
This chapter explains what steps are recommended after discovery and confirmation of a candidate omics-based test (as described in Chapter 2) and before conducting studies to assess the test for clinical utility and use (as described in Chapter 4). Figure 3-1 highlights omics-based test validation, the second component of the committee’s recommended omics-based test development and evaluation process. The committee defines an omics-based test as consisting of both the data-generating assay and the fully specified computational procedures used for analysis of the assay data. Both components of omics-based tests used to direct patient management in a clinical trial setting should be validated during the test validation phase using several steps (Recommendation 2). The optimal validation process for translation of the candidate omics-based test from the discovery phase (see Recommendation 1, Chapter 2) into a validated test ready for use in a clinical trial setting is the same validation process used for other new tests performed in clinical laboratories.
This chapter references the current standards used for regulation of clinical laboratories and clinical tests, specifically the Clinical Laboratory Standards defined under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) and the Food and Drug Administration (FDA) regulatory standards for devices. Issues are noted for both of these standards. Under CLIA, which was designed, enacted, and implemented from 1988 to 1992, with updates over the years, the current regulations do not specify the mechanisms for validation of a test in a clinical laboratory (referred to as laboratory-developed tests or LDTs) and do not have specific requirements for omics-based tests. However, CLIA is a minimal standard for the operation
of a clinical laboratory, which is more stringent and provides a baseline level of quality assurance compared to a research laboratory setting. A report by the Secretary’s Advisory Committee on Genetics, Health, and Society in 2008 delineated concerns with the current CLIA standards and oversight of clinical laboratories and made recommendations for improving the oversight of clinical laboratories (SACGS, 2008). The second standard referenced in this chapter is oversight by FDA. FDA has not enforced its authority for oversight of LDTs, which is one pathway for the translation of an omics discovery into a clinical test, and FDA has not yet defined its regulatory approach to oversight of omics-based tests, especially when developed as an LDT. Implementation of the committee’s recommendation could lead to an increased workload for FDA, with an impact on the finite resources of FDA. This issue is not addressed by this report. FDA’s responsibility for assuring the safety and effectiveness of any medical device, including omics-based tests, does not require demonstration of clinical utility, which is a standard required by payers. However, FDA is the federal regulatory agency with authority for oversight of testing and should be seen as a partner in the test validation phase. Therefore, the committee uses the CLIA and FDA regulatory standards as a minimum for assuring the quality of a test prior to use in a clinical trial to direct patient management.
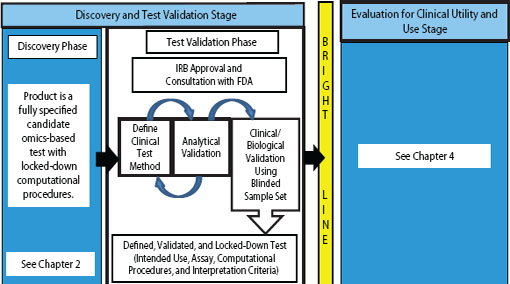
FIGURE 3-1 Omics-based test development process, highlighting the test validation phase. The first stage of omics-based test development is comprised of two phases: discovery and test validation. In the test validation phase, the omics-based test undergoes analytical and clinical/biological validation. Statistics and bioinformatics validation occurs throughout the discovery and test validation stage as well as the stage for evaluation of clinical utility and use. The bright line signifies the point in test development where a fully defined, validated, and locked-down clinical test method is necessary. Changes to the test after the bright line is crossed require a return to the test validation phase, approval by the Institutional Review Board (IRB), and possibly consultation with the Food and Drug Administration (FDA).
In addition to federal oversight of clinical laboratories and clinical tests, several professional societies also develop guidelines and best practices for clinical laboratories. The College of American Pathologists (CAP) has laboratory accreditation standards that meet and exceed the CLIA standards, sets practice standards, and provides proficiency testing samples for molecular pathology clinical laboratories and other types of clinical laboratories. A second professional society that works to establish best practices for molecular laboratories is the Association for Molecular Pathology (founded in the 1995). A third society is the American College of Medical Genetics, which also develops and publishes guidelines and best practices in molecular genetic testing.
