
On June 26, 2000, two biologists—Francis Collins of the international Human Genome Project and Craig Venter of Celera Genomics—stood side by side with President Clinton in the East Room of the White House and announced that they had finished sequencing the first draft of the human genome. All of a sudden, the molecular code that makes us human seemed like an open book. The genome was subsequently published in Science magazine.
Though it was heralded as a breakthrough in biology—and rightfully so—the cracking of the human genetic code also owed a great deal to the mathematical sciences. The Human Genome Project had begun in 1990 and was originally expected to take at least 15 years. However, around 1998 advances in the new discipline of bioinformatics—which combines biology with computer science, statistics, linear algebra, combinatorics, and even geometry—dramatically accelerated the project, turning it from a marathon into a 2-year sprint to the finish.
In the years since 2000, genetic sequencing has become even more dependent on mathematical science techniques. Next-generation sequencers have reduced the cost of reading an entire human genome from $300 million to $30,000 and the time from years to weeks. Further improvements, including the “$1,000 genome,” are anticipated soon. The lower cost and quicker turnaround trend are continuing. The speed of information processing has now become the rate-limiting factor.
How did scientists assemble the human genome? The process is often compared to putting together a jigsaw puzzle. The analogy is a good one, but it is incomplete. In genomics, many of the pieces don’t match, and some are duplicates. Also, many of the pieces come in pairs with a string glued to each piece, so you know roughly how
far apart they are supposed to be in the puzzle. These complications present both opportunities and challenges for mathematical analysis.
Human DNA is a long molecule that is shaped like a spiral staircase, in which each step contains a pair of amino acids that fit together like a tongue and groove joint. Adenine (A) fits together with thymine (T), and cytosine (C) fits together with guanine (G). Each chemical fits only one of the others, so that the sequence of letters along one side of the staircase (GATTCC…) uniquely determines the corresponding sequence on the other side (CTAAGG…), which is conventionally read in the opposite direction (… GGAATC). Like a photographic negative, one strand is a template for duplicating the other (see Figures 19 and 20).
In all, human DNA contains about 3 billion “base pairs,” or rungs of the staircase. The goal of the Human Genome Project was to list all of them in order. Unfortunately, chemists can sequence only a few hundred base pairs at a time. To sequence the whole genome, scientists had to chop it into millions of shorter pieces, sequence those pieces, and reassemble them.
The publicly funded Human Genome Project and the privately funded Celera Genomics adopted two different strategies, both of which eventually led to the same mathematical problem. You have millions of short (500-base) overlapping puzzle pieces that have been completely scrambled by the chopping process. There are enough pieces to cover the length of the genome seven or eight times over, so there are many overlaps between pieces. You want to use these overlaps as a guide to assemble the pieces into the longest possible sequence of contiguous regions.

19 / Human DNA can be extracted from biological tissue such as skin and blood and a unique genetic sequence of amino acids can be determined. Image courtesy of the National Institutes of Health. /
20 / The structure of DNA: a double helix with matching base pairs of CG and AT. Image from U.S. Department of Energy Genomic Science Program. /
If all of the sequence reads were perfectly accurate, matching the overlapping ones would be routine. However, about 1 percent of base pairs were misread, and this meant the overlapping jigsaw puzzle pieces would not match. The approach then became to find a good match rather than the best match (see Figure 21).
Another issue, somewhat more subtle, was the problem of repeats. Human genomes include many sequences that repeat identically in many places. These repeats were a big headache for genome sequencers because when a contiguous region ended with a pattern that occurred in many places, they had no idea which puzzle piece should come next.
The way around the problem, it turned out, was to take a longer snippet of DNA—say, several thousand base pairs long—and sequence both ends. Even though you can’t sequence the middle, you can at least get a few hundred base pairs at each end and estimate how many base pairs lie between them. This gives you strings that link two jigsaw puzzle pieces together, including some that are on opposite sides of a gap or a repeat. These tethers create a scaffold to hang the “contigs” onto. Finally, the scaffolds could be wheeled into proper position by using the Human Genome Project’s high-level map of the genome.
