
Outcomes of Written Requests,
Requirements, Studies, and
Labeling Changes
One measure of the accomplishments that have been achieved under the Best Pharmaceuticals with Children Act (BPCA) and the Pediatric Research Equity Act (PREA) is simply the number of labeling changes attributed to these policies since they or their predecessor policies went into effect. From July 1, 1998, through October 25, 2011, the Food and Drug Administration (FDA) approved 425 labeling changes attributed to studies or analyses requested under BPCA or required under PREA.1 FDA attributed approximately half (54 percent) of the changes to studies required under PREA and approximately one third (35 percent) to studies requested under BPCA; the remaining changes (11 percent) were attributed to both laws.2 Almost 10 percent (n = 39) of the changes were not based on data from new pediatric studies. For example, for a 2009 change in pediatric dosing for zidovudine (Retrovir) for the treatment of HIV infection, FDA approved the change on the basis of the sponsor’s reanalysis of existing data (Alivisatos, 2008).
![]()
1 One further labeling change was posted for December 2011. As of the end of January 2012, FDA indicated that eight more changes for 2011 were yet to be posted (personal communication, Catherine Lee, Office of Pediatric Therapeutics, FDA, January 27, 2012). The first labeling change in FDA’s listing (February 10, 1998, for naratriptan [Amerge]) is attributed to the 1994 Pediatric Rule (personal communication, Robert Nelson, Office of Pediatric Therapeutics, FDA, January 10, 2011).
2 Some products have had more than one labeling change (e.g., for different indications or additional pediatric age groups). A labeling change can involve either the addition of pediatric information to the existing label for a previously approved product or the new labeling of a product not previously approved.
As described in Appendix A, FDA’s listings of labeling changes related to BCPA and PREA (and their predecessor policies) are not complete. Specifically, they do not include changes for biologics that were made prior to September 27, 2007. FDA could not supply the committee with the missing information. Thus, the list provided to the committee understates to an unknown extent the number of labeling changes made as a result of studies of biologics that were required under PREA or the Pediatric Rule.
Figure 7-1 shows the time trend of labeling changes attributed by FDA to BPCA and PREA through October 25, 2011. From 1998 through 2004, the general pattern is one of yearly increases in the number of changes
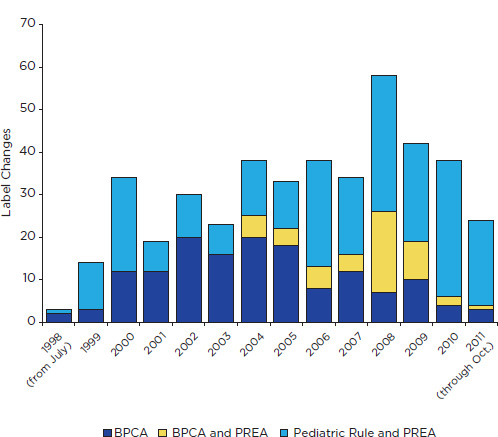
FIGURE 7-1 Changes in drug labeling associated with BPCA, PREA (including the Pediatric Rule), or both, July 1998 through October 2011. The figure excludes changes for biologics regulated under the Public Health Service Act that were approved before September 27, 2007. It includes changes for some products (e.g., contraceptives) that were excluded from the committee’s analysis as well as one change that is attributed to the 1994 Pediatric Rule.
SOURCE: Compiled from information periodically updated in an Excel file downloadable at http://www.accessdata.fda.gov/scripts/sda/sdNavigation.cfm?sd=labelingdatabase.
attributed to BPCA. Although the pattern after that is uneven, most subsequent years show a decrease in changes attributable to BPCA alone. Since 2005, pediatric studies required under PREA have accounted for most labeling changes. Some of these changes are for products studied from the outset in at least one pediatric age group.
A few labeling changes that are attributed to PREA might be more appropriately linked to other policies. One such policy is FDA’s unapproved drugs initiative (FDA, 2006a). That initiative has led to pediatric studies and the approval of three previously unapproved but long-marketed pancreatic enzyme replacement products for use by children and adults (see, e.g., Giuliano et al., 2011). When it approved these products in 2009 and 2010, FDA imposed a deferred PREA requirement for the development of a formulation suitable for the youngest and lowest-weight patients (see, e.g., Beitz, 2009a).
To cite a different example, the labeling of pralidoxime chloride (Protopam) for pediatric use in 2010 (attributed to PREA, with no new studies submitted) might be credited to efforts of the child health advocates and others concerned about children’s emergency access to this treatment for exposure to organophosphate pesticides and chemicals (e.g., nerve agents) (Krug et al., 2011). The drug was originally approved in 1964 and was listed by the National Institute of Child Health and Human Development as a priority for a systematic literature review in 2006 (71 FR 23931). The 2011 to 2016 strategic plan of the Biomedical Advanced Research and Development Authority (a unit within the Department of Health and Human Services) includes “supporting the development of medical countermeasures suitable for use in special populations such as children” (BARDA, 2011, p. 11).
Some pediatric studies conducted and submitted to FDA under BPCA have not yielded labeling changes. With its list of products with labeling changes related to BPCA and PREA, FDA also supplied the committee with a list of 14 active moieties for which requested studies were conducted and exclusivity was granted without information from the studies being added to the label. In addition, it is possible that some requests have led to studies for which FDA neither approved a labeling change nor granted exclusivity. FDA may deny exclusivity if submitted studies do not meet the terms of the written request.
Twelve of the 14 grants of exclusivity without labeling changes were approved before September 2007. For five of these, no information about the study results is posted. For the remaining seven, short summaries are available (consistent with the requirements of BPCA of 2002). Some of these summaries reveal that FDA concluded that no labeling change was necessary because the studies had not demonstrated efficacy but did not raise new safety signals. In one case, a summary reveals FDA’s concern that inclusion of any information from a requested study (of the pharmacoki-
![]()
3 Unless otherwise noted, the data discussed in this section are compiled from FDA sources. The agency lists the moieties for which requests have been made and posts statistics on written
netics of topotecan [Hycamtin]) could be interpreted to imply approval for pediatric use even if the label noted that safety and efficacy had not been established (Hirschfeld, 2003).
In addition, in a presentation to the Institute of Medicine (IOM) committee, FDA staff explained that in the early years of BPCA and PREA (and the Pediatric Rule), pediatric studies were sometimes submitted in sponsor’s annual reports (not as part of a New Drug Application [NDA] or Biologics License Application [BLA]), were not reviewed, and did not lead to labeling changes (Mathis and Jain, 2011). Moreover, the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER) traditionally did not amend labels to reflect efficacy findings that did not support pediatric use. For example, the labeling for fluconazole (Diflucan) still does not note that the product was studied (by request) for the treatment of tinea capitis in children and that the studies found that the product did not work better than an already approved product (griseofulvin) (Mathis and Jain, 2011).
In the Food and Drug Administration Amendments Act of 2007 (FDAAA), Congress required that information from studies conducted under PREA and BPCA be incorporated in product labeling, whether or not the results supported pediatric use or raised new safety signals. Congress also directed that FDA post the clinical, clinical pharmacology, and statistical reviews for these studies. Both actions have increased the value to the public of the studies requested under BPCA or required under PREA.
Nevertheless, two products (bivalirudin [Angiomax] and gatifloxacin [Zymar]) that were granted exclusivity after the passage of FDAAA did not have associated labeling changes. The clinical reviews for these products are posted, but the recommendations on regulatory action and all or part of the risk-benefit assessments are redacted (Ayache, 2009; Nevitt, 2009), making it difficult to assess why no labeling change was made.
The rest of this chapter starts with a discussion of written requests and PREA requirements. Later sections discuss the committee’s assessment of pediatric studies (as reviewed by FDA staff) and labeling changes. (Appendix A discusses how the committee selected its sample.)
