
The Interpretation of Genomic Data
Important Points Highlighted by Individual Speakers
• For genomic testing to be accepted, it should have not only analytical validity but also clinical and social utility.
• Genomic testing should be used as a tool that is integrated with traditional tests for making a disease diagnosis and guiding therapy.
• Human genetic diversity and genetic differences between maternally and paternally derived chromosomes need to be considered when interpreting genomic data.
The interpretation of genomic data is even more difficult than generating and curating the data, said Muin Khoury of the Centers for Disease Control and Prevention (CDC), who moderated the workshop session on interpretation of genomic data. The genome does not mean the same thing to each person at every point in time. The significance of particular variants can depend on age, health status, and other contextual factors during different life stages. The interpretation of genomic data also raises many difficult questions: Will different vendors use the same data to offer different interpretations? How will the results be verified? Who will generate second opinions? How will non-genomic information such as epigenetic data be incorporated into interpretations? And how will interpretation services be regulated?
DATA INTEPRETATION FROM A CLINICIAN’S PERSPECTIVE
A genetic test can have different types of utility, including clinical utility and social utility, according to Robert Nussbaum of the University of California, San Francisco. Clinical utility is a measure of how valuable a test result is to a patient and a clinician in making decisions about whether to do further diagnostic testing or end the “diagnostic odyssey” as well as in deciding how treatment is managed or how lifestyle should be altered. A test that leads to a diagnosis may have tremendous utility for a patient and clinician, but a third-party payer wants the test to result in an action that makes a measurable difference in health. Individual patients may have the option to pay for a test out of pocket if third-party payers refuse to provide reimbursement, but in that case, Nussbaum observed, many people will be excluded because they cannot afford to pay for the test.
In addition to clinical utility, a test must have social utility if private and government insurers are to be willing to pay for it. The insurers need to be convinced that a new test, when compared with current standards of care, would lead to improved health by reducing the need for less successful therapy and would decrease costs by preventing more costly outcomes. “You have to convince them that it is worth their paying for it as opposed to paying for other things,” Nussbaum said.
Critical Assessments of Genomic Testing for Prevention
Nussbaum outlined six areas in which whole-genome sequencing could be used in a clinical setting and described the common criticisms that arose during discussions with his colleagues. First, whole-genome testing could have clinical use for the identification of carriers of Mendelian disorders prior to conception. At this point it is far less expensive to test for most of the common autosomal recessive conditions than to perform complete genome sequencing. Although the sequencing coverage is not complete for the tests currently used to detect autosomal recessive genes, current standard practice probably continues to be cost-effective compared to the use of whole-genome sequencing for the same indication, Nussbaum said.
A second area, the use of genomics in prenatal and pre-implantation testing, raises many issues, Nussbaum said. “A very serious decision has to be made under severe time pressure with unclear genotype and phenotype correlation.” Pre-implantation testing has advantages over prenatal testing, but it is often limited by the amount of tissue available. This could change with the development of epiblast biopsies to take the place of single-cell testing.
Identification of personal risk for Mendelian disorders is a third area that also raises unanswered questions, especially with regard to clinical
validity. This application of genomics raises a variety of questions, including which variants are responsible for a particular phenotype, what mutations are pathogenic, and what the penetrance is of a known pathogenic mutation. “There is a real gap in being able to tie the genotype to the phenotype,” Nussbaum said, and progress needs to be made in that regard.
A fourth area in which genomics can be used in the clinic is pharmacogenetic testing. In some cases it already has clear clinical validity and proven clinical and social utility. One well-established application, for example, is human leukocyte antigen typing to prevent idiosyncratic adverse reactions for drugs such as abacavir, an antiviral for treatment of human immunodeficiency virus (HIV) infection (Hughes et al., 2008). But the clinical and social utility of tests for common variants that affect the pharmacokinetics or dynamics for drugs such as warfarin, clopidogrel, irinotecan, codeine, and 6-thiopurine is still unclear (EGAPP, 2009; Gong et al., 2011; Ned, 2010; Teml et al., 2007; Zhou, 2009). Part of the reason for the lack of clarity, Nussbaum said, is that there may be replacement drugs used instead, such as dabigatran and rivaroxaban for warfarin or prasugrel instead of clopidogrel (Brandt et al., 2007). Furthermore, Nussbaum said, it is too late to perform a complete sequencing when a drug is about to be prescribed; the information generated by the sequencing needs to be available in advance.