Two primary routes are used to develop omics-based tests intended for clinical use. The first is partnership with industry to develop the test for commercial production of test kits with clearance or approval as a device through FDA or proprietary performance of the test by the company within a specific CLIA-certified clinical laboratory. The second is partnership with a commercial or local academic CLIA-certified laboratory to validate the discovery for performance as an LDT in the CLIA-certified laboratory. The committee’s recommendations apply to both pathways. Regardless of the route pursued, the committee recommends that the candidate omics-based test and its intended use should be discussed with FDA prior to initiation of validation studies (Recommendation 2a). As discussed in Chapter 4, if a test will be used to direct patient management in a clinicaltrial,
then an investigational device exemption1 (IDE) application must be filed with FDA (Gutierrez, 2011) (see Recommendation 3b), then the test should be validated in a CLIA-certified clinical laboratory, and the clinical trial that uses the test should be approved by an Institutional Review Board. If the clinical trial will not use the results of the omics-based test to direct patient management, then an IDE is not required and the testing could be performed in a research laboratory setting. However, if the ultimate goal of the clinical study of the new test is to produce evidence to support use of the omics-based test in medical care, then consulting regulatory authorities, such as FDA, for guidance in the appropriate validation and clinical performance of the omics-based test is recommended to ensure complete assessment of the performance of the test before evaluation for clinical use. It is never too early to confer with FDA about the omics-based test development process, and the agreements at a pre-IDE meeting may be crucial to the development process because they will provide a degree of certainty about subsequent test development steps and data collection.
An abbreviated description of the process for test validation in a clinical laboratory is provided in this chapter and serves as the committee’s recommended best practices for translation of the candidate omics-based test into a test suitable for use in a clinical trial. Test validation includes steps to confirm the performance characteristics of the assay method, the fully specified and locked-down computational procedures from the discovery phase (defined in Chapter 2 as the series of computational steps performed in processing the raw data, as well as the mathematical formula or formulas used to convert the data into a prediction of the phenotype of interest), and all assay data handling methods required to reach a final test result.
Translation of a candidate omics-based test from the discovery phase into a clinical test requires confirmation of the research findings using a defined assay and fully specified computational procedures performed in the setting of a clinical laboratory certified under CLIA through the Centers for Medicare & Medicaid Services (CMS) or an accreditation organization with deeming authority under CLIA. This step is necessary whether the test will be developed into an in vitro diagnostic test kit with approval or clearance by FDA, or performed as an LDT within a CLIA-certified laboratory. The CLIA regulations were established in 1988 to ensure that minimum personnel and quality requirements were followed in all clinical laboratories.
1 An FDA designation that allows an investigational device to be used in a clinical study to collect safety and effectiveness data supporting a pre-market approval application or a pre-market notification submission.
Today, compliance with CLIA regulations is required for any laboratory performing testing on human specimens for clinical care, especially when seeking payment under the Medicare and Medicaid programs (CDC, 2004). If the laboratory does not report patient-specific results for the diagnosis, prevention, or treatment of a disease or for the assessment of the health of individual patients, then the laboratory does not need to comply with CLIA requirements when testing human specimens. However, if any test result, including an omics-based test result, is used for patient care, even in the setting of a clinical trial, the test must be performed in a CLIA-certified laboratory and must meet all the regulatory requirements for performance of a clinical test under CLIA regulations. These regulations state that a clinical laboratory must validate the test to define the test method and the performance characteristics of the test, and then the validated test must be performed and regularly assessed using specific quality standards. Validation of the omics-based test in a CLIA-certified laboratory setting before use for patient management decisions in a clinical trial is necessary to ensure that clinical quality standards are used for the test validation and performance to minimize the potential for patient harm.
An assessment for clinical utility and use of an omics-based test can be accomplished through clinical trial designs that do not use the test results to direct patient management, which would allow the test to be performed in a research laboratory setting (see Chapter 4, Evaluation for Clinical Utility and Use Stage). However, if the goal is to generate data to support the clinical use of the omics-based test, performance of the test in compliance with CLIA regulations in a CLIA-certified laboratory would ensure proper definition of the test and its performance characteristics, and facilitate later transition of the test into clinical use following completion of the clinical trial. Therefore, the best practice for use of an omics-based test in a clinical trial setting, regardless of the trial design, is performance of the test in compliance with CLIA regulations in a CLIA-certified clinical laboratory.
