
Important Points Highlighted by Individual Speakers
• The mobilization of patient communities can spur research and lead to the creation of new and more effective therapies.
• Increased collaboration both within FDA and between FDA and other stakeholders has hastened the approval of new drugs and diagnostics.
• The blending of pre- and postmarket environments could combine considerations of safety and efficacy with considerations of clinical effectiveness.
The fourth session of the workshop featured case studies of organizations and initiatives that have furthered genomic-based approaches to drug discovery and development. A prominent theme of these case studies was the importance of collaboration in building the relationships necessary for timely advances. Collaborations can exist both within an organization, such as within FDA, or among organizations. They also can remain within a single sector or span sectors. As is discussed further in the next chapter, collaborative work will be essential to the creation of new paradigms in drug discovery and development.
FOUNDATIONS AND DRUG DEVELOPMENT: AN EXAMPLE
The Multiple Myeloma Research Foundation (MMRF), which was founded in 1998, is the largest private funder of multiple myeloma research, raising over $190 million to date. It funds research around the world, builds community among people affected by multiple myeloma, and partners with the Multiple Myeloma Research Consortium (MMRC), which supports research in 16 institutions to determine which drugs can move from preclinical testing into Phase I and Phase II development and which was founded in 2004. Walter Capone of the MMRF and the MMRC described the organizations and the vision behind their approach.
Multiple myeloma is the second most common blood cancer, affecting 64,000 people in the United States and causing more than 10,000 deaths annually. It occurs largely among older adults, often African American, and predominantly male. In 1998 the average survival period with the disease was 3 years and no drugs were in the pipeline. Today, the average survival period is 7 years, 4 drugs for the disease have been approved, and 9 drugs are in Phase III trials.
MMRF’s success has been based on increasingly complex collaborative models to drive drug development. By developing strategic plans and building community, MMRC has accelerated trials and built a tissue bank that includes more than 3,500 samples. Through the Multiple Myeloma Genomics Initiative, more than 80 of these samples have been sequenced and are available through an open-access portal, with plans to sequence 250 samples by the end of 2012. More than 1,000 researchers have accessed the data, and more are expected in the future. Interestingly, while expected mutations were found through the genomic analysis, 4 percent of patients also had activating BRAF mutations, compelling the foundation to examine the use of vemurafenib for treatment of multiple myeloma.
This effort in turn has supported the Multiple Myeloma Personalized Medicine Initiative, which seeks to more fully characterize the range of disease subtypes to enable the development of targeted therapies and potentially curative approaches for patients. Spread across 50 centers and including industry partners, the project combines a 1,000-patient longitudinal study with a companion genomics study that will comprehensively assess the molecular profiles of patient’s tumors throughout disease progression and be correlated to clinical interventions, including treatment regimens. The study design allows 3 years for enrollment and includes 5 years of follow-up, and data will be open access with no intellectual property restrictions.
The MMRF is continuing to expand its programs as the network of stakeholders in the field grows more complex. Today, not just academia, industry, and patients are involved but also regulators, physicians, payers, and diagnostic and platform companies. By working with clinicians,
researchers, and a dedicated validation and basic science team, MMRC has increased the success rate of new drugs in Phase I to between 35 and 40 percent, well above the 10 percent success rate in the pharmaceutical industry as a whole. It has partnered with industry to open 37 trials with 20 novel agents, has sped the time to the opening of trials by 60 percent, has reduced the time between Phase I and Phase II trial development and completion by a third through common agreements and dispensing with contracting, and has increased enrollment by 14 percent. MMRC also has expanded its clinical reach well beyond its 16 sites and it has launched an early-access program for the drugs that have gone into the last stages of regulatory review.
The Multiple Myeloma Personalized Medicine Initiative also has taken a collaborative approach. Linking research, clinical, and community activities, it is a multi-year observational study with tissue banking and matching as well as in-depth sequencing work. Information generated by the study is openly available for researchers to identify new targets and biomarkers and to connect researchers with the patient community. The study is putting information in clinicians’ and patients’ hands, Capone said. “Combining clinical and genomic data in a single platform [will] compel and initiate scientific discoveries that are not possible today.”
