
Important Points Highlighted by Individual Speakers
• The development of drugs and companion diagnostics requires the collection of rigorous and reliable chemical, pharmaceutical, and biological data.
• Candidate gene association studies can identify genetic markers associated with treatment response, generating hypotheses for further testing.
• Small-sample randomized clinical trials can generate valuable data that can be aggregated to generate information similar to that from a large clinical trial.
• Drug and diagnostic development can proceed quickly if the appropriate patient populations can be identified.
The second session of the workshop featured case studies of genomic-based drug discovery and development, two which have gained FDA approval and a third that is still in development. The first example was the use of crizotinib for non-small-cell lung cancer, the second was the use of pomaglumetad methionil for schizophrenia, and the third was the use of ivacaftor for cystic fibrosis. Each of these drugs emerged from a somewhat different development process, depending on the data available, the state of biological understanding, and the nature of the disease. But, together, these three examples demonstrate that genomic-based approaches
can yield new drugs and diagnostics that have substantial benefits for human health.
THE DEVELOPMENT OF CRIZOTINIB FOR TREATMENT OF NON-SMALL-CELL LUNG CANCER
The drug crizotinib provides an excellent example of how a diagnostic test can be used to identify patients who will benefit from a treatment, said Steffan Ho of Pfizer Inc. Crizotinib, which was originally known as PF-02341066 and has the trade name XALKORI, is a small molecule that binds to the catalytic site of kinases and competes with ATP, thereby inhibiting kinase activity. Its primary targets are the receptor tyrosine kinases known as c-MET, ALK, and ROS. It was approved for use by the FDA on August 26, 2011.
As stated in the indications and usage notes for crizotinib, “XALKORI is a kinase inhibitor indicated for the treatment of patients with locally advanced or metastatic non-small-cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test.” The related diagnostic device, which was simultaneously approved for use with crizotinib, is the Abbott Vysis ALK Break Apart FISH Probe Kit, which is described in the package insert as “a qualitative test to detect rearrangements involving the ALK gene via fluorescence in situ hybridization (FISH) in formalin-fixed paraffin-embedded (FFPE) non-small-cell lung cancer tissue specimens to aid in identifying those patients eligible for treatment with XALKORI (crizotinib).” The device is a Class III diagnostic test, requiring the highest level of rigor and scrutiny, because of the risks associated with its use to inform physicians about how to treat or not treat a patient.
The approval of crizotinib was conditional, Ho noted, because it was based on the response rate. At the time of approval, no data were available that demonstrated improvement in patient-reported outcomes or survival with crizotinib. Additional Phase III clinical studies were under way at the time of the workshop to investigate the hypothesis that crizotinib both improves the response rate and provides a survival advantage.
From the treatment of the first patient deemed to be ALK-positive, approval took just 4 years—a “remarkable accomplishment,” Ho said (Figure 3-1). While uncommonly fast compared to most drugs, this timeframe has been fairly common in oncology with targeted therapeutic agents, he added. The development of vemurafenib, imatinib, and trastuzumab—other oncology drugs in which targeted patient populations are identified—also went quickly. “It supports the concept that once we can identify the right population, the clinical efficacy is very clear,” he said.
In oncology drug development, the datasets that provide the confidence to move into the clinic include not only the chemical and pharmaceutical
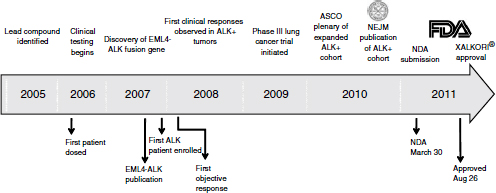
NOTE: ASCO, American Society of Clinical Oncology; EML4-ALK, echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase; FDA, U.S. Food and Drug Administration; NDA, new drug application; NEJM, New England Journal of Medicine.