WRITTEN REQUESTS AND PREA REQUIREMENTS
Written Requests
Status of Written Requests
By October 2011, FDA had issued 340 written requests since BPCA became effective on July 1, 1998.3 Of these requests, FDA had subse-
quently granted exclusivity for 176 active moieties (and 185 products).4 Thus, roughly half of the written requests issued to date have led to the submission of pediatric studies for which exclusivity was granted, although at least 14 of these did not lead to changes in product labeling. Although FDA does not identify them, some written requests have been declined by sponsors and other requests are still open, with studies planned, under way, or submitted but not yet evaluated. Some sponsors have submitted some of the requested studies, but an exclusivity determination will not be made until all the requested studies are submitted and evaluated. More grants of exclusivity and labeling changes can therefore be expected for previously issued written requests.
Figure 7-2 shows trends in the issuing of written requests and the granting of exclusivity. Written requests peaked in 1999 and then dropped off sharply, with a relative leveling off more recently. Although FDA sometimes issues written requests for studies that are under way or already completed (see discussion of nitric oxide in Chapter 6), studies initiated in response to written requests usually take years to plan, conduct, complete, analyze, and submit. Thus, the peak in grants of exclusivity comes in 2008, several years after the peak for written requests.
The early surge in written requests is not surprising, given that neither incentives nor requirements for pediatric studies had previously been in place and that a substantial number of already approved drugs had not been studied in children (see Table 1-1 in Chapter 1). Once FDA had issued requests for many obvious candidates (e.g., drugs widely used off-label by children, blockbuster drugs with possible pediatric use, and drugs with pediatric studies already planned or under way), a subsequent decline is likewise not surprising. Also, with the passage of time, a reduction in written requests could be expected in part because of the growth in the number of products for which studies had been required under PREA and in part because of the loss of eligibility for popular older products as existing patents or other exclusivity expired.
Despite its declining role, BPCA has continuing value because its incentives are not limited to the indications covered by an application for the approval of a new drug. For example, written requests may take into account advances in knowledge since a determination about required studies of an indication was made under PREA. New data may show that a condition
![]()
requests and exclusivity at http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm049867.htm.
4 Of the 138 earliest written requests that were issued as of September 2000 (Appendix B in FDA, 2001a), FDA had approved labeling changes for 78 (56 percent) of the active moieties by October 2011. Some of these written requests may still be open. For example, in 2010, an FDA advisory committee was asked for its views on the advisability of an amendment to a 2001 written request for studies of the drug sildenafil (Revatio) for treatment of pulmonary arterial hypertension (Temple, 2010).
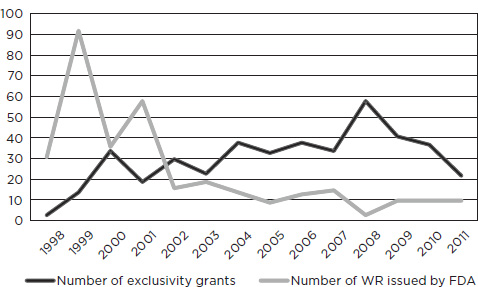
SOURCE: Personal communication, Catherine Lee, Office of Pediatric Therapeutics, FDA, November 3, 2011.
is more common in children than previously believed or new research may suggest a promising new use in children.
Most written requests are proposed by sponsors rather than initiated by FDA, although FDA may significantly alter those proposals. Overall, sponsor proposals are associated with approximately 80 percent of the written requests that FDA had issued as of October 31, 2011. FDA initiated the other 20 percent of requests. By October 2011, FDA had received approximately 700 sponsor proposals for written requests. The high rate of requests related to sponsor proposals may explain why almost 90 percent of written requests issued since January 1, 2008, have been accepted by sponsors (personal communication, Catherine Lee, Office of Pediatric Therapeutics, FDA, November 30, 2011).
Changes in Written Requests
The committee’s sample of 46 FDA actions included 27 products for which written requests were issued, including one product for which the written request covered two indications. Some requests were not amended; others had four or more amendments. The committee (or consultants) also reviewed additional written requests for studies of migraine, hypertension, and gastroesophageal reflux disease (GERD). Most of these additional requests and any amendments dated from the late 1990s to the mid-2000s
and thus were not reviewed by the Pediatric Review Committee (PeRC), as provided for in the reauthorization of BPCA in 2007 (see Chapter 3).
The requests that the committee reviewed generally followed a standard template similar to the one presented in Chapter 3. Depending on the drug and indication, FDA might also have developed a more substantive, product-specific template. It has done so, for example, for studies of pediatric hypertension, migraine in adolescents, and GERD. Box 7-1 summarizes major substantive changes in written requests for studies of migraine. Chapter 6 summarizes the changes in written requests for studies of proton pump inhibitors to treat GERD in neonates and infants, and Appendix E provides a more detailed analysis of changes in written requests for studies of pediatric hypertension.
The substantive details in written requests have tended to become somewhat more specific over time. Changes in the basic request template or in specific requests have often
• added precision (e.g., by specifying a period for safety follow-up or a minimum percentage for age or racial subgroups in a sample);
• required more rigor in trial designs (e.g., by dropping the option for a trial with no placebo and only alternative doses of the test drug or by increasing the statistical power of trials to detect a clinically meaningful effect);
• required more accommodation of developmental variability (e.g., by requiring sponsors to try to develop age-appropriate formulations, if needed); or
• incorporated the legislative requirements for greater public access to information about study results (e.g., by requiring that sponsors submit NDA supplements to add information from trials—whether negative or positive—to the label).
Although FDA’s letters that describe amendments in particular written requests often do not explain the reason for changes, subsequent clinical reviews of submitted studies suggest that some changes have come after a sponsor encountered difficulties with conducting the studies as requested. For example, if studies with an older age group identified serious safety concerns (e.g., as in studies of the anesthetic desflurane), the agency might amend a request to eliminate a study with a younger age group. Similarly, if a sponsor encountered serious difficulties in enrolling children, an amendment might reduce the specified sample size.
Potential of Requests to Generate Useful Information
On the basis of the requests in its sample, the committee concluded that most written requests had reasonable potential to generate useful inform-
Zolmitriptan (Zomig)
The written request for zolmitriptan was issued in March 1999. It specified four studies in adolescents: (1) a short-term safety and tolerability study (sample size not specified), (2) a pharmacokinetic study (sample size not specified), (3) a well-powered efficacy study (sample size not specified), and (4) a long-term safety study with 300 subjects. There were no substantive amendments.
Sumatriptan (Imitrex)
The original written request for sumatriptan was issued in June 1999. It specified three studies: (1) a single-dose pharmacokinetic study with adolescents (sample size not specified), (2) a well-powered efficacy study (sample size not specified), and (3) a long-term safety study with 300 subjects. The amended written request in May 2000 changed the entry criteria in the efficacy and safety studies from subjects with an average of one to six migraines per month (with “migraines” defined by the International Headache Society) to subjects with an average of two or more migraines per month, as requested by the sponsor. The amendments also allowed the sponsor to compare pharmacokinetic results from adolescents with those from historical adult controls.
Almotriptan (Axert)
The original written request for almotriptan was issued in October 2001. It specified three studies: (1) a single-dose pharmacokinetic study with 18 to 50 adolescents, (2) a well-powered efficacy study (sample size not specified), and (3) a long-term safety study with 300 adolescents. The amended written request in February 2005 dropped the request for the pharmacokinetic study and decreased the sample size of the third study to 200 participants. Inclusion criteria for the efficacy trial did not change between the original and the amended request.
