
IOM Staff Paper
Context and Glossary of Select Terms Associated with the Clinical Trials Enterprise
Rebecca English Institute of Medicine, Forum on Drug Discovery, Development, and Translation
CONTEXT
The following is a glossary of clinical research terms drawn from planning discussions and conversations that took place at the November 2011 workshop summarized in this report. These definitions were not formally presented at the workshop but have been compiled retrospectively by Institute of Medicine (IOM) staff to clarify some of the terms used at the workshop. The glossary was developed to provide greater background information to the reader who may be unfamiliar with the terms but is not intended to be an exhaustive or complete list of terms relevant to the clinical trials enterprise. These are practical working definitions to help the reader understand the terms used.
All clinical trials are designed to answer one or more specific questions. They can vary by the study population chosen (number of subjects, as well as criteria to enter the study) and the type of question(s) posed. For example, clinical trials to gain U.S. Food and Drug Administration (FDA) approval for a new drug are designed to show its safety and efficacy over the course of a specified period of time—traditionally using a randomized controlled clinical trial design (RCT) (IOM, 2010a). These trials seek to answer narrowly defined questions related to safety and efficacy in a carefully selected group of study participants most likely to experience the intended effects of the drug. Clinical trials conducted without the goal of regulatory approval might test a drug or intervention in a diverse group of study participants, include a longer time frame for follow-up of study subjects, and address a broader set of questions.
Although RCTs, the primary focus of the Forum’s workshop series on clinical trials, will continue to play an important role in the development of new drugs and therapies, many believe the approach is not always feasible, ethical, or practical to answer the wide array of research questions facing the nation today. The scientific value of RCTs is well established, but the high cost and time commitment required for these studies, combined with the often limited applicability of results to patient populations that differ from those in the original study, can disassociate this type of research from the reality of medical practice. A primary theme of the workshop is the need to bridge the divide between clinical research and clinical practice— i.e., to effectively bring clinical research into the community. Doing so would involve partnerships among researchers, physicians, and patients and facilitate the development of studies focused on answering meaningful questions for clinical practice, which in turn could inform the development of new, targeted therapies for the changing health needs of the population.
Workshop participants discussed a broad range of research methods that would engage patients and clinicians and go beyond determinations of efficacy (i.e., can it work under ideal circumstances?) to evaluate clinical effectiveness (i.e., can it work in real world, clinical practice settings?). It was noted at the workshop that the full battery of clinical research methods will be necessary to improve our understanding of the questions that emerge through the lifecycle of a medical product. For instance, Figure 1 suggests a timeline of medical products research and the corresponding development of various types of evidence.
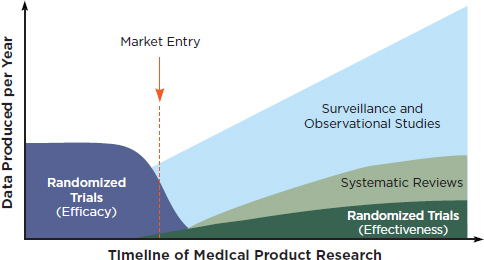
FIGURE 1 Evidence development in the learning health system over the course of a medical product’s lifetime.
SOURCE: IOM, 2010b. Redesigning the Clinical Effectiveness Research Paradigm.
GLOSSARY
Adaptive trial design: experimental study design in which the treatment allocation ratio of an RCT is altered based on a prespecified plan defining prescribed changes to study end points, and other criteria, over the course of the study based on collected data and treatment responses of prior participants. Bayesian or Frequentist analyses are based on the accumulated treatment responses of prior participants and used to inform adaptive designs by assessing the probability or frequency, respectively, with which an event of interest occurs (e.g., positive response to a particular treatment) (IOM, 2010b).
Clinical effectiveness research: a type of research that builds on safety and efficacy determinations necessary for regulatory approval to develop research results to guide the delivery of appropriate care to individual patients (IOM, 2010b). Clinical effectiveness research includes a broad range of research designs and data sources—including post-marketing data for medical products.
