
2
The Importance of Innovative Regulatory Science
Key Messages
• Collaboration among federal agencies offers a particularly productive venue for developing a regulatory science workforce.
• Creating a supportive academic environment and training a new generation of researchers who are skilled in such areas as clinical pharmacology can contribute greatly to the development of regulatory science as an accepted discipline.
• Regulatory science is more than just developing new methods for understanding and assessing risk; it also includes consideration of cultural and societal issues relating to how individual patients and society view the tradeoff between reward and risk.
Different stakeholders in the therapeutics development enterprise may have somewhat different perspectives on regulatory science. In the opening session of the workshop, representatives of the four key “locuses” of the development and practice of innovative regulatory science for therapeutics development (FDA, NIH, the pharmaceutical industry, and academia) described these perspectives. All four emphasized the importance of developing regulatory science as an academic discipline to address the multiple challenges they face. At the same time, different perspectives create opportunities for productive collaboration, though
as some participants cautioned, also potentially create opportunities for misunderstandings.
Vicki Seyfert-Margolis, Senior Advisor for Science Innovation and Policy, Office of the Commissioner, FDA, presented FDA’s rationale for regulatory science and the strategic plan the agency has developed to pursue this science. Story Landis, Director, National Institute of Neurological Disorders and Stroke (NINDS), NIH, described some of the collaboration ongoing between FDA and NIH. Andrew Dahlem, Vice President and Chief Operating Officer, Lilly Research Laboratories, Eli Lilly & Co., summarized some of the challenges and needs facing industry in a radically changed drug development environment. Ralph Snyderman, Chancellor Emeritus, Duke University, discussed therapeutics development in the broader context of personalized medicine. Ellen Sigal, Chair and Founder, Friends of Cancer Research, provided a patient perspective on the importance of regulatory science and moderated a panel discussion among the keynote speakers.
PERSPECTIVE FROM THE FOOD AND DRUG ADMINISTRATION1
Addressing the Product Development Ecosystem
Seyfert-Margolis noted that FDA can bring data, insight, and knowledge to the therapeutics development ecosystem. In the months preceding the workshop, FDA had been examining the medical products ecosystem to determine how the agency can facilitate innovation. The agency’s innovation initiative will include regulatory science as a key component.2 According to Seyfert-Margolis, a major finding from this review was that all of the various players in this arena are experiencing new stresses and challenges. The scientific and global market environments are moving away from the traditional blockbuster drug development model. In aca-
________________
1This section is based on the presentation by Vicki Seyfert-Margolis, Senior Advisor for Science Innovation and Policy, Office of the Commissioner, FDA.
2FDA released a report on innovation, Driving Biomedical Innovation: Initiatives to Improve Products for Patients, on October 5, 2011. The report defines the “medical product ecosystem” in the following context:
Translating a new idea from a discovery into a medical product is a complex process involving an entire ecosystem consisting of academia, industry, small businesses, payors, physicians, government agencies, and patient and consumer groups. Each member of the ecosystem has an important role to play in bringing a new medical product to market, and each piece of the ecosystem is currently under stress, putting America at risk of losing its competitive edge as the leader in scientific innovation.
For more information, see http://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/ucm274333.htm (accessed November 28, 2011).
demia, the demands of tenure and recognition do not always align well with the team-oriented imperatives of clinical research and development. At the same time, academics confront difficulties in moving their ideas into the clinic given the challenges of securing funding.
A number of interesting models are emerging to deal with these challenges, said Seyfert-Margolis. Some of these models are being supported directly by academic institutions, while others involve venture philanthropy. Small business plays an important role in the product development ecosystem, but small businesses today are often undercapitalized and are struggling with the complexities of the therapeutic product development cycle. FDA has been engaged in discussions with companies along the size spectrum—from small biotechnology to large pharmaceuticals—to provide advice on how to move ideas forward successfully. Seyfert-Margolis noted that the calculus of risks and benefits is an important discussion to have as a scientific community and as a society and needs to be taken into account in consideration of potential new regulatory pathways such as conditional or progressive approval strategies.
