutritional resources.
Interestingly, the genome contains clusters of homologues to LuxR homologues—32 divided into two major clusters (Fig. A21-2) (Duchaud et al., 2003). In Vibrio fischeri, LuxR and LuxI participate in a quorum sensing circuit that enables bacteria to communicate (Waters and Bassler, 2005). LuxI is an enzyme that produces a small molecule (AI-1) that freely diffuses through the membrane. AI-1 accumulates and, when a threshold is crossed, it activates the response regulator LuxR. Many bacteria contain multiple orthogonal pairs of LuxI/LuxR homologues, which forms a sort of “language” by which cells can communicate (Fig. A21-5). Oddly, P. luminescens has many LuxR homologues, but no corresponding LuxI homologues (Duchaud et al., 2003). It has been postulated that the cluster of LuxR proteins may be there to sense many of the bacteria that would be competing for the nutrients available in the diseased insect. In addition, some of these sensors may have evolved to respond to host hormones, such as insect juvenile hormone, to determine the identity and developmental state of the insect (Wilkinson et al., 2009).
It is useful to be able to program communication between cells for a variety of applications biotechnology (Brenner et al., 2008). Quorum sensing provides a language by which this can be achieved, where each chemical signal represents a channel for communication (Waters and Bassler, 2005).The LuxI enzyme is a “sender device” that produces the signal and LuxR is a “receiver device” that responds to the signal (Basu et al., 2005). When these devices are separated between cells, this enables cells to communicate. This has been used to program cells to form patterns, including bull’s eyes (Fig. A21-5) (Basu et al., 2005) and to implement an edge detection algorithm (Tabor et al., 2009). More direct applications in biotechnology have been proposed for quorum sensing, including controlling the density of a population of bacteria in a fermentor and killing cancer cells once an invading population crosses a threshold density (Brenner et al., 2008; Anderson et al., 2006).
7. Conclusions
In this review, we have attempted to capture the diversity of functions that are encoded in gene clusters. All of these have many potential applications in biotechnology. Achieving this potential will require methods that enable the reliable re-engineering of clusters. To date, this has been challenging because of the size of clusters, the number of genetic parts that are involved, and the complexity of the genetic regulation. Recently, progress in genetic engineering has increased
the scale of projects that are achievable. Synthetic biology has emerged as a field and libraries have been populated with genetic “parts” that can carefully control transcription and translation and “devices” that encode regulatory sensors and circuits. Methods have been developed for the rapid assembly of these parts on the scale of gene clusters, and methods exist to ultimately combine multiple clusters into a host. Together, these advances will enable the bottom-up assembly of synthetic gene clusters to simplify and optimize their function. Beyond functions that are naturally organized in clusters, it will be interesting if the design principles from studying these systems could be applied to other functions—such as the flagellum—to create similarly transferable units. This may move genetic engineering to an era of genome design where simplified gene clusters are combined in order to pull together functions from many diverse organisms to build a synthetic one.

References
Alberti, M., Burke, D. H., Hearst, J. E., Structure and sequence of the photosynthesis gene cluster, in: Blankenship, E., Madigan, M. T., Bauer, C. E. (Ed.), Anoxygenic Photosynthetic Bacteria, Kluwer Academics, Netherlands 1995, pp. 1083–1106.
Aldor, I. S., Keasling, J. D., Process design for microbial plastic factories: Metabolic engineering of polyhydroxyalkanoates. Curr. Opin. Biotechnol. 2003, 14, 475–483.
Anderson, J. C., Clarke, E. J., Arkin, A. P., Voigt, C. A., Environmentally controlled invasion of cancer cells by engineered bacteria. J. Mol. Biol. 2006, 355, 619–627.
Baltz, R. H., Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr. Opin. Chem. Biol. 2009, 13, 144–151.
Basu, S., Gerchman, Y., Collins, C. H., Arnold, F. H., Weiss, R., A synthetic multicellular system for programmed pattern formation. Nature 2005, 434, 1130–1134.
Bayer, E. A., Morag, E., Lamed, R., The cellulosome—a treasure-trove for biotechnology. Trends Biotechnol. 1994, 12, 379–385.
Becker, A., Katzen, F., Puhler, A., Ielpi, L., Xanthan gum biosynthesis and application: a biochemical/genetic perspective. Appl. Microbiol. Biotechnol. 1998, 50, 145–152.
Benders, G. A. et al., Cloning whole bacterial genomes in yeast. Nucleic Acids Res. 2010, 38, 2558–2569.
Bhattacharjee, R. B., Singh, A., Mukhopadhyay, S. N., Use of nitrogen-fixing bacteria as biofertiliser for non-legumes: Prospects and challenges. Appl. Microbiol. Biotechnol. 2008, 80, 199–209.
Blouzard, J.-C., Coutinho, P. M., Fierobe, H-P., Henrissat, B. et al., Modulation of cellulosome composition in Clostridium cellulyticum: Adaptation to the polysaccharide environment revealed by proteomic and carbohydrate-active enzyme analyses. Proteomics 2010, 10, 541–544.
Brenner, K., You, L., Arnold, F. H., Engineering microbial consortia: A new frontier in synthetic biology. Cell 2008, 26, 483–489.
Brouers, M., Hall, D. O., Ammonia and hydrogen production by immobilized cyanobacteria. J. Bacteriol. 1986, 3, 307–321.
Challis, G. L., Mining mircobial genomes for new natural products and biosynthetic pathways. Microbiology 2008, 154, 1555–1569.
Chen, J.-S., Toth, J., Kasap, M., Nitrogen-fixation genes and nitrogenase activity in Clostridium acetobutylicum and Clostridium beijerinckii. J. Ind. Microbiol. Biotechnol. 2001, 27, 281–286.
Cheng, Q., Perspectives in biological nitrogen fixation. J. Integr. Plant Biol. 2008, 50, 786–798.
Clarke, D. J., Photorhabdus: a model for the analysis of pathogenicity and mutualism. Cell. Microbiol. 2008, 10, 2159–2167.
Cogdell, R. J., Gall, A., Kohler, J., The architecture and function of the light-harvesting apparatus of purple bacteria: from single molecules to in vivo membranes. Q. Rev. Biophys. 2006, 39, 227–324.
Cornelis, G. R., The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825.
Czar, M. J., Anderson, J. C., Bader, J. S., Peccoud, J., Gene synthesis demystified. Trends Biotechnol. 2008, 27, 63–73.
Desvaux, M., The cellulosome of Clostridium cellulyticum. Enzyme Microb. Technol. 2005, 37, 373–385.
Ding, S. Y., Rincon, M. T., Lamed, R., Martin, J. C. et al., Cellusomal scaffoldin-like proteins from Ruminocuccus flavefaciens. J. Bacteriol. 2001, 183, 1945–1953.
Dixon, R., Kahn, D., Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2004, 2, 621–631.
Dixon, R., Postgate, J. R., Genetic transfer of nitrogen fixation from Klebsiella pneunomia to Escherichia coli. Nature 1972, 237, 102–103.
Dixon, R., Cheng, Q., Shen, G-F., Day, A., Dowson-Day, M., Nif gene transfer and expression in chloroplasts: Prospects and problems. Plant Soil 1997, 194, 193–203.
Duchaud, E., Rusniok, C., Frangeul, L., Buchrieser, C. et al., The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat. Biotechnol. 2003, 21, 1307–1313.
El-Naggar, M. Y., Gorby, Y. A., Xia, W., Nealson, K. H., The molecular density of states in bacterial nanowires. Biophys. J. 2008, L10–L12.
Endy, D., Deese, I., Wadey, C., Adventures in synthetic biology. Nature 2005, 438, 449–453.
Fan, C., Cheng, S., Liu, Y., Escobar, C. M. et al., Short N-terminal sequences package proteins into bacterial microcompartments. Proc. Natl. Acad. Sci. USA 2010, 107, 7509–7514.
Fischbach, M. A., Walsh, C. T., Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496.
Fischbach, M. A., Walsh, C. T., Clardy, J., The evolution of gene collectives: How natural selection drives chemical innovation. Proc. Natl. Acad. Sci. USA 2008, 105, 4601–4608.
Fischer, H.-M., Genetic regulation of nitrogen fixation in rhizobia. Microbiol. Rev. 1994, 58, 352–386.
Flynn, C. E., Lee, S-W., Peelle, B. R., Belcher, A., Viruses as vehicles for growth, organization and assembly of materials. Acta Mater. 2003, 51, 5867–5880.
Fontes, M. G. A., Gilbert, H. J., Cellulosomes: Highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 2010, 79, 655–681.
Fortin, P. D., MacPherson, I., Neau, D. B., Bolin, J. T., Eltis, L. D., Directed evolution of a ring-cleaving dioxygenase for polychlorinated biphenyl degradation. J. Biol. Chem. 2005, 280, 42307–42314.