Eletriptan (Relpax)
The written request for eletriptan was issued in July 2004. It specified two studies: (1) a well-powered efficacy study (specific sample size not specified) and (2) a long-term safety study with 200 adolescents. There were no amendments. (Note that in August 2005 this written request was declined by the sponsor and was referred to the Foundation of the National Institutes of Health in accord with provisions of BPCA of 2002 [71 FR 6081; see Chapter 3 for a discussion of these provisions].)
![]()
SOURCE: Personal communication, P. Brian Smith, Department of Pediatrics, Duke University Medical Center, June 22, 2011.
tion for clinicians who care for children. These requests include those for studies of drugs that
• had a new mechanism of action compared with existing medications approved for pediatric use (e.g., aripiprazole for schizophrenia) or were improvements over first-generation drugs in the class (e.g., many second-or later-generation antibiotics);
• offered a possible treatment for a serious or life-threatening condition for which few or no treatments had been demonstrated to be safe and effective (e.g., irinotecan hydrochloride for treatment of solid tumors and bisphosphonates for treatment of osteogenesis imperfecta);
• were commonly used off-label with no controlled studies of pharmacokinetics, dosing, safety, or efficacy (e.g., antibiotics for various infections and proton pump inhibitors for GERD);
• lacked important information on safety (e.g., desflurane for the maintenance of anesthesia in nonintubated children);
• would potentially allow safe and effective use of the drug in a new pediatric population if a new formulation was developed (e.g., terbinafine hydrochloride for tinea capitis); or
• had new safety concerns suggested by new data (e.g., remifentanyl hydrochloride for anesthesia).
Some requests had elements that, in the committee’s judgment, could limit the potential of the requested studies to yield strong information to guide the care of children. Box 7-2 provides examples.
To focus on one therapeutic area, details of the written requests for pediatric studies of a number of drugs used to treat hypertension in adults—and the resulting trials—have been criticized for a number of reasons. An analysis by Benjamin and colleagues reached the following conclusions:
Successful studies showed large differences in doses, with little or no overlap between low, medium, and high doses; failed trials used narrow dose ranges with considerable overlap. Successful trials also provided pediatric formulations and used reduction in diastolic, not systolic, blood pressure as the primary endpoint. Careful attention to pediatric pharmacology and selection of primary endpoints can improve trial performance. We found poor dose selection, lack of acknowledgment of differences between adult and pediatric populations, and lack of pediatric formulations to be associated with failures. (Benjamin et al., 2008, p. 834)
The authors have also suggested that for these trials feasibility was more important to the sponsors than strong trial design, particularly since exclusivity can be granted regardless of whether studies demonstrate ef-
Toxicity profile of drug. One request involved a drug (stavudine [Zerit]) with a known toxicity profile that made its use for HIV-exposed or-infected neonates or young infants unlikely given available alternative treatments.
Timing of pharmacokinetic study. For a drug (levalbuterol tartrate [Xopenex HFA]) to treat asthma, a request did not specify that pharmacokinetic studies be performed early enough to guide the efficacy and safety trial.
Failure to request pharmacokinetic study. FDA requested safety and efficacy studies but not a pharmacokinetic study for a drug to treat asthma (albuterol sulfate inhalation aerosol [Ventolin HFA]) in children ages birth up to 2 years and 2 years up to 4 years. The clinical reviewer concluded that the studies did not show efficacy and that the dose chosen for the studies might not have been optimal (Wang, 2008).
Failure to request relevant studies. For a drug (inhaled nitric oxide [INOmax]) to prevent bronchopulmonary dysplasia in newborns, FDA did not request pharmacokinetic data, despite the diverse gestational ages of infants to be studied, and did not specify safety endpoints other than those associated with prematurity.
Need to tailor studies to age groups. As specified in a request for studies of esomeprazole magnesium [Nexium]), a study design with a run-in treatment stage followed by randomized, placebo-controlled withdrawal, although appropriate for older age groups, may not be optimal in a study of the treatment of GERD in infants. If the initial run-in phase effectively heals erosive esophagitis, withdrawal is not likely to show a Significant difference between the placebo treatment and the proton pump inhibitor treatment. An amendment to the written request eliminated the efficacy study, although the drug is widely used by infants.
Selection of endpoints inappropriate for age group. In a study of a drug (salmeterol xinafoate/fluticasone propionate [Advair]) for treatment of asthma in children ages 4 to 11 years, the requested endpoint of forced expiratory volume in 1 second (FEV1) did not adequately recognize the inability of the youngest children to perform the necessary breathing maneuvers.
Insufficient definition or scope of intervention. For a requested study of an anesthetic agent (desflurane [Suprane]), the request specified management by either a face mask or a laryngeal mask airway device, leaving the choice of approach to the anesthesiologist rather than defined as part of the trial design.
ficacy. As outlined in Appendix E, FDA has amended the template for requests for these studies by specifying stronger trial design options, which are more likely to provide useful information about efficacy.
Chapter 6 notes that some requests for studies with neonates yielded little information, in part because of uncertainty about the nature and
means of assessment of the condition in children. At the same time, the chapter noted the shortage of studies of drugs often prescribed off-label for neonates. In addition, certain categories of requests, for example, repeated requests for studies of similar (“me too”) drugs, were generally not compelling, although studies of such drugs might still provide some useful pharmacokinetic and dosing information. For the variety of drugs used for the treatment of AIDS, often in new combinations, pediatric studies (sometimes requested, sometimes required) provide reassurance about safety and appropriate dosing in children.
Except for sponsors, who may propose pediatric study requests, no organized process currently exists to obtain broader public input. Moreover, neither Congress nor FDA has spelled out the criteria to be considered in assessments of the health benefits of a request, and written requests usually contain little or nothing by way of rationale for the request. It is not clear that the magnitude or importance of the expected benefit matters. Particularly for requests that follow several other requests for studies of drugs in the same class, especially when the initially requested studies do not find efficacy, it would be in the public interest for FDA to discuss whether the expected benefit of a drug proposed for a written request would exceed that of existing therapies for all or some subgroups of children (e.g., because the drug allowed less frequent dosing or had a safer formulation).
In addition, although it did not consider alternatives to the current period of exclusivity, the committee was troubled by the disparity in effort associated with more demanding requested studies that lead to the same reward as less demanding studies. Six months of exclusivity is the reward whether the requested studies primarily involve small pharmacokinetic, pharmacodynamic, and safety studies or larger, well-controlled studies of safety and efficacy. These concerns do not imply the need to change the current policy that allows the granting of exclusivity for both studies with positive results and studies with negative results, as long as they meet the terms of the written request.
The committee concluded that, in general, changes in the basic template for written requests and the amendments to specific written requests have improved requests. Although the committee examined few written requests that were issued after the PeRC initiated its reviews and many of these requests are not yet public, it expects that the additional pediatric and methodologic expertise provided by these reviews are improving the quality of requests (and subsequent studies). As described in Chapter 6, the lack of availability of expertise in neonatal research and clinical care, including for the specification of appropriate written requests for studies of the youngest pediatric patients, remains a concern.
Written Requests and NIH
As explained in Chapter 3, BPCA of 2002 created a role for the National Institutes of Health (NIH) and the Foundation for the NIH in supporting pediatric drug studies for both on-patent and off-patent drugs. The Government Accountability Office (GAO) has reported that one sponsor accepted a written request for study of an off-patent drug between 2002 and the end of 2005 (GAO, 2007) and that no sponsor has accepted a written request for an off-patent drug since then (GAO, 2011).
According to the director of BPCA activities at the National Institute of Child Health and Human Development (NICHD), FDA has referred to NIH 9 written requests for studies of on-patent drugs and 17 requests for studies of off-patent drugs (involving 14 products) (personal communication, Anne Zajicek, Chief, Obstetric and Pediatric Pharmacology Branch, NICHD, June 29, 2011, and December 1, 2011).5 The results of at least five NIH-funded studies have been submitted to FDA.