Physicians and patients have their own concerns regarding new technologies and therapeutics. Society’s tolerance for risk is low, Seyfert-Margolis observed, so it is important to communicate information and concepts about risks and rewards more effectively to patients and physicians. Payers are focusing on the concepts of evidence- and value-based medicine to align their reimbursement structures with the actual real-world performance of drugs, which is usually less than that seen in clinical trials. Once in the real world, the risk equation shifts as comorbidities and other influences on drug performance become apparent.
The Rationale for Regulatory Science
According to Seyfert-Margolis, there are four major reasons why regulatory science is needed.
First, major investments and advances in basic sciences are not fully translating into products to benefit patients. In part, this is because of the so-called “valley of death” (Figure 2-1). The valley of death refers to the gap in funding between, on the one hand, NIH, state, and foundation grants that support discovery, preclinical development, and the earliest stages of clinical development and, on the other hand, pharmaceutical company involvement that supports the later stages of clinical development and commercialization. This funding gap has a critical, negative impact on the translation of discoveries into medical products and new medicines. Seyfert-Margolis emphasized that regulatory science can play an important role in helping to bridge this gap.
Second, product development is increasingly costly and uncertain
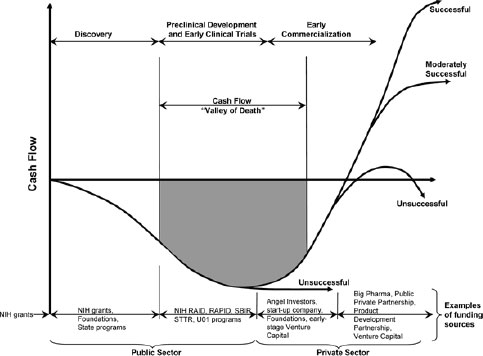
NOTE: RAID, Rapid Access to Interventional Development; RAPID, Rapid Access to Preventive Intervention Development; SBIR, Small Business Innovative Research; STTR, Small Business Technology Transfer; U01, Cooperative Agreement Research Project Awards.
SOURCE: Seyfert-Margolis, 2011. Presentation at IOM workshop on Strengthening a Workforce for Innovative Regulatory Science in Therapeutics Development.
while success rates remain low. For example, according to Seyfert-Margolis, the need for cost containment has driven clinical trials to other countries; now, approximately 50 percent of the clinical trials data included in regulatory submissions to FDA is generated in India or China. These populations differ from the average U.S. population in terms of nutrition, genetics, environment, lifestyle considerations, and other factors. As a result, data from those populations may not fully translate into the average U.S. population, said Seyfert-Margolis, while noting that this issue needs further study. FDA believes that uncertainties associated with differences among populations lie more in the scientific than in the policy realm, pointing to the need for regulatory science.
Third, development and evaluation tools and approaches have not kept pace with and have not incorporated emerging technologies. Examples include the new fields of nanotechnology, where tools to evaluate
the safety and toxicity of nanoscale products and genetically engineered foods are lacking. Shortcomings in scientific capacity in this area also affect the ability of FDA to educate and communicate with the public about the risks and benefits of new technologies.
Fourth, without regulatory science the economic health of the biotechnology and medical products industry is at risk. This is of particular concern given the importance to the U.S. economy of maintaining and building on the nation’s leadership role in this important technological sector.
FDA now has several programs in place, including a formal partnership with NIH, to advance regulatory science. For the first time in its history, FDA has a budgetary line item for regulatory science and has issued direct funding solicitations for projects in reproductive toxicology and biomarker research and qualification.
The FDA Strategic Plan for Regulatory Science
FDA has issued a strategic plan for regulatory science that was crafted with input from all of its centers yet takes an agency-wide, rather than center-specific, perspective (FDA, 2011). The strategic plan’s vision statement says that “FDA will advance regulatory science to speed innovation, improve regulatory decision-making, and get safe and effective products to people in need. Twenty-first-century regulatory science will be a driving force as FDA works with diverse partners to protect and promote the health of our nation and the global community.”
The strategic plan lays out eight priority areas:
1. Modernize toxicology to enhance product safety.
2. Stimulate innovation in clinical evaluation and personalized medicine.
3. Support new approaches to improve product manufacturing and quality.
4. Ensure FDA readiness to evaluate emerging technologies.
5. Harness diverse data through information sciences to improve health outcomes.
6. Enable a prevention-focused food safety system.
7. Facilitate development of medical countermeasures (MCMs) to protect U.S. and global health and security.
8. Strengthen social and behavioral science to help consumers and professionals make informed decisions.
Seyfert-Margolis briefly described FDA’s intentions in each of these eight areas (see Box 2-1).