Fujii, I., Heterologous expression systems for polyketide synthases. Nat. Prod. Rep. 2009, 26, 155–169.
Galen, J. E., Pasetti, M. F., Tennant, S., Ruiz-Olvera, P. et al., Salmonella enterica serovar Typhi live vector vaccines finally come of age. Immunol. Cell Biol. 2009, 87, 400–412.
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C. et al., Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345.
Gibson, D. G., Glass, J. I., Lartigue, C., Noskov, V. N. et al., Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56.
Gorby, Y. A., Yanina, S., McLean, J. S., Rosso, K. M. et al., Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms. Proc. Natl. Acad. Sci. USA 2006, 103, 11358–11363.
Gupta, N., Balomajumder, C., Agarwal, V. K., Enzymatic mechanism and biochemistry for cyanide degradation: A review. J. Hazard. Mater. 2010, 176, 1–13.
Hansen-Wester, I., Chakravortty, D., Hensel, M., Functional transfer of Salmonella pathogenicity island 2 to Salmonella bongori and Escherichia coli. Infect. Immun. 2004, 72, 2879–2888.
Hecker, M., Pane-Farre, J., Volker, U., SigB-dependent general stress response in Bacillus subtilis and related Gram-positive bacteria. Annu. Rev. Microbiol. 2007, 61, 215–236.
Hooshangi, S., Thiberge, S., Weiss, R., Ultasensitivity and noise propagation in a synthetic transcriptional cascade. Proc. Natl. Acad. Sci. USA 2005, 102, 3581–3586.
Horvath, P., Barrangou, R., CRISPR/Cas, the immune system of bacteria and archea. Science 2010, 327, 167–170.
Howard-Jones, A. R., Walsh, C. T., Staurosporine and rebeccamycin aglycones are assembled by the oxidative action of StaP, StaC, and RebC on chromopyrrolic acid. J. Am. Chem. Soc. 2006, 128, 12289–12298.
Igarashi, R. Y., Seefeldt, L. C., Nitrogen fixation:The mechanism of the Mo-dependent nitrogenase. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 351–384.
Igoshin, O. A., Brody, M. S., Price, C. W., Savageau, M. A., Distinctive topologies of partner-switching signaling networks correlate with their physiological roles. J. Mol. Biol. 2007, 369, 1333–1352.
Jogler, C., Schuler, D., Genomics, genetics, and cell biology of magnetosome formation. Annu. Rev. Microbiol. 2009, 63, 501–521.
Jones, D. T., Shirley, M., Wu, X., Keis, S., Bacteriophage infections in the inudstrial acetone butanol (AB) fermentation process. J. Mol. Micrbiol. Biotechnol. 2000, 2, 21–26.
Jones, M. R., The petite purple powerpack. Biochem. Soc. Trans. 2009, 37, 400–407.
Karaolis, D. K. R., Somara, S., Maneval, D. R. Jr., Johnson, J. A., Kaper, J. B., A bacteriophage encoding a pathogenicity island, a type-IV pilus and a phage receptor in cholera bacteria. Nature 1999, 399, 375–379.
Karlin, S., Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 2001, 9, 335–343.
Kerfeld, C. A., Heinhorst, S., Cannon, G. C., Bacterial microcompartments. Annu. Rev. Microbiol. 2010, 64.
Komeili, A., Molecular mechanisms of magnetosome formation. Annu. Rev. Biochem. 2007, 76, 351–366.
Korbekandi, H., Iravani, S., Abbasi, S., Production of nanoparticles using organisms. Crit. Rev. Biotechnol. 2009, 29, 279–306.
Kubori, T., Matsushima, Y., Nakamura, D., Uralil, J. et al., Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 1998, 280, 602–605.
Kubori, T. A., Sukhan, S., Aizawa, I., Galan, J. E., Molecular characterization and assembly of the needle complex of Salmonella typhimurium type III protein secretion system. Proc. Natl. Acad. Sci. USA 2000, 97, 10225–10230.
Lawrence, J. G., Roth, J. R., Selfish operons: Horizontal transfer may drive the evolution of gene clusters. Genetics 1996, 143, 1843–1860.
Li, N., Cannon, M., Gas vesicle genes identified in Bacillus megaterium and functional expression in Escherichia coli. J. Bacteriol. 1998, 180, 2450–2458.
Lilly, M., Fierobe, H-P., van Zyl, W. V., Volschenk, H., Heterologous expression of a Clostridium minicellulosome in Saccharomyces cerevisiae. FEMS Yeast Res. 2009, 9, 1236–1249.
Lindell, D., Sullivan, M. B., Johnson, Z. I., Tolonen, A. C. et al., Transfer of photosynthesis genes to and from Prochlorococcus virus. Proc. Natl. Acad. Sci. USA 2004, 101, 11013–11018.
Logan, B. E., Regan, J. M., Electricity-producing bacterial communities in microbial fuel cells. Trends Microbiol. 2006, 14, 512–518.
Los, M., Czyz, A., Sell, E., Wegrezyn, A. et al., Beacteriophage contamination: is there a simple method to reduce its deleterious effects in laboratory cultures and biotechnological factories? J. Appl. Genet. 2004, 45, 111–120.
Lovley, D. R., Bug juice: harvesting electricity with microorganisms. Nat. Rev. Microbiology Microbiol. 2006, 4, 497–508.
Lu, T. K., Collins, J. J., Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202.
Lu, Y., Xu, J., Liu, B., Kong, J., Photosynthetic reaction center functionalized nanocomposite films: Effective strategies for probing and exploiting the photo-induced electron transfer of photosensitive membrane protein. Biosens. Bioelectron. 2007, 22, 1173–1185.
Madison, L. L., Huisman, G. W., Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol. Mol. Biol. Rev. 1999, 63, 21–53.
Marles-Wright, J., Lewis, R. J., The stressosome: Molecular architecture of a signaling hub. Biochem. Soc. Trans. 2010, 38, 928–933.
Marles-Wright, J., Grant, T., Delumeau, O., van Duinen, G. et al., Molecular architecture of the “stressosome,” a signal integration and transduction hub. Science 2008, 322, 92–96.
Marraffini, L. A., Sontheimer, E. J., CRISPR interference: RNA-directed adaptive immunity in bacteria and archea. Nat. Rev. Genet. 2010, 11, 181–190.
Martin, V. J., Pitera, D. J., Withers, S. T., Newman, J. D., Keasling, J. D., Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat. Biotechnol. 2003, 21, 796–802.
Matsunaga, T., Suzuki, T., Tanaka, M., Arakaki, A., Molecular analysis of magnetotactic bacteria and development of functional bacterial magnetic particles for nano-biotechnology. Trends Biotechnol. 2007, 25, 182–188.
McDaniel, R., Thamchaipenet, A., Gustafsson, C., Fu, H. et al., Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc. Natl. Acad. Sci. USA 1999, 96, 1846–1851.
Menzella, H. G., Reid, R., Carney, J. R., Chandran, S. S. et al., Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 2005, 23, 1171–1176.
Moshiri, F., Kim, J., Fu, C., Maier, R., The FeSII protein of Azotobacter vinelandii is not essential for aerobic nitrogen fixation but confers significant protection to oxygen mediated inactivation of nitrogenase in vitro and in vivo. Mol. Microb. 1994, 14, 104–114.
Mukhopadhyay, A., Redding, A. M., Rutherford, B. J., Keasling, J. D., Importance of systems biology in engineering microbes for biofuel production. Curr. Opin. Biotechnol. 2008, 19, 228–234.
Nakano, H., Omura, S., Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J. Antibiot. (Tokyo) 2009, 62, 17–26.
Naylor, G. W., Addlesee, H. A., Gibson, L. C. D., Hunter, C. N., The photosynthesis gene cluster of Rhodobacter sphaeroides. Photosyn. Res. 1999, 62, 121–139.
Noodleman, L., Case, D. A., Han, W.-G., Lovell, T. et al., Scientific Report, Scripps 2004.
Nordon, R. E., Craig, S. J., Foong, F. C., Molecular engineering of the cellulosome complex for affinity and bioenergy applications. Biotechnol. Lett. 2009, 31, 465–476.
Ntarlagiannis, D., Atekwana, E. A., Hill, E. A., Gorby, Y., Microbial nanowires: Is the subsurface “hardwired”? Geophys. Res. Lett. 2007, 34, L17305.
Ochman, H., Lawrence, J. G., Groisman, E. A., Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304.
Pawelek, J. M., Sodi, S., Chakraborty, A. K., Platt, J. T. et al., Salmonella pathogenicity island-2 and anticancer activity in mice. Cancer Gene Ther. 2002, 9, 813–818.