The fifth area is direct tissue typing for transplantation. This technique is, at the moment, still less costly to conduct than whole-genome sequencing, although delays in receiving the results may affect transplantation for some indications. A possible advantage of whole-genome sequencing in this case is that it may be possible for tissue typing information to be incorporated into networks for organ sharing, which would provide better and more rapid identification of donor–recipient matches.
The sixth area is the identification of alleles, whether rare or common, that increase the risk for common disorders. Such identification, at the present, has very limited clinical validity and utility, Nussbaum said, suggesting that this use of genetic testing currently “is more in the realm of entertainment than medicine.” The few people who make the effort to have their genomes tested do not necessarily consider it an essential component of their medical care. Furthermore, this information generally cannot clearly distinguish people who will suffer from a disease from those who will not. For example, results from a panel of 13 SNPs show that people in the top quintile of risk for coronary artery disease have a risk that is 1.7-fold higher than those in the lowest quintile (Ripatti et al., 2010). But the distributions of people at different levels of risk overlap extensively, Nussbaum said, and the test offers “very little discrimination between those who have coronary artery disease and those who do not.”
Genomic Testing for Diagnosis
“Could whole-genome sequencing be a cost-effective replacement for candidate gene panels?” Nussbaum asked. It now costs more than $3,000 to have two genes tested, so in the future, sequencing the entire genome could be reasonable by comparison, but, he said, “I don’t think we are anywhere near there yet.” Discovery of the genetic reasons for undiagnosed hereditary diseases has yielded some remarkable success stories, but in other cases extensive searches have not been successful. It remains to be seen whether the genes that have been uncovered in the recent past, such as those for Miller syndrome (Ng et al., 2010), are exceptions or the rule, Nussbaum said. As technology is applied to larger pools of patients and families, the success rate will go down.
With cancer genomes, sequencing is revealing a tremendous amount of information about the variants that can be used for classification, prognosis, and therapeutic management. Thus, it is reasonable to ask whether in the future the most cost-effective and efficient assay with the most predictive power will be whole-tumor-genome sequencing or simply sequencing a few key variants. An important research question will be whether every cancer is different, which, if true, would make it necessary to scan large amounts of genomic data to understand each person’s disease.
Identifying a Path Forward
Nussbaum stressed that he was not trying to discourage the discussion with his criticisms. “The analytical validity of whole-genome sequencing is improving, the costs are coming down, and the poor state of genotype– phenotype correlation is a recognized problem.”
Several points need to be emphasized in moving forward, he said. First, the potential advantages of having complete sequences need to be recognized. “Knowing this element or that element is not the same as knowing the periodic table.” Because of the huge amounts of data involved, bioinformatics will be essential in building a genomic basis for clinical work. Second, a few demonstration projects are needed as part of an overall health assessment throughout all stages of life that would integrate genomic data into ongoing health care. A good candidate organization to carry out such a project would be a large health plan that is also a provider and is willing to establish a partnership. Third, interpretation should be ongoing, with genome sequencing becoming more like a subscription service in which technologies, software, and information constantly improve and knowledge is exchanged. Patients expect their health care providers to be talking with each other. Furthermore, a subscription implies a continuing relationship among bioinformaticists, providers, and patients and their families
in interpreting genomic data. Nussbaum added that efforts to standardize nomenclature are under way, which should enhance collaboration. “As the databases grow and become more and more useful,” he said, “there is going to be constraint on the way people report things, and they will come to a common reporting. I am actually fairly optimistic about it.”
Finally, it will be important to develop software to interpret variants and provide decision support. In part, this will require establishing partnerships among laboratories, clinics, and the institutions that create and maintain electronic medical records that are functional and allow for interpretation but not necessarily storage of genomic data. “It is useless to think about dumping all the sequence data into an electronic medical record,” said Nussbaum. “It has to live someplace else in a way that makes sense, and the interpretation and the re-interpretation have to come into the medical record in a way that is valuable.”