Application of PREA Requirements
As described above, PREA requirements have become increasingly important as a source of pediatric studies. As noted earlier, the scope of such requirements is limited to the indication covered in a New Drug Application (NDA) or BLA submission. Nonetheless, even when a required study is limited to only the most recent of several indications for which the product has been approved for adults, a pediatric study of use of the drug for that indication may increase understanding of the drug’s pharmacokinetics and safety profile and thereby provide information relevant to off-label use for other indications.
To help understand how FDA specified requirements for pediatric studies, the committee examined, when possible, the FDA letters to sponsors that originally set forth requirements. The committee also reviewed some approvals letters for biologics, as will be discussed in Chapter 8. In addition, to get a sense of how FDA is now applying PREA requirements, the committee examined several approval letters issued in 2011.
In some cases, the committee found approval letters (mostly issued several years ago) that did not mention the requirement for pediatric studies. In some cases, that was an oversight, and studies were required. In other cases, the sponsor had an orphan drug designation for the product or an indication in question or FDA did not consider the submission to involve a new active ingredient, indication, new dosage form, new dosing regimen,
![]()
5 The requests are posted at http://bpca.nichd.nih.gov/clinical/requests/index.cfm.
or new route of administration.6 The lack of explicit discussion of PREA requirements is, nonetheless, an oversight.
One difficulty for the committee and others seeking to assess activities performed under PREA is that the FDA has no easily used, comprehensive public database of product-specific PREA requirements covering the period from the Pediatric Rule to the present. (As described below, the agency does have a website that allows searches for studies required under PREA.) It sometimes required examination of several sources to identify FDA’s determinations about waivers or deferrals of PREA requirements for particular products, and information for biologics was sometimes lacking altogether.
Rationales for Waived or Deferred Studies
With respect to PREA requirements stated in FDA letters, the committee concluded that statements about waivers or deferrals have become more specific and somewhat more informative to the public over time. Some early letters did not mention requirements of the Pediatric Rule or merely noted that required studies had been waived or deferred. They often provided no rationale for a waiver or deferral and no information about the kind of deferred study that would be expected. In some cases, the decision about deferral or waiver was itself deferred. Recent letters are generally specific about the applicability of PREA requirements and the rationale for determinations.
In an analysis required by Congress in 2007, FDA’s PeRC analyzed the extent to which FDA approval letters or other documents were citing appropriate rationales for waiving or deferring pediatric studies under PREA (PeRC, 2010). The retrospective review of actions taken between January 1, 2004, and September 27, 2007, found that 22 percent of the waivers were granted for reasons other than those specified in PREA of 2003 and another 10 percent incorrectly applied the specified criteria. Examples of incorrect rationales included explanations that no appropriate formulation was available. (PREA allows FDA to direct a sponsor to try to develop such a formulation if necessary for pediatric studies.)
The most common basis for waivers was that studies would be impossible or highly impracticable. Other waivers were based on safety concerns. In the PeRC sample, no waivers were based on the rationale that the product did not represent a meaningful therapeutic benefit over existing
![]()
6 For example, in June 2011, FDA approved a tamper-proof tablet form of an opioid product (oxycodone HCl [Oxecta]) for which no studies were required under PREA because it considered this not to be a new form (Rappaport, 2011b). It has considered extended-release tablets, among other types of tablets, to be a new dosing form (see Appendix C in FDA’s Orange Book [FDA, 2011b]).
therapies and would not be expected to be used by a substantial number of pediatric patients. The committee identified some examples of such waivers, for example, for studies of benzyl alcohol (Ulesfia) to treat head lice in children ages 1 to 6 years (Beitz, 2009b).
In contrast to the pattern for waivers, nearly all (98 percent) of the rationales for deferrals were consistent with the law. The most common reason for deferral was that a product was ready for approval for adults.
In addition to the findings about waivers and deferrals, PeRC identified problems with scientific quality, particularly in the early years following the enactment of PREA in 2003. Consistent with the shortcomings described above for written requests, these included inadequate processes for selection of the study dose and inadequate numbers of subjects for realistic statistical assessment, both of which are problems that could have contributed to the failure of studies to find efficacy. The report concluded that a more detailed pediatric plan or more specific recommendations from the review division might have avoided the problems.
Specifications for Types of Studies Required
As noted above, some approval letters, particularly early letters, did not mention PREA requirements, whereas others noted a requirement for deferred studies but provided no specifics beyond those for the age group involved. The most recent letters tend to be considerably more detailed than earlier letters in specifying the kinds of studies required, although they are less detailed than a written request.
The 2011 letter approving an NDA for albumin (human) (Kedbumin) provides an example of a more detailed specification. It described a requirement for a “prospective, randomized, multicenter, controlled open label study to evaluate the safety of Kedbumin 25% compared to normal saline solution in the treatment of post-surgical hypovolemia associated with hypoalbuminemia in pediatric patients undergoing major elective surgery” that would “enroll a total of 100 subjects, 50 subjects in each treatment cohort, with approximately equal numbers of subjects in the following subpopulations”: 1 day up to 2 years, 2 up to 6 years, and 6 up to 12 years (Malarkey and Epstein, 2011, unpaged). Other recent letters specify more than one required study. In one example involving a product for topical treatment of plaque psoriasis, the agency specified pharmacokinetic/dynamic studies for children ages 2 through 11 years and ages 12 through 16 years and also specified a safety and efficacy study for the younger age group (Walker, 2010b).
Sponsor plans for the study of products for pediatric use are not made public, so the committee could not assess the plans submitted for products included in its sample. In conversations, FDA staff described the
pediatric research plans submitted by sponsors as variable, ranging from well thought out to perfunctory. In the one example that the committee found online (BPL, 2009), the pediatric research plan was approximately as detailed as many of the written requests that the committee reviewed. It included a proposed clinical study approach with a description of the proposed design, the age groups to be studied and number in each group to be studied, the entry criteria, the primary and secondary efficacy endpoints, the safety variables, the timing of various assessments, and the general types of statistical analyses to be provided.
Status of Deferred Studies
The timely initiation, completion, and reporting of studies required under PREA are important to meeting the objectives of the law. Likewise, FDA’s timely assessment of NDAs or BLAs submitting studies required under PREA is important. Responding to concerns that postmarket studies required by FDA were not being adequately monitored or completed in a timely fashion (or at all), Congress in 1997 required that FDA monitor and make public information on the status of studies that have been agreed to by sponsors (21 USC 356b).
Beginning in 2007, Congress also required studies undertaken in response to a PREA requirement to be submitted in a supplemental NDA or BLA that required approval (see Chapter 3). Prior to that provision, sponsors might submit reports in general correspondence or other forms that did not trigger explicit FDA response and labeling determinations, thereby undermining a key objective of the law.
FDA established a website that allows individuals to inquire about postmarket study requirements or commitments, including those required under PREA.7 FDA also posts summary reports on studies deferred, waived, and completed under PREA. Table 7-1 presents information on the status of deferred studies in the years from September 2007 through 2010.8
In 2010, FDA counted 63 (15 percent) deferred studies as delayed; 262 (63 percent) as pending but not defined as delayed; and 36 as ongoing. In
![]()
7 The site does not generate an easily used comprehensive listing of those requirements. The descriptions of the studies vary in specificity (e.g., identification of age groups). As the committee conducted its assessment of studies conducted under PREA, it discovered some products that should have been but were not included in the database and notified FDA so the agency could add the information. After one year, the agency drops listings for fulfilled or released study commitments; thus, the site does not provide complete information on the status of these commitments.