In the same manner, by mobilizing the multiple myeloma community through a dedicated online portal, the MMRF aims to accelerate and enable personalized therapies. Key features of the online community include groups based on common molecular profiles, the ability to connect with similar patients, a health metrics tracker, tools to help manage the disease, access to educational materials and data, live Web discussions, and clinical trial recruitment tools.
Finally, this approach does not apply just to myeloma, Capone said. It offers a way of bringing information together worldwide from multiple organizations into a common platform that can drive progress for many different diseases. The foremost challenge, Capone said, is collecting and tracking large numbers of patients who have a particular disease or problem. Even in the case of multiple myeloma, only a few thousand patients are being followed. “What if all the patients who were afflicted with a disease were able to become part of and contribute to the community that’s going to drive toward a cure ultimately, and in the process be fully engaged in advocating for their own care by having and understanding the latest advances in the field?”
GENOMICS AND REGULATORY SCIENCE
The mandate of FDA is to protect and promote public health, noted Michael Pacanowski of the FDA’s Center for Drug Evaluation and Research
(CDER). However, a tension often exists between protection and promotion, between risk aversion and innovation, and between regulation and flexibility while still ensuring that safe and effective drugs are brought to market. However, personalized medicine is changing old ways of thinking about these issues and FDA has been on the leading edge of these changes, Pacanowski said.
Genomic-based drug development creates both promise and challenges. Of the approximately 30 drug approvals in 2011, Pacanowski said, at least a dozen had some type of genomic information included in their clinical development, ranging from dosing based on drug metabolism to exploratory analyses for known biomarkers to co-developed drugs. He noted that several recent drug approvals exhibited features that are likely to become increasingly prominent in the future. For example, the approvals of ivacaftor and crizotinib were very fast, taking just 3 months rather than the more common 6 to 10 months. The approval of ivacaftor also took advantage of partnerships with the Cystic Fibrosis Foundation, which greatly helped in bringing the drug to market quickly.
Drug regulation has been described as the progressive reduction of uncertainty, Pacanowski observed. While genomics may alter the current paradigm, it will not change the need to satisfy the same evidentiary standards that currently exist. In that respect, the advantage of genomic-based drug development is not that it requires fewer data, but that it often has the potential for a higher probability of success. Drug development will shift toward a “quick win, fast fail” model, Pacanowski predicted.
One early way in which CDER has stimulated innovation in the genomic sciences is through the Voluntary Exploratory Data Submission program. This program allowed companies to share data informally without regulatory consequences; to obtain feedback on trial designs, methodologies, and data interpretation; to gain insights into evolving regulatory practices; to provide experience to facilitate policy development; to discuss data elements used to streamline new drug applications; to educate FDA scientists on emerging data and innovative approaches; and to forge partnerships among scientists from different sectors. The agency also prepared guidance on genomic data submissions, which helped companies navigate the drug application process, and established a Biomarker Qualification Program, which promoted the development of biomarkers that are broadly applicable to multiple drug developers. Furthermore, recent negotiations over the Prescription Drug User Fee Act have created the potential for funding to enhance the agency’s biomarker and genomic teams.
Internal changes at FDA have spurred these advances. Since 2008, CDER and the Center for Devices and Radiological Health (CDRH) have greatly increased their communication and have harmonized their procedures. In addition, new guidances have been issued on such topics as
developing companion diagnostics and early-stage and clinical pharmacogenomic studies. Currently under development is guidance on enrichment strategies when using selected populations.
Partnerships have been and will continue to be critical at FDA, as emphasized in its most recent strategic plan, Pacanowski said (FDA, 2011). Areas where this is particularly true include the effective development of qualified tools and surrogate biomarkers, creating a drug safety research infrastructure, and carrying out comparative effectiveness research. Such partnerships can take many forms, including industrial consortia, academic collaborations, government-catalyzed partnerships, or contracts with payers to do postmarketing research. Pacanowski noted that developing partnerships with clinical practice societies will be of importance to the agency because these groups will play a large role in determining what is considered standard of care for personalized medicine.
In the past, precompetitive collaborations have been an elusive goal, but barriers are being overcome to establish such partnerships. Successful examples include the international Serious Adverse Event Consortium and the Predictive Safety Testing Consortium from C-Path. “It is possible to put together these partnerships and have effective outputs,” Pacanowski said.