This chapter will briefly describe the process of test validation and performance under CLIA regulations, with a focus on important overarching concepts underpinning the validation steps; the specific details of the process will vary considerably depending on the test method and the intended use. There are detailed guidelines, guidance documents, and published reviews providing extensive detail on validation in various scenarios, as cited in this chapter. Description of the CLIA-certified laboratory processes for test validation and performance will assist investigators at the discovery phase and clinical trial stage of omics-based test development research to understand the requirements for clinical test performance in preparation for assessment of the new omics-based test in a clinical trial setting (Chapter 4).
Considerations Prior to Analytical and Clinical/Biological Validation
The committee recommends that test validation should be performed in a CLIA-certified clinical laboratory, beginning with a defined candidate omics-based test from the discovery phase (Recommendation 2b). Ultimately, the goal of the investigator is to generate high-quality evidence that warrants assessment of the test for its intended clinical use. Therefore, the candidate omics-based test from the discovery phase should be converted into an analytically validated and clinically practical test. Once an omics-based discovery is defined and confirmed using an independent, blinded set of samples in the research setting (see Chapter 2), investigators can work with the director and staff of a CLIA-certified laboratory to transition the test to the clinical laboratory setting in preparation for studies to evaluate the test for clinical use. The investigators should be able to provide the clinical laboratory with the defined assay method and the fully specified and locked-down computational procedures used to interpret the omics measurements in the research setting. In addition, the investigators should work with the clinical laboratory director and staff to define the intended use and performance requirements of the test, including the purpose of the test (e.g., risk assessment, screening, diagnosis, prognosis, predicting response to therapy, or monitoring); the patient population to be tested (e.g., stage II colon cancer patients, low-birth-weight infants less than 6 months of age, etc.); the specimen type (e.g., blood, urine, fresh tumor tissue, paraffin-embedded formalin-fixed tumor tissue, etc.); the specimen handling requirements (e.g., immediately frozen tissue for RNA-based testing, separation of serum from blood cells within 4 hours of collection for viral load testing, etc.); required or ideal test performance characteristics (e.g., sensitivity, accuracy, specificity, etc.); and the required turnaround time for test results based on clinical decision requirements (e.g., within 1 hour of specimen collection, same day, within 1 week).
Specimens comparable to those on which the omics-based test will eventually be applied in the clinic should be identified for the analytical and clinical/biological validation of the test. Obtaining appropriate specimens can take a significant amount of time and can delay the validation process if not initiated early. Availability of appropriate specimens is essential for the successful analytical and clinical/biological validation of a test; strategies for sharing of available specimen sets are essential because this is a barrier identified by test developers. Specimens with defined characteristics that provide a range of test results (e.g., positive and negative results, or a distribution across a quantitative range of results, etc.) are needed for the analytical validation of the test. This validation requires a large volume
of a small number of specimens representing the range of expected test results. The large volume of these specimens is needed for the repeated testing of the same specimens as the test method is optimized and defined, and for repeated testing to establish the technical variability of the test when performed on specimens handled in different ways prior to testing, and as the test is performed at different times, by different technologists, or on different instruments. Clinical specimens are optimal for analytical validation, but in some cases commercial sources of control analytes may be spiked into negative specimens of the same type that will eventually be used for the test.
A set of clinical or biological specimens in sufficient numbers to provide statistical assessment of the test’s performance characteristics is needed for the clinical/biological validation of the test. While a specific number of specimens cannot be generically defined for all tests, the New York State Department of Health has defined a minimum of 30 positive and 10 negative samples of each specimen type plus all controls used for infectious disease tests using amplification methods (Wadsworth Center, 2009). Clearly, the correct number of specimens depends on the complexity of the test itself, the range of expected results, the range of specimen types, and other factors. Specimens should come from patient populations similar to those on which the test will be used clinically, and should be available in sufficient volume to allow for several repetitions of the test. Sources of specimens include biobank specimens with related clinical information, residual specimens in clinical laboratories, or commercially available cell lines or pathogens with known characteristics that could be spiked into the relevant clinical specimen type. Ideally, a new set of specimens not used in the discovery phase should be used for the analytical and/or clinical/ biological validation of the test in the CLIA-certified laboratory setting, although in some cases, that may not be possible.