INTEGRATING GENOMIC DATA WITH PATHOLOGY
In 2009 Mark Boguski of Harvard Medical School and two colleagues published a paper that laid out a futuristic scenario for cancer care in the year 2020 (Boguski et al., 2009). The process they described begins when a patient presents with symptoms and needs to rely on the involvement of a clinical laboratory for care (Figure 4-1). In addition to conventional analyses of formalin-fixed, paraffin-embedded tissues, such as hematoxylin and eosin and immunohistochemical staining, genome sequencing is also performed. In this situation the pathology report is not just a textual description of what is seen through a microscope and a diagnostic code. It is a dataset and a collection of therapeutic recommendations that includes the parameters under which the modeling was conducted. The oncologist and the rest of the clinical care team then can accept the report’s simulations or develop their own to administer precision targeted therapy. This is a model in which advances in sequencing, systems biology, and other areas make it possible to reverse engineer disease pathways, to annotate disease networks and drug targets, and to simulate therapeutic interventions with virtual drugs or combinations of virtual drugs.
In 2009 this seemed improbable, Boguski said. A year later, however, a paper published in Genome Biology demonstrated as a proof of concept every conceptual step in the scenario (Jones et al., 2010).
Integration of Genomic Data and Cancer Pathology
Boguski described a case study from the paper that demonstrates what is possible. A 78-year-old man with no prior history of cancer presented with a sore throat. A biopsy of a lump on the back of his tongue revealed
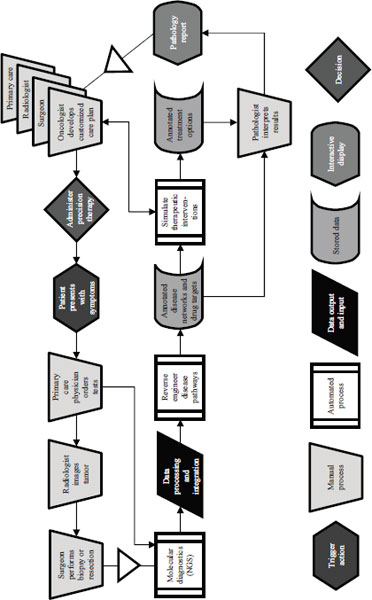
NOTE: NGS = next-generation sequencing.
SOURCE: Adapted from Boguski et al., 2009.
a papillary adenocarcinoma, which probably originated in a minor salivary gland. A lymph node dissection revealed that the tumor had spread beyond his tongue and was present in 3 of 21 lymph nodes in his neck. In response, he was given adjuvant radiation therapy.
Four months later he returned for a follow-up visit, and a scan revealed that the tumor had metastasized to both of his lungs. Because this was a relatively rare tumor, no standard chemotherapy was available. A pathology review indicated that the tumor was positive for epidermal growth factor receptor (EGFR), and the man was started on targeted therapy with erlotinib. However, the tumor continued to grow.
Both genome and transcriptome sequencing analyses were performed on the patient’s tissue sample as a part of his clinical care. In this case the transcriptome analysis was more important than the genome analysis, as it showed an absence of EGFR expression. This finding indicated that treatment with a different chemotherapeutic agent, sunitinib, might be beneficial. After switching the patient to the new treatment, the disease progression stabilized for 4 to 6 months.
Boguski described a second case in which genomic data was used to guide therapy. A 60-year-old man with a long history of alcohol and tobacco abuse presented with difficulty breathing and heart palpitations. Physical examination of the patient revealed palpable right supraclavicular lymph nodes, and a biopsy revealed metastatic squamous cell carcinoma originating in his esophagus. Standard cytotoxic chemotherapy was initiated. The patient’s tumor genome was then sequenced along with DNA from his peripheral blood cells. Following genomic analysis, cytotoxic chemotherapy was discontinued, and the patient was started on imatinib, a targeted therapy that disrupts tyrosine kinase signaling, and the tumor responded.
Cancers can continue to mutate after treatment has begun, Boguski noted, and this makes them difficult to treat. In the first case above, the patient eventually became resistant to sunitinib, and a second genomic and transcriptome analysis was done, and the drug was changed. Again the patient stabilized, and today the patient is still alive. “Had the genome analysis not been done, I doubt that would have been the outcome,” Boguski said, “but this is one of those stories that … is quite dramatic when you see potentially what kind of cost avoidance and precision diagnosis can be achieved with genome and transcriptome analysis.”
A lesson drawn from the first case study is that whole-genome analysis will consist of a variable package of genome or exome sequencing with or without transcriptome analysis, depending on the clinical indication and diagnostic goals. In the future, such analyses could also include epigenetic measurements or other data. It is still unclear, Boguski said, “whether the genome or transcriptome or some combination of both is going to be most efficacious for certain kinds of cancers.”