BOX 2-1a
FDA Strategic Plan for Regulatory Science
Modernize toxicology to enhance product safety. FDA plans to develop better models of human adverse response, identify and evaluate biomarkers and end points that can be used in nonclinical and clinical evaluations, and use and develop computational methods and in silico modeling.
Stimulate innovation in clinical evaluation and personalized medicine. FDA seeks to develop and refine clinical trial designs, end points, and analysis methods; leverage existing and future clinical trial data; identify and qualify biomarkers and study end points; increase the accuracy and consistency, and reduce interplatform variability, of analytical methods to measure biomarkers; and develop a “virtual physiologic patient.”
Support new approaches to improve product manufacturing and quality. FDA plans to enable development and evaluation of novel and improved manufacturing methods, develop new analytical methods, and reduce the risk of microbial contamination of products.
Ensure FDA readiness to evaluate emerging technologies. FDA will stimulate development of innovative medical products while concurrently developing novel assessment tools and methodologies, develop assessment tools for novel therapies, assure safe and effective medical innovation, and coordinate regulatory science for emerging technology product areas.
Harness diverse data through information sciences to improve health outcomes. FDA plans to enhance information technology infrastructure development and data mining; develop and apply simulation models for product life cycles, risk assessment, and other regulatory science uses; analyze large-scale clinical and preclinical data sets; incorporate knowledge from FDA regulatory files into a data-base integrating a broad array of data types; and develop new data sources and innovative analytical methods and approaches.
Enable a prevention-focused food safety system. The agency will establish and implement centralized planning and performance measurement processes, improve information sharing internally and externally, maintain mission-critical science capabilities, and cultivate expert institutional knowledge.
Facilitate development of MCMs to protect U.S. and global health and security. FDA will develop, characterize, and qualify animal models for MCM development; modernize tools to evaluate MCM product safety, efficacy, and quality; develop and qualify biomarkers of diseases or conditions; and enhance emergency communication.
Strengthen social and behavioral science to help consumers and professionals make informed decisions. FDA seeks to know its audience, reach that audience, ensure audience understanding, and evaluate the effectiveness of communication about regulated products.
________________
aBased on the presentation by Vicki Seyfert-Margolis, Senior Advisor for Science Innovation and Policy, Office of the Commissioner, FDA, which drew directly from FDA’s recent strategic plan for regulatory science (FDA, 2011).
Seyfert-Margolis also noted that applications were under review for a new National Capitol Region Center for Excellence in Regulatory Science and Innovation (CERSI). The goal of the new center is “to advance the field of regulatory science (including laboratory, population, behavioral, and manufacturing sciences) and the Critical Path Initiative toward more effective and efficient product development and evaluation. CERSI efforts will focus on promoting innovation in support of the development and evaluation of safe and effective products through training, applied collaborative science, professional development and scientific exchanges.”3
PERSPECTIVE FROM THE NATIONAL INSTITUTES OF HEALTH4
Landis illustrated the promise of regulatory science through the case example of advancements over the past 15 years in the development of human embryonic stem cells and new techniques allowing dedifferentiation and differentiation, with extraordinary potential for application to regenerative medicine. She commented that there is a need for regulatory science in thinking about how to understand, regulate, and enable clinical trials using human embryonic stem cells. She noted that FDA has embraced this innovative technology and enabled it to move forward, with three clinical trials currently being conducted in the United States.
Landis also described an innovative new initiative in which NIH is partnering with FDA and the Defense Advanced Research Projects Agency (DARPA) to develop embryonic stem cells as microphysiological systems—organs on a chip—that could be used instead of laboratory animals to screen for safe and effective drugs. This partnership has received 5 years of funding worth $140 million, a substantial investment that addresses goals of modernizing toxicology and adopting new innovative technologies.