Penn, J., Li, X., Whiting, A., Latif, M. et al., Heterologous production of daptomycin in Streptomyces lividans. J. Ind. Microbiol. Biotechnol. 2006, 33, 121–128.
Pfeifer, F., Gas vesicles of archea and bacteria, in: Shively, J. M. (Ed.), Complex Intracellular Structures in Prokaryotes, Springer, Berlin 2006, pp. 115–140.
Pieper, D. H., Aerobic degradation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 2005, 67, 170–191.
Pieper, D. H., Seeger, M., Bacterial metabolism of polychlorinated biphenyls. J. Mol. Microbiol. Biotechnol. 2008, 15, 121–138.
Pohlmann, A., Fricke, W. F., Reinecke, F., Kusian, B. et al., Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 2006, 24, 1257–1262.
Reguera, G., McCarthy, K. D., Mehta, T., Nicoll, J. S. et al., Extracellular electron transfer via microbial nanowires. Nature 2005, 435, 1098–1101.
Revil, A., Mendonca, C. A., Atekwana, E. A., Kulessa, B. et al., Understanding biogeobatteries: Where geophysics meets microbiology. J. Geophys. Res. 2010, 115, 1–22.
Richter, H., McCarthy, K., Nevin, K. P., Johnson, J. P. et al., Electricity generation by Geobacter sulfurreducens attached to gold electrodes. Langmuir 2008, 24, 4376–4379.
Rodriguez, E., Hu, Z., Ou, S., Volchegursky, Y. et al., Rapid engineering of polyketide overproduction by gene transfer to industrially optimized strains. J. Ind. Microbiol. Biotechnol. 2003, 30, 480–488.
Rubio, L. M., Ludden, P. W., Maturation of nitrogenase: a biochemical puzzle. J. Bacteriol. 2005, 187, 405–414.
——., Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 2008, 62, 93–111.
Sabathe, F., Soucaille, P., Characterization of the CipA scaffolding protein and in vivo production of a minicellulosome in Clostridium acetobutylicum. J. Bacteriol. 2003, 185, 1092–1096.
Sabirova, J. S., Ferrer, M., Lunsdorf, H., Wray, V. et al., Mutation in a “tesB-like” hydroxyacylcoenzyme A-specific thioesterase gene causes hyperproduction of extracellular polyhydroxyalkanoates by Alcanivorax borkumensis SK2. J. Bacteriol. 2006, 188, 8452–8459.
Sa-Correia, I., Fialho, A. M., Videira, P., Moreira, L. M. et al., Gellan gum biosynthesis in Sphingomonas paucimobilis ATCC 31461: genes, enzymes and exopolysaccharide production engineering. J. Ind. Microbiol. Biotechnol. 2002, 29, 170–176.
Salas, J. A., Mendez, C., Indolocarbazole antitumour compounds by combinatorial biosynthesis. Curr. Opin. Chem. Biol. 2009, 13, 152–160.
Sanchez, C., Mendez, C., Salas, J. A., Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045.
Scherlach, K., Hertweck, C., Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009, 7, 1753–1760.
Scheuring, S., Rigaud, J-L., Sturgis, J. N., Variable LH2 stoichiometry and core clustering in native membranes of Rhodospirillum photometricum. EMBO J. 2004, 23, 4127–4133.
Schmidt-Dannert, C., Umeno, D., Arnold, F. H., Molecular breeding of carotenoid biosynthetic pathways. Nat. Biotechnol. 2000, 18, 750–753.
Schraidt, O., Lefebre, M. D., Brunner, M. J., Schmied, W. H. et al., Topology and organization of the Salmonella typhimurium type III secretion needle complex components. PLoS Pathog. 2010, 6, e1000824.
Schuler, D., Frankel, R. B., Bacterial magnetosomes: Microbiology, biomineralization, and biotechnological applications. Appl. Microbiol. Biotechnol. 1999, 52, 464–473.
Seedorf, H., Fricke, W. F., Veith, B., Bruggemann, H. et al., T he genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc. Natl. Acad. Sci. USA 2008, 105, 2128–2133.
Slater, S., Mitsky, T. A., Houmiel, K. L., Hao, M. et al., Metabolic engineering of Arabidopsis and Brassica for poly(3-hydroxybutyrate-co-3-hydroxyvalerate) copolymer production. Nat. Biotechnol. 1999, 17, 1011–1016.
Sremac, M., Stuart, E. S., Recombinant gas vesicles from Halobacterium sp. displaying SIV peptides demonstrate biotechnology potential as a pathogen peptide delivery vehicle. BMC Biotechnol. 2008, 8, 9.
Staunton, J., Weissman, K. J., Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 2001, 18, 380–416.
Sticher, P., Jasper, M. C. M., Stemmler, K., Harms, H. et al., Development and characterization of a whole cell bioluminescent sensor for bioavailable middle-chain alkanes in contaminated groundwater samples. Appl. Environ. Microb. 1997, 63, 4053–4060.
Straight, P. D., Fischbach, M. A., Walsh, C. T., Rudner, D. Z., Kolter, R., A singular enzymatic mega-complex from Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 305–310.
Suenaga, H., Ohnuki, T., Miyazaki, K., Functional screening of a metagenomic library for genes involved in microbial degradation of aromatic compounds. Environ. Microbiol. 2007, 9, 2289–2297.
Sulakvelidze, A., Alavidze, Z., Morris, J. G., Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659.
Sundararajan, A., Ju, L., Glutaraldehyde treatment of proteinaceous gas vesicles from cyanobacterium Anabaena flosaquae. Biotechnol. Prog. 2000, 16, 1124–1128.
Szczepanowski, R., Braun, S., Riedel, V., Schneiker, S. et al., The 120 592 bp IncF plasmid pRSB107 isolated from a sewage-treatment plant encodes nine different antibiotic-resistance determinants, two iron-acquisition systems and other putative virulence-associated functions. Microbiology 2005, 151, 1095–1111.
Tabor, J. J., Salis, H. M., Simpson, Z. B., Chevalier, A. A. et al., A synthetic genetic edge detection program. Cell 2009, 137, 1272–1281.
Temme, K., Salis, H., Tullman-Ercek, D., Levskaya, A. et al., Induction and relaxation dynamics of the regulatory network controlling the type III secretion system encoded within Salmonella pathogeneity island 1. J. Mol. Biol. 2008, 377, 47–61.
Umeno, D., Tobias, A. V., Arnold, F. H., Diversifying carotenoid biosynthetic pathways by directed evolution. Microbiol. Mol. Biol. Rev. 2005, 69, 51–78.
van Beilen, J. B., Funhoff, E. G., Alkane hydroxylases involved in microbial alkane degradation. Appl. Microbiol. Biotechnol. 2007, 74, 13–21.
van Beilen, J. B., Panke, S., Lucchini, S., Franchini, A. G. et al., Analysis of Pseudomonas putida alkane-degradation gene clusters and flanking insertion sequences: Evolution and regulation of the alk genes. Microbiology 2001, 147, 1621–1630.
van der Ploeg, J. R., Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of aquired immunity against infection by M102-like bacteriophages. Micr-biology 2009, 155, 1966–1976.
van Hamme, J. D., Singh, A., Ward, O. P., Recent advances in petroleum microbiology. Microbiol. Mol. Biol. Rev. 2003, 67, 503–549.
van Keulen, G., Hopwood, D.A., Dijkhuizen, L., Sawers, R. G., Gas vesicles in actinomyces: old buoys in novel habitats? Trends Microbiol. 2005, 13, 350–354.
Voigt, C. A., Genetic parts to program bacteria. Curr. Opin. Biotechnol. 2006, 17, 548–557.
Wackett, L. P., Frias, J. A., Seffernick, J. L., Sukovich, D. J., Cameron, S. M., Genomic and biochemical studies demonstrating the absence of an alkane-producing phenotype in Vibrio furnissii M1. Appl. Environ. Microb. 2007, 73, 7192–7198.
Walsby, A. E., Gas vesicles. Microbiol. Rev. 1994, 58, 94–144.
Walsh, C. T., Fischbach, M. A., Natural products version 2. 0: Connecting genes to molecules. J. Am. Chem. Soc. 2010, 132, 2469–2493.
Waters, C. M., Bassler, B. L., Quorum Sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346.
Widmaier, D. M., Tullman-Ercek, D., Mirsky, E. A., Hill, R. et al., Engineering the Salmonella type III secretion system to export spider silk monomers. Mol. Syst. Biol. 2009, 5, 309.
Wilkinson, P., Waterfield, N. R., Crossman, L., Corton, C. et al., Comparative genomics of the emerging human pathogen Photorhabdus asymbiotica with the insect pathogen Photorhabdus luminescens. BMC Genomics 2009, 10, 302.