8 For these and earlier years, FDA has also published in the Federal Register annual status reports for several types of required postmarket studies. Before 2008, these reports did not break out information on studies required under PREA.
TABLE 7-1 Progress of Pediatric Studies Deferred Under PREA, 2007 to 2010
|
|
||||||||
| No. (%) of Studies | ||||||||
|---|---|---|---|---|---|---|---|---|
| Study Status | 9/27/2007 | 2008 | 2009 | 2010 | ||||
|
|
||||||||
| Pending | 188 | (83) | 180 | (60) | 219 | (60) | 262 | (63) |
| Ongoing | 8 | (4) | 26 | (9) | 32 | (9) | 36 | (9) |
| Submitted | 16 | (7) | 26 | (9) | 33 | (9) | 24 | (6) |
| Fulfilled | 2 | (1) | 17 | (6) | 14 | (4) | 14 | (3) |
| Released | 1 | (<1) | 12 | (4) | 18 | (5) | 12 | (3) |
| Delayed | 11 | (5) | 35 | (12) | 46 | (13) | 63 | (15) |
| Terminated | 1 | (<1) | 3 | (1) | 3 | (<1) | 3 | (<1) |
| Total studies | 227 | (100) | 299 | (100) | 365 | (100) | 414 | (100) |
| Total products | 190 | 230 | 263 | 267 | ||||
|
|
||||||||
NOTES: Pending indicates that the study has not been started but it is not considered delayed. Ongoing indicates that the study is on or ahead of the original schedule. Submitted indicates that the applicant has concluded or terminated the study and has submitted a final report but that FDA has not notified the applicant in writing that its study commitment has been fulfilled or released. Fulfilled indicates that the applicant has submitted the final study report and that FDA has determined that the applicant has met its study commitment. Released indicates that FDA has released the applicant from its obligation because the study is either no longer feasible or no longer useful. Delayed indicates that the study is behind the original study schedule. Terminated indicates that the applicant ended the study before completion but has not yet submitted a final study report. Percentages may not add to 100 due to rounding. SOURCE: Compiled from information at http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/PediatricTherapeuticsResearch/UCM195000.pdf.
the same year, 24 deferred studies were submitted to FDA; another 14 were judged by FDA to fulfill requirements; and sponsors were released from requirements for 12 studies. From 2008 to 2010, the number of pending studies grew by almost 50 percent while the number of delayed studies increased by more than 80 percent. Without information that is not public, it is hard to evaluate these numbers.
An FDA-commissioned analysis of the backlog of postmarket studies (not limited to those required under PREA) provides some perspective. It found that PREA studies accounted for a somewhat larger share of delayed studies than of total studies in the backlog (Booz Allen Hamilton, 2010). Of the 220 PREA studies in the backlog, 6 had been issued without a specified completion date. An earlier analysis that excluded PREA studies found that difficulty with patient enrollment was the most common reason that a study had been categorized as delayed (Booz Allen Hamilton, 2008). Such difficulties are also likely to be a factor in delayed PREA studies as well as in the release of sponsors from requirements for studies.
The committee did not locate a report that charted the status of PREA requirements by year, for example, how many studies that were originally specified in 2004 were pending, fulfilled, or otherwise categorized as of the end of 2010. Chapter 3 explained that FDA has limited leverage to compel completion and submission of a required study and suggested that Congress provide the agency with more flexibility to impose sanctions, including monetary penalties, for unreasonably delayed pediatric studies.
PEDIATRIC DRUG STUDIES AND FDA REVIEWS
As described in Chapter 5, the committee did not have direct access to the voluminous submissions by study sponsors of study findings and other information (and would not, in any case, have had the resources to review them). Rather, the committee relied on the reviews of these submissions by FDA staff, which generally included the clinical review, the clinical pharmacology review (if any), and the statistical review (if any). In some cases, for example, when there was disagreement about conclusions, the initial clinical review was supplemented by memoranda from the review team leader or division director (or both) commenting on some aspect of the review. Some reviews cited discussions by FDA advisory committees, and summaries of those discussions were consulted if available.
In several reviews that the committee examined, the redaction of significant sections created problems for the committee’s analyses, especially when the redactions covered the reviewer’s overall conclusions and recommendations (see Chapter 5). In conversations, FDA staff explained that the criteria for redaction were related not only to confidential or proprietary information but also to issues involving negotiation with sponsors (e.g., about labeling language) or agency deliberations (e.g., reviewer judgments that were not upheld as they went through levels of organizational review). Chapter 4 of this report suggests that Congress ask for an independent assessment of the extent to which redactions in reviews of pediatric studies are appropriate.
Quality of FDA Reviews
For the most part, the committee judged the FDA reviews, especially the more recent reviews, to be of good quality. As described in Chapter 5, recent reviews that follow the Center for Drug Evaluation and Research template help the reader identify important information and conclusions about safety and efficacy. CBER has not formally adopted such a template. Heavily redacted reviews were of limited use, but that issue was not under the control of the reviewers.
Some reviews included little discussion of developmental variability when the committee judged such discussion to be warranted, for example, for findings (or absence of findings) for neonates or adolescents. The committee did not attempt to identify whether reviewers had pediatric training or experience.
Chapter 5 also notes that clinical reviewers typically did not say much, if anything, about the appropriateness or validation of alternative endpoints used in efficacy studies and that it would be desirable for such discussion to be added to reviews (and written requests). Likewise, the justification for the use of extrapolation could be expanded, although the law requires only brief documentation.
The PeRC report cited above commented that implementation of PREA, including the level of detail used in reviewing protocols for pediatric studies, varied across divisions. The committee did not attempt to assess variability across divisions but expects that variability across divisions likewise exists for clinical reviews. A recent IOM report (2010) noted variability in FDA evaluations of studies submitted under the Orphan Drug Act and recommended that the agency investigate the extent to which such variability is appropriate.
Types of Studies Supporting Labeling Changes
In 2007, Congress required FDA to begin reporting certain characteristics of studies conducted under BPCA and PREA, including the types of studies submitted by sponsors to support labeling changes or pediatric exclusivity determinations. Of the requested or required studies reported since then, FDA has classified two-thirds (229 of 346) as efficacy and safety studies (Table 7-2). Studies are labeled by their primary purpose, although pharmacokinetic studies typically yield some information about a drug’s safety. Similarly, some findings relevant to efficacy may be reported in these studies, and pharmacokinetic studies may be a component of efficacy and safety studies.
More than 70 percent of all studies (219 of 346) for the time period covered were associated solely with PREA requirements; another 12 percent (68 of 346) were associated with both BPCA requests and PREA requirements, and 17 percent were linked to BPCA requests. About 75 percent of studies that were conducted only under PREA requirements were categorized as efficacy and safety studies as were about 60 percent of the studies associated solely with BPCA requests. For reasons that are not obvious, the studies that were related to both BPCA requests and PREA requirements were more likely to involve safety and pharmacokinetics rather than safety and efficacy.
|
|
||||||||
| No. (%) of Studies | ||||||||
|---|---|---|---|---|---|---|---|---|
| Type of Study | BPCA | BPCA + PREA | PREA | Total | ||||
|
|
||||||||
| Efficacy/safety | 36 | (61) | 28 | (41) | 165 | (75) | 229 | (66) |
| PK/safety | 10 | (17) | 30 | (44) | 17 | (8) | 57 | (16) |
| PK/PD | 5 | (8) | 7 | (10) | 3 | (1) | 15 | (4) |
| Safety | 8 | (14) | 3 | (4) | 23 | (10) | 34 | (10) |
| Other | 0 | (0) | 0 | (0) | 11 | (5) | 11 | (3) |
| Total | 59 | (100) | 68 | (100) | 219 | (100) | 346 | (100) |
|
|
||||||||
NOTE: These studies were associated with 130 different products, 13 of which were vaccines (personal communication, Catherine Lee, Office of Pediatric Therapeutics, FDA, July 26, 2011). The table does not necessarily include Phase I or Phase II studies and thus likely undercounts pharmacokinetic studies. Also some pharmacokinetic studies may be incorporated in efficacy/safety studies (personal communication, Catherine Lee, Office of Pediatric Therapeutics, FDA, October 5, 2011). PK = pharmacokinetic; PD = pharmacodynamic. Percentages may not add to 100 due to rounding.
