
Scientific Challenges in Developing Investigational Combination Therapies
Participants identified numerous scientific challenges to developing investigational combination therapies, including the need for:
• Better animal models and validated preclinical tests;
• Better dosing and treatment schedules to avoid toxicity, yet be effective;
• Better benchmarks, endpoints, and clinical trial designs for combination therapies;
• A way to prioritize which combinations to test;
• A way to select patients most likely to respond to combinations; and
• More basic research on the molecular mechanisms that underpin cancer and how they interact.
IMPROVING PRECLINICAL DEVELOPMENT OF INVESTIGATIONAL COMBINATION THERAPIES
Standard preclinical development of drugs involves assessing the effects of varying concentrations of experimental compounds in in vitro or animal models and using those results to determine initial doses to test in clinical trials. Such preclinical development presents numerous challenges that may be exacerbated in the development of combination therapies, including cell lines or animal models that do not adequately mimic the tumor, tumor microenvironment, or the propensity to develop
resistance, and a lack of biomarkers for efficacy. In addition, many animal models do not adequately mimic the immune response to tumors, so it is difficult to assess how immunotherapies are working in those models. There are also challenges that are unique to the development of combinations. For example, animal models appropriate for one therapeutic class might not be appropriate for another class with which they are being combined. These challenges were discussed at the workshop, as well as ways to address them.
Key Suggestions for Improving Preclinical Development of Combinations from Various Workshop Participants
• Funds to develop novel animal models that better mimic human cancer
• Use of non-cancer animal models (e.g., as autoimmune or infectious disease models) as surrogate efficacy models for anticancer immunotherapies
• Strategies to ensure target engagement and inhibition
• Innovative approaches to maximize dose and schedule of combinations
• Better ways to distinguish on-target versus off-target toxicities
• Greater use of animal models to identify resistance mechanisms
• Greater use of statistical modeling
Inadequate Models of Human Tumors and Tumor Microenvironment
Dr. Lewis Cantley, professor of medicine at Harvard Medical School and director of the Beth Israel Deaconess Hospital Cancer Center, noted that cell lines do not adequately model the diversity of tumor types, but rather those tumor cells that can grow in a petri dish or under other common laboratory conditions. “Once you establish these cell lines, they have been selected for and evolved to grow on plastic and they are not selected to grow in vivo. They have clearly evolved away from the original tumor from which they arose and do not represent what you see in the disease,” he said. Consequently, he suggested that the most appropriate models in which to test cancer therapies are mouse explant models in which the tumor cells are growing within the animal, which ideally should be
a “humanized” mouse.1 Alternatively, he suggested researchers mutate the same targets that are altered in the human cancer in the same tissue at the same time of development in the animal model. “Both of these are more powerful approaches than what we currently do,” Dr. Cantley said.
But Dr. Kurt Bachman, Head of Translational Medicine and Biology for the Cancer Metabolism discovery unit at GlaxoSmithKline (GSK), pointed out that multiple tumor explants are needed to capture the diversity of tumor types. The tumor cells available for explants may not have the tumor subtype likely to respond to the combination therapy being tested preclinically. “We want to target K-ras mutant lung cancer, but those explants may not have K-ras mutations,” said Dr. Bachman. It is also more expensive to test therapies in explant animal models than in numerous cell lines, Dr. Stern added. Dr. James Zwiebel, chief of the Investigational Drug Branch in the NCI Cancer Therapy Evaluation Program (CTEP), noted that NCI recently launched the Center for Advanced Preclinical Research, which will serve as a national resource for comprehensive preclinical testing of anti-tumor efficacy and selectivity, biodistribution, and metabolism in early-stage candidate drugs using genetically engineered mouse models (NCI, 2011a). “That’s an approach that hopefully will gain traction,” he said, and added that Dr. Terry Van Dyke is coordinating this effort and looking for interested parties to participate in it.
Dr. Bachman stressed that understanding how the tumor microenvironment affects growth of the tumor is crucial to improving cancer therapy and pointed out that his laboratory is starting to grow tumor cell lines in different microenvironments to see how that influences the action of inhibitors they’ve developed. He noted that effects seen in cell lines grown in three-dimensional cultures are different from when they are grown in standard culture conditions. “We are doing a lot of experiments to see if our culture conditions shift anything so the cell line looks more like a primary tumor that we can use to better predict what we are going to see in the clinic,” he said.
Inadequate Models of Human Immune Responses to Cancer
Dr. Nils Lonberg, senior vice president of Biologics Discovery at Bristol-Myers Squibb, noted that immunotherapy combinations cannot be tested in standard tumor models in which tumors are grafted onto immunodeficient mice. Dr. Haleh Saber, supervisory pharmacologist in the FDA Office of Oncology Drug Products, concurred, adding that she
![]()
1 Humanized mice have become an important research tool for the in vivostudy of human cells and tissues. Humanized mice are immunodeficient mice engrafted with human hematopoietic cells or tissues, or mice that transgenically express human genes.
wanted to test a treatment for leukemia that combined genetically modified human immune cells with a small molecule that was designed to activate a particular gene in the cells. “There was no in vivo model so we couldn’t do the animal pharmacology and toxicology studies,” she noted. In addition, models appropriate for one type of therapy—a vaccine, for example—might not work for another type, such as a small molecule, noted Dr. Ramzi Dagher, vice president for Worldwide Regulatory Strategy and regulatory head for the Oncology Business Unit at Pfizer, Inc. Thus, finding the appropriate model in which to test their combination can be challenging.
Dr. Lonberg added that animal cancer models tend to be limited in how well they mimic the full spectrum of interactions between the host and the tumor that are key to assessing how well combination immunotherapies are working. He suggested using surrogate efficacy models, such as autoimmune or infectious disease models, to assess the effects of combinations of agents in immunotherapy. For example, the NOD2 autoimmune mouse model can show synergy between immune system molecules by revealing a heightened autoimmune response, such as diabetes, when both molecules are combined compared to when they are given singly. Some researchers, such as Dr. Rafi Ahmed at Emory University, have also used chronic viral infection models, particularly the LCMV3 mouse model, to reveal interaction between various components in the host immune system and the effects of that interaction on the viral load of infected cells (Kim and Ahmed, 2010). Alternatively, researchers, such as Dr. James Allison at Memorial Sloan Kettering Cancer Center, have tested combination immunotherapies preclinically by creating animal versions of the human antibodies or other immunotherapies that have been developed, and testing those in animals with intact immune systems.