Yakimov, M. M., Timmis, K. N., Golyshin, P. N., Obligate oil-degrading marine bacteria. Curr. Opin. Biotechnol. 2007, 18, 257–266.
Yeats, T. O., Crowley, C. S., Tanaka, S., Bacterial microcompartment organelles: Protein shell structure and evolution. Annu. Rev. Biophys. 2010, 39, 185–205.
Barbara M. Bakker,100,* R. Luise Krauth-Siegel,101Christine Clayton,102Keith Matthews,103Mark Girolami,104Hans V. Westerhoff,105Paul A. M. Michels,106Ranier Breitling,107and Michael P. Barrett108
Summary
African trypanosomes have emerged as promising unicellular model organisms for the next generation of systems biology. They offer unique advan-
_______________
99 Reprinted with permission from Bakker, B. M., R. L. Krauth-Siegel, C. Clayton, K. Matthews, M. Girolami, H. V. Westerhoff, P. M. Michels, R. Breitling, and M. P. Barrett. 2010. The silicon trypanosome. Parasitology 137:1333-1341. Reprinted with permission.
100 Department of Pediatrics, Center for Liver, Digestive and Metabolic Diseases, University Medical Center Groningen, University of Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands.
101 Biochemie-Zentrum der Universität Heidelberg, 69120 Heidelberg, Germany.
102 Zentrum für Molekulare Biologie der Universität Heidelberg, ZMBH-DKFZ Alliance, D69120 Heidelberg, Germany.
103 School of Biological Sciences, University of Edinburgh, Ashworth Laboratories, Edinburgh EH9 3JT, United Kingdom.
104 University of Glasgow, Department of Computing Science & Department of Statistics, Glasgow, G12 8QQ, United Kingdom.
105 Department of Molecular Cell Physiology, VU University Amsterdam, 1081 HV Amsterdam, The Netherlands; and Manchester Centre for Integrative Systems Biology, Manchester Interdisciplinary BioCentre, The University of Manchester, Manchester M1 7ND, United Kingdom.
106 Research Unit for Tropical Diseases, de Duve Institute and Laboratory of Biochemistry, Université catholique de Louvain, Brussels, Belgium and Faculty of Biomolecular and Life Sciences, University of Glasgow, Glasgow, G12 8QQ, UK.
107 Groningen Bioinformatics Centre, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, 9751 NN Haren, The Netherlands.
108 Faculty of Biomolecular and Life Sciences and Wellcome Centre of Molecular Parasitology, University of Glasgow, Glasgow Biomedical Research Centre, Glasgow G12 8TA, United Kingdom.
* Corresponding author: B.M. Bakker, Tel: +31 (0) 50361 1542. E-mail: b.m.bakker@med.umcg.nl Key words: Trypanosoma brucei, metabolism, gene expression, differentiation, Silicon Cell.
tages, due to their relative simplicity, the availability of all standard genomics techniques and a long history of quantitative research. Reproducible cultivation methods exist for morphologically and physiologically distinct life-cycle stages. The genome has been sequenced, and microarrays, RNA-interference and high-accuracy metabolomics are available. Furthermore, the availability of extensive kinetic data on all glycolytic enzymes has led to the early development of a complete, experiment-based dynamic model of an important biochemical pathway. Here we describe the achievements of trypanosome systems biology so far and outline the necessary steps towards the ambitious aim of creating a ‘Silicon Trypanosome’, a comprehensive, experiment-based, multi-scale mathematical model of trypanosome physiology. We expect that, in the long run, the quantitative modelling enabled by the Silicon Trypanosome will play a key role in selecting the most suitable targets for developing new anti-parasite drugs.
The Ambition of Systems Biology
Systems biology seeks to understand how functional properties of living systems, such as biological rhythms, cellular differentiation or the adaptation of organisms to changes in their environment, emerge from interactions between the components in the underlying molecular networks (Bruggeman and Westerhoff, 2007). In the case of parasites with multiple hosts, differentiation and adaptation to drugs may be particularly relevant. Current systems biology is to a large extent (but not exclusively) focused on single-cell systems. These are more amenable to global molecular analysis than multicellular organisms. This is partly because high-throughput post-genomic technologies (transcriptomics, proteomics and metabolomics) make it relatively easy to measure many components of a homogeneous population of cells simultaneously. Furthermore, dynamic measurements of the response of a cell population to a shared stimulus allow insight into the functional connectivity between components (Richard et al., 1996; Hynne et al., 2001; Nikerel et al., 2006, 2009; Schmitz et al., 2009).
Mathematical methods that enable quantitative descriptions of the dynamic interplay between the molecules in living cells are being developed and, for the first time, it is possible to envisage a comprehensive molecular description of the functional circuitry of cellular systems. The Silicon Cell project (Snoep et al., 2006; Westerhoff et al., 2009) involves an international consortium of researchers aiming at a mathematical description of life at the cellular level on the basis of complete and quantitative genomic, transcriptomic, proteomic, metabolomic and phenotypic information. So far, the most ambitious whole-cell modelling efforts have targeted the model organisms Escherichia coli and Saccharomyces cerevisiae. The advanced state of understanding and the enormous amount of data relating to these organisms have made them obvious candidates for such a comprehensive description. Yet, the variety of organisms in the ‘JWS Online’ model repository (www.jjj.bio.vu.nl) demonstrates that the silicon cell initiative is not limited to these organisms.
The Unique Advantages of Trypanosome Systems Biology
The African trypanosome, Trypanosoma brucei, the causative agent of human African trypanosomiasis and Nagana cattle disease (Barrett et al., 2003), has emerged as a front runner in systems biology analysis. The relative simplicity of the energy metabolism of its bloodstream form and the early availability of a comprehensive and uniform set of kinetic data of the enzymes involved, were crucial factors for the successful construction of a detailed computer model of trypanosome glycolysis (Bakker et al., 1997). The obvious questions for this model were initially in the realm of drug-target selection; the first studies analysed in depth how sensitive the pathway overall would be to varying extents of inhibition of each enzyme (Bakker et al., 1999a). Another important factor stimulating further development of trypanosome systems biology was the possibility of reproducible in vitro cultivation, first of the procyclic insect stage, but later also of the long slender bloodstream form (Hirumi and Hirumi, 1989; Haanstra, 2009). Transitions between distinct life-cycle stages can be studied in a tractable and synchronous differentiation system (Fenn and Matthews, 2007). Moreover, the complete genome of T. brucei has been sequenced and annotated, and a metabolic pathway database has been developed (Berriman et al., 2005; Chukualim et al., 2008).
An extension of the scope of trypanosome systems biology to include gene expression is facilitated by the absence of transcriptional regulation in trypanosomes. This implies that, unlike in most other organisms, the gene-expression cascade is regulated only post-transcriptionally. The genes of African trypanosomes – as well as those of the other kinetoplastids – are arranged in polycistronic transcription units which can be hundreds of kilobases long (Berriman et al., 2005; Siegel et al., 2009). All evidence so far indicates that RNA polymerase II transcribes constitutively, without intervention of regulatory factors (Lee et al., 2009; Palenchar and Bellofatto, 2006). Individual mRNAs are excised by a trans-splicing complex which places identically capped 39 nt leaders at the 5k end of every mRNA (Liang et al. 2003); this splicing is co-ordinated with polyadenylation of the RNA located immediately upstream. Indeed, regulation of mRNA biogenesis may well be restricted to the processing steps (Lustig et al., 2007; Stern et al., 2009), while steady-state levels are further influenced by the rate of mRNA degradation (Clayton and Shapira, 2007). In fact, the majority of evidence concerning regulation of gene expression has implicated mRNA decay as the dominant factor (Clayton and Shapira, 2007; Haanstra et al., 2008b), and this is the only step for which mechanistic details of the regulation are available.
After the complete sequencing of the trypanosome genome (Berriman et al., 2005), mRNA microarray analyses of the differentiation from the bloodstream to the procyclic form have demonstrated that the expression of whole sets of mRNAs is coordinately regulated (Queiroz et al., 2009). When gene-expression is studied during synchronous differentiation, accurate time profiles of extremely homogenous cell populations can be obtained (Kabani et al., 2009). Most results
so far suggest that such regulation is mediated by RNA-binding proteins that bind to specific sequences in the 3k-untranslated regions of mRNAs (Archer et al., 2009; Clayton and Shapira, 2007; Estévez, 2008). The rate of mRNA translation and protein turnover are other factors influencing the steady-state protein levels. Here too, key regulatory proteins have been identified (Paterou et al., 2006; Walrad et al., 2009).