Clinical trial: a medical research study in humans to evaluate the effects of one or more interventions/treatments. Clinical trials typically evaluate the effects of an intervention by comparing the outcomes of a group receiving the intervention and a group receiving standard care (when there is no standard of care, a placebo is traditionally used). Data on adverse events are typically collected systematically during such trials. Clinical trials (also called “research studies”) are used to determine whether new drugs or treatments are safe and effective. See definitions below of randomized controlled clinical trials and non-randomized clinical trials for information on the merits of each design.
Clinical trials enterprise (CTE): a broad term that encompasses the full spectrum of clinical trials and their applications. The CTE includes the processes, institutions, and individuals that eventually apply clinical trial findings to patient care.
ClinicalTrials.gov: a publicly funded and available registry (clinicaltrials.gov/) of completed and ongoing clinical trials. The database, which was mandated in the Food and Drug Administration Modernization Act (FDAMA) of 1997, is intended to serve as a centralized information source for clinical trials including studies on products regulated by FDA. The registry was developed in 2000, is administered by the NIH’s National Library of Medicine (NLM), and includes federally and privately supported clinical trials. Study sponsors are responsible for submitting timely and accurate information to clinicaltrials.gov/ about their trials
before the trial is started, while it is ongoing (should there be changes), and after the trial is completed. The number of trials registered increased substantially in 2005 as a result of a requirement by the International Committee of Medical Journal Editors (ICMJE) that studies published in ICMJE-associated journals be registered on ClinicalTrials.gov or another equivalent publicly available trial registry.
Cluster Randomized Controlled Trial: an experimental study design in which groups (e.g., individuals or patients from entire clinics, schools, communities, or physician practices), instead of individuals, are randomized to a particular treatment or study arm. This design is useful for a wide array of clinical effectiveness topics and may be required in situations in which individual randomization is not feasible. (IOM, 2010b). For an example of physician practices in the UK participating in a cluster randomized controlled trial, see the Discussion Paper in Appendix J, Novel Ways to Get Good Trial Data: the UK Experience.
Continuous improvement: quality improvement activities, typically conducted by health care delivery organizations, to assess processes of providing care to meet patient needs. These activities usually compare the relative effectiveness of delivering existing approved therapies or diagnostics. IOM reports in 2000 and 2001 found that most medical errors resulted from faulty processes and systems, not individuals. Quality improvement efforts to identify and correct errors and inefficiencies in the processes of delivering patient care have become common practice within health care delivery organizations. Today, the concept of continuous improvement in health care includes more than just measurements of quality and could also incorporate innovation and a strong scientific foundation to inform improvement efforts. Several workshop participants suggested that to pursue the goal of further integrating clinical research and practice, policy change or clarification be taken up that would expressly permit inclusion of routine evidence generation activities of health care organizations that pose no more than a minimal risk to patients under the concept of continuous improvement activities, which would have the effect of exempting these activities from the traditional regulatory framework applied to experimental research activities (e.g., Common Rule, HIPAA). They noted that incorporating low-risk research into the concept of continuous improvement could improve the ability of health care systems to engage in research efforts. For a discussion of the distinctions between continuous improvement—including quality and clinical effectiveness assessments—and human subjects research that requires formal oversight by an Institutional Review Board (IRB), see Selker et al., 2011.
Coverage with Evidence Development (CED): an insurance coverage and simultaneous evidence-development strategy under which the Centers for Medicare and Medicaid Services (CMS) issues a National Coverage Determination (NCD) requiring, as a condition of paying for a particular treatment under federal programs, prospective collection of additional patient data to supplement standard claims data. Under a CED decision, in order for Medicare to pay for a particular treatment patients must agree to participate in a clinical trial or registry to further evaluate the risks and benefits of that product. Although the term “coverage with evidence development” was first coined in a draft guidance issued by the CMS in April 2005, the policy of linking Medicare coverage with clinical research began in 1995 (Tunis and Pearson, 2006).