________________
3According to the CERSI Request for Application (RFA), the CERSIs should be academic, M.D. and Ph.D. degree–granting institutions with both strong life science and clinical medical science activities. FDA also specified that they should be located within a 50-mile radius of FDA’s Silver Spring, Maryland, campus to facilitate and enable training, research, scientific exchanges, and other collaboration among CERSI institution staff, students, and trainees and FDA staff scientists. See http://grants.nih.gov/grants/guide/rfa-files/RFA-FD-11-033.html (accessed November 28, 2011). The CERSI awards, totaling $2 million, were announced on October 26, 2011. The centers, which will be located at the University of Maryland and Georgetown University, will focus on strengthening science and training needed to modernize and improve the ways drugs and medical devices are reviewed and evaluated. See http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm277267.htm (accessed November 28, 2011).
4This section is based on the presentation by Story Landis, Director, NINDS, NIH. Landis also serves as one of the co-chairs of NIH’s regulatory science initiative and is a member of the NIH-FDA Joint Leadership Council.
NIH-FDA Collaborative Efforts and Regulatory Science Funded Projects
Landis noted that the NIH and FDA have a history of innovations in therapeutics development, for example, the creation of the exploratory Investigational New Drug (IND) application. The exploratory IND came about through a partnership between the National Cancer Institute (NCI) and FDA and aimed to address the fact that the behavior of anticancer agents seen in animal models does not translate well into humans by developing innovative strategies to screen possible therapeutic candidates for cancer in humans. The exploratory IND is designed to inform decision making on target selection and dose. It assesses pharmacokinetics, biodistribution, and drug engagement with the appropriate target using very small doses of drug and with no therapeutic intent.
The formation of the NIH-FDA Joint Leadership Council in 2010 greatly enhanced the partnership between NIH and FDA, Landis noted. The council has six working groups that review proposals for collaboration in areas that include regulatory science, training, and education. In addition, the leadership council created cooperative research grants to advance translational regulatory science, and four projects totaling approximately $9 million over 3 years have been funded under this initiative:
• Creating an organ on a chip that functions as a heart-lung micromachine model to test the safety and efficacy of drugs
• Designing innovative, adaptive clinical trials for evaluating drugs and devices used in the emergency care of patients with acute neurological illness or injury
• Developing an understanding of how nanoparticles interact with the complement system and developing a model that can predict which nanoparticles might activate the complement cascade
• Developing a novel strategy to predict ocular irritancy
NCATS and NIH Training Initiatives
The NIH-proposed National Center for Advancing Translational Sciences (NCATS), if appropriated and stood up, would assume responsibility for the agency’s regulatory science initiatives, in a shift from the distributed function currently residing in NINDS and the National Institute of Diabetes and Digestive and Kidney Diseases.5 The vision and mission of NCATS have evolved over the year that the proposed new center
________________
5NCATS was formally established by Congress in the FY 2012 Omnibus Appropriations Bill, signed into law on December 23, 2011 (after the date of the workshop). The new center has a budget of $575 million.
has been in the planning stages; although there may have initially been a notion that NCATS was envisioned as the “center for cures,” as the mission has been clarified, the center will be focused on “the science of how you do translation better.” The center will be charged with advancing the discipline of translational science to catalyze the development and testing of novel diagnostics and therapeutics across a broad range of diseases and conditions. In this pursuit the proposed NCATS will approach the drug development pipeline as a “scientific problem ripe for intervention, experimentation, and process engineering, looking for bottlenecks and creating solutions.” NCATS will also have a training function in translational science, including clinical pharmacology.
Landis noted that NIH currently carries out a training function through the institutes and centers. The broad training environment created by NIH-funded programs operated by the institutes and centers represents a real opportunity for producing scientists and physician-scientists who can think about regulatory science, she said. She cited three examples of training programs of relevance to regulatory science:
• Pre- and postdoctoral fellows programs that often feed trainees into positions at FDA
• Programs in the National Institute for General Medical Sciences for physician-scientists, together with institute-specific initiatives targeted at physician-scientists
• The Clinical and Translational Science Awards (CTSAs) (currently residing within the National Center for Research Resources; slated to move to the new NCATS)
PERSPECTIVE FROM THE PHARMACEUTICAL INDUSTRY6
Views about the division of labor among NIH, academia, and industry in drug discovery and development have changed dramatically over the past 20 years, observed Dahlem. Historically, NIH has had responsibility for basic research, academia discovered potential new targets, and industry looked for the commercial potential of those targets, with FDA providing regulatory oversight of new drug applications (NDAs). In a shift, today, Dahlem said, NIH has become increasingly interested in drug discovery and development, while researchers in academia have joined their industry colleagues in searching for new drugs rather than limiting their pursuits to basic discovery.