SOURCE: This information is periodically updated and is available online at http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm190622.htm.
Issues in Pediatric Studies Submitted for FDA Review
Many studies that the committee reviewed generated valuable information, including, in some cases, negative information about unexpected adverse events or lack of efficacy. Examples of informative labeling changes resulting from these studies are discussed in the next section.
This section focuses on studies that did not reach their potential. To the extent that original or amended written requests failed to specify appropriate trial design and associated measures and methods, the resulting studies may have corresponding weaknesses. For example, if the written request has shortcomings in the specification of endpoints, dose-finding strategy, study design, or sample adequacy (including pediatric subgroups), then the studies as conducted and submitted to FDA are likely to suffer unless appropriate amendments to the request change the terms.
Similar problems may arise with studies conducted under PREA, especially if the protocol review process for studies is limited or lacking in appropriate pediatric expertise. As observed in the PeRC report cited earlier “[w]here there was evidence of specific discussion and documentation of the studies needed to fulfill the PREA requirements before commencement and/or submission of the studies, the PREA assessments [i.e., the studies conducted by the sponsor] generally were of higher quality” (PeRC, 2010, p. 10).
In a few cases, however, problems appeared to arise as much or more from the execution of requested or required studies as from the specifications for the studies. Box 7-3 provides examples of aspects of study planning or execution that may have limited the usefulness of the information submitted. (Chapter 6 discussed problems with written requests and studies that stem from uncertainties about the nature of GERD and bacterial conjunctivitis in neonates.)
In one instance, the committee found unusual labeling language that conveyed FDA’s dissatisfaction with the sponsor’s design and conduct of a study of a drug to reduce mortality and morbidity in neonates and infants with cyanotic congenital heart disease palliated with a systemic artery-to-pulmonary artery shunt. The reviewer particularly cited deficiencies in the sponsor’s approach to selecting the dose for study, which the reviewer and others at FDA concluded was too low to have the desired antiplatelet effect (Rose, 2010; Grant, 2011). Other problems included the concomitant use of aspirin and the late initiation of therapy. After noting the study results and these likely contributing factors, the label goes on to state that “[i]t cannot be ruled out that a trial with a different design would demonstrate a clinical benefit in this patient population” (BMS/SPP, 2011).
In addition to problems with various aspects of study design, studies may not be completed to the standard desired—or at all—because sponsors encounter difficulties with enrollment of sufficient numbers of children, despite reasonable efforts. This challenge was highlighted in Chapter 1. In the committee’s sample, one example of such enrollment problems involved a study comparing leflunomide (Arava) to methotrexate for treatment of juvenile rheumatoid arthritis. Enrollment shortfalls prompted the amendment of the written request to specify a superiority trial with 94 participants instead of the originally requested noninferiority or equivalence trial with 120 participants (Yancey, 2004). The eventual findings for the randomized, double-blind trial favored the active comparator.
Another example of enrollment difficulties involved a combined pharmacokinetic, safety, and efficacy trial testing pegfilgrastim (Neulasta) to reduce episodes of febrile neutropenia in children with sarcoma. Of 50 eligible study sites, only 18 agreed to participate in the trial; of these, only 10 enrolled any children (Summers, 2008). A likely contributing factor was that pefilgrastim was already marketed and available, so parents may have been reluctant to have their child participate in a trial comparing this drug, which involved a single injection, to neupogen, which required daily injections. Some pharmacokinetic information was added to the labeling. FDA judged the sponsor to have made diligent effort to fulfill PREA requirements and noted that the Children’s Oncology Group (COG; which is centrally involved in the conduct of most pediatric cancer trials in the United States) had indicated to the sponsor that the conduct of an additional efficacy study
of the drug was not a priority. For another pediatric cancer drug study, which was ended early for lack of evidence of test drug activity, a different reviewer noted that because relatively few children are diagnosed with cancer compared with the number of adults, “COG prioritizes its trials to study the most promising agents first” (Honig, 2002, unpaged).
PEDIATRIC STUDIES AND CHANGES IN LABELING
Types of Labeling Changes
All but one of the products in the committee’s sample had labeling changes that resulted from the studies conducted under BPCA or PREA. Three labeling changes involved one product. Of the 45 labeling changes in the sample, 17 involved the extension of age limits for an indication that had already been approved in adults or another pediatric age group. Another 10 changes involved approval of a new product with pediatric labeling or a new indication that had not previously been approved for adults. Thus, 60 percent of labeling changes in the sample resulted from analyses that found efficacy and safety. (A few changes occurred without requirements for efficacy studies.) The addition of an indication to labeling was generally accompanied by information on dosing, pharmacokinetics, and safety. As described in Chapter 6, five products studied with substantial numbers of neonates did not have a labeling change that incorporated any information from these studies.
For the labeling changes that did not involve the addition or expansion of a pediatric indication, changes generally included the addition of some information about safety and pharmacokinetics. For changes that followed from studies that did not show efficacy, the presentation of that information varied. Some labels added statements to the effect that use of the product was not indicated or recommended, whereas most stated that safety and efficacy had not been established for all or some pediatric age groups. The latter language is rather imprecise, in itself not making clear whether studies have not been conducted and submitted to FDA or whether studies have been submitted and did not show safety and efficacy. Additional text in the label may clarify the situation, but the key summary sentence is still ambiguous.
FDA has not evaluated information added to the label as a result of studies required under PREA, but FDA staff have published two articles that have reviewed labeling changes associated with BPCA (Roberts et al., 2003; Rodriguez et al., 2008). The most recent article presents data from an analysis of labeling changes from July 1998 through October 2005 (Rodriguez et al., 2008). For the 108 drugs with labeling changes resulting from studies conducted under BPCA, 77 changes extended the age limits
Questions about participant characteristics and dosing issues. For the pivotal study of omalizumab (Xolair) for the treatment of moderate to severe persistent asthma in children ages 6 to 11 years (inclusive), the children enrolled in studies had, on average, normal pulmonary function (determined from the forced expiratory volume in 1 second [FEV1]). As summarized by the clinical reviewer, an FDA advisory committee was concerned that “the applicants had not studied patients for whom the drug is intended, namely the most severe asthmatic patients who are not responding to alternative therapies” and was “very concerned that the applicants had not explored any dose ranging” for this age group (Starke, 2009, p. 95). Taking the results for all efficacy endpoints and safety data into account, the advisory committee concluded that the risk-benefit assessment did not favor approval of the product. Almost all of the overview of the risk-benefit section of the review was redacted.
Questions about adequacy of dosing. In requested studies of leflunomide (Arava), children with juvenile rheumatoid arthritis receiving this drug showed less improvement than children in the active comparator (methotrexate) control arm of the trial (68 versus 89 percent) (Yancey, 2004). On the basis of questions about the adequacy of the dosing used for lower-weight children, the drug was labeled in 2004 as having not been fully evaluated. The label included information about pharmacokinetics and safety and a summary of the trial results.
Problems with data quality. In analyzing a submission of studies of zolmitriptan (Zomig) for treatment of migraine in adolescents, the statistical reviewer described “extreme difficulties in analyzing the data due to poor data quality, missing information (information not entered in the data by the sponsor), poor organization of the data, and various errors” (Yan, 2008, p. 4). The reviewer also noted problems with poor patient compliance and with the deviations from the statistical analysis plan in the sponsor’s imputation of efficacy values. The reviewer concluded that no statistically significant difference existed between the test drug and the placebo for either 1-hour headache response or 2-hour sustained headache response.
