
Written Requests for Studies of
Pediatric Hypertension: Longitudinal
Changes in FDA Specifications
Jennifer Li*
The analysis presented here examines written requests for clinical studies issued by the Food and Drug Administration (FDA) to investigate potential drug treatments for pediatric hypertension. It begins with a summary of key elements in the written requests issued in the first 30 months after pediatric exclusivity provisions became effective in July 1998. The subsequent summaries describe key elements that either modified specifications (e.g., by more precisely describing safety follow-up) or added to them (e.g., by creating requirements for interim analyses). Some changes were required by legislation (e.g., registration of trials at ClinicalTrials.gov or documentation of a failed attempt to develop a new formulation).
FDA began with a basic template for the written requests for clinical studies to investigate drug treatments for pediatric hypertension. In general, the changes in elements of the template for both new and amended requests tended to have a few common purposes. They might
• add precision (e.g., by specifying a 1-year period for safety follow-up or by specifying minimum percentages of individuals of particular age or racial subgroups enrolled in trials);
• require more rigor in trial designs (e.g., by dropping the option for a trial with no placebo and only alternative doses of the test drug or by increasing the statistical power of trials to detect a clinically meaningful effect);
![]()
* Jennifer Li, M.D., is a member of the study committee.
• require more accommodation of the developmental variability of children (e.g., by requiring sponsors to try to develop age-appropriate formulations, if needed); or
• increase transparency (e.g., by requiring that sponsors submit New Drug Application supplements to add to the label information—whether negative or positive—from clinical trials).
KEY ELEMENTS SPECIFIED IN WRITTEN REQUESTS/
AMENDMENTS ISSUED FROM 1998 TO 2000
• Requested trials:
– Dose-ranging trial with hypertensive pediatric patients
– Trial of pharmacokinetics (PKs) in children in four pediatric age groups (infants and toddlers, preschool-age children, school-age children, and adolescents)
– Safety data from a controlled trial with an open treatment phase following the trial or from some other comparable database with a summary of all available information on the safety of the drug in pediatric patients
• Race: Ensure a mixture of black and nonblack patients.
• Formulation: If no suspension/solution is available, a solid dosage form suspended in food could be used, if it has been shown to have acceptable bioavailability in adults.
• Trial design: Randomized, double-blinded observation of parallel dose groups (it need not be successful, but it must be interpretable).
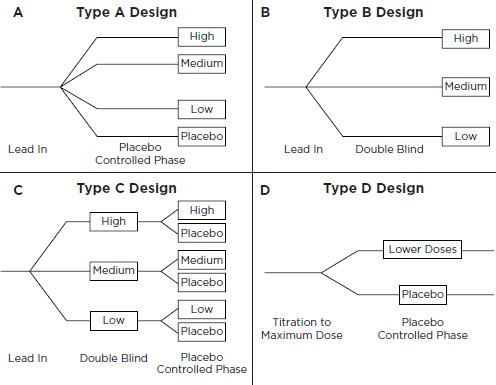
• Four design options: A, B, C, and D (Figure E-1)
– Trial Design A: Each patient is randomized to placebo or to one of three doses ranging from slightly less than the lowest approved adult dose to slightly greater than the highest approved adult dose. After 2 weeks of treatment, the trial would be analyzed by looking for a significantly positive slope of the placebo-corrected change in blood pressure from baseline as a function of dose. If the slope of this line is not differentiable from 0, the trial would be unsuccessful but it would be interpretable.
– Trial Design B: Design B is similar to Design A, but without a placebo arm. If analysis revealed a significantly positive slope to the dose-response line, the trial would be successful. If, however, no dose-response is detected, the trial will be considered not interpretable and not responsive to the written request.
– Trial Design C: To avoid the possibility of uninterpretable find-
SOURCE: Reproduced with permission from Smith et al. (2008). See also Benjamin et al. (2008).
ings, Design C consists of Design B modified to include a randomized withdrawal phase. Patients would be recruited and treated like those in the trial with Design B. At the end of a 2-week treatment period, patients would be rerandomized in a blinded fashion to continue to their assigned treatments or be withdrawn to placebo. A slope analysis would be used for the first phase and then, if the dose-response curve is flat, an analysis of the second phase would determine whether a blood pressure effect existed. The result would be considered interpretable no matter what the outcome, so long as the sample size for the withdrawal phase was adequate.
– Trial Design D: Design D uses randomized withdrawal. Patients would be force-titrated to maximal tolerated doses and then randomly withdrawn to lower doses, including placebo.