But even these animal models may not fully mimic how the human immune system interacts with the tumor, according to Dr. Lonberg. He pointed out that the initial immune response to a tumor is an elimination phase in which the host immune system attacks the tumor. But then an equilibrium ensues. During this equilibrium phase, tumor cells express immunoevasion molecules that enable them to survive in equilibrium with the host immune system, with occasional tumor cells escaping immune defenses.
![]()
2 Non-obese diabetic (NOD) mice exhibit a susceptibility to spontaneous development of autoimmune insulin-dependent diabetes mellitus.
3 Lymphocytic choriomeningitis virus (LCMV): This mouse model has been useful for examining mechanisms of viral persistence and the basic concepts of virus-induced immunity and immunopathology.
Dr. Allison’s animal model only mimics the initial elimination phase of an immune response. “You don’t have time in a tumor model like that to look at equilibrium and escape,” Dr. Lonberg said. So any advantages or disadvantages a combination immunotherapy might have in that regard cannot be predicted in preclinical testing in such animal models, he said.
One participant stressed that it is critical that the therapeutic mechanism targeted by a treatment is present in the animal model in which it is tested, and is relevant to human disease. For example, immunotherapy that acts as a CTLA-44 blockade does not work in a lot of animal models, he said, although ipilimumab, a monoclonal antibody targeting CTLA-4, has recently been approved by the FDA5 for patients with advanced melanoma in first- and second-line treatment.
Dr. Cantley suggested using well-designed mouse models in which researchers can verify that each drug had adequately hit its target and had the desired downstream effects, that is, blocked the pathways that foster tumor growth. Evidence of those blocked pathways can then be gathered from the repeat biopsies taken from patients being clinically tested with the drug combination.
Given current deficiencies, Dr. Stern suggested that there be better access to animal models for combination therapies or funds to develop them. “For wet bench investigators, the bottleneck is often moving from cell biology to animals,” he said.
Combined Toxicity
“Sometimes [drug] synergy is going to take us in the direction of enhanced toxicity,” Dr. Flaherty noted. For example, Dr. Engelman described a combination therapy that was highly effective when tested in vitro, but when he gave the maximum tolerated dose of each of those drugs to mice simultaneously, they killed every mouse tested. “You want to shut down these pathways [in tumors], but these are very important pathways for lots of cellular processes. It was only when we started playing with different schedules and doses that we were able to find the sweet spot where the mice lived and the tumors shrank,” he said.
Dr. Engelman suggested being more creative and innovative in how combination therapies are scheduled and dosed. “Lots of these thera-
![]()
4 CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4) is a protein that plays an important regulatory role in the immune system. It is a member of the immunoglobulin superfamily, which is expressed on the surface of Helper T cells and transmits an inhibitory signal to T cells.
5 See http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm1193237.htm (accessed December 14, 2011).
pies are going to require three or four drugs, and a patient cannot be on all of them ad infinitum. They have to be pulsed or sequenced—we can’t just give them everything every day and only dose-reduce when they experience too much toxicity.” One regimen he suggested testing was giving monotherapy with periodic pulses of an additional treatment aimed at killing off those tumor cells that have become resistant to the monotherapy.
Divergent Effects Depending on Dose or Sequence
Dr. Patricia LoRusso, professor of internal medicine at Wayne State University Medical School and director of the Center for Experimental Therapeutics at Karmanos Cancer Institute, and Dr. Lutzker gave an example of the extensive preclinical testing of combination targeted cancer therapies done by Genentech. This preclinical testing of an MEK inhibitor combined with a PI3K inhibitor, which took about a year, not only assessed additivity versus synergy in various genetically diverse cancer cell lines, but also tested a wide range of daily dosing versus intermittent dosing in animal models that aided subsequent clinical trial design. “Genentech did an excellent job in trying to figure out how best to dose escalate. In a first-in-patient study [Shapiro et al., 2011], we were able to conduct multiple arms simultaneously so that we could more efficiently define the combination of each of the drugs leading the pack, which has helped us in the final outcome of this study,” Dr. LoRusso said. “It is important when you get in the clinic to make sure you have drugs that can actually achieve the types of pharmacodynamic effects that you want or you hope to see in patients,” Dr. Lutzker added.
For combinations that include immunotherapies, dose scheduling is key, Dr. Schlom pointed out. He noted that studies have found that tumor vaccines given after chemotherapy regimens are not as effective as those given prior to chemotherapy. Dr. Lonberg added that in one of his studies of two immunotherapy drugs, he found that when the drugs were given sequentially, there was a much more modest effect than when they were given together.
Finding the appropriate dose of an immunotherapy is also critical, Dr. Schlom added, because many immune modulators have dual functionality, depending on dose, including many immune stimulants that have no effect at high doses. He noted that these potential therapeutics have been shelved merely because they showed no effects and were toxic at the maximum tolerated dose in Phase I studies, but they might have some useful effects at lower doses and in combination with other treatments. “It’s not only a matter of drug interaction, but it’s also a matter of
what biologically makes sense in terms of sequencing and combining,” summed up Dr. Canetta.
Differing Pharmacokinetics and Pharmacodynamics Between Mice and People
Dr. Cantley stressed that the pharmacokinetics and pharmacodynamics of drugs are dramatically different in mice and in humans. “We need to get beyond this fear that if a combination kills mice it’s therefore going to stop a clinical trial, because those mice data don’t mean anything,” he asserted. Dr. Saber added that all oncology drugs are toxic and “the question is, can you monitor those toxicities? Most of the time we can and we adjust the dose so you’re good to go.”
But Dr. Engelman noted that although pharmacokinetic and pharmacodynamic data do not directly transfer from the mouse to human, they can suggest a framework for how to reduce dosing to counter toxicity. For example, researchers can use the mouse to test the effects of reducing both drugs on both targeting and toxicity versus reducing the dose of just one drug, or keeping the dose of both drugs, but increasing the duration between doses.
Dr. Donald Berry, professor of biostatistics at the MD Anderson Cancer Center, suggested going from bench to bedside and back to the bench by doing Bayesian statistical modeling of mouse preclinical test results the same way one would do for a clinical trial. “If there is no relationship between the mouse results and the human results, then we will just focus on the clinical aspects,” he said, “but there is a tremendous opportunity to augment the one with the other. You can do it with a statistical model.”