Direct-to-patient trial: a new research approach that recruits, administers therapeutics, and monitors clinical trial participants via the internet. Also called a “virtual” clinical trial, this could result in faster clinical trials as the potentially eligible patient population includes anyone with internet access—as opposed to the current model that relies on recruiting patients who live in close proximity to clinical trial sites. Trial sponsors could also realize lower clinical trial costs using this model because individual “brick and mortar” trial sites would not need to be set up across the country. The first FDA approved “virtual” clinical trial is being conducted by Pfizer to evaluate the safety and efficacy of an overactive bladder drug.
Electronic data capture (EDC): a technological tool that facilitates the collection of data electronically from clinical trial sites. EDC can reduce errors in data sent from trial sites to the coordinating center and trial sponsor and can help make clinical trial results available faster (Kush, 2006). Use of EDC in clinical trials was discussed during the workshop by some participants who noted that other risk-management practices of clinical trial sponsors, such as having a monitor travel to a clinical trial site to compare hard copies of patient EHRs and EDC printouts instead of correlating this information from electronic archive systems, can nullify the originally intended efficiencies of EDC.
Electronic Health Record (EHR): an electronic tool that captures a wide range of information related to a patient’s health, entered by health care providers in various settings, and aggregates the data to serve different needs. According to the Healthcare Information and Management Systems Society (HIMSS), an electronic health record is defined as a longitudinal electronic record of patient health information generated by one or more encounters in any care delivery setting. Included in this information are patient demographics, progress notes, problems, medications,
vital signs, past medical history, immunizations, laboratory data, and radiology reports. The data in the EHR is used to generate a complete record of a clinical patient encounter, as well as supporting other care-related activities directly or indirectly via interface, including evidence-based decision support, quality management, and outcomes reporting. Beyond patient-specific medical data, EHRs include clinical decision support tools, computerized provider order entry systems, and e-prescribing systems (IOM, 2012). As discussed during the workshop, use of the EHR for clinical research purposes could play a key role in bridging the current divide between clinical research and practice.
Equipoise: the point at which a rational, informed person would not have a preference between the therapies in each arm of a trial (Lilford and Jackson, 1995). In clinical trials, the ethical concept of equipoise is satisfied when genuine physician and patient uncertainty exists as to the comparative benefits of the therapies in each treatment group of the trial. The concept and applicability of equipoise have been debated, as differences between the uncertainties and preferences of an individual (individual equipoise) and society (collective equipoise) can vary (Chard and Lilford, 1998).
Good Clinical Practice (GCP): an international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects (ICH, 1996). Satisfying GCP requirements provides assurance that the data and reported results of the clinical trial are credible and accurate and that the rights, integrity, and confidentiality of trial subjects are protected (ICH, 1996). GCP guidelines are developed by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), which brings together regulatory authorities and pharmaceutical industry of Europe, Japan, and United States.
Non-randomized clinical trial: a clinical trial design in which patients are not assigned by chance to different treatment groups—instead the patient or his/her practitioner might choose her or his treatment group or be assigned to a group by a researcher. In either scenario, the assignment to a treatment group is not left to chance.
Observational study: a type of research in which the investigator observes the measure of interest and no interventions or treatments are provided as part of the study. Patient registries and EHRs are examples of data sources for an observational study. When it is not considered ethical or feasible to answer a research question using a controlled trial, an observational study
could develop information to answer the research question or advance a hypothesis, which could, in turn, be tested by a future RCT. Because observational studies have less internal validity (i.e., greater opportunity for the introduction of bias in the study) than an RCT, the results are not always sufficient to base a decision to change medical practice. However, valuable information can be gathered through observational studies about the implementation and use of medical treatments in clinical practice. Because observational studies can feasibly collect data on large populations over a long period of time, they are the most reliable way to gather data on rare medical events.
Patient-centered outcomes research (PCOR): PCOR in the United States, being further developed and funded by the independent Patient-Centered Outcomes Research Institute (PCORI), aims to help people and their care-givers make informed health care decisions and allows their voice to be heard in assessing the value of health care options (PCORI, 2012). PCOR could involve clinical effectiveness research methods as well as head-to-head comparisons to evaluate the relative strengths and weaknesses of various interventions. PCOR is similar to what is called “patient-oriented research” in Canada’s current strategy to improve health outcomes and enhance patient’s health care experience based on translation of research outcomes into the health care system (see Discussion Paper in Appendix H for more information on the effort in Canada).