In the more than a decade since the completion of the human genome, the landscape has changed in at least two important ways. First, because a “reference” human genome is now available (in fact, many such reference genomes are), no human genome has to be sequenced from scratch. If you have a patient with cancer or with a genetic disease, you can zero in on the 0.1 percent of the genome that is different from the reference version and ignore the 99.9 percent that is the same. Thus the problem is not one of assembling the genome but of looking up sequences in the reference genome that are similar to (but slightly different from) your patient’s.
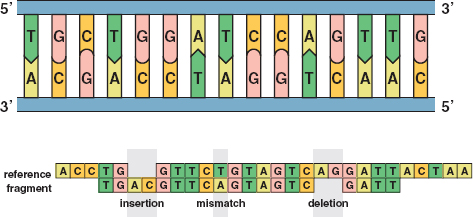
21 / The top image illustrates the sequence of C’s, G’s, T’s, and A’s in an untwisted segment of DNA. The bottom image shows typical errors in DNA sequencing: insertions, mismatches, and deletions. Reprinted with permission from the American Mathematical Society. /
Once again, the solution to this lookup problem came from far outside biology. In 1994, a “transform” was devised that dramatically speeds up the search for strings of text in a long file. Researchers created a table in which each row is a copy of the string, shifted either left or right. The rows are in alphabetical order. To search for a string like ATCTTG, you look for all the rows beginning with A, then for all the ones beginning with AT, and so on. Instead of searching through a linear string of 3 billion characters, you descend a tree with (in this case) six layers of branches. Once you get to the bottom, a transform identifies all the places where ATCTTG appears in the original string. Thanks to such indexing techniques, the reference genome can be searched in a fraction of a second.
A second transformation of genomics was the introduction of commercial next-generation gene sequencers, around 2004. Thanks to new advances in chemistry, biologists can now read hundreds of thousands of DNA snippets simultaneously. But the technology comes at a cost: The snippets have to be much shorter. One popular commercial sequencer can read fragments of only 50 to 75 base pairs, and another can manage only 100 to 150. The short reads are a double whammy for biologists. First, they need to collect much more data—typically the new machines will sequence enough snippets to cover the genome 30 times over. Second, a tiny 50-base read is more likely than a 500-base read to fall right into the middle of a repeated motif. The methods used by the first generation cannot deal with this increase in ambiguity.
Again, the right mathematics was already in existence and ready to be used, but it was unknown to biologists. The idea is to create a network in which nodes represent substrings of the genome and edges represent overlapping substrings. The first-generation methods amount to finding a path through a network that passes through each node once. This problem is known to take a hopelessly long time to
solve. However, a better approach is to find a path that passes through each link of the network exactly once. (Note that it may visit some nodes repeatedly. These correspond to repeated strings in the genome.) This problem, called the Eulerian path problem, has a computationally efficient solution, which makes next-generation sequencing practical (especially for other animal species, which have no reference genome to consult).
The application of the mathematical sciences to the genome stands to have a great impact on society
The application of the mathematical sciences to the genome stands to have a great impact on society. A Battelle Memorial Institute study in 2011 concluded that the economic impact of the Human Genome Project has nearly reached $800 billion—quite a return on the U.S. government’s $3 billion investment. And that doesn’t even begin to account for the impact on humans, which is just beginning.
For example, in 2010 a 39-year-old patient was admitted to Barnes-Jewish Hospital in St. Louis with leukemia. There were two possible diagnoses with two different treatments. The patient had symptoms of a form of leukemia called APL, which responds well to chemotherapy. At the same time she had symptoms of another condition that would require a stem cell transplant, a risky procedure that can itself be fatal.
The standard genetic test for APL looks for two regions on chromosomes 15 and 17 that are swapped. That test came back negative. As an experiment, the patient’s doctors spent $40,000 and six weeks to sequence her genome, comparing her skin cells to her cancer cells. In the cancer cells, they found about 77,000 base pairs of her chromosome 15 had been inserted into the chromosome 17—too small a fragment for the standard test to detect but easily detectable by genomics. Since then, they have identified similar genetic alterations in two other leukemia patients.
The doctors went on to treat the patient with the anti-APL drug instead of the dangerous stem cell transplant. It eliminated her cancer cells, and she was still in remission 15 months later. This is exactly the sort of individualized medicine that doctors dreamed the Human Genome Project would make possible. Though such medicine is still experimental today, with continued progress in bioinformatics it will probably be common a generation from now.