Another conclusion drawn by Boguski’s paper is that whole-genome characterization will become a routine part of cancer pathology. Furthermore, it will be done not once but multiple times during the course of the disease for tumor subtyping, monitoring response to therapy, and diagnosing the reasons for recurrence and therapeutic failures. “2020 is here,” Boguski said. “This will become [the] standard of care for certain cancers sooner than we think. I am absolutely convinced of that… . If I had that choice as a patient, I would certainly [have my genome sequenced].”
Increased integration of genomic medicine in routine care also opens up the potential for increased disparities in health care access and services. Nicholas Schork from the Scripps Translational Science Institute suggested that disparities could well be exacerbated in the short term. But when sequencing becomes routine, he added, disparities will not be as severe. Boguski made the same point, observing that in the first part of the 20th century a major source of health care costs was hospitalization because of infectious diseases, whereas today such diseases are mostly treated with generic drugs. Similarly, a major therapy for gastric ulcers in the 1960s was removal of part of the stomach, but the realization that ulcers are caused by an infectious agent has led to the disease being managed with a 10-day course of antibiotics. “This is the history of medicine,” Boguski said. “[Genomic medicine] will eventually become democratized and available to a larger portion of people.”
Overcoming Obstacles
Genomics is the pathologist’s new microscope, Boguski said. A torrent of data will emerge from high-technology platforms. To pathologists these technologies will largely be black boxes. The important factors will be cost, accuracy, and turnaround time.
Interpretation of whole-genome analysis could be costly, Boguski said, noting that some have talked of the $1,000 genome and a $1 million interpretation. But technologies will likely drive down the cost of interpretation. In particular, data annotation will increasingly be outsourced and automated. What is ultimately needed, Boguski said, is a clinically actionable knowledge base that any pathologist, genetic counselor, or medical geneticist can rely on to make a decision.
Workforce issues could be a more severe constraint, Boguski said. Today the United States has about 1,000 medical geneticists, 3,000 genetic counselors, and 17,000 pathologists. Many more people in these specialties will be needed in the relatively near-term future if the potential of genomic medicine is to be realized.
A paper focused on workforce issues that emerged from an October 2010 meeting (Green and Guyer, 2011) called for several “Blue Dot” pilot
projects within 2 to 20 months that would establish a nationwide program for residency training by July 2012, define the concept of the “primary care pathologist” in genomic-era medicine, and establish by December 2011 a prototype “clinical-grade” disease variant database for one disease area. The other projects are to compile and analyze current genetic, newborn, and molecular pathology tests and create a whole-genome analysis “replacement map,” to identify and validate operations models for whole-genome analysis, to formulate the regulatory guidelines to conduct whole-genome analysis test accreditation, and to address reimbursement issues.
Progress has been made on several of the Blue Dot projects, including the development of a program called Training Residents in Genomics, which is a collaborative project of pathology organizations, the National Society for Genetic Counselors, and the National Coalition for Health Professional Education in Genetics. The group is creating a modular transportable curriculum for training pathology residents in personalized genomic medicine that will be introduced into a third of U.S. pathology residency programs on a pilot basis by 2012.
Boguski concluded by saying that the current cost of a genome sequence has decreased significantly from the initial cost of sequencing the first human genome, which exceeded $2 billion. With the cost of genome sequencing nearing that of routine clinical tests, the implications of such a capability are going to be revolutionary, not evolutionary, Boguski said. “Next-generation sequencing and whole-genome analysis is a disruptive technology.”
USING A BIOINFORMATICS MODEL FOR INTERPRETATION
Individual genomes contain about 4 million variants that are not in reference genomes, with 50,000 to perhaps 150,000 that have not been seen before, Schork said. Many of these variants influence phenotypic expression, but the question is which ones. If a variant is in an exon or an intron, it can be studied to determine if it is likely to disrupt the functioning of the gene, but this is not a useful approach for novel variants because they have not previously been examined by a functional assay. Instead, determining the likely functional effect of those variants requires bioinformatics.