Although a candidate omics-based test will have been developed and confirmed in the research setting using certain instruments and assay methods, those same methods may not be appropriate in the clinical laboratory setting based on the clinical requirements for the test. Clinical laboratories prefer to use existing instruments and methods familiar to the laboratory staff for financial and workflow reasons. When the clinical laboratory does not have appropriate instruments for a new omics-based test, either a new method can be developed and performed on instruments already available in the clinical laboratory, the appropriate instruments can be purchased for the clinical laboratory, or the research instruments can be brought under CLIA standards and used by the clinical laboratory for test validation and eventual performance of the test in a clinical trial setting. The financial support for the translation of an omics-based test into the clinical laboratory setting in preparation for clinical trials should be considered when seeking
research support for either the omics discovery phase or the clinical trial phase of the translation pathway, if the test will be validated in the academic setting. If the test is validated through an industry partnership, the discovery should be validated as a clinical test by the company prior to use of the test in a clinical trial setting.
The committee recommends that the CLIA-certified laboratory should design, optimize, validate, and implement the omics-based test under current clinical laboratory standards (Recommendation 2c). The clinical laboratory, in collaboration with the discovery investigators, should design the clinical test methods based on the intended use and clinical requirements for the test. While the use of a test may change once it reaches clinical implementation, the intended use of the candidate test should be defined prior to the test validation process to assure that the proper types of specimens are used for test validation. Using control analytes and clinical or biological specimens relevant to the intended use of the test, the initial test methods will be assessed for their performance characteristics, and adjusted as needed to achieve predefined required performance characteristics. The defined test method should include the fully specified computational procedures that were locked down at the end of the discovery phase, which will be used for analysis of the assay results to yield the final test result that is used as the interpretive criteria for the omics-based test. Once the test method, including the interpretive criteria, is established and documented, the methods can be assessed for their analytical and clinical performance characteristics in a test validation process.
Analytical and Clinical/Biological Validation
The test validation process includes the steps used to establish the analytical and clinical performance characteristics and the test limitations of a test prior to clinical use (Jennings et al., 2009; Mattocks et al., 2010). Test validation is defined under ISO 9000:20052 to mean “confirmation, through the provision of objective evidence, that the requirements for a specific intended use or application have been fulfilled,” which has been interpreted as “doing the correct test.” This is in contrast to test verification, which is defined under ISO 9000:2005 to mean “confirmation, through the provision of objective evidence, that specified requirements have been fulfilled,” which has been interpreted as “doing the test correctly” (Jennings et al., 2009; Mattocks et al., 2010). This distinction is critical in the validation of a new clinical test because the intended use of the test should be defined
2 These standards are published by the International Organization for Standardization (ISO). ISO 9000:2005 provides information on the fundamentals and vocabulary used in quality management systems.
at a very early stage of test development to ensure that the proper specimen types from appropriate patients with specific clinical characteristics are used for the test validation before clinical use of the new test.
The test validation process has been defined for molecular tests (Jennings et al., 2009; Mattocks et al., 2010), but not for complex omics-based tests, which are based on large datasets that must be interpreted using a computational model. All aspects of the test validation process should be applied not only to the assay that generates the data, but also to the fully specified computational procedures used to analyze the data, assuring the analytical and clinical performance of both aspects of the complete test. For omics-based tests, the usual interpretive criteria used to produce a final test result will generally be replaced by these computational procedures, although the specific cutoff values and potentially definition of indeterminate results may need to be defined as the test method is developed within the CLIA-certified laboratory. Finally, for test validation processes that warrant publication because of the novel or new aspects of the test, the appropriate guidelines for reporting of biomarker study results should be considered at the planning phase of the test validation process and be used to ensure consistency in the reporting of the test validation process results (Surinova et al., 2011). As described in Appendix D, several reporting guidelines have been developed for different types of biomarker research, or different aspects of the test development process, including BRISQ (Moore et al., 2011), REMARK (Altman et al., 2012a,b; McShane et al., 2005, 2012), and STARD (Bossuyt et al., 2003).