Milestones of Trypanosome Systems Biology
A quantitative mathematical model of energy metabolism in the long slender form of the trypanosome (i.e. the form that replicates in the mammalian bloodstream) has been developed (Bakker et al., 1997) and iteratively updated after experimental testing (Bakker et al., 1999a,b; Albert et al., 2005; Haanstra et al., 2008a). This model yields quantitative predictions of the flux through glycolysis, the concomitant ATP production flux, and the concentrations of glycolytic metabolites, at steady state as well as following a perturbation. Input data for the model are kinetic equations and parameters of enzymes and their concentrations. Through this model, the effects of drugs on the glycolytic pathway can be assessed quantitatively starting from their effects on the individual enzymes.
Free-energy metabolism in the bloodstream form of T. brucei has been a logical starting point for the ‘bottom-up’ construction of a ‘Silicon Trypanosome’. Bloodstream forms of the parasite depend exclusively on substrate-level phosphorylation for ATP production through glycolysis, which proceeds as far as pyruvate (Flynn and Bowman, 1973) (Fig. A22-1). Pyruvate is the end product and is secreted from the cell. Many of the glycolytic enzymes differ in terms of allosteric regulation from their mammalian counterparts, and this probably relates to the fact that in T. brucei the first seven enzymes of the pathway reside within membrane-bounded, peroxisome-like organelles called glycosomes (Opperdoes and Borst, 1977; Parsons, 2004; Michels et al. 2006; Haanstra et al. 2008), which isolate most of the glycolytic pathway from the rest of the metabolic network involved in consumption of ATP and NAD(H). Even in growing and dividing trypanosomes, virtually all glucose is converted to pyruvate, as the amount required for biosynthesis is quantitatively negligible (Haanstra, 2009). This finding supports the initial choice to model the glycolytic pathway without any branches other than the one to glycerol. Glycerol production is crucial under anaerobic conditions (Fairlamb et al., 1977).
Since the publication of the first version of the glycolysis model (Bakker et al., 1997), there have been two major updates (Helfert et al., 2001; Albert et al., 2005). Both of these involved updates and extensions of the enzyme kinetic dataset, e.g. the explicit inclusion of individual enzymes that were previously grouped into a net multi-step conversion. In the second update (Albert et al., 2005) the enzyme expression levels (Vmax) were adapted to reflect the concentrations observed in trypanosomes obtained from controlled state-of-the-art in vitro
FIGURE A22-1 The glycolytic pathway in Trypanosoma brucei. Reaction numbers indicate: 1. glucose transport; 2. hexokinase; 3. phosphoglucose isomerase; 4. phosphofructokinase; 5. aldolase; 6. triose-phosphate isomerase; 7. glyceraldehyde-3-phosphate dehydrogenase; 8. phosphoglycerate kinase; 9. phosphoglycerate mutase; 10. enolase; 11. pyruvate kinase; 12. pyruvate transport; 13. glycerol-3-phosphate dehydrogenase; 14. glycerol-3-phosphate oxidase (a combined process of mitochondrial glycerol-3-phosphate dehydrogenase and trypanosome alterative oxidase; 15. glycerol kinase; 16. combined ATP utilization; 17. glycosomal adenylate kinase; 18. cytosolic adenylate kinase. Question marks indicate uncharacterized transport processes. Abbreviations of metabolite names: Glc-6-P: glucose 6-phosphate; Fru-6-P: fructose 6-phosphate; Fru-1,6-BP: fructose 1,6-bisphosphate; DHAP: dihydroxyacetone phosphate; Gly-3-P: glycerol 3-phosphate; GA-3-P: glyceraldehyde 3-phosphate; 1,3-BPGA: 1,3-bisphosphoglycerate; 3-PGA: 3-phosphoglycerate; 2-PGA: 2-phosphoglycerate; PEP: phospho-enolpyruvate.
cultivation. Key missing pieces of information remain the mechanism and kinetics of the transport of glycolytic metabolites across the glycosomal membrane. The identification of semi-selective pores in peroxisomal membranes suggests that the smaller metabolites equilibrate across the glycosomal membrane, while bulkier molecules like ATP or NADH require specific transporters (Grunau et al., 2009; Rokka et al., 2009). This idea justifies, with hindsight, the choice to model the transport of a number of small intermediates as rapid-equilibrium processes.
A number of basic and applied biological questions have been addressed using the glycolysis model. For example, it was predicted and then experimentally confirmed (Bakker et al., 1999a,b) that the uptake of glucose across the plasma membrane was a major flux controlling step and therefore an interesting drug target. Enzymes that have been suggested to control glycolysis in mammalian cells, like hexokinase, phosphofructokinase and pyruvate kinase (Schuster and Holzhütter, 1995), exerted little control in trypanosomes, according to the model (Bakker et al., 1999a; Albert et al., 2005). Experiments, in which the expression of these enzymes was knocked down, confirmed this prediction qualitatively. However, the enormous overcapacity of some enzymes, which was predicted by the model, was shown to be exaggerated (Albert et al., 2005). This suggests that there are in vivo regulation mechanisms affecting these enzymes in a currently unknown fashion. Protein phosphorylation may contribute, since a glycosomal phosphatase has been identified in developmental signalling (Szoor and Matthews, unpublished data). The inhibition of anaerobic glycolysis by glycerol was also reproduced by the model, first qualitatively and then quantitatively (Bakker et al., 1997; Albert et al., 2005).
An interesting biological feature that was revealed by the model was the relationship between compartmentation of glycolysis in glycosomes and the virtual absence of allosteric regulation of the glycolytic enzymes. Glycolysis models predict that glycolytic intermediates accumulate readily due to the investment of ATP at the beginning of the pathway (Teusink et al., 1998; Bakker et al., 2000). This risky ‘turbo’ effect can be avoided either by allosteric feedback regulation of hexokinase or by compartmentation of the pathway in glycosomes (Fig. A22-2). Compartmentation prevents the accumulation of intermediates, because the net ATP production occurs outside the glycosome and this excess of ATP cannot activate the first enzymes of glycolysis. This model prediction was recently confirmed experimentally (Haanstra et al., 2008a), providing a clear example of model-driven experimental design and hypothesis-driven systems biology. According to model predictions the glycolytic intermediates glucose 6-phos-phate, fructose 6-phosphate and fructose-1,6-bisphosphate should accumulate on addition of glucose if the glycolytic enzymes are not properly located in the glycosome. Indeed, accumulation of glucose 6-phosphate could be measured in a PEX14-RNAi mutant in which protein import into the glycosomes is disturbed. A similar phenotype was observed on glycerol addition, which led to accumulation of glycerol 3-phosphate, both in the model and in the PEX14-RNAi cells.
FIGURE A22-2 A. The positive feedback from the ATP produced by glycolysis to the initial kinase reactions can lead to toxic accumulation of hexose phosphates. In many organisms this is prevented by a negative feedback from the hexose phosphates to hexokinase. B. In trypanosomes, the compartmentation of glycolysis prevents the positive feedback. This renders the negative feedback unnecessary, and indeed there is no evidence for such feedback in trypanosomes.
Also in accordance with model predictions, a down-regulation of the expression of the genes encoding hexokinase and glycerol kinase rescues the PEX14-RNAi cells on glucose and glycerol, respectively (Kessler and Parsons, 2005; Haanstra et al., 2008a).
More recently, a model of the gene-expression cascade, based on quantitative knowledge of transcription, RNA precursor degradation, trans-splicing and mRNA degradation for phosphoglycerate kinase (PGK) has been generated (Haanstra et al. 2008b). The model allowed a quantitative analysis of the control and regulation of the expression of the PGK isoenzymes. It was shown that regulation of mRNA degradation explains 80–90% of the regulation of mature mRNA levels, while precursor degradation and trans-splicing make only minor contributions.
In spite of the success of the model, it covers to date only a small part of trypanosome metabolism. This relates, for instance, to the fact that even the
compartmentalised glycolysis does branch into other pathways, for example towards the biosynthesis of glycoconjugates and the pentose phosphate pathway. Although the fluxes into these branches may be small, they are vital for trypanosomes. Sufficient kinetic data have become available to enable extension of the model to include the pentose phosphate pathway which provides NADPH for reductive biosyntheses and also reducing equivalents to sustain cellular redox balance. Since redox balance is intimately related to the biosynthesis of trypanothione (from polyamine and glutathione precursors), a natural next step in a bottom-up systems biology approach to trypanosome metabolism would be the inclusion of the trypanothione–pentose phosphate pathway and related areas of redox metabolism (Fig. A22-3).
Growth Stages of Building a Silicon Trypanosome
Our current level of knowledge of trypanosome redox metabolism, as well as its biological importance (Krauth-Siegel and Comini, 2008), render it a natural choice for a next model extension (Fig. A22-3). The inclusion of redox metabolism is particularly interesting as trypanosome redox metabolism is sufficiently different from its human counterpart to offer perspectives for drug discovery. The unusual polyamine–glutathione conjugate trypanothione or bis(glutathionyl) spermidine (Fairlamb and Cerami, 1992) takes on the majority of roles served by glutathione in most other cell types. In addition, work in the last few years revealed that the enzymes involved in the synthesis and reduction of trypanothione are essential for the parasite (Krauth-Siegel and Comini, 2008).