SOURCE: Ho, workshop presentation, March 21, 2012. Copyright © 2012 Pfizer Inc. All rights reserved.
properties of the drug but also data relevant to the biological setting, Ho said. In the case of crizotinib, more than 700 tumor cell lines were screened for sensitivity to growth inhibition. The results indicated that the drug was active against gastric, esophageal, and lung cancer cell lines that exhibited c-MET amplification; in fact, the drug was originally developed with c-MET as the primary kinase target. It also was found to be active against neuroblastoma with ALK mutation or amplifications, anaplastic large-cell lymphoma with an NPM-ALK fusion, and NSCLC with ALK and ROS alterations. In addition, preclinical tumor models exhibiting dysregulation of c-MET or ALK were highly sensitive to crizotinib. The use of cell models as model systems has limitations, Ho noted, but the regression of tumor cell lines implanted as xenografts into mice provided confidence that the compound could be successful.
In addition, biological understanding of the function of ALK supported the movement of crizotinib into the clinic. Based on the kinase’s role in lymphoma, it was known that when ALK undergoes gene rearrangement, it can function as a transforming driver oncogene. In 2007 a team of researchers led by Hiroyuki Mano used a functional genomics screen to demonstrate that ALK is also relevant in NSCLC, being capable of inducing transforming events in 3T3 mouse fibroblast cell lines, tumor growth in mouse models, and malignant transformation in transgenic mice containing the translocated or rearranged ALK gene (Soda et al., 2007).
This study was also followed shortly thereafter by an independent study using a global phosphorylation approach that confirmed the result (Rikova et al., 2007). These basic science results were critical in motivating the development of the drug and shifting the focus of the Phase I development strategy, Ho said.
ALK translocation in lung cancer is a relatively low-frequency alteration, occurring in only about 6 percent of cases (Kris et al., 2011; Riess and Wakelee, 2012). This represents a very low frequency for a stratified medicine approach, Ho observed. For example, with trastuzumab, HER2 amplification is present in about 25 percent of patients. Once the relevant alteration was identified, the clinical trial of crizotinib quickly began to screen for patients who had the ALK translocation. The first patient who was ALK-positive was entered into the trial at the end of 2007. Within a month, the patient’s tumor was shrinking. In clinical use, patients have reported symptom relief after just a few doses of crizotinib, suggesting that their tumors began to shrink as soon as the drug reached therapeutic levels in the blood. Furthermore, the overwhelming majority of ALK-positive patients exhibited some level of tumor shrinkage when treated with crizotinib. “Very rapidly we were able to determine that we had targeted the correct oncogene and that we had an active drug,” Ho said.
With the demonstration of clinical efficacy, the companion diagnostic test, which initially was laboratory developed, needed further development on a rapid time scale. Within just a few years Abbott Molecular brought forward a test that was suitable for broad clinical use and sufficiently rigorous to enable submission of a PMA application and ultimately to obtain approval as a Class III diagnostic test (Figure 3-2). The development of the diagnostic proceeded in parallel with the clinical trials, so that the data package brought forward to support efficacy included data from patients identified by the final diagnostic test. In this way, the data supported not only the drug approval, but also the diagnostic approval.
Just as with drug candidates, the development of a candidate companion diagnostic test requires rigorous, data-driven evaluation, Ho said. High-quality predictive markers need to be defined that warrant clinical testing, and clinical assays need to be of sufficient quality to test the hypothesis. The transition to the investigational use only (IUO) phase needs to be anticipated, and the decision to pursue IUO development needs to be based on data.