One bioinformatics approach to determining the functionality of a variant relies on evolutionary conservation. If a nucleotide or nucleotide sequence is conserved across species, a variant in that sequence or at that position in humans is likely to be functional because otherwise it would have been seen in other species. Analogous strategies for identifying functionally important sites can also be used for determining variants that disrupt regulatory elements in exons, introns, silencers, and promoters (Torkamani and Schork, 2008). But there are problems with just using sequence conservation for determining the functional effects of variants,
Schork observed, as a recent analysis showed that structural information about a protein is more effective than conservation information for determining which variants are functional (Torkamani et al., 2008).
A recent paper described a program called Variant Annotation, Analysis, and Search Tool, which uses bioinformatic techniques to identify variants that are likely to be correlated with idiopathic conditions (Rope et al., 2011). The program compares a patient’s genome with reference genomes to rule out variants seen in other individuals who do not have the disease. Any novel variants that are identified in functional elements are then considered as potential causative candidates for the idiopathic condition. Annotations and predicted functional effects then can help prioritize variants.
Schork and his colleagues have applied this approach to every variant in public domain databases, including those from the 1000 Genomes Project, dbSNP, and the Online Mendelian Inheritance in Man variants. Many novel variants in each individual’s genome were predicted to be functional. Not all of these variants cause disease, however, even where the coding variants that affect the proteins are predicted to be damaging, which makes interpretation difficult.
Commenting about using these types of bioinformatics tools, Schork said that students need to become more computer savvy. Even among students at the Scripps Translational Science Institute, he said, many could improve their knowledge about how to conduct basic BLAST searches or annotate variants against dbSNP. Exposure to such tools is absolutely critical, even if they are not used on a day-to-day basis. Furthermore, exposure to concepts in systems biology is important to build understanding of how genes work in concert rather than separately.
Genomic Diversity
In a clinical context it will be essential to take into account the genomic diversity of the human population, Schork said. This will be important, for example, in comparing individuals with a disease from one part of the world with individuals without the disease from another part of the world. Some variants would appear to be more frequent in the diseased individuals than in the non-diseased individuals, but these would reflect false-positive results because of population stratification. Similarly, if a reference panel from one population were to be used to draw inferences about the novelty of variants from a patient from a different population, the resulting conclusions could be highly misleading (Bustamante et al., 2011).
Human genetic diversity is greater in Africa than elsewhere in the world because only a subset of the variation present in Africa made its way through the Middle East into Asia, Europe, and the Americas as modern humans migrated out of Africa. As a result of this bottleneck,
particular genomic positions might have greater homozygosity in European populations than in African populations (Lohmueller et al., 2008). Using the bioinformatics tools described earlier, Schork’s group found more functional variation in African genomes than in European, Asian, or Native American genomes because of the increased diversity in Africa. Similarly, African genomes have more novel functional variants. However, the standard human “reference” genome is made up largely of contemporary European DNA, which presents a misleading view of global variation in the genome. Reference panels need to be larger, Schork said, in order to determine what is novel and what is not.
Considering the Complexity of the Diploid Genome
The DNA sequencing community often ignores the fact that humans are diploid. Yet variants that differ between the maternally and paternally derived chromosomes can have a critical effect on health (Tewhey et al., 2011). If the maternally derived homolog can compensate for mutations in a paternally derived chromosome, for example, gene function can be normal. If it cannot, the result is haploinsufficiency. Similarly, the presence of different mutations or polymorphisms within the same gene but on separate alleles, such as in the coding region of the maternal homolog and in the regulatory region of the paternal homolog, can yield a phenotype unique to a diploid organism called compound heterozygosity. “Merely knowing that this individual was heterozygous at these two sites wouldn’t be enough,” Schork said. “You would have to know that one damaging variant [which impairs protein function] was on the paternal homolog and [a separate function-impairing] variant was on the maternal homolog.” Copy number variations and other insertions and deletions also can have different effects depending on parental origins.
In order to identify maternally and paternally derived homologs, it is necessary to sequence families to determine which variant was inherited from whom, or the assembly of DNA sequences can be used to recover the two chromosomes that an individual inherited. Collecting phase information, however, is not a simple matter, especially with new sequencing technologies, since the short stretches of DNA that they generate make assembly of different genomes difficult.
Another way to generate phase information is to use chemical or molecular tweezers to pull apart chromosomes during metaphase and sequence them separately (Bansal et al., 2011). This approach probably could not be routinely adopted in clinics, Schork said, but other approaches may be able to distinguish between maternally and paternally derived homologs and deal with issues like compound heterozygosity.