• Ages: Adolescents and at least 50 percent of subjects 6 to 12 years of age or ≤Tanner 3
• Statistical considerations: 80 percent power to detect a treatment effect of conventional statistical significance (p = 0.05)
• PKs from infants and toddlers, preschool-age children, school-age children, and adolescents: Traditional or sparse sampling can be chosen, and for the parent drug and each metabolite, estimate bioavailability (area under the concentration-time curve), half-life, maximum concentration of drug in plasma (Cmax), and time to Cmax in the various age groups.
• Labeling change: Appropriate sections of the label may be changed to incorporate the findings of the studies.
CHANGES ADDED OR ELEMENTS MODIFIED IN SOME OR ALL
NEW REQUESTS OR AMENDMENTS FROM 2001 TO 2003
• Safety data: One-year follow-up is specified, with all available information (published and unpublished) to include information on adverse events, growth (change in head circumference, weight, length, or height), and development (milestones, school performance, neurocognitive testing) at baseline and 1 year.
• Age groups: 25 percent of participants should be infants to preschool age.
• Race: Black enrollment is specified to be 40 to 60 percent of total enrollment.
• Age-appropriate formulation: An age-appropriate formulation or documentation of an attempt to obtain an age-appropriate formulation, if the attempt was unsuccessful, is required.
• Statistical considerations: The ability to detect a 3-mm-Hg blood pressure change with 90 percent power is required.
• Efficacy endpoints: For the trial designs other than randomized withdrawal from active drug (see above), the primary efficacy measurement should be the change in blood pressure from baseline to the end of the treatment period plus the interdosing interval (trough). For randomized withdrawal trial designs, the primary efficacy measurement should be the change in blood pressure from the last on-treatment visit to the end of the withdrawal period.
CHANGES ADDED OR ELEMENTS MODIFIED, 2006
• Interim analyses allowed to assess variability according to a prespecified rule to adjust the sample size to achieve the specified
target power: This interim analysis must be performed with >90 percent of the initially planned enrollment. Options for estimating variability are (1) a blinded, pooled analysis of all groups, (2) a blinded analysis of one group, or (3) a partially unblinded analysis within each group (performed by an independent third party).
• Dissemination of information: Summaries of medical and clinical pharmacology reviews are posted on the FDA website.
CHANGES ADDED OR ELEMENTS MODIFIED, 2009
• Trial design: Two types
– Type A: randomized, double-blind parallel, placebo and two doses
– Type B: two active doses with randomized withdrawal (same as Trial Design C described above but with two doses)
• Statistical considerations: The primary endpoint must be either absolute or the percent change in systolic or diastolic pressure. The statistical approach used will depend on the specific trial design; but broadly, the sponsor can allocate alpha to each active arm in the placebo-controlled comparison or to some combination of treatment arms (highest, two doses), or the sponsor can look for a positive slope in the dose-response relationship.
• Sample size: The trial program must have a total of no less than 200 patients in the 6-to 16-year-old age groups and no less than 50 patients in the 1-to 5-year-old age groups.
• Formulation: If reasonable attempts to develop a commercially marketable formulation have failed, the sponsor must develop and test an age-appropriate formulation that can be compounded by a licensed pharmacist, in a licensed pharmacy, from commercially available ingredients. The sponsor must document attempts and reasons that attempts failed. If the reasons are accepted and studies are conducted with the compounded formulation product, the label must include detailed compounding information.
• Dissemination of information: The written request and medical, statistical, and clinical pharmacology reviews will be posted on the FDA website, and the trial will be registered at ClinicalTrials.gov.
• Labeling: Regardless of whether the studies demonstrate that the drug is safe and effective or whether the results of such studies with the pediatric population are inconclusive, the sponsor must submit labeling to include information about the results of the studies.
REFERENCES
Benjamin, D. K., Jr., P. B. Smith, P. Jadhav, J. V. Gobburu, D. Murphy, V. Hasselblad, C. Baker-Smith, R. M. Califf, and J. S. Li. 2008. Pediatric Antihypertensive Trial Failures: Analysis of End Points and Dose Range. Hypertension 51(4):834-840. http://hyper.ahajournals.org/content/51/4/834.full.pdf+html (accessed December 7, 2011).
Smith, P. B., J. S. Li, D. Murphy, R. M. Califf, and D. K. Benjamin, Jr. 2008. Safety of placebo controls in pediatric hypertension trials. Hypertension 51(4):829-833. http://hyper.ahajournals.org/cgi/reprint/51/4/829 (accessed December 7, 2011).