Analytical Validation
The test method should be optimized to achieve an analytically validated test, including the assay and the fully specified computational procedures. Once the test methods have been defined and have achieved the predefined performance characteristics for the test, the next step should be rigorous testing of the analytical performance of the test. This analytical validation identifies and quantifies the technical variations of a test performed on patient specimens. The test parameters for analytical validation are extensive and vary depending on the purpose of the test, such as qualitative or quantitative, and include, for example, accuracy, precision, reproducibility, linearity, reportable range, analytical sensitivity and specificity, and limit of detection. Analytical validation is performed using specimens comparable to the patient specimens on which the test will eventually be used, with known or expected characteristics related to the test being validated. If necessary, an alternative to using a limited supply of valuable clinical specimens with known or expected test results is the use of control materials that will provide known or expected test results and can be spiked into negative clinical specimens of the same type to be used
for the test. Different amounts of the control material can be introduced into the negative specimens to assess the linearity, sensitivity, cut-off values, and other performance characteristics of the test during analytical validation of the test.
The different analytical performance characteristics to be established depend on the purpose of the test. The types of performance characteristics to be established during analytical validation of a test are described by Jennings et al. (2009). For an omics-based test, the precisely defined computational procedures used for interpretation of the assay results must be included in the validation process to ensure that it performs well on the assay results yielded by the test method in the clinical laboratory. Also, all data management steps and processes should be defined and tested to ensure that no errors are introduced at the data transfer or interpretation steps of the test.
Clinical/Biological Validation
The optimized test should be evaluated for clinical/biological validity using an independent specimen set, consistent with the intended use for the test. The expected or previously determined test results for the validation set of specimens and clinical information about the patients from which the specimens were derived should not be available to the individuals performing and interpreting the test results, to reduce the chance that results will be affected by conscious or unconscious bias. This process of blocking this information from the tester is called blinding of the specimens. Clinical/ biological validation provides evidence that the test results are linked to disease states, and is guided by the intended use of the test. Clinical/biological validation includes but is not limited to reference range, clinical sensitivity, and clinical specificity (Jennings et al., 2009). Clinical/biological validation is performed using specimens comparable to the patient specimens on which the test will eventually be used, and a sufficient number of specimens to allow statistical assessment of the test performance in relation to the specimen characteristics. If clinical specimens are not available, then appropriate biological specimens, such as cell lines approximating the eventual clinical specimens can be used, but this is not ideal because the clinical specimens will introduce variability not seen in the biological specimens. The number of specimens needed for the clinical/biological validation of a test depends on many factors, including whether a comparative test is available for comparison of results, the test performance characteristics required for clinical use (e.g., accuracy, sensitivity, etc.), the prevalence of the disease, and the confidence level required for good medical practice (Jennings et al., 2009).
As noted above, the identity and expected results of the specimens used for clinical/biological validation of the omics-based test should be
blinded to the individuals performing and interpreting the test results during clinical/biological validation of the test. Generating the results in a blinded fashion will reduce bias during the clinical/biological validation and improve the odds of successful assessment of clinical use when the test is applied to specimens collected in prospective trials or is used to direct patient management in a clinical trial (see Chapter 4). If specimens from the omics discovery phase are used for the clinical/biological validation of the test, then the specimens can be provided in tubes labeled numerically, while maintaining a master key to the identity of the specimens. Once results are completed, the key can be used to compare the test results to results obtained during the discovery research phase, or to the clinical outcomes of the patients from whom the specimens were derived. Again, the use of specimens from the discovery phase is not optimal, because it does not provide an independent assessment of the performance of the test.