Inadequate enrollment of relevant age groups. One of two studies described in the written request for propofol (Diprivan), which anesthesiologists use in all age groups, was a randomized, open-label trial comparing 1 percent propofol versus standard anesthetic technique for induction and maintenance of general anesthesia in children from birth to 3 years of age (Raczkowski, 1999). The request specified “substantial representation” of three age groups, including children from birth to 2 months of age. In reviewing the study as conducted, the clinical reviewer concluded that “the only age group not adequately covered was the birth to <2 month age group” (Hartwell, 2000, p. 66); only one neonate was in the propofol arm, whereas four were in the standard anesthetic arm. The labeling states that the product is not recommended for maintenance of anesthesia in this age group because safety and effectiveness have not been established.
Safety concern not addressed. In a study of sotalol (Betapace) for treatment of arrythmias, the reviewer noted higher peak concentrations of the drug in neonates and infants than older children and attributed some of the difference to differences in renal function (Karkowsky, 2000). The studies enrolled fewer neonates than planned (6 rather than 20). In general, the reviewer notes that the studies provided no information about dosing of children who have diminished renal function.
Questions about pediatric subgroup. Guanfacine hydrochloride (Intuniv) was studied for treatment of attention deficit hyperactivity disorder in children ages 6 to 17 years of age. For the 13-to 17-year-old age group, the studies did not find a statistically significant different result for the study drug than for the placebo. The clinical reviewer noted that the sponsor used fixed rather than flexible, weight-based doses in the trials and concluded that “it is highly likely that one contributing factor [to the study results] was the lower serum guanfacine exposures observed in the Intuniv clinical program” (Levin, 2007, p. 43) The product was approved for the entire age group with a weight-based dosing regimen, labeling that described the study results, and a postmarket commitment for an additional study with adolescents to confirm efficacy.
Weak trial design. Etodolac (Lodine XL) was studied for treatment of juvenile rheumatoid arthritis in children 6 to 12 and 12 to 16 years of age in an open-label uncontrolled trial to assess pharmacokinetics, safety, and efficacy. The clinical reviewer concluded that “especially without some arm for comparison, it is difficult to understand how any of this information can be placed into a proper context short of historical controls either in an adult or pediatric population” (Witter, 1999, p. 17). The pharmacometrics reviewer concluded “that no statistical comparison can be made on pediatric and adult PK [pharmacokinetics] based on the studies submitted” (Wang, 2000, p. 14). The pediatric use section of the label approved in 2000 read, “If a decision is made to use Lodine XL for patients six years of age or older, as with other NSAIDs [nonsteroidal anti-inflammatory drugs], such patients should be monitored periodically” (http://www.accessdata.fda.gov/drug satfda_docs/nda/2000/20-584S005_Lodine_prntlbl.pdf). By 2005, however, that section of the label had been amended to state that safety and effectiveness in patients 6 to 16 years of age were supported by extrapolation from adult trials and by safety, pharmacokinetic, and efficacy data from an open-label trial with children in that age group (http://www.accessdata.fda.gov/drugsatfda_docs/label/2005/020584s004,006,007lbl.pdf). It is not clear what prompted that change, which is not recorded in FDA’s overview table of BPCA-and PREA-related labeling changes.
Problems with administration of test and control drugs. In a trial of fluticasone inhalation aerosol (Flovent) involving children 6 to 23 and 24 to 47 months of age, the report for the pharmacokinetic study revealed detectable levels of the study drug in some participants in the placebo control arm. Further investigation also showed that some participants in the active drug arm had no detectable levels of the test drug. The reviewer concluded that “the studies could not be meaningfully interpreted, and no conclusions may be drawn regarding either efficacy or safety from the clinical studies” (Starke, 2003, p. 5).
for an approved indication; 19 changes added information about lack of efficacy. Of the other changes, 23 involved dosing or pharmacokinetic information, 34 involved safety, and 12 described a new pediatric formulation. The discussion and examples focused on the changes related to new information on pharmacokinetics or dosing.
The analysis by Rodriguez and colleagues (2008) stressed the importance of studies requested under BPCA to generate knowledge important for safe and effective dosing. It noted that the results of studies were not necessarily predictable on the basis of weight differences and data from adults.
For the sample that the committee examined, Box 7-4 presents examples of informative changes to labeling resulting from requested or required pediatric studies. Most changes supported the use of the drug with children but some did not. Some changes reflected safety findings for children that differed from findings for adults.
As discussed in Chapter 5, FDA sometimes requests only pharmacokinetic and safety information and expects to extrapolate efficacy on the basis
BOX 7-4
Examples of Informative Labeling Changes
Vinorelbine tartrate injection (Navelbine) (2002). Requested studies did not show activity of the drug against recurrent malignant solid tumors, which is important information for clinicians. Labeling noted that toxicities were similar to those in adults. Recent studies suggest that the drug may have value against other cancers; the clinical review is not publicly posted by FDA but includes pharmacokinetic data that could be useful to investigators.
Remifentanil (Ultiva) (2004). Requested studies with infants from birth to 2 months of age showed high variability in the drug’s phamacokinetics in neonates, which led FDA to recommend careful titration of individual doses. Given concerns about possible negative neurodevelopmental effects of anesthetics in young children, the information about an ultra-short-acting opioid without suspected neurotoxic effects is valuable.
Desflurane (Suprane) (2006). Requested studies clarified the risks from use of this anesthetic, which is approved for maintenance of anesthesia in pediatric patients with intubation and after induction with another agent. The studies led to stronger safety information in the labeling stating that the product is not approved for maintenance of anesthesia in nonintubated children. The warning now appears at the front of the labeling, a change made possible by the switch in 2010 to the structured labeling format that FDA has been phasing in since 2006.
Aripiprazole (Abilify) (2007, 2008). This drug has a different mechanism of action than other antipsychotic medications available at the time that written requests
of efficacy studies with adults, absent unexpected safety findings. In the case of sotalol (Betapace), FDA requested pharmacokinetic, pharmacodynamic, and safety information to guide pediatric use but did not request efficacy studies and did not extrapolate safety and efficacy from adults. The labeling for the product notes that safety and efficacy have not been established, but it includes pediatric dosing and pharmacokinetic information (for children more than 2 years of age and children younger than that) based on two requested studies (FDA, 2001b).
Box 7-5 presents examples of committee concerns about the labeling changes that followed pediatric studies. Most involve how labels presented information about pediatric studies that did not demonstrate efficacy.
In some cases, labeling seemed to convey contradictory information, as illustrated in the first example in Box 7-5. Such labeling may stem from the dilemma faced by FDA in labeling of products that it expects may have continued off-label use, despite studies that do not demonstrate efficacy. It may also stem from FDA concerns about the shortcomings of efficacy studies (e.g., enrollment problems) that might have limited the possibility
were issued. Studies led to labeling for pediatric use for the treatment of schizophrenia and mania associated with bipolar disorder. Under PREA, the drug has also been approved for treatment of irritability associated with autism.
Adalimumab (Humira) (2008). Required studies of children with juvenile idiopathic arthritis demonstrated efficacy. They also found several safety signals that had not been identified in adults, including elevations of creatine phosphokinase, a higher rate of immunogenicity, and a higher rate of nonserious hypersensitivity reactions.