Dr. LoRusso stressed that “we need to have much higher standards as to what we are considering effective combinations preclinically. I don’t know that the models have failed us. I think the way we are interpreting the models is what’s really failing us.” Dr. Engelman added that seeing a treatment response that is greater than a control response in preclinical tests does not necessarily mean that the treatment will cause clinical responses, but rather that the treatment has a biological effect. It is more important that the treatment causes significant tumor shrinkage in preclinical tests, he said. “If we can’t see tumor regressions in a simple 200 mm cubed tumor—which is the most homogeneous sensitive model—than what’s the likelihood that Mr. Jones, who has a huge amount of cancer that is heterogeneous, is going to benefit?” he said.
But Dr. Lutzker countered that most human tumors do not grow as fast as tumors in mice. “I’m not prepared to give up on a combination just
because in a xenograft6 it didn’t shrink the tumor,” he said. Dr. Engelman agreed and said he would view a lack of tumor growth in a xenograft model as a positive indicator, but added that “maybe as a community, we have been too accepting of seeing a biological effect and assuming that would translate into a therapeutic benefit in the clinic.”
On-Target Versus Off-Target Effects
Dr. Cantley suggested determining whether the limiting toxicity seen in animal models stems from how the combination affects the target, or alternatively whether it is due to how one or both drugs affect something other than the target. “If it’s on-target combined toxicity, then you have done the best you can. If it’s off-target, that means you try another combination, another PI3K or MEK inhibitor, for example. Fortunately, we have 18 of one and 7 of the other, so the probability that all combinations are going to have the same toxicity is unlikely,” he said. He noted that often doses of the combination hit the targets hard enough before toxicity is seen either in the mouse or the human. “Sometimes you don’t need to reach the mean toxicity because the toxicity is not on target,” he said.
Dr. Saber suggested basing dose selection on data, when available, from Phase I clinical trials with the single agents that researchers plan to use in combination. Often sponsors will test a few doses of the single agents in people before combining them. But Dr. Roy Herbst, professor of medicine and chief of the Medical Oncology Section at Yale Comprehensive Cancer Center, said it is possible that lower doses of the two drugs combined might be more effective than the same dose of either agent used singly.
Identifying Resistance Mechanisms
Dr. Cantley described in his presentation how he often goes back and forth from bench to bedside. He uses animal models to determine what causes resistance to targeted treatments, and thus what treatments should be combined. He does this by doing a mutational analysis of the tumors removed from drug-resistant mice. For example, through this procedure he has discovered that resistance to a PI3K inhibitor can occur through amplification of MET. Armed with this information, he biopsies patients who have not responded to a PI3K inhibitor to see if their tumors also have MET amplification or produce high levels of MET protein. If that is
![]()
6 A xenograft is a surgical graft of tissue from one species (in this case, a human) to an unlike species (in this case, a mouse).
the case, he will consider entering these patients into a trial that tests a combination of a MET inhibitor with a PI3K inhibitor.
PRIORITIZING COMBINATIONS TO TEST
Strategies for Prioritizing Which Combinations to Test in the Clinic as Suggested by Various Workshop Participants
• Using stricter preclinical benchmarks for effectiveness, such as tumor shrinkage, and demonstrating consistent effects in multiple animal models
• Demonstrating adequate pharmacokinetics and evidence of target activity at clinically relevant doses
• Doing high-throughput in vitro screening of drug combinations to detect synergy
• Subprioritizing so there is testing of the best drugs of each class
• Using genetic analyses and response biomarkers
• Testing combinations that optimize the benefit of already approved drugs
The growing number of targeted therapies that could be tested in combination, as well as the limited government and industry resources for such testing and the finite number of patients in whom combinations can be tested, suggests the need for a better way to prioritize which combinations get tested in clinical trials, several participants pointed out. Such prioritizing is key to developing a focus for patient advocates, federal agencies, and pharmaceutical chief executive officers (CEOs), said Dr. Michaele Christian, former NCI CTEP director, so everyone knows what the high-priority combinations are. But such prioritization can be challenging. As Dr. June noted, even in restricting combination therapy to combinations of immunotherapies, there is “a menu that is much too large to test in a combinatorial approach without some way of prioritization.” Dr. Engelman added, “We are going to have more combinations than we have patients.”
Dr. Engelman suggested using stricter preclinical benchmarks for effectiveness when deciding which combinations to test in the clinic. One of those benchmarks should be seeing tumors shrink in animals, as opposed to blocking tumors from forming or from growing. “Lots of times we get excited about a biological effect, yet the tumor still grows slowly or the cells still grow slowly and that does not predict for efficacy in the clinic,” he said. Dr. Engelman added that he would prefer to see
synergy versus additivity in preclinical tests, but the most important effect is seeing the tumor shrink, regardless of whether it is caused by synergy or additivity. He also noted that he has been impressed with a web-based system7 that Drs. William Pao and Mia Levy at Vanderbilt University have built to disseminate information on patients’ tumor mutations and responses to various therapies to enable a genetically-informed approach to cancer medicine. The My Cancer Genome website is an international collaboration of contributing physicians and physician scientists that compiles information on the mutations influencing cancer progression and growth, potential therapies that may be effective against specific mutations, and available clinical trials that target specific mutations. These data can be used to prioritize which combinations of targeted cancer therapies should be tested in the clinic, and can inform clinicians at the point of care about tumor mutations and possible targeted therapies.
Dr. Bachman said that when testing combinations in cell lines, he also aims for finding synergistic, not just additive, results.
Dr. James Doroshow, deputy director for Clinical and Translational Research at NCI, suggested that combinations be tested clinically only if they work in at least three xenografts, and that they be based on a biological mechanism for which there is an assay. Before testing a combination clinically, Dr. Helen Chen, associate branch chief of the Investigational Drug Branch at NCI, said that the agents in the combination should have already demonstrated adequate pharmacokinetics and some evidence of activity or target engagement at clinically relevant doses and exposures. Ideally the individual targets should be validated, and priority should be given for combinations that have shown a higher degree of efficacy, such as those that have converted growth inhibition to a tumor cell kill, she said.