The trypanocidal drug eflornithine exerts its trypanocidal activity as an irreversible inhibitor of the enzyme ornithine decarboxylase (Bacchi et al., 1980), an enzyme involved in trypanothione biosynthesis (enzyme 10 in Fig. A22-3). A significant amount of information is available on kinetic parameters of that pathway, too. Preliminary attempts to model trypanothione metabolism have been made (Xu Gu, University of Glasgow PhD thesis, unpublished). Information available on the abundance of key metabolites measured in bloodstream form T. brucei grown in vitro (Fairlamb et al., 1987) and in vivo (Xiao et al., 2009), before and after exposure to eflornithine, was used to determine whether predicted behaviour under those perturbed conditions emulated the measured behaviour. The scarcity of kinetic data describing the whole pathway, however, has presented many challenges to constructing a model that captures observed behaviour. The acquisition of new kinetic data and the implementation of new mathematical tools to fill gaps in the data (Nikerel et al., 2006; Smallbone et al., 2007; Resendis-Antonio, 2009) should improve this.
An extension of the glycolysis model to include the pentose phosphate pathway (Hanau et al., 1996; Barrett, 1997; Duffieux et al., 2000) and trypanothione metabolism should be a suitable next step in the modular approach that we envisage towards a complete Silicon Trypanosome. Initial efforts in this direction (not
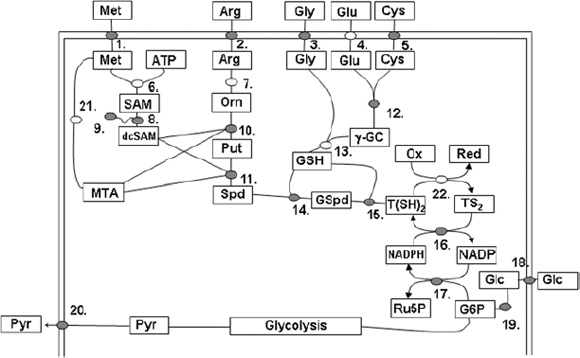
FIGURE A22-3 The glycolytic and trypanothione pathways are linked through the oxidative pentose phosphate pathway. Metabolites are presented in abbreviated form within rectangles. Enzymes and transporters are circles. Kinetic data is available for those shaded grey. Met = methionine; Arg = arginine; Gly = glycine; Glu = glutamate; Cys = cysteine; ATP = adenosine triphosphate; SAM = S-adenosylmethionine; dcSAM = decarboxylated S-adenosylmethionine; MTA = methylthioadenosine; Orn = ornithine; Put = putrescine; Spd = spermidine; c-GC = c-glutamylcysteine; GSH = glutathione; GSpd = glutathionylspermidine; T(SH2) = reduced trypanothione; TS2 = oxidised trypanothione; NADP = nicotinamide adenine dinucleotide phosphate; NADPH = reduced nicotinamide adenine dinucleotide phosphate; Glc = glucose; G6P = glucose 6-phosphate; Ru5P = ribulose 5-phosphate; Ox = oxidised cellular metabolites; Red = reduced cellular metabolites. 1. = methionine transport; 2. = arginine transport; 3. = glycine transport; 4. = glutamate transport; 5. = cysteine transport; 6. = methionine adenosyltransferase; 7. = arginase (N.B., a robust arginase gene orthologue has not been annotated in the T. brucei genome project, raising the possibility that arginine does not serve as a source of ornithine in these cells); 8. = S-adenosylmethionine decarboxylase; 9. = prozyme; 10. = ornithine decarboxylase; 11. = spermidine synthase; 12. = c-glutamylcysteine synthetase; 13. = glutathione synthetase; 14. = c-glutathionylspermidine synthetase; 15. = trypanothione synthetase/ amidase (in T. brucei 14. & 15. are catalysed by a single protein); 16. = trypanothione reductase; 17. oxidative pentose phosphate pathway (glucose 6-phosphate dehydrogenase, 6-phopshogluconolactonase & 6-phosphoglconate dehydrogenase); 18. = glucose transporter; 19. = hexokinase (this enzyme links the redox pathway to glycolysis); 20. = pyruvate transporter; 21. = methionine cycle enzymes; 22. The pathway of electrons from reduced trypanothione for final acceptance on oxidised cellular metabolites or macromolecules is complex, involving transfers via other redox active intermediates including tryparedoxin (thioredoxin-like proteins) and peroxidoxin.
published) have indicated the importance of the compartmentation of the pentose phosphate pathway. Although most of the enzymes of the pathway have a peroxisome targeting sequence (PTS1), a significant fraction of their activity is often found in the cytosol (Michels et al., 2006; Heise and Opperdoes, 1999; Duffieux et al., 2000). A correct localisation of the enzymes as well as good estimates of the transport of intermediates across the glycosomal membrane will be key to a good model of the pentose phosphate pathway.
Challenges of Trypanosome Systems Biology
The first initiatives in systems biology of trypanosomes as well as of other organisms dealt with enzymatic sub-systems, such as glycolysis. The models have depended largely on kinetic data for isolated enzymes. However, the abundance of these enzymes can, in principle, be controlled by the rates of transcription, RNA processing, translation, protein modification and turnover. These processes themselves may be regulated through complex signalling networks in response to both internal and external conditions (Westerhoff et al. 1990).
To include gene expression in a Silicon Trypanosome requires a dramatic increase in model complexity – as well as the acquisition of new types of data on a large scale. Fortunately, the absence of transcriptional control of most individual open reading frames makes trypanosome gene expression simpler than that of yeast or even E. coli, rendering it uniquely amenable to mathematical modelling.
It may well be possible to describe much of trypanosome mRNA metabolism using the following parameters: the rate constant of processing of the precursor RNA, i.e. of trans-splicing; the rate constant of degradation of the precursor (which competes with its trans-splicing); the rate constant of polyadenylation; and the rate constant of mRNA degradation. The rates of degradation of the precursor and the mature mRNA can be measured by inhibiting splicing and transcription. To measure the rate of mRNA processing two approaches are possible. First, one can inhibit transcription alone, and assay precursor decay; this approach is, however, compromised by practical constraints since splicing is very rapid. Second, the splicing rate can be calculated based on the steady-state abundance of the precursor mRNA, and the half-life and abundance of the products. This methodology has already been applied to the mRNA encoding PGK and it was demonstrated that splicing occurred within less than one minute; mRNA decay was the primary determinant of mRNA abundance (Haanstra et al., 2008b).
Previous microarray studies with yeast have yielded estimates of the half-lives and polysomal loading of many mRNAs (e.g. Grigull et al., 2004). Deep sequencing technology – being more sensitive – should allow measurement of the abundances of all mRNAs and precursors on a genome-wide scale and to the accuracy required for the modelling; from these data, it should be possible to derive the steady-state abundances and half-lives of all RNAs, revealing regulated or inefficient processing. This – combined with global polysome profiling
– will provide quantitative data which allow quantifying the regulation of the processing, degradation and translation of each mRNA (Daran-Lapujade et al. 2007). The next challenge would then be to integrate such measurements with metabolic modelling in order to provide a complete model of pathways, from DNA to metabolic end-products.
Anticipated Outcomes from a Silicon Trypanosome
So far, the systems biology approach to trypanosomes has contributed to a fundamental understanding of cellular regulation (Bakker et al., 1999a; Haanstra et al., 2008a), as well as to improvements in the drug-target selection process (Bakker et al., 1999a,b; Albert et al., 2005; Hornberg et al., 2007). Since the initial systems biology analysis only addressed processes associated with less that 1% of the organism’s genome, we would expect many more new insights to lie ahead.
Drugs currently used against human African trypanosomiasis are unsatisfactory for a number of reasons, including their extreme adverse effects in the patient and the emergence of resistant parasites. New drugs are urgently needed and there is hope that a better understanding of the control points of the metabolic network can guide the selection of optimal drug targets. This has already been achieved for enzymes of the glycolytic pathway (Hornberg et al., 2007). This information can be used alongside enhanced chemoinformatics (Frearson et al., 2007) in order to determine those components of the trypanosome that are most amenable to drug targeting.