PREPARATION FOR INVESTIGATIONAL USE OF THE VALIDATED TEST
All requirements for the performance of clinical tests in a CLIA-certified laboratory should be met for the defined and validated omics-based test. The final step of test validation is the implementation of the new test in the workflow and quality management system of the CLIA-certified laboratory. This final step in the test validation process is essential for assuring that the test is performed under a quality management system designed to comply with best practices for clinical laboratories. Test implementation requires a written standard operating procedure (SOP) for the test that describes the specimens and specimen handling requirements, assay methods, data handling procedures, fully specified computational procedures used to interpret the assay results in order to yield clinically actionable results, and methods for reporting results. The SOP also should describe the procedures for quality control and quality assurance, including issues related to the preanalytical, analytical, and postanalytical aspects of testing; quality indicators to be monitored such as turnaround time, documentation, and investigation of test failures; and trends in test results, as well as ongoing calibration processes for the instruments and the test. The SOP also is used for training technologists who will perform the test, with documentation of the success of initial training and of continued competency of all personnel performing the test. Because proficiency testing programs for the new test almost certainly will not be available, procedures internal to the laboratory can be used for ongoing assessment of the quality of the test performance. If the test will be used in a clinical trial setting, mechanisms for reporting of the test results with delivery to the clinical team should be defined and tested.
The committee recommends that if the omics-based test will be performed
in more than one CLIA-certified laboratory for a clinical trial, analytical validation and CLIA requirements for the same omics-based test should be met by each laboratory, working with the primary laboratory (Recommendation 2d). The test validation process described above covers aspects of test validation in a single laboratory, but clinical trials often are performed at multiple clinical sites. Sometimes testing still can be performed in a single laboratory, even with multiple clinical trial sites, if the specimen handling requirements and the turnaround times required for clinical care can be accommodated with specimen transport to the single laboratory. If testing is to be performed in laboratories at each clinical site, then each laboratory must complete a test validation process, and a plan to compare findings from the different laboratories should be defined. However, if the test is validated as described above as a new test by one laboratory, then the other laboratories can use the test methods, specimens, and test results available from the first laboratory to complete a more abbreviated test validation process, and use the primary laboratory’s SOP and quality control and assurance procedures with modifications as required by each laboratory site. If different test methods are used by the laboratories at different sites, then a full test validation process as described in this chapter should be completed by each laboratory. When different laboratories are used, comparison of the test results between sites should be included in the study design to assess bias introduced by laboratory differences.
FUNDING FOR VALIDATION OF A CANDIDATE OMICS-BASED TEST IN A CLIA-CERTIFIED CLINICAL LABORATORY
Validation of a candidate omics-based test in a CLIA-certified clinical laboratory requires funding for the reagents, supplies, controls, and personnel time, as well as submission of the IRB protocol for access to and use of appropriate clinical specimens, and potentially for new instrumentation required for the selected test method. For academic investigators, two options are available for completing the test validation phase of the translational pathway; either partnering with industry through the technology transfer offices of academic institutions or continuing the development within the academic environment. While the discovery phase of omics-based test development (described in Chapter 2) is routinely funded by the National Institutes of Health (NIH), grant funding historically has not been available for validation of a candidate omics-based test in a CLIA-certified laboratory (described in this chapter) prior to assessment in a prospective clinical trial (described in Chapter 4). In a clinical setting, the costs of validation of a new clinical test for immediate clinical use are supported by the clinical funding for the laboratory in anticipation of clinical billing and reimbursement for the test; however, the costs of validation of a candidate
omics-based test in preparation for assessment in a clinical trial will not be supported by the clinical funding. As an alternative to a CLIA-certified laboratory in an academic medical center, commercial clinical research organizations are available for development and validation of a candidate omics-based test in a CLIA-certified laboratory setting, but also will require financial support. Investigators need to include this phase of test development and validation in their grant proposals, and as recommended in Chapter 5, funding agencies should provide appropriate support for this phase of test development that addresses analytical and clinical/biological validation, in anticipation of assessment of the test in a prospective clinical trial to direct patient management.
Any test that is used for patient management decisions, whether in a clinical trial or patient care setting, should be performed in a clinical laboratory certified under CLIA through CMS or an accreditation organization with deeming authority under CLIA. CLIA certification ensures that the clinical laboratory operates under standards for quality control, quality assurance, and quality management. Validation and performance of a test in a CLIA-certified laboratory provides an overall quality management system that ensures best clinical laboratory practices are used and reduces the risk for patient harm due to improper test validation or performance. At the end of the test development phase described in this chapter, the clinical test method should be locked down (fully defined and not changed in the next development stage), just as the computational procedures were locked down in the discovery phase in Chapter 2.