Ho also observed that the approval of crizotinib was not linked on the label to a specific diagnostic, which opens up the possibility that other diagnostics could be used. But questions would arise in bringing forward a second diagnostic. Data could be generated to prove that the second diagnostic was sufficiently equivalent to the original, but to demonstrate that a second diagnostic was detecting patients who would benefit but were not
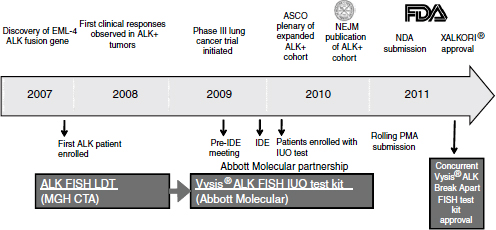
NOTE: ASCO, American Society of Clinical Oncology; EML4-ALK, echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase; FDA, U.S. Food and Drug Administration; FISH, fluorescence in situ hybridization; IDE, investigational device exemption; IUO, investigational use only; LDT, laboratory developed test; MGH CTA, Massachusetts General Hospital clinical trial assay; NDA, new drug application; NEJM, New England Journal of Medicine; PMA, premarket approval.
SOURCE: Ho, workshop presentation, March 21, 2012. Copyright © 2012 Pfizer Inc. All rights reserved.
detected by the original diagnostic, new clinical trials would be required. Nevertheless, patients will likely be treated with crizotinib based on results from other than the approved diagnostic test, certainly in other parts of the world. But there will be no formal data supporting those uses. “It raises a lot of interesting questions as far as further development of diagnostic tests.”
Ho mentioned the issue of biologic heterogeneity within a patient and within a population. The success of targeted therapy will depend on the source of this heterogeneity. In NSCLC, the population exhibits heterogeneity, which allows population subgroups to be identified. But there is also significant heterogeneity within a tumor, even if one driver mutation within the tumor as a whole is playing a critical role in the cancer. The same situation may apply in other diseases, though this question remains largely unanswered.
The underlying biology of human malignancy is very different from that of other therapeutic indications, Ho said, which has implications for
the potential of stratified medicine. In many cases, today’s understanding of the biology of disease is severely limited. As a result, new treatments sometimes move into the clinic in response to hypotheses that are not sufficiently supported. Model organisms provide an opportunity to develop data to support a hypothesis about drug efficacy, whereas moving into the clinic too soon has created challenges for the industry.
“Ultimately, it all requires data,” Ho concluded. “One has to have good data to drive those decisions, and good data requires well-designed studies.”
USE OF GENETICS TO INFORM DRUG DEVELOPMENT FOR THE TREATMENT OF SCHIZOPHRENIA
Just as genetics is useful for informing drug development for cancers, it is also relevant for other disease states, said Laura Nisenbaum of Eli Lilly and Company. Like cancer, these other diseases can be polymorphic and arise through complex pathways. Diseases like schizophrenia also are polygenic and heritable, and patients have differential responses to treatments.
Schizophrenia is a chronic disabling psychiatric disorder with mortality rates two to three times higher than those in the general population. Even after five decades of modern pharmacotherapy, the clinical management of patients with schizophrenia remains challenging, Nisenbaum said. The efficacy of the currently available drugs to treat the symptoms associated with schizophrenia is still very limited, leading to poor outcomes for these patients, including suicide. Drugs with a greater level of efficacy are needed to increase compliance, reduce adverse effects, and give patients hope for the future.
Recent studies have begun to uncover both common and rare genetic variants that are associated with the disease. But there is as yet no clear understanding of the biological mechanisms that contribute to the disease, Nisenbaum said. Thus, instead of using knowledge of the molecular genetics of the disease, as has been done with cancer, Eli Lilly and Company researchers used knowledge of drug mechanisms to formulate a strategy for the discovery of drug-response markers.
The drug currently being developed for the treatment of schizophrenia by Eli Lilly and Company is called pomaglumetad methionil (hereafter referred to as pomaglumetad). It is an agonist of several group II metabotropic glutamate receptors, in contrast to other available antipsychotics, which target the dopamine D2 receptor, Nisenbaum said. It is thought to work by suppressing excitatory neurotransmission in brain neurocircuits that are dysregulated in schizophrenia.