Dr. Cantley pointed out that sometimes agents used singly do not have a significant effect because they are not tested at high enough doses, and subsequent combination therapies using those same agents do show an effect. “We have learned that you really have to hit these targets hard,” he said. In addition, some immunotherapeutic agents only work in combination and not singly, several experts in this field pointed out.
Dr. Chen also suggested assessing whether the synergism of the combination is seen consistently across all preclinical models, and if not, whether a predictive marker can be identified to choose those patients likely to respond to the synergistic interaction. Dr. Lutzker added that “in order to do small clinical trials, it is critical to try to understand which patient you want to test the combination in,” and suggested not doing any clinical testing unless there is a biomarker test that can be done simulta-
![]()
7 See www.mycancergenome.org (accessed December 14, 2011).
neously to assess which types of patients respond or do not respond to the treatment.
To systematically assess which combinations should be tested clinically, Dr. Stern is collaborating with Dr. Marcus Bosenberg to conduct high-throughput screening of 40 compounds at 3 concentrations on 30 tumor cell lines that model common human combinations of mutations. The tested drugs were heavily weighted toward drugs that target tumor cell growth signaling, but included conventional cytotoxic therapies as well. This research has revealed numerous additive or synergistic interactions in various combinations for specific tumor genotypes, some of which revealed novel pathway interactions. Drs. Stern and Bosenberg are currently linking these functional results to phosphoproteomic data as well as exome sequencing so researchers can use it to predict combination drug sensitivity according to genotype, such as by BRAF status, ras8 status, and other genetic aspects of a tumor.
Dr. Barrett suggested a more personalized approach to determining which combinations of therapeutics should be tested in patients. Such an approach can be taken by assessing the genomes of patients’ tumor, identifying which genetic mutations are driving the growth of those tumors, and then devising combinations that block those drivers. To do this, Dr. Barrett uses a small amount of tumor tissue that can be obtained from standard or needle biopsies. Then he uses flow cytometry to separate tumor cells based on the duplications or deletions of chromosomes or other characteristics that can be measured by examining individual cells with a laser. He then uses comparative genomic hybridization to genetically profile these subpopulations of tumor cells, noting that multiple populations of tumor cells can be present in a single biopsy. From this profiling he said it is possible to detect more than 100 chromosomal aberrations, which are then quantified and ranked according to how likely they are to be influencing the development or growth of the patient’s tumor. Based on this information, statistical and bioinformatics techniques are then used to depict what he calls a “wiring diagram” of the activated pathways that are fueling the tumor. This is then used to determine the most appropriate combination therapy. “There is lots of heterogeneity, but we find all the populations and can purify them out and often find convergence on these pathways,” Dr. Barrett noted. “What we need to do is identify the concurrent aberrations and mutations in each tumor cell population if genomics is really going to help advance the development of these targeted therapies, particularly combination therapies.”
As Dr. LoRusso noted, there are a lot of drugs of similar class. For
![]()
8 The ras family of genes code for proteins involved in cell signaling, cell growth, and apoptosis. Mutations in ras genes can lead to cancer.
example, there are at least a half dozen MEK inhibitors and PI3K inhibitors. She suggested that perhaps combination therapies should be limited to combining a few of the best in each class. Dr. Bachman also suggested subprioritizing which compounds within a class should be tested in combination because not all inhibitors are the same. “It’s important, when we start to think about prioritizing combinations, that we subprioritize, say PI3[K]/MEK combinations depending on what the inhibitors are telling us, what their potencies are,” she said.
Dr. Robert Iannone, section head of Clinical Oncology and cochair for the Pediatric Development Committee at Merck Research Laboratories, suggested developing more biomarkers that predict response to monotherapies as well as additional predictive biomarkers for when they are used in combination to indicate which combinations are most promising. “We can do the cell line work and the xenograft work, but still you get to the clinic and you find that your efficacy is not as good as you had hoped based on those preclinical tests. So we really need to go one step further to understand: What are the predictive biomarkers for these monotherapies and combination therapies?” Dr. Iannone asked. Dr. Lutzker added that both predictive markers for patient selection as well as pharmacodynamic markers that show modulation of the pathway of interest are critical for combination therapies.
Dr. Lutzker noted that Genentech’s initial strategy for prioritizing combinations has been to focus on drugs that were already approved and in the clinic, so as to maximize the benefit of those drugs to patients. More recently, the company has moved this rational combination development strategy earlier in the drug development process to drugs that are still in Phase I clinical testing. These strategies include combining compounds that have different mechanisms of action on the same target, such as combining two different antibodies to HER2 (antibodies that target two different epitopes on HER2). Genentech has also tested drugs for their ability to enhance the effects of bevacizumab.9 Such drugs are thought to sensitize tumors to the effects of bevacizumab or prevent or alleviate resistance to this drug. They have also tested a combination of erlotinib,10 which targets EGFR, and an anti-MET antibody to prevent the development of resistance to erlotinib by activation of the MET backup pathway. “These rational combination strategies have started to bear some fruit in the clinic,” Dr. Lutzker said.
![]()
9 Bevacizumab (trade name Avastin), is a drug that blocks angiogenesis, the growth of new blood vessels. It is used to treat various cancers, including colorectal, lung, and kidney cancers.
10 Erlotinib (trade name Tarceva) is a tyrosine kinase inhibitor drug that acts on the epidermal growth factor receptor. It is used to treat non-small cell lung cancer, pancreatic cancer, and several other types of cancer.
He added that not only should combinations have a strong scientific underpinning, but that they should also be composed of pharmacologically compatible molecules. “If one drug has a very long half-life and the other one has a very short half-life, and a toxicity develops, what can you do in terms of trying to maintain safety for patients? Do you have to stop both drugs? There are a whole number of issues that need to be thought through,” he said.
BUILDING ON THE BASIC KNOWLEDGE BASE
Key Suggestions to Build on the Basic Knowledge Base by Various Workshop Participants
• Government and industry support for academic explorations in basic research
• Gathering more information on gene expression, signaling perturbations, and DNA damage in tumors
• Developing tools to examine genotype/phenotype relationships
• Better understanding of the mechanisms of action of targeted therapies
Additional basic information has to be understood at the molecular level for combination therapies to be effective, several participants pointed out. They suggested that researchers gather more information on gene expression and the feedback and network responses to signaling perturbations and DNA damage. More information is also needed on the non-genetic effects that influence treatment, including the microenvironment of the tumor, the host immune response, and the proteins made by the tumor and surrounding cells.