Tenofovir disoproxil fumarate (Viread) (2010). Several factors complicated the required study of this drug’s efficacy for treatment of HIV infection in adolescents, but the pharmacokinetic and safety data combined with adult data allowed the extrapolation of efficacy to this pediatric age group. Although the drug has been used in adolescents on the basis of a favorable toxicity profile in adults and pharmacokinetics that allow once-a-day dosing, the studies provided reassurance for such use on the basis of the safety and pharmacokinetic results. (Based on these studies and studies with adults suggesting adverse bone effects, FDA required a postmarket clinical trial to further investigate the drug’s effects on bone in pediatric patients.)
Candesartan (Atacand) (2009). Requested studies of children ages 1 to 17 years showed safety and efficacy of the drug for the treatment of hypertension. The pharmacokinetic data provided the basis for dosing recommendations for children ages 1 up to 6 years and children ages 6 up to 17 years. Other data for children less than 1 year of age led FDA to drop the requested study with children in this age group and to specifically warn in the label that the product must not be used by this age group.
BOX 7-5
Concerns About Clarity of Labeling Changes
Information appears to be contradictory. The labeling for zoledronic acid (Zometa) states that it is not indicated for use in children but also states (as in the previous label) that “[b]ecause of long-term retention in bone, Zometa should only be used in children if the potential benefit outweighs the potential risk” (NPC, 2012, unpaged). That advice applies to the use of any medication by children or adults.
Lack of efficacy is downplayed. The pediatric use section of the labeling for buspirone (Buspar) does not state that efficacy has not been demonstrated. Rather, it describes safety and pharmacokinetic data from two placebo-controlled trials and that the trials found “no significant differences between buspirone and placebo with regard to the symptoms of GAD [generalized anxiety disorder] following doses recommended for the treatment of GAD in adults” (BMS, 2010, p. 11).
Lack of efficacy in an age group not explicitly stated. The pediatric use section of the labeling for olmesartan (Benicar) notes that it was studied in children ages 1 to 16 years and that it was generally well tolerated and had an adverse experience profile similar to that for adults. It does not explicitly state that studies did not show efficacy in the younger age cohort studied (ages 1 to 5 years).
Lack of advantage of higher dose could have been clearer. In the highlights section of prescribing information for aripiprazole (Abilify) for treatment of schizophrenia in adolescents, the dosing chart lists a maximum dose without noting that it was not shown to be more effective than the recommended dose. The discussion of dosing later in the labeling notes this. The discussion of adverse events does not discuss the effects of the higher dose on adverse events (e.g., somnolence and extrapyramidal effects).
Relevant data about dosing were not highlighted. Studies of mometasone furoate (Asmanex) yielded convincing data that twice-a-day administration of the 110-mg dose to children ages 4 to 11 years was more efficacious than once-a-day dosing for severe asthma. These data do not have a prominent place on the label.
Placement of information is unexpected. Data on the pharmacokinetics of irinotecan hydrochloride (Camptosar) are included in the precautions section of the labeling rather than in the section on clinical pharmacology. The latter section does not provide a cross-reference to the precautions section, which begins by explaining that studies had not demonstrated effectiveness for the treatment of solid tumors in pediatric patients.
of finding statistically significant positive findings for a drug that is, in fact, efficacious.
Aside from specific language in labeling, another concern stems from the incomplete transition from the old labeling format to a new format, which was introduced in 2006, as described in Chapter 3. Of the 45
labeling changes in the committee’s sample, the labeling for 15 products remained in the old format at the time that it was consulted. That is, FDA has not required the sponsor to revise the label to meet current standards for new labeling that, in particular, requires an initial highlights section that summarizes key information about approved uses and age groups (ideally), warnings, and use by special populations.
Reformatting can significantly clarify information. For example, when the labeling for desflurane (Suprane) was reformatted, the highlights segment on pediatric use stated that for safety reasons the product was not recommended for induction of anesthesia in or for maintenance of anesthesia in nonintubated children. In the old format, the indications and usage section did not explicitly state that it was not recommended for the latter use.
Although the committee was not asked to evaluate the efforts by FDA or others to disseminate information from pediatric studies and labeling changes, it recognized that these efforts are important. The committee was aware that clinicians often do not consult a product’s labeling. They instead rely on intermediary sources, as described in Appendix B. Nonetheless, to the extent that labels still in the old format are consulted by clinicians or others, including parents searching the Internet for additional information on a child’s treatment, the format hinders the identification of key information about efficacy and safety. To acknowledge the importance of getting information to clinicians, the committee commissioned the background paper that appears in Appendix B. It underscores the challenges of getting up-to-date information to clinicians who care for children.
Pediatric studies conducted under BPCA and PREA are yielding important information to guide clinical care for children. The information generated varies by medical condition and age group. As discussed in Chapter 6, studies with neonates are a particular challenge. Findings from pediatric studies sometimes support and sometimes run counter to expectations about the efficacy, safety, and pharmacokinetics of a drug in children of different ages.
Some studies requested under BPCA or required under PREA do not achieve their full potential. Reasons vary. Some problems stem from the use of weak study designs and underpowered samples, the lack of dose-ranging studies to guide efficacy trials, and the omission of relevant information from labels. Other problems stem from sponsor difficulties enrolling sufficient numbers of children in clinical trials. One persistent need is for strict and consistent attention by FDA, sponsors, and investigators to dose selection for evaluation in pediatric drug studies.
The committee concluded that the steps that Congress and FDA have initiated appear to be improving the quality of requests and requirements for pediatric studies. These steps include increased review by pediatric experts, increased specificity in the template for written requests and amendments to specific written requests, and earlier specificity about deferred studies required under PREA. In addition, as suggested in Chapter 4, FDA could more clearly articulate the health benefits expected of requested studies so that children do not participate in research of minimal value. Chapter 5 suggested similar articulation of the rationales for the acceptance of extrapolation and the use of alternative endpoints.
Although FDA now monitors, analyzes, and reports more information about the status of studies (e.g., required studies that are pending or delayed and clinical areas represented by written requests), some information is not readily available. If FDA creates a formal system for tracking pediatric drug applications through the submission and review process as recommended by GAO, it would be helpful for the system to track pediatric studies by age group, including neonates specifically.
The organization and highlighting of key information in the current structured labeling format are substantial improvements over the previous version. Transitions to the new format provide FDA with the opportunity to clarify inadequately described, ambiguous, or contradictory information in older labeling.
The committee recognizes that FDA faces some dilemmas when submitted studies do not show efficacy but the agency expects that physicians will continue to use the drug off-label. If the agency includes pharmacokinetic and safety data in labeling, it is important that the label be clear that the provision of information about pediatric dosing does not mean that the product is recommended for pediatric use.
FDA likewise faces a dilemma when off-label use of a medication is common but controlled studies of efficacy are not or may not be feasible. The agency may have to weigh competing risks. If it requests or requires sponsors to conduct only pharmacokinetic and other studies to guide dosing decisions, it risks encouraging increased use of a product that has not been demonstrated to be effective. If it does not seek this information in the absence of more comprehensive investigations, it leaves physicians without data that could potentially reduce the harm or increase the benefit from off-label use.
In the future, FDA’s efforts to strengthen regulatory science (e.g., methods for evaluating drugs and biologics) should support further improvements as should a number of activities the agency has undertaken to analyze specific challenges in pediatric trial design and analysis and propose innovative strategies to meet these challenges. Examples include the analyses of pediatric hypertension trials described in this chapter and the assessment
of pediatric studies of analgesic medication and other pain prevention and alleviation strategies described in Chapter 6. To improve pediatric studies of drugs and biologics and their evaluation, it is important for FDA to continue and expand initiatives to strengthen the science base for its work, analyze shortcomings in pediatric studies, and develop innovative strategies to meet the specific challenges of pediatric trials.
This page intentionally left blank.






