RECOMMENDATION 2: Test Validation Phase
An omics-based test consists of both the data-generating assay and the fully specified computational procedures used for analysis of the assay data. Both components of omics-based tests used to direct patient management in a clinical trial setting should be validated during the test validation phase using the following steps:
a. The candidate omics-based test and its intended use should be discussed with FDA prior to initiation of validation studies.
b. Test validation should be performed in a CLIA-certified clinical laboratory, beginning with a defined candidate omics-based test concept from the discovery phase.
c. The CLIA-certified laboratory should design, optimize, validate, and implement the omics-based test under current clinical laboratory standards.
d. If the omics-based test will be performed in more than one CLIA-certified laboratory for a clinical trial, analytical validation and CLIA requirements for the same omics-based test should be met by each laboratory, working with the primary laboratory.
Altman, D. G., L. M. McShane, W. Sauerbrei, and S. E. Taube. 2012a. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. BMC Medicine 10:51.
Altman, D. G., L. M. McShane, W. Sauerbrei, and S. E. Taube. 2012b. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. PLoS Medicine 9(5):e1001216. doi:10.1371/journal.pmed.1001216.
Bossuyt, P. M., J. B. Reitsma, D. E. Bruns, C. A. Gatsonis, P. P. Glasziou, L. M. Irwig, J. G. Lijmer, D. Moher, D. Rennie, H. C. de Vet, and Standards for Reporting of Diagnostic Accuracy. 2003. Towards complete and accurate reporting of studies of diagnostic accuracy: The STARD initiative. Standards for Reporting of Diagnostic Accuracy. Clinical Chemistry 49(1):1-6.
CDC (Centers for Disease Control and Prevention). 2004. Subpart A—General Provisions. http://wwwn.cdc.gov/clia/regs/subpart_a.aspx#493.3 (accessed October 13, 2011).
Gutierrez, A. 2011. Discussion with the Science and Technology Working Group of the IOM Committee to Review Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials, Conference Call with Alberto Gutierrez, Washington, DC, August 19.
Jennings, L., V. M. Van Deerlin, and M. L. Gulley. 2009. Recommended principles and practices for validating clinical molecular pathology tests. Archives of Pathology and Laboratory Medicine 133(5):743-755.
Mattocks, C. J., M. A. Morris, G. Matthijs, E. Swinnen, A. Corveleyn, E. Dequeker, C. R. Muller, V. Pratt, A. Wallace, and EuroGentest Validation Group. 2010. A standardized framework for the validation and verification of clinical molecular genetic tests. European Journal of Human Genetics 18(12):1276-1288.
McShane, L. M. 2012. Statistical challenges in the development and evaluation of marker-based clinical tests. BMC Medicine 10:52.
McShane, L. M., D. G. Altman, W. Sauerbrei, S. E. Taube, M. Gion, and G. M. Clark. 2005. Reporting recommendations for tumor marker prognostic studies (REMARK). Journal of the National Cancer Institute 97(16):1180-1184.
Moore, H. M., A. B. Kelly, S. D. Jewell, L. M. McShane, D. P. Clark, R. Greenspan, D. F. Hayes, P. Hainaut, P. Kim, E. A. Mansfield, O. Potapova, P. Riegman, Y. Rubinstein, E. Seijo, S. Somiari, P. Watson, H.-U. Weier, C. Zhu, and J. Vaught. 2011. Biospecimen reporting for improved study quality (BRISQ). Cancer Cytopathology 119(2):92-102.
SACGHS (Secretary’s Advisory Committee on Genetics, Health, and Society). 2008. U.S. System of Oversight of Genetic Testing: A Response to the Charge of the Secretary of Health and Human Services; a Report of the Secretary’s Advisory Committee on Genetics, Health, and Society. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_oversight_report.pdf.
Surinova, S., R. Schiess, R. Huttenhain, G. Cerciello, B. Wollscheid, and R. Aebersold. 2011. On the development of plasma protein biomarkers. Journal of Proteome Research 10(1):5-16.
Wadsworth Center. 2009. Submission guidelines for nucleic acid amplification tests for infectious agents, Rev 7/1/2009. http://www.wadsworth.org/labcert/TestApproval/forms/proposedguidelines.pdf (accessed February 2, 2012).