No specific genetic data were available at the outset of the program to guide the development of the drug, Nisenbaum said. However,
pomaglumetad was known to be active in neurocircuits within the brain, and this information was used to develop the pharmacogenetic strategy to generate hypotheses that could be tested in the clinic. Furthermore, it was felt that any new therapeutic would need to differentiate itself from the available generic and branded competition, leading to significant interest in identifying response markers that could be used to identify patients who would respond better to treatment.
In the original proof-of-concept study, both pomaglumetad and the existing standard-of-care treatment olanzapine significantly decreased the number of symptoms experienced by patients relative to placebo treatment (Patil et al., 2007). However, pomaglumetad did not distinguish itself, based on efficacy, from the standard of care. The developers therefore investigated whether a segment of the population might respond differently to this new type of treatment of schizophrenia. Fortunately, the proof-of-concept study included optional DNA collection for patients, and the collection rate was roughly 70 percent, Nisenbaum stated. Using these DNA samples, a candidate gene association study revealed 16 genetic variants in the serotonin 2A receptor that were associated with differential response to pomaglumetad. In particular, patients who were either homozygous for the rare allele or were heterozygous for a particular single nucleotide polymorphism (SNP) in the receptor had a greater response than patients who were homozygous for the common allele. Similar results were observed in a second clinical trial. This kind of discovery is relatively rare in psychiatric genetics, Nisenbaum said, in that few studies have repeated a finding prospectively in a second clinical trial.
Eli Lilly and Company researchers are now trying to validate the marker in larger registration studies. “You’ll have to stay tuned to see how the story plays out,” Nisenbaum said.1 “But we are very excited at the prospect of potentially being able to help tailor something in the psychiatric space where we know that the response rate for these types of drugs is modest.”
As genetic markers related to the serotonin 2A receptor are considered for further use in clinical trials, it is necessary to understand additional factors regarding receptor expression, Nisenbaum said. The serotonin variants identified in the proof-of-concept study are all located within a large intron of the serotonin 2A receptor, and the variants do not have an obvi-
![]()
1 Eli Lilly and Company announced results from the first of these studies, H8Y-MC-HBBM, subsequent to the workshop on July 11, 2012. Results indicated that the primary efficacy endpoint had not been met and that neither pomaglumetad nor the active control used, risperidone, had separated from placebo “in either the overall or predefined genetic subpopulation” for the two doses that had been investigated. Further trials are ongoing. Details of the announcement can be found at http://newsroom.lilly.com/releasedetail.cfm?ReleaseID=690836 (accessed August 7, 2012).
ous impact on protein coding. Interestingly, all of the SNPs identified in Caucasian patients lie in tight linkage disequilibrium to one another, and an antisense nested gene is located in this region of the chromosome as well, though the function of the nested gene is not yet known.
An important limitation of the research done to date is that the marker has been identified in one population—non-Hispanic Caucasians—but the genetics of the region are different in African Americans. Researchers now need access to samples from other populations to determine whether the marker is useful for identifying patients from other races and ethnicities.
Because of the limited biological understanding, there was no a priori hypothesis for genetic-based drug discovery and development in this case, Nisenbaum concluded. Rather, hypotheses needed to be generated in Phase II, and these hypotheses then needed to be replicated and validated in Phase III. Also, as was the case with crizotinib, if Phase III results support the need for a companion diagnostic, the development of that diagnostic will need to be timed appropriately so that it does not become the rate-limiting factor for the drug approval.
A GENETIC APPROACH TO THE TREATMENT OF CYSTIC FIBROSIS
Cystic fibrosis is an orphan disease, which differentiates it from cancer and schizophrenia, said Peter Mueller of Vertex Pharmaceuticals. The disease is linked to a genetic defect that leads to an impairment in the ability of cystic fibrosis transmembrane conductance regulator (CFTR) channels to pump chloride and other ions across cell membranes due to either incorrect localization of the protein in the cell or production of nonfunctional proteins. This lack of ion transport causes a variety of adverse outcomes, including the accumulation of a sticky mucus which is characteristic of cystic fibrosis and which eventually leads to chronic infections and death.