“The problem is that the drugs target function—phenotype—whereas the measurement tools we have mainly query genotype, and the genotype/phenotype connections have not yet entirely been solved,” said Dr. Stern, and this is slowing the progress in developing molecularly targeted therapies. He added that on a detailed level, “there is a fundamental lack of knowledge on how even the most effective targeted therapies work. We know trastuzumab [Herceptin] works through a number of means, but I don’t think anyone here can tell us what the balance is of down-modulation, partial activation, [or how] the immune system is involved. Much basic science remains [to be elucidated]. We need to know how the target pathways interact, patterns of drug resistance and
response, and how to interpret transcriptional phenotypes so we can link what we can measure to where we can intervene.”
Dr. Munos added, “Biological modeling is not predictive because we have huge knowledge gaps. Forty percent of the human genome is still [uncharacterized]. Obviously, some of that stuff does something important, and unless we figure out what it does, it’s not going to work.”
Dr. Chen noted that the molecular pathways that drive some tumors are exceptionally complex, and researchers continue to discover new feedback loops and other mechanisms that tumors use to bypass blocked molecular pathways. “The real question is whether we are able to win the battle over such a highly adaptive tumor. Even if we can inhibit two or even three targets, the tumor may still find a way to escape,” she said.
Dr. Doroshow pointed out, “We know almost nothing at the molecular level about the toxicology of combination molecularly targeted agents.” To counter that lack of knowledge, NCI is currently developing a laboratory to study the toxicology of targeted combinations at the molecular level, he said.
Dr. Stern suggested that government and industry provide more support for academic explorations in the basic research areas that are so integral to fostering more effective cancer therapeutics.
IMPROVING CLINICAL TRIALS FOR COMBINATION THERAPIES
Suggestions from Various Workshop Participants on How to Improve Clinical Trials for Combination Therapies
• Having assays to select likely patient responders
• Using adaptive trial designs to determine the best combinations, dosing, and patient selection biomarkers as the trial progresses
• Using appropriate endpoints and setting a higher bar for effectiveness
• Establishing a single Institutional Review Board of record for multi-institutional trials
• Repeat biopsies of patient’s tumors to assess therapeutic effectiveness
• Developing a precompetitive venue for testing drug combinations in a limited number of patients
Patient Selection
Several speakers stressed the need to have biomarkers for patient selection, given that most of the agents being tested in combination therapies target highly specific molecular differences. “Almost every person who has cancer has an orphan disease, because there are really thousands of different subsets of cancer. Hopefully, we won’t need thousands of therapies, but until we divide these cancers, we’re not going to conquer them,” Dr. Cantley said.
Biomarker assays for key molecular differences that researchers can use to select patients will be crucial to such dividing and conquering, he added. Dr. LoRusso noted that genetic profiling of patients might enable researchers to determine what drug ratios to test on them in combination Phase I trials. Dr. Chen added that patient selection will be important not only to improve clinical trial efficiency, but to find those patients who can fit into a narrow window of therapeutic effectiveness because they are so sensitive to a drug’s effect to a target that a low enough dose can be effective without causing toxicity. This may be more important for combination therapies, she said, when significant dose reduction is often required to avoid adverse effects.
Although patient profiling would be expensive, it could save money in the long run by directing patients to the most appropriate therapies, Dr. LoRusso said. But she noted that “for the majority of cases, we lack the appropriate interrogation tools and assays for patient profiling, and even when they are available, many times we don’t take advantage of them.” Dr. Christian concurred and pointed out that clinical testing of drugs often precedes the development of patient selection biomarkers or biomarkers that indicate whether a target has been adequately hit.
Dr. LoRusso regretted that in her own Phase I study of breast cancer patients given a combination therapy that targets two different aberrant genes in stem cells, she did not determine if patients had those aberrant genes prior to testing them with the combination, although she plans to assess this in posttreatment biopsies. She noted that there is a concern that patient profiling and selection could slow down patient recruitment, but countered that when such profiling is done well, “it could actually help expedite rather than slow down the big picture.”
Dr. Barrett added that patient profiling can be done in real time, and does not necessarily slow down the patient recruitment process. For a trial funded by Stand Up To Cancer,11 in which he is involved (see Appendix A), “We can get a sample in on a Monday and by Thursday we can generate a report that is a rank list of what we believe the targets of the therapy
![]()
11 See http://www.standup2cancer.org/su2c/about_us (accessed December 14, 2011).
should be,” Dr. Barrett said, adding that “even 2 weeks can be almost a lifetime for some of these patients.” Dr. Perlmutter concurred, saying, “For some patients that 2-week wait is extra scary. We have to not only get better and cheaper in our testing, but we also have to get faster in our testing.” To do more detailed whole-genome sequencing is more time consuming, Drs. Engelman and Barrett noted, and currently is not practical for patient selection, although whole-exome sequencing has led to the discovery of a feasible number of exons—600–800—that could be assessed within 3 weeks and be potentially clinically useful, Dr. Engelman added.
Dr. LoRusso gave a positive example of patient selection in a Phase II clinical trial of a MEK inhibitor tested in combination with a BRAF inhibitor by Jeff Infante of the Sarah Cannon Research Institute in Nashville, Tennessee. She said Dr. Infante preselected his patients based on the presence of BRAF mutations in their tumors, and preliminary results suggest a better response rate with the combined therapy than what was observed with the single agents (Infante et al., 2011). The trial is still ongoing, with the majority of enrolled patients continuing in the study. Dr. Lutzker added that he preselected patients in a trial that tested a MET monoclonal antibody plus erlotinib. Such preselection was done using an assay for high-level expression of MET by immunohistochemistry. This Phase II study showed strong efficacy in this patient group in terms of progression-free and overall survival, he said. Dr. Cantley noted his team of researchers spent a lot of time discovering and testing biomarkers for early response that were quantitative, predicted clinical outcomes, and worked well across institutions. These biomarkers included those that could be evaluated in positron emission tomography (PET) scans.