Risk Predictions and Perceptions
Schork concluded his remarks by talking about the utility of genomic information compared with other measures that clinicians use. For example, to predict diabetes, an individual could be typed at all known diabetes susceptibility loci, or else a family history, body mass index, glucose and insulin responses, and other indicators could be collected. Comparisons of these two approaches reveal that genetic information does not provide a benefit over clinical information, Schork said (Lyssenko et al., 2008). However, genotyping did prove to be better at making long-term predictions, as compared with the clinical predictors, which were better at predicting who would become diabetic within 1 or 2 years. Thus, if long-term predictions were of interest, “there might be utility in using genetic information over and above clinical information,” Schork said.
Schork has been involved in an investigation to study the behavior of 3,000 individuals who received genetic information after undergoing consumer-oriented genome-wide testing (Bloss et al., 2011). The study found that such testing did not result in any measurable short-term changes in physiological health (e.g., anxiety), diet or exercise behavior, or the use of screening tests. Furthermore, other studies have shown that people respond best when they are involved in social networks or have some other kind of support to change their behaviors.
ENVISIONING CLINICAL GENOMICS IN 2020
Each of the three speakers commented on how genomic data will be interpreted in the year 2020 if research and development continue to progress on their current trajectory. Interpretation will focus on patterns in a genome that point to biological pathways subject to perturbation rather than on single mutations that might explain a particular disease, Schork predicted. It will be necessary to integrate information from many parts of the genome using a greater understanding of systems biology in order to derive actionable conclusions, he said.
Boguski emphasized the role of empowered patients and participatory medicine in making actionable conclusions. When a patient comes into an emergency room with a stroke, the emergency room personnel will probably not ask the patient for the password to a commercial genome testing website to check on warfarin sensitivity, but they may check a patient’s status on an online site where such information is routinely posted. “In some cases it is going to be easier to get medical information out of a person’s Facebook profile than legacy EMR systems,” Boguski said. For example, the Association of Cancer Online Resources has about 18,000 cancer patients who share information on diagnoses and therapeutic inter-
ventions. “If the medical profession is not going to be capable of doing this, it will be crowd sourced. That is my prediction for 2020.”
Nussbaum predicted that instead of a single solution there will be multiple solutions based on partnerships. Patient empowerment is important, he said, “but at the same time I firmly believe that this has to be embedded in traditional medical care in some way.” Research into genomic interpretation is now progressing on a broad front, but clinical and family information still will be needed to understand the effects of genetic variants. Furthermore, this information needs to be in the public domain so that people can use it.
How Clinical Trials Will Be Affected by Genomics
Each participant in this section also commented on how the realization of genomic medicine will change the role of clinical trials. Nussbaum suggested that the era of randomized controlled trials is fading. Cohort sizes are too small for such trials, because every cancer patient has a unique set of genetic variants, making large-scale trials impractical. Instead, clinical trial designs will need to be adaptive and engage patients during Phase IV1 of the clinical trial. “Once a drug is out there, we have to be collecting information from [patients] about efficacy and side effects.” Furthermore, genomic information will need to be re-interpreted throughout a patient’s life. Physicians will gather clinical information and write an order for updated interpretation of the genome based on the validated information that has accrued since the patient’s last encounter with the health care system.
Boguski added that clinical trials may still exist, but they will not be organized by drug companies. Instead, they will be organized by other organizations once a new drug becomes available. Patients will be much more active in contributing observations to organizations that collect and act on their data. “We have to expand our notion of what a clinical trial is, and I think it is going to involve post-marketing surveillance and so-called Phase IV use of those drugs.”
Schork emphasized that trials for individual patients can be done as a part of the standard of care. “If you can show that treating each patient based on the genomic profile is the way to go rather than standard care, then despite the fact that you are treating each patient individually, you vetted the whole concept.” This becomes more complicated with rare diseases, however, as a surrogate endpoint is needed to measure the efficacy of treatments.
_________________
1 During Phase I clinical trials, researchers evaluate the safety of a drug or treatment, establish a dosage range, and identify side effects. In Phase II, efficacy of the drug or treatment is evaluated. In Phase III, drug or treatment effectiveness is confirmed, side effects are further observed, comparisons to current treatments are performed, and safe-use information is obtained. In Phase IV, post-marketing studies are performed to gather additional evidence