________________
6This section is based on the presentation by Andrew Dahlem, Vice President and Chief Operating Officer, Lilly Research Laboratories, Eli Lilly & Co.
Challenges Faced by Industry
Dahlem noted that drug development expenditures have increased while approvals have decreased. The number of drugs that will go off patent is increasing, which will cause companies to become even more cost-conscious in the coming years. This will affect not only industry hiring (and contribute to the shedding of jobs) but also the amount of money industry will provide to academia to support research. This evolving framework provides a backdrop for thinking about the workforce needs and capacity.
Industry is acting to reduce the failure rate of drugs in the development pipeline, said Dahlem. For example, in 1988, nearly 40 percent of Lilly’s drugs failed because of pharmacokinetic problems; today that number has been reduced to about 3 percent through advancements in science. Now, the most common reason for a drug to fail in clinical trials is that data from animal models are not good enough predictors of success in humans.
Dahlem also noted that, in any exercise to define and ascertain how to develop a workforce, one must keep in mind that, from any student’s perspective, a training program must provide opportunities that allow trainees to generate sufficient income to repay debts assumed during education and training.
Dahlem provided several observations about industry needs for the regulatory science process. First, industry needs timely and predictable reviews of NDAs, which could be achieved through enhanced collaboration among industry, academia, and FDA. Second, Dahlem called for unbiased third-party assessments of benefits and risks, especially when drug candidates move from the preclinical setting to human clinical trials. Such a mechanism could address real and perceived biases that may color industry assessment of risk and benefit. Third, harmonization of global regulatory expectations is also a critical need, as industry files for regulatory approvals around the world. Fourth, industry needs researchers and regulators who are trained to make both science- and judgment-based decisions. This is a particular concern today because of the increasing number of experienced regulators and researchers—individuals who have seen multiple iterations of problems and issues and who understand not just the rules but potential exceptions to those rules that they have uncovered over the course of their careers—who are retiring or leaving the industry because of consolidations and mergers. Dahlem emphasized that there is a critical need to capture the experiences of these individuals and to convey, perhaps using information technology–based tools, their experiences to new generations of researchers and regulators. Fifth, there is a huge need to better educate the public about risks and benefits to reduce public misperceptions about medications and vaccines.
Dahlem also listed specific scientific challenges that could be addressed through collaborative regulatory science initiatives:
• Scientists who are trained to understand the preclinical-to-clinical transition. In particular, he said, funding needs to be restored for training scientists in the classical disciplines of whole-animal pharmacology and physiology.
• Toxicology advances to provide better predictions of human clinical outcomes. This will involve better selection of animal models to ensure human safety, new methods to provide better correlations between whole-animal studies and human disease outcomes, and improved in silico modeling.
• In the clinical area, industry needs better clinical trial designs as well as an improved understanding of clinical trial data, particularly of the placebo effect. The biological origins of phenotypic responses need to be better understood, as do the effects of drugs in special populations.
The magnitude of change that has occurred in medicine over the past century is staggering, Snyderman observed. The first major transformation occurred when science brought to medicine the understanding that at least some diseases could be defined as being caused by specific agents such as particular types of bacteria. This mechanism-based understanding of disease led to what he called the “find and fix it” philosophy that currently guides medicine. Today, the application of genomics, proteomics, metabolomics, systems biology, informatics, and micro- and nanoprocessing is creating a “predict it and personalize it” philosophy, one guided by understanding the complexity of both health and disease.