Dr. LoRusso questioned the relevance of the genetic profiling being done in metastatic tumors to determine appropriate treatment combinations to patients with non-metastatic disease, who might be more likely to benefit from combination therapy. “What are the risks that are involved if we are studying these combinations in the wrong patient population at the wrong clinical stage?” she asked, especially if negative findings in a metastatic patient population led to combinations being rejected for further testing in patients with early-stage disease.
Dr. Sharon Murphy, scholar in residence at the Institute of Medicine, suggested conducting more combination therapy trials in pediatric cancer populations. She noted that there are extensive tissue banks of pediatric tumors that are clinically well annotated and could serve as valuable resources for investigators. “When we think about combination targeted therapies or targeted treatments, we should think about childhood cancer, which arguably is a better model because genetically it’s simpler than many adult forms of cancer. There are fewer signaling pathways, and children need these drugs too,” she said. She added that investigators
should not have to wait to test promising combination therapies in pediatric populations until after they show promise in adult Phase I trials. Dr. Chen responded that CTEP has been doing a lot of Phase I and II testing of investigational cancer treatment combinations in the pediatric population. Dr. Samuel Blackman, Director of the Oncology Early Development Unit at GSK, agreed that pediatric populations should be engaged to achieve early proof of concept, and for some subtypes of cancers such testing is easier to do in the pediatric than the adult population because pediatric patients with these tumors tend to be grouped according to the genetic drivers of their tumors and treated in disease-specific programs offered in major pediatric cancer centers.
Adaptive Trial Designs
According to Dr. Berry, adaptive clinical trial designs are especially suited for answering the numerous questions that combination therapy raises, such as which of several possible drug combinations, patient selection biomarkers, doses, and dosing schedules are the safest and most effective. He said that an adaptive design enables researchers both to answer questions as well as to raise new questions and test new hypotheses during the course of a trial. Adaptive trials use Bayesian statistics to model and predict during a trial which option is most likely to be beneficial based on the results to date. Researchers use these predictions, while the trial progresses, to increase the number of patients being tested in the arms showing the most promise, and reduce or drop the number of patients being treated in those arms generating poor results.
For example, Dr. Berry designed an adaptive Phase I/II trial for a two-drug combination therapy for leukemia in which the admissible doses expanded or contracted during the trial depending on toxicity and effectiveness. For trials of two agents given separately or together, patients are randomized to each possibility, but “as you are learning, for example, that agent 1 is not doing as well as you might have hoped, you might give it a lower probability [and assign less patients to receive this agent],” he said, adding, “At some point we make a decision as to what is going to be the confirmatory stage [for the agent that is having the best results]” (see Figure 3-1).
Another example of an adaptive trial is I-SPY 2 TRIAL (Investigation of Serial studies to Predict Your Therapeutic Response with Imaging And moLecular analysis 2; see Figure 3-2 and Appendix A), which tests various treatments for breast cancer used singly or in combination while simultaneously assessing which patient selection biomarkers are most appropriate for each treatment. This trial has five experimental arms in which new treatments are “plugged in” to be tested once other tested
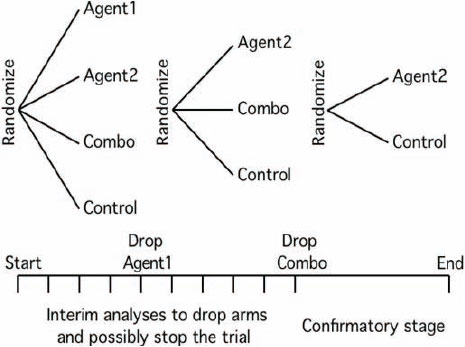
SOURCE: IOM, 2010b.
agents progress to confirmatory trials or fail to show favorable results, Dr. Berry said. It has a factorial design in which single agents plus standard of care are tested against combinations of agents plus standard of care. As the trial progresses, single agents may be dropped because the results are more favorable when they are used in combination, “but meanwhile we have some experience in the single agents and some notion of synergy or additivity,” Dr. Berry pointed out.
The I-SPY 2 TRIAL is innovative in that there is an adaptive design with regard to both treatment and the patient selection biomarker for the treatment. Dr. Berry stressed this is critical given that researchers continue to uncover new biomarkers for patient selectivity. “We have to figure out ways that we can update that information and use additional markers to understand who benefits from treatment. The only way to do it is to build it into our clinical trial structure and learn as we go,” he said. Dr. Iannone highlighted that trials can be more efficient if patients in a clinical trial of
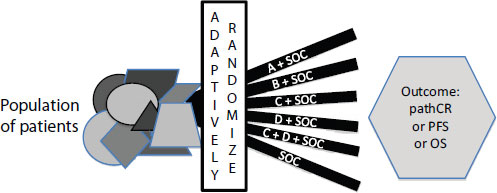
NOTE: OS = overall survival; pathCR = pathological complete response; PFS = progression-free survival; SOC = standard of care.
SOURCE: Berry presentation (June 13, 2011).
combination therapies can easily move from one arm to another, based on some early measure of success or failure through a specific pharmacodynamic response biomarker. Efficiency is especially improved if there is a high negative predictive value and patients can be quickly assigned to another treatment arm without needing another baseline biopsy. “There is an efficiency for investigators, but there is a huge efficiency and potential upside for patients as well,” he pointed out.
Dr. Larry Rubinstein, statistician at the NCI Biometric Research Branch, agreed that the adaptive trial designs Dr. Berry presented were well suited to trials of investigational drug combinations because they avoid the problem of setting the maximum tolerated dose prematurely, which often occurs with standard Phase I trial designs that have a small
number of patients. Adaptive trial designs also address the ethical imperative of aiming to benefit patients by focusing on the dose combinations that are most promising, he said. The randomization of patients to biomarker tests is also an important way to assess whether the biomarker indicates if a patient is likely to do better when given a particular treatment, versus whether a patient is likely to do better regardless of which treatment he or she receives, that is, it enables researchers to distinguish between predictive and prognostic markers.
But in addition to assessing the clinical toxicity of varying doses of experimental agents, Dr. Rubinstein suggested introducing in vivo pharmacodynamic assays for efficacy or toxicity, or even using these assays during the course of a clinical trial to assess whether the individual agents are hitting their targets and otherwise working mechanistically as expected. “This means you may end up terminating escalation for an agent, not on the basis of toxicity, but because you appear to have reached the limit of its efficacy,” he said. Such pharmacodynamic assays could be a part of an adaptive clinical trials design, he noted.