More than 1,700 mutations have been linked to impairment of the CFTR channel, which makes gene therapy difficult, Mueller said. Instead, his company has sought to develop small molecules that are orally bioavailable and that improve CFTR function, thereby reducing and halting the progression of the disease.
The mutations responsible for cystic fibrosis can be divided into three categories. People with CFTR gating mutations express channels on the surface of their cells that do not function properly. People with residual CFTR function express a minimal number of channels on their cell surface that do not work optimally. And the largest group consists of people who have almost no CFTR function due to a failure to express channels on cell surfaces.
Understanding the underlying genetics and biology behind the loss of
protein function is essential for progress, Mueller said. Linking the genetics involved with the observed phenotypes pointed to two distinct functional consequences that would need to be corrected to reverse or halt the effects of cystic fibrosis: a lack of surface expression and a lack of transport activity by the CFTR channel. Accordingly, Vertex investigated both potentiators that increase channel activity and correctors that increase the delivery, or trafficking, of CFTR protein to the cell surface. A search of about 10,000 molecules turned up a particular molecule, ivacaftor, that restored function and removed mucus in patient cells with a particular mutation known as G551D. This result was strong enough to take the drug into the clinic.
After negotiations with regulatory agencies in different countries, four studies were conducted: a study in patients 12 and older with the G551D mutation, a similar study in children 6 to 11 years old, a safety study in subjects homozygous for a common cystic fibrosis mutation, and a rollover extension trial of patients who completed two previous trials.
The outcome was “stellar,” according to Mueller (Figure 3-3). After just 2 weeks, patients demonstrated a 10 to 12 percent improvement on average in their lung function which persisted throughout the trial (Ramsey et al., 2011). “That never has been seen with any other drug in the respiratory field,” Mueller said. “Normally you get about 3 or 4 percent, and then you are really happy.” At the same time, sweat chloride concentrations dropped to almost normal levels, indicating an increased function of
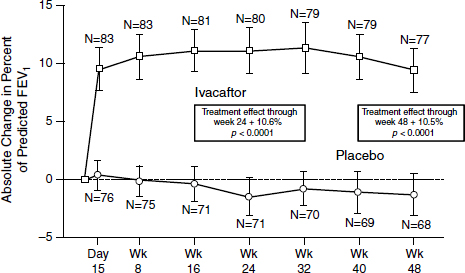
SOURCE: Ramsey et al., 2011.
the channels systemically. Sweat chloride is an indirect rather than a direct measure of what is happening in the lungs, Mueller noted, but more work is being done on validating the results.
Treatment also had other benefits. Most important, it allowed patients to gain weight, which is difficult for cystic fibrosis sufferers because of the high metabolic rate they need to support their breathing. Also, the number of pulmonary exacerbations dropped substantially. Finally, patients taking ivacaftor had fewer adverse events than the patients taking a placebo, and no important safety concerns were identified for the patients on the drug. Even after 3 years on the drug, Mueller noted, patients report that they are still doing well, and these long-term benefits have been confirmed by a new lung imaging technology that uses hyperpolarized helium to measure airflow. Given these results, the drug was approved in just 3 months and 2 days. “There was work on both ends to make it really happen, [but] it’s doable. Everybody wanted to get it to the patients.”
Only about 5 percent of patients with cystic fibrosis have a gating mutation such as G551D (Figure 3-4). The first need for people with other
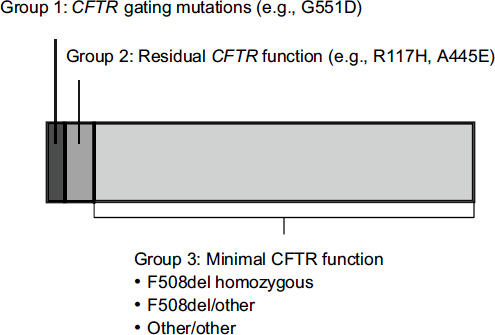
FIGURE 3-4 Approximately 5 percent of people with CFTR mutations have gating mutations such as G551D.