As a consortium we have embarked on the construction of a Silicon Trypanosome. In this review we have discussed the current status and future directions of trypanosome systems biology that form the context of this endeavour. Our ambition is to achieve a comprehensive, quantitative description of the flow of information from gene, through transcript and protein, to metabolism and back. This will allow prediction of how the parasite responds to changes in its environment, with respect to nutrients, temperature and/or chemical inhibitors. It will also assist the deciphering of complex phenotypes generated by genetic perturbations in the laboratory or in the field. Thus, model predictions will improve our biological understanding of the differentiation and adaptation of the parasite as well as stimulate the discovery of inhibitors that attack processes which control trypanosome growth. The latter should contribute to the development of new optimised drugs for trypanocidal chemotherapy. Pioneering efforts have focused on energy metabolism and recently started to include adaptations of the parasite via gene expression (Haanstra, 2009).
The construction of a complete Silicon Trypanosome, which integrates metabolism, gene expression and signal transduction is an ambitious project. Clearly the route towards this objective will be long, and many challenges will emerge as the datasets required to build such a model are collected and analysed. How-
ever, the emergence of methods to allow collection of massive datasets, at every level, suggests that we may, in time, be able to generate a reasonably complete mathematical description of trypanosome cellular biology. Even if completion is not feasible, the evolving description will always represent the best conceivable dynamic representation of our knowledge of trypanosome biology. As a result, drug development programmes will have at their disposal a predictive model of the trypanosome to help identify those parts of metabolism most amenable to targeting by novel drugs and to controlling vital functions of the parasite. The project will be strengthened by parallel world-wide systems biology projects of human metabolism, in which some of us will be involved. After all, killing trypanosomes is easy. The difficulty is to kill the trypanosome without harming its host (Bakker et al., 2002). A careful comparison of the behaviour of our Silicon Trypanosome to quantitative knowledge of the control of human metabolism, will allow the identification of selective targets.
Acknowledgements
The work of BMB was funded by NWO Vernieuwingsimpuls and by a Rosalind Franklin Fellowship. RB was supported by an NWO-Vidi fellowship. HVW thanks BBSRC and EPSC for support through the MCISB grant (http://www.systembiology.net/support/). MPB is grateful to the BBSRC for their support of the BBSRC-ANR “Systryp” consortium. The Silicon Trypanosome consortium is supported by a grant from SysMO2 (www.sysmo.net).
References
Albert, M. A., Haanstra, J. R., Hannaert, V., Van Roy, J., Opperdoes, F. R., Bakker, B. M., and Michels, P. A. M. (2005). Experimental and in silico analyses of glycolytic flux control in bloodstream form Trypanosoma brucei. Journal of Biological Chemistry 280, 28306–28315.
Archer, S. K., Luu, V. D., de Queiroz, R., Brems, S. and Clayton, C. E. (2009). Trypanosoma brucei PUF9 regulates mRNAs for proteins involved in replicative processes over the cell cycle. PLoS Pathogens 5, e1000565.
Bacchi, C. J., Nathan, H. C., Hutner, S. H., McCann, P. P. and Sjoerdsma, A. (1980). Polyamine metabolism: a potential therapeutic target in trypanosomes. Science 210, 332–334.
Bakker, B. M., Aßmus, H. E., Bruggeman, F., Haanstra, J., Klipp, E. and Westerhoff, H. V. (2002). Network-based selectivity of antiparasitic inhibitors. Molecular Biology Reports 29, 1–5.
Bakker, B. M., Mensonides, F. I. C., Teusink, B., Michels, P. A. M. and Westerhoff, H. V. (2000). Compartmentation protects trypanosomes from the dangerous design of glycolysis. Proceedings of the National Academy of Sciences, USA 97, 2087–2092.
Bakker, B. M., Michels, P. A. M., Opperdoes, F. R. and Westerhoff, H. V. (1997). Glycolysis in bloodstream form Trypanosoma brucei can be understood in terms of the kinetics of the glycolytic enzymes. Journal of Biological Chemistry 272, 3207–3215.
——. (1999a). What controls glycolysis in bloodstream form Trypanosoma brucei? Journal of Biological Chemistry 274, 14551–14559.
Bakker, B. M., Walsh, M. C., ter Kuile, B. H., Mensonides, F. I., Michels, P. A. M., Opperdoes, F. R. and Westerhoff, H. V. (1999b). Contribution of glucose transport to the control of the glycolytic flux in Trypanosoma brucei. Proceedings of the National Academy of Sciences, USA 96, 10098–10103.
Barrett, M. P. (1997). The pentose phosphate pathway and parasitic protozoa. Parasitology Today 13, 11–16.
Barrett, M. P., Burchmore, R. J., Stich, A., Lazzari, J. O., Frasch, A. C., Cazzulo, J. J. and Krishna, S. (2003). The trypanosomiases. Lancet 362, 1469–1480.
Berriman, M., Ghedin, E., Hertz-Fowler, C., Blandin, G., Renauld, H. et al. (2005). The genome of the African trypanosome Trypanosoma brucei. Science 309, 416–422.
Bruggeman, F. J. and Westerhoff, H. V. (2007). The nature of systems biology. Trends in Microbiology 15, 45–50.
Chukualim, B., Peters, N., Hertz-Fowler, C. and Berriman, M. (2008). TrypanoCyc – a metabolic pathway database for Trypanosoma brucei. BMC Bioinformatics 9 (Suppl 10), P5.
Clayton, C. and Shapira, M. (2007). Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Molecular and Biochemical Parasitology 156, 93–101.
Daran-Lapujade, P., Rossell, S., van Gulik, W. M., Luttik, M. A. H., de Groot, M. J. L., Slijper, M., Heck, A. J. R., Daran, J. M., de Winde, J. H., Westerhoff, H. V., Pronk, J. T. and Bakker, B. M. (2007). The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proceedings of the National Academy of Sciences, USA 104, 15753–15758.
Duffieux, F., Van Roy, J., Michels, P. A. M., Opperdoes, F. R. (2000). Molecular characterization of the first two enzymes of the pentose-phosphate pathway of Trypanosoma brucei. Glucose-6phosphate dehydrogenase and 6-phosphogluconolactonase. Journal of Biological Chemistry 275, 27559–27565.
Estévez, A. (2008). The RNA-binding protein TbDRBD3 regulates the stability of a specific subset of mRNAs in trypanosomes. Nucleic Acids Research 36, 4573–4586.
Fairlamb, A. H. and Cerami, A. (1992). Metabolism and functions of trypanothione in the Kinetoplastida. Annual Review of Microbiology 46, 695–729.
Fairlamb, A. H., Henderson, G. B., Bacchi, C. J. and Cerami, A. (1987). In vivo effects of difluoromethylornithine on trypanothione and polyamine levels in bloodstream forms of Trypanosoma brucei. Molecular and Biochemical Parasitology 24, 185–191.
Fairlamb, A. J., Opperdoes, F. R. and Borst, P. (1977). New approach to screening drugs for activity against African trypanosomes. Nature 265, 270–271.
Fenn, K. and Matthews, K. R. (2007). The cell biology of Trypanosoma brucei differentiation. Current Opinion in Microbiology 10, 539–546.
Flynn, I. W. and Bowman, I. B. (1973). The metabolism of carbohydrate by pleomorphic African trypanosomes. Comparative Biochemistry and Physiology B 45, 25–42.
Frearson, J. A., Wyatt, P. G., Gilbert, I. H. and Fairlamb, A. H. (2007). Target assessment for antiparasitic drug discovery. Trends in Parasitology 23, 589–595.
Grigull, J., Mnaimneh, S., Pootoolal, J., Robinson, M. and Hughes, T. (2004). Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Molecular and Cellular Biology 24, 5534–5547.
Grunau, S., Mindtho, S., Rottensteiner, H., Sormunen, R. T., Hiltunen, J. K., Erdmann, R. and Antonenkov, V. D. (2009) Channel-forming activities of peroxisomal membrane proteins from the yeast Saccharomyces cerevisiae. FEBS Journal 276, 1698–1708.
Haanstra, J. (2009). The power of network-based drug design and the interplay between metabolism and gene expression in Trypanosoma brucei. PhD thesis Vrije Universiteit Amsterdam, ISBN 978-90-8659-331-6.
Haanstra, J. R., Stewart, M., Luu, V. D., van Tuijl, A., Westerhoff, H. V., Clayton, C. and Bakker, B. M. (2008b). Control and regulation of gene expression: quantitative analysis of the expression of phosphoglycerate kinase in bloodstream form Trypanosoma brucei. Journal of Biological Chemistry 283, 2495–2507.
Haanstra, J. R., van Tuijl, A., Kessler, P., Reijnders, W., Michels, P. A. M., Westerhoff, H. V., Parsons, M. and Bakker, B. M. (2008a). Compartmentation prevents a lethal turbo-explosion of glycolysis in trypanosomes. Proceedings of the National Academy of Sciences, USA 105, 17718–17723.
Hanau, S., Rippa, M., Bertelli, M., Dallocchio, F. and Barrett, M. P. (1996). 6-Phosphogluconate dehydrogenase from Trypanosoma brucei. Kinetic analysis and inhibition by trypanocidal drugs. European Journal of Biochemistry 240, 592–599.