Also transformational has been the growing understanding that all diseases develop over time. Some develop rapidly, but the major chronic diseases typically develop over a long period of time. For those diseases, intervention typically occurs late, when the underlying pathology makes reversibility difficult and therapy expensive. Today, new technologies are seeking to enable earlier interventions, before primary symptoms develop. For example, genomics provides an understanding of baseline risks through the interaction of biology with the environment. This new knowledge will lead to new products that will require a new way of looking at regulation. FDA’s strategic plan for regulatory
________________
7This section is based on the presentation by Ralph Snyderman, Chancellor Emeritus, Duke University.
science is timely because it seeks to develop the regulatory science that will be needed to deal with these new technologies and products, noted Snyderman.
Snyderman reinforced the observation that, whereas 20 years ago academic medical centers were focused largely on basic discovery work, today scientists at these institutions participate in the entire range of drug discovery and development activities. Moving forward, he said, these activities will include regulatory science and the development of new tools for measuring the safety and efficacy of not only drugs but also the accompanying diagnostics that are being developed.
Snyderman analogized the development of a discipline of regulatory science to efforts to advance translational research and the conduct of clinical trials within academic medical centers. There is now a need to approach the regulatory sciences in a concerted, organized way, to define the discipline and competencies associated with its conduct, and to define it as an innovative science that is a valid career path for young scientists.
Snyderman listed the following as necessary elements in the fostering of the regulatory scientist-investigator:
• Recognition and role models within the academic institution
• Training and research training programs
• Appropriate tenure tracks
• Research infrastructure where scientists reside
• Means of support, including federal funding
• Respect of their colleagues—regulatory science being viewed as “worthwhile”
Snyderman made the following recommendations for the development and advancement of regulatory science:
• Recognize it as a discipline
• Define the discipline
• Define the qualifications
• Define educational needs
• Create academic homes and promotion/tenure tracks
Sigal reminded the workshop participants that the ultimate responsibility of regulatory science is to meet the needs of the patient. Among
________________
8This section is based on remarks given by Ellen Sigal, Chair and Founder, Friends of Cancer Research.
the questions patients ask are the following: Will this new drug work for me? What are my risks? Sigal also noted that, until recently, FDA was not perceived to be a science-based agency. It had the burden of approving drugs and diagnostics, but it did not have the ability to do the science needed to guide decision making for the benefit of patients, which was instead assumed to be the province of NIH.
Sigal remarked that a greater sense of urgency is needed to reinforce that regulatory science is crucial to delivering therapies for patients. She remarked that the agency has not had sufficient resources to accomplish the science needs and added that the resources that are needed to advance regulatory science include not just a budget line item but also a supportive scientific ecosystem to continue the collaborative advancements, notwithstanding a heavily resource-constrained environment.
In a panel discussion, the keynote speakers and audience members identified what they saw as the key needs for strengthening regulatory science. The discussion included individual observations about the principal barriers as well as the most promising opportunities. This section provides an integrated summary of their remarks and discussions with workshop participants during the panel and should not be construed as reflecting consensus or endorsement by the workshop participants, the planning committee, the Forum, or the National Academies:
• Among the biggest barriers to advancement of the science is a shortage of experienced regulatory scientists and the lack of experience among the scientists now being trained.
• A key element of the practice of regulatory science is an understanding of societal and personal tolerance for risk and how society and individuals experience the benefits of new drugs and technologies. Participants called for a more developed approach to benefit-risk assessment that takes patient perspectives into account.
• It is important for FDA scientists to be able to participate as scientists in academic settings or at NIH. It also is important that academic and NIH scientists be allowed and encouraged to work at FDA.
• Having an academic culture that views regulatory science as an inherently collaborative enterprise and that recognizes the need for development of people in academia to contribute to the science would contribute greatly to development of the workforce.
• Safe harbors could support and protect collaborations not only among the federal government and academia but also including the pharmaceutical industry and patients.
• Although progress has been made in providing opportunities for continuing formal interactions between industry and FDA during the drug development process, more work is needed to be done to enhance and increase access to resources at FDA, including increased informal interactions with FDA scientists and staff.
• Some participants called for definition as to what each sector (academia, industry, the agencies) needs to do to improve and enhance the support and practice of regulatory science.
• A next–level, precise definition of regulatory science—and the components of its practice—would spur academic medical centers to create programs in this area. This exercise would include identification of specific regulatory science competencies and definition of collaborative mechanisms established to connect academic training programs to relevant programs at NIH and FDA.