NOTE: CFTR, cystic fibrosis transmembrane conductance regulator.
SOURCE: Mueller, workshop presentation, March 21, 2012; data derived from the Cystic Fibrosis Foundation Annual Patient Registry Report, 2009.
kinds of mutations, Mueller said, is for in vitro data on specific mutations, which can provide the basis for new clinical trials, although he noted that carrying out clinical trials in many cases becomes more complicated since there may only be a few patients with a particular mutation. This applies as well to people who have residual function of CFTR channels. Though their disease tends to be milder, models showing that channel function can be improved can be used to move forward. In some of these cases, combination drugs may be required that enhance channel trafficking as well as conductance. “The hope is, at the end of the day, that almost everybody can benefit from at least the combination regimen and go back to a level that is almost nonsymptomatic,” he said. “That’s our ultimate goal.”
Mueller also noted that ivacaftor was chosen to be extremely selective for the CFTR channel. But this channel plays a role in other conditions, from bronchitis to problems with sperm maturation, which raises the possibility that it could be used in other settings.
Bringing people with cystic fibrosis almost back to normal produces tremendous savings in terms of hospitalization and co-medications, Mueller said. Furthermore, these people are able to go back to work and participate in daily life. Personalized medicine has the potential to make a dramatic difference in a person’s life, which creates powerful incentives to create and use such therapies.
One lesson from this experience, Mueller said, is that regulators have a strong interest in bringing the right therapy to the right patients. It is best to involve them early, sometimes across divisions of FDA, and to have a constructive and not adversarial dialogue. Also, regulations and regulators in other parts of the world differ from those in the United States. Harmonized regulatory procedures around the world would bring effective medicines to people faster.
Another important lesson involves the registration of people with a disease. Because cystic fibrosis is an orphan disease, Mueller said, people with the disease are registered, which means they have already been diagnosed and can be approached for the collection of biological samples. It is important for this registry process to be harmonized across different countries, which would help to standardize the data that are gathered and would provide for the wider collection of samples. Data collection and standardization are also occurring through such mechanisms as the Cancer Genome Atlas, Ho said, which is doing multidimensional profiling of a large number of cancer samples. Michelle Penny of Eli Lilly and Company added that during the development of pomaglumetad methionil, the company made the collection of DNA samples mandatory where local regulations and IRB approval allowed, with consents ranging from candidate gene study to whole-genome sequencing. Only by having these
samples available could the company collaborate with partners to do the needed research.
Finally, Mueller made the observation that it was important in cystic fibrosis research to translate results from in vitro systems into the clinic. However, the large number of different mutations complicates the process of finding patients who can benefit from particular therapies. Genotypic and phenotypic strategies need to be combined to facilitate this process.
Small-sample randomized clinical trials known as N-of-1 trials could be a way of generating valuable data (Lillie et al., 2011). First used in the 1960s for behavior research, N-of-1 trials rely on randomized, placebo-controlled, repeated crossovers in a single individual. Remote clinical phenotyping, including ambulatory and home monitoring, has greatly increased the practicality of such trials, and methodologies now exist to aggregate multiple N-of-1 trials to generate information similar to that generated by a large clinical trial. The result would be probability characteristics for markers to use for therapeutic benefit, and discussions are under way with regulatory agencies to enable the use of such information.
“The normal, standard stuff doesn’t work when you have only three patients in the world that have one SNP,” Mueller said. “We have to be creative and go a new way.” New paradigms are needed that can bring benefits to small groups of people. “That’s where the world will go, and we will be part of it.”