Heise, N. and Opperdoes, F. R. (1999). Purification, localisation and characterisation of glucose-6phosphate dehydrogenase of Trypanosoma brucei. Molecular and Biochemical Parasitology 99, 21–32.
Helfert, S., Bakker, B. M., Michels, P. A. M. and Clayton, C. (2001). An essential role of triosephosphate isomerase and aerobic metabolism in trypanosomes. Biochemical Journal 357, 117–125.
Hirumi, H. and Hirumi, K. (1989). Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. Journal of Parasitology 75, 985–989.
Hornberg, J. J., Bruggeman, F. J., Bakker, B. M. and Westerhoff, H. V. (2007). Metabolic control analysis to identify optimal drug targets. Progress in Drug Research 64, 173–189.
Hynne, F., Danø, S. and Sørensen, P. G. (2001). Full-scale model of glycolysis in Saccharomyces cerevisiae. Biophysical Chemistry 94, 121–163.
Kabani, S., Fenn, K., Ross, A., Ivens, A., Smith, T. K., Ghazal, P. and Matthews, K. (2009). Genome-wide expression profiling of in vivo-derived bloodstream parasite stages and dynamic analysis of mRNA alterations during synchronous differentiation in Trypanosoma brucei. BMC Genomics 10, 427.
Kessler, P. S. and Parsons, M. (2005). Probing the role of compartmentation of glycolysis in procyclic form Trypanosoma brucei: RNA interference studies of PEX14, hexokinase and phosphofructokinase. Journal of Biological Chemistry 280, 9030–9036.
Krauth-Siegel, R. L. and Comini, M. A. (2008). Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochimica et Biophysica Acta 1780, 1236–1248
Lee, J. H., Jung, H. S. and Gunzl, A. (2009). Transcriptionally active TFIIH of the early-diverged eukaryote Trypanosoma brucei harbors two novel core subunits but not a cyclin-activating kinase complex. Nucleic Acids Research 37, 3811–3820.
Liang, X., Haritan, A., Uliel, S. and Michaeli, S. (2003). Trans and cis splicing in trypanosomatids: mechanism, factors, and regulation. Eukaryotic Cell 2, 830–840.
Lustig, Y., Sheiner, L., Vagima, Y., Goldshmidt, H., Das, A., Bellofatto, V. and Michaeli, S. (2007). Spliced-leader RNA silencing: a novel stress-induced mechanism in Trypanosoma brucei. EMBO Reports 8, 408–413.
Michels, P. A. M., Bringaud, F., Herman, M. and Hannaert, V. (2006). Metabolic functions of glycosomes in trypanosomatids. Biochimica et Biophysica Acta 1763, 1463–1477.
Nikerel, I. E., van Winden, W. A., van Gulik, W. M. and Heijnen, J. J. (2006). A method for estimation of elasticities in metabolic networks using steady state and dynamic metabolomics data and linlog kinetics. BMC Bioinformatics 7, 540.
Nikerel, I. E., van Winden, W. A., Verheijen, J. and Heijnen, J. J. (2009). Model reduction and a priori kinetic parameter identifiability analysis using metabolome time series for metabolic reaction networks with linlog kinetics. Metabolic Engineering 11, 20–30.
Opperdoes, F. R. and Borst, P. (1977). Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: the glycosome. FEBS Letters 80, 360–364.
Parsons, M. (2004). Glycosomes: parasites and the divergence of peroxisomal purpose. Molecular Microbiology 53, 717–724.
Palenchar, J. B. and Bellofatto, V. (2006). Gene transcription in trypanosomes. Molecular and Biochemical Parasitology 146, 135–141.
Paterou, A., Walrad, P., Craddy, P., Fenn, K. and Matthews, K. (2006). Identification and stage-specific association with the translational apparatus of TbZFP3, a ccch protein that promotes trypanosome life cycle development. Journal of Biological Chemistry 281, 39002–39013.
Queiroz, R., Benz, C., Fellenberg, K., Hoheisel, J. and Clayton, C. (2009). Transcriptome analysis of differentiating trypanosomes reveals the existence of multiple post-transcriptional regulons. BMC Genomics 10, 495.
Resendis-Antonio, O. (2009). Filling kinetic gaps: dynamic modeling of metabolism where detailed kinetic information is lacking. PLoS One 4, e4967.
Richard, P., Teusink, B., Hemker, M. B., van Dam, K. and Westerhoff, H. V. (1996). Sustained oscillations in free-energy state and hexose phosphates in yeast. Yeast 12, 731–740.
Rokka, A., Antonenkov, V. D., Soininen, R., Immonen, H. L., Pirilä, P. L., Bergmann, U., Sormunen, R. T., Weckström, M., Benz, R. and Hiltunen, J. K. (2009). Pxmp2 is a channel-forming protein in Mammalian peroxisomal membrane. PLoS One 4, e5090.
Schmitz, J. P., Van Riel, N. A., Nicolay, K., Hilbers, P. A. and Jeneson, J. A. (2009). Silencing of glycolysis in muscle: experimental observation and numerical analysis. Experimental Physiology 95, 380–397.
Schuster, R. and Holzhütter, H. G. (1995). Use of mathematical models for predicting the metabolic effect of large-scale enzyme activity alterations. Application to enzyme deficiencies of red blood cells. European Journal of Biochemistry 229, 403–418.
Siegel, T., Hekstra, D., Kemp, L., Figueiredo, L., Lowell, J., Fenyo, D., Wang, X., Dewell, S. and Cross, G. (2009). Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes & Development 23, 1063–1076.
Smallbone, K., Simeonidis, E., Broomhead, D. S. and Kell, D. B. (2007). Something from nothing: bridging the gap between constraint-based and kinetic modelling. FEBS Journal 274, 5576–5585.
Snoep, J. L., Bruggeman, F., Olivier, B. G. and Westerhoff, H. V. (2006). Towards building the silicon cell: a modular approach. Biosystems 83, 207–216.
Stern, M., Gupta, S., Salmon-Divon, M., Haham, T., Barda, O., Levi, S., Wachtel, C., Nilsen, T. and Michaeli, S. (2009). Multiple roles for polypyrimidine tract binding (PTB) proteins in trypanosome RNA metabolism. RNA 15, 648–665.
Teusink, B., Walsh, M. C., Van Dam, K. and Westerhoff, H. V. (1998). The danger of metabolic pathways with turbo design. Trends in Biochemical Sciences 23, 162–169.
Walrad, P., Paterou, A., Acosta-Serrano, A. and Matthews, K. R. (2009). Differential trypanosome surface coat regulation by a CCCH protein that co-associates with procyclin mRNA cis-elements. PLoS Pathogens 5, e1000317.
Westerhoff, H. V., Kolodkin, A., Conradie, R., Wilkinson, S. J., Bruggeman, F. J., Krab, K., Van Schuppen, J. H., Hardin, H., Bakker, B. M., Moné, M. J., Rybakova, K. N., Eijken, M., Van Leeuwen, H. J. and Snoep, J. L. (2009). Sytems biology towards life in silico: mathematics of the control of living cells. Journal of Mathematical Biology 58, 7–34.
Westerhoff, H. V., Koster, J. G., Van Workum, M. and Rudd, K. E. (1990). On the control of gene expression. In Control of Metabolic Processes (ed. Cornish-Bowden, A. and Cardenas, M.-L.), pp. 399–412. Plenum Press, New York.
Xiao, Y., McCloskey, D. E. and Phillips, M. A. (2009). RNA interference-mediated silencing of ornithine decarboxylase and spermidine synthase genes in Trypanosoma brucei provides insight into regulation of polyamine biosynthesis. Eukaryotic Cell 8, 747–755.






































































































































































































































































































































































