Dr. Doroshow emphasized the importance of demonstrating a mechanism of action early in a clinical trial to validate one’s presumptions in this regard. “We have an enormous number of presumptions going into first-in-human studies, and often those presumptions are wrong,” he said. “It’s critical to get this proof-of-mechanism information for the subsequent development of which combination to utilize. If you don’t have an assay to demonstrate target inhibition, it’s almost impossible to develop an appropriate schedule, in terms of relating systemic exposure to the targeting effect.” Thus, NCI is currently developing more than 50 assays for evaluating the mechanisms of action of molecularly targeted agents, Dr. Doroshow reported (see Appendix A).
Dr. Steven Piantadosi, director of the Samuel Oschin Comprehensive Cancer Institute at Cedars-Sinai Medical Center, stressed that some sort of factorial clinical trial design must be used to investigate the interactions of agents when they are used in combination, and that there be enough sampling points in the “two-dimensional dose space” from which researchers can reap adequate information about how the response changes over that two-dimensional surface. Dr. Lutzker added that “modeling the dose–response curve is really critical in that aspect.”
Repetitive Tumor Biopsies
As a means for assessing whether drug agents are hitting their targets in patients, several conference participants suggested conducting assays on repetitive biopsies of patients’ tumors. Dr. Cantley also suggested examining repeat biopsies from clinical trial patients to assess not only
whether each target of a combination therapy has been hit individually, but that “you have hit something that you know should be a consequence of inhibiting both,” he said.
In addition to analyzing patient tumor samples prior to a clinical trial, Dr. Engelman noted that his research group biopsies every patient who becomes resistant to a tested targeted therapy. As many as five biopsy cores are taken in one procedure, at no greater risk to patients than a single biopsy, because these cores are removed through a single transducer needle that makes only one puncture to access tissue specimens from the site. “We could be more aggressive about getting tissue for lots of studies,” he said. Dr. Engelman added that patients are more than willing to have such biopsies performed.
Dr. Perlmutter agreed about the general willingness of patients to have needle biopsies performed, but noted some situations in which a biopsy may not be feasible. “I don’t think you would ask a brain cancer patient to give you a biopsy, but many cancers are biopsied,” she said. “Patients are often more than happy to provide multiple biopsies, but doctors often do not request them,” she added, stressing that a trial should get as much data as possible and patients generally recognize that providing specimens is in their best interests. Dr. Cantley added that the combination of clinicians and patient advocates stressing the importance of the biopsies required in the clinical trials he has been involved with has led to patient willingness to have these biopsies performed and to enroll in protocols in which such biopsies are mandatory.
Dr. LoRusso stressed that serial biopsies of patients are the best way to assess “not so much what went right, but more importantly what went wrong” in a clinical trial. She said within the previous year, her research team used full-time technicians to biopsy at least 300 out of 500 patients. “I feel we are still relying too heavily on surrogates and I haven’t found many surrogates that have led me down the appropriate path of taking that drug forward into the appropriate patient subset,” she said. She added that imaging results are also inadequate surrogates. She uses imaging, such as PET or DCE-MRI (dynamic contrast enhanced-magnetic resonance imaging), to assess treatment effectiveness during the course of a clinical trial, but these are very expensive, she said, “for the amount of information that we are not getting because of the variability of multiple factors, including the heterogeneous patient population in a Phase I trial.”
Determining Appropriate Dose and Schedule
Several participants noted that it can be challenging to determine appropriate dosing because of the variability in how patients respond to different agents. Dr. LoRusso raised the question of whether “all doses of
combinations are created equal or do we need to personalize the doses of the individual drugs relative to the mutational status and changes in the tumor,” which she said no one has explored yet, but added “it’s not an insignificant issue and I don’t think we can forget it as we are developing these combinations.” Dr. Doroshow suggested that NCI’s toxicogenomics program (see Appendix A) should help researchers find correlations between pharmacokinetics and systemic exposure with the genomic profiles of various tumors according to the class of drug being tested. This information is being made public as it is gathered.
Dr. Chen suggested that if the goal of the therapeutic outcome is to achieve a sustained major response or cure, then one should consider giving short but intensive doses that are lethal to the tumors. If that goal is not achievable and continuous therapy exposure is required, she suggested that lighter, less intensive therapy may have to be given so it can be tolerated, or that combination therapy be given sequentially rather than concurrently.
Dr. Chen added that hundreds of clinical trials testing combinations of these targeted agents reveal they can be quite toxic. Often there is an increase in the severity and frequency of the known toxicity of the single agents used in combination, although sometimes new toxicities arise. In some cases there appeared to be synergistic toxicity, perhaps due to the nonspecific targets of these molecules, and significant dose reductions were required. Some combinations, such as the VEGF (vascular endothelial growth factor) inhibitor sunitinib and the mTOR inhibitor temsirolimus, had to be abandoned because of their combined toxicity.
According to Dr. Chen, combinations of agents that target parallel pathways are less likely to have overlapping toxicity and are better tolerated, as are agents with more specificity. She added that combinations appear to be less tolerable if they target the more downstream elements of signaling pathways. In all cases, the maximum tolerated dose based on cycles one and two do not appear to predict long-term tolerability. Dr. Chen stressed that what she has learned from all these clinical trials is that full doses of each individual agent are often not tolerable in combination, and that the adverse effects on normal tissues may limit the spectrum and degree of duration of combined target inhibition. This raises numerous questions about the best way to develop a combination dosing strategy to reduce toxicity. These questions are probably best addressed with more intensive preclinical studies to determine the optimal dose and schedule, she said. Such dosing will probably be based on the pharmacodynamics or pharmacokinetics required for synergism, keeping in mind that the dose required for synergism may not be the same as that required for single-agent activity, she said.
Dr. LoRusso pointed out that the maximum tolerated doses of the
drugs used in a combination are not necessarily meaningful. “There are various trial designs that could actually hurt you sometimes more than help you, depending on which is the most important drug, and how the ratios need to be defined in the clinical scenario,” she said. She added that often toxicities, such as rashes, are expected to be worse in combinations of drugs if each drug has shown such a side effect in Phase I trials. Dr. Herbst noted that about half of the combination targeted cancer therapies currently being tested clinically have shown dose-limiting skin toxicity. However, sometimes the side effect is not seen in the Phase I trial of the combined drugs. Also, additional toxicities not predicted by single-agent studies can surface when the drugs are tested in combination. “What we sometimes can predict or theorize based on preliminary monotherapy data may not actually come true when we do the combination,” Dr. LoRusso said.