Protocol: an extensive “blueprint” for the clinical trial including the trial objective, design, methodology, and organization. Protocols are required to be submitted to the relevant institutions and organizations that provide ethical and regulatory approval for clinical trials. Protocol design plays a crucial role in the success of a trial as it outlines exactly what the trial will entail, the eligibility criteria for those who can participate, and the frequency of the procedures and treatments that trial participants will be expected to receive over the course of the study. In recent years, protocols have become more complex and demanding for clinical trial sites to implement. One study found that the number of unique procedures and the frequency of procedures per clinical trial protocol have increased at an annual rate of 6.5 percent and 8.7 percent, respectively, from 1999 to 2005 (Getz et al., 2008). Researchers found that over this same time period, study conduct performance worsened—patient recruitment and retention rates lowered, more amendments to the clinical trial protocol were initiated, and more serious adverse events were observed (Getz et al., 2008).
Randomized controlled clinical trial (RCT): an experimental study design in which patients are randomly allocated to treatment groups
in a trial. Analysis estimates the size of difference in predefined outcomes, under ideal treatment conditions, between treatment groups. “Controlled” refers to the standard that allows experimental observations to the evaluated—the control group of patients receives standard treatment or placebo to provide the basis for comparing the effect of the experimental treatment given to the other group of patients. Randomly assigning patients to receive the intervention addresses concerns that known or unknown differences in intervention groups might affect the outcome of the trial. RCTs are characterized by a focus on efficacy, internal validity, maximal compliance with the assigned regimen (see definition of “protocol” above), and typically, complete patient follow up. When feasible and appropriate, trials are “double-blind”—i.e., both patients and trialists are unaware of treatment assignment throughout the study (IOM, 2010b). Not all clinical trials are randomized (see the above definition of non-randomized clinical trial).
REFERENCES
Chard, J. A. and R. J. Lilford. 1998. The use of equipoise in clinical trials. Social Science & Medicine 47(7):891-898.
ClinicalTrials.gov. http://clinicaltrials.gov/ct2/info/glossary.
Getz, K. A., J. Wenger, R. A. Campo, E. S. Sequine, and K. I. Kaitin. 2008. Assessing the impact of protocol design change on clinical trial performance. American Journal of Therapeutics 15(5): 450-457.
Institute of Medicine (IOM). 2010a. Transforming clinical research in the United States: Challenges and opportunities. Workshop Summary. Washington, DC: The National Academies Press.
IOM. 2010b. Redesigning the clinical effectiveness research paradigm: Innovation and practice-based approaches. Workshop Summary. Washington, DC: The National Academies Press.
IOM. 2012. Health IT and patient safety: Building safer systems for better care. Washington, DC: The National Academies Press.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). 1996. Guidance for industry E6 good clinical practice: Consolidated guidance. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm065004.htm (accessed February 15, 2012).
Kush, R. 2006. Electronic data capture—pros and cons. BioExecutive International 2(6):S48-S52.
Patient-Centered Outcomes Research Institutes (PCORI). 2012. Patient-Centered Outcomes Research. http://www.pcori.org/patient-centered-outcomes-research/ (accessed April 9, 2012).
Selker, H., C. Grossman, A. Adams, D. Goldmann, C. Dezii, G. Meyer, V. Roger, L. Savitz, and R. Platt. 2011. The common rule and continuous improvement in health care: A learning health system perspective. Discussion Paper, Institute of Medicine, Washington, DC. http://www.iom.edu/Global/Perspectives/2012/CommonRule.aspx (accessed February 3, 2012).
Tunis, S. R. and S. D. Pearson. 2006. Coverage options for promising technologies: Medicare’s coverage with evidence development. Health Affairs 25(5):1218-1230.