Dr. June added that the T cells used in cell-based immunotherapies are living and often long lasting and self-replicating, so they have different pharmacologic and pharmacokinetic parameters than drugs for which simple clearances can be assessed. Because of this, a clinical trial design quite different from a standard Phase I approach is needed. For example, in a Phase I clinical trial of a cell-based immunotherapy, his research group tries to identify an optimal biologic dose rather than the more standard maximum tolerated dose.
Appropriate Endpoints and Other Study Measures
Researchers are finding that immunotherapies such as Provenge and various tumor vaccines used to treat cancer often extend survival without delaying time to progression, Dr. Schlom pointed out. These therapies often stabilize rather than diminish the size of tumors and may also extend survival without diminishing the growth rate of metastatic cancers. These findings suggest that traditional endpoints may not be appropriate for clinical trials of immunotherapies, according to Dr. Schlom, and that overall survival might be the best indicator of their effectiveness. It is not clear whether this applies only to combination immunotherapies or also to treatments that combine an immunotherapy with standard chemotherapy or targeted treatments. However, he noted that preliminary data from one study (NCI, 2011b) showed that a tumor vaccine combined with docetaxel did extend time to progression over the docetaxel treatment given singly. Dr. June added that for many immunotherapies, determining the optimal biologic dose is the most appropriate aim of Phase I studies, as opposed to determining the maximum tolerated doses. Especially for immunotherapies that apply live cells, such as modified T cells, the typical dose-escalation Phase I clinical trial design is not appropriate, he said.
A few participants suggested there should be higher standards for clinical response in trials of combination therapies. “Given the fact that we will be running out of patients and resources [to test combination treatments], we need to be setting our bars way higher than we are,” Dr. LoRusso said.
Dr. Wendy Demark-Wahnefried, associate director for Cancer Prevention and Control at the University of Alabama at Birmingham Comprehensive Cancer Center, encouraged researchers to include lifestyle factors, especially measures of energy balance and obesity, when assessing the effectiveness of combination therapies because obesity has been shown to affect some of the same molecular pathways targeted by certain cancer drugs. Breast and endometrial cancers, for example, are hormonally driven cancers that are affected by obesity, she said, and collecting body mass index data at baseline and at follow-up of patients with these cancers being treated with combination therapies could provide useful information. Dr. Cantley agreed, adding that prostate, colorectal, and pancreatic cancers are also affected by obesity, presumably through its effects on IGF1, and that he has suggested to industry to modulate those effects by using the diabetes drug metformin in clinical trials. “This is something we’re very much aware of,” he said.
Dr. Chen stressed that though many targeted therapies show evidence of being therapeutic in the clinic when used singly, many of those treatments fail clinical trials when they are used in combination. For example, VEGF and EGFR inhibitors showed no effect when given with chemotherapy to treat several cancer types, including colon, pancreatic, kidney, and breast cancers, she pointed out. Combinations that target mTOR have also failed Phase II or III trials. “Do we have a failure of the hypothesis or a failure of the clinical trials because we did not use the right dose or choose the right patients? All these possibilities are possible for different scenarios,” she said.
Speeding Up the Collaborative Clinical Trial Process
Dr. Vassiliki Papadimitrakopoulou, professor of medicine in the Department of Thoracic/Head and Neck Medical Oncology at MD Anderson Cancer Center, pointed out that multiple steps need to be satisfied to have several pharmaceutical companies and academic institutions collaborate in combination therapy trials of investigational anticancer agents, including coordinating Institutional Review Board (IRB) reviews, data sharing and analysis, intellectual property agreements, Investigational New Drug (IND) applications, etc. It can take years to accomplish all those steps so that a collaborative, multisite clinical trial of combination therapy can begin. There is concern that during that lengthy start-up
time, scientific advances will occur that might indicate that the combinations in the trial are no longer the most promising ones to test, she pointed out. “We need to speed things up,” she said.
Dr. Cantley added that a major time impediment is acquiring the IRB approvals from multiple institutions. He suggested that presenting a strong trial concept initially to the IRBs can help speed things up, as can having regular face-to-face meetings and teleconferences, and having investigators with clinical trial experience on a research team, in addition to the Principal Investigators, to provide valuable advice and help others to benefit from their experience. Dr. Papadimitrakopoulou suggested that patient advocates can help speed up the process by putting more pressure on academia to make their IRBs more expedient. Dr. Perlmutter suggested that every multicenter trial have a single IRB12 and noted that for the I-SPY 2 TRIAL there are 15 different versions of informed consent. “It certainly adds expense and confusion that is totally unnecessary,” she said. Dr. John Hohneker, senior vice president and global head of development of the Integrated Hospital Care Franchise at Novartis Pharma AG, added that many institutions are afraid to commit to and execute an agreement without having their own IRBs approve it.
Dr. Flaherty suggested that time and resources could be saved in the long run if there were a precompetitive venue for testing drug combinations in a limited number of patients—less than 20—to more rapidly sift out combinations likely to be effective in the clinic. Sponsors would have an incentive to contribute their drugs to such a system because it would be an efficient way of triaging combinations that they do not have the resources and the time to test, according to Dr. Flaherty. “We need to create some kind of mechanism for cranking through these combinations in relatively small patient numbers in a much more rapid fashion than we currently have the capacity to do,” he said.
Drs. Christian and Flaherty also called for strong patient advocacy to support a list of vetted important targets and combinations that should have priority status for clinical tests. “Figuring out a way of having a rolling, ongoing dialogue about prioritization is absolutely critical for the early combinations,” Dr. Flaherty said. Dr. Christian added, “There are all these patient advocacy groups and we just need to figure out how to make them talk to each other about this most important topic.”
![]()
12 On July 26, 2011, the U.S. Department of Health and Human Services announced that the federal government is contemplating various ways of enhancing the regulations overseeing research on human subjects, as described in an Advance Notice of Proposed Rulemaking (HHS, 2011).
This page intentionally left blank.


