FDA has been and will continue to be committed to personalized medicine and individualized therapeutics, Pacanowski concluded. “It is part and parcel to rational and sound drug development and will probably be applied in almost every scenario in the coming decades.”
PHARMACY BENEFIT MANAGEMENT AND PHARMACOGENOMICS
In the current paradigm of drug discovery and development, the premarket environment and the postmarket environment are separate and distinct (Figure 5-1). Companies try to get regulatory approval for a drug and then hope that patients and providers will use it and that payers will pay for it. Increasingly, there are examples in which there is regulatory success but commercial failure, said Felix Frueh of the Medco Research Institute.
This paradigm will change, Frueh predicted. In the future, the premarket environment and postmarket environment will be blended (Figure 5-2). Companies will receive information from payers early in the drug development process. Considerations of efficacy and safety will interact with considerations of not just clinical utility but clinical effectiveness. The logistics of deploying a new therapy will be a factor—including, for example, off-label uses. The stakeholders in the drug development process will be confronted with a new set of questions, Frueh said, and it will be necessary to assess how this new paradigm will influence drug development.
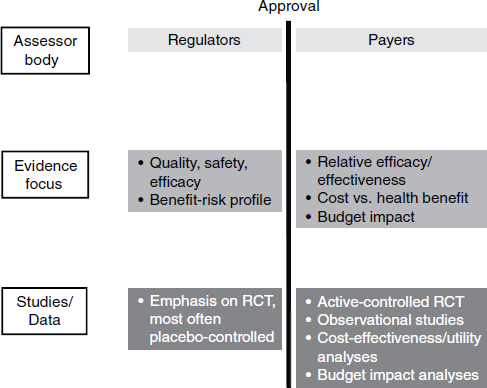
NOTE: RCT, randomized control trial.
SOURCE: Adapted from Eichler et al., 2010.
These changes will affect regulators and payers as much as they will industry and government, Frueh said. Today, regulators are increasingly interested in comparative data and outcomes research. The demand for more safety data cannot be met entirely by randomized controlled trials, so regulators in the United States and Europe have set up sentinel networks to assess postmarket data. Reimbursement bodies are calling for value-based pricing that is tied to the demonstration of comparative effectiveness in the real world. In Germany, for example, the Federal Joint Committee requires drug makers to demonstrate greater efficacy for a new compound before they can charge more.
Strategic partnerships are also emerging to generate and access postmarket data. Pfizer, for example, has teamed with Medco to use large patient databases to perform both retrospective and prospective research on personalized therapies. In this way clinical trials can be designed to answer not only regulatory questions but questions that are relevant for the payer,
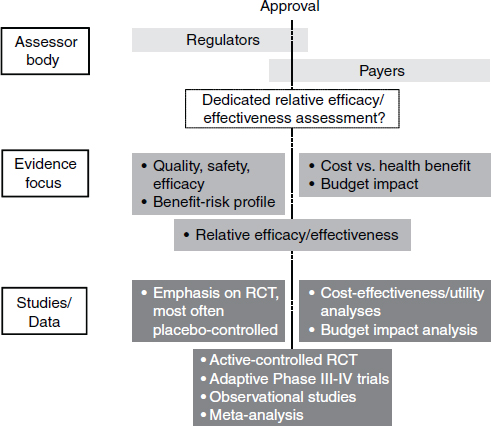
NOTE: RCT, randomized control trial.
SOURCE: Adapted from Eichler et al., 2010.
such as whether the right clinical endpoint has been selected. Similarly, questions of comparative effectiveness can be addressed, such as whether an older drug (for example, clopidogrel) that is about to go off patent is safer and more effective than a new and more expensive drug (for example, prasugrel) for people with particular genetic backgrounds. While not the primary concern of payers, economic questions also take on significance in these analyses, Frueh said. All of this information can also be important for drug developers who need to make decisions about whether and how to proceed with the development of a particular compound. Companies could utilize a personalized medicine methodology to identify an unmet medical need, for example. Developers could approach payers early about creating programs to identify patients who are unlikely to benefit from a drug already on the market but could benefit from a newly developed drug.
Frueh concluded with several provocative thoughts emphasizing the role of payers:
• Comparative-effectiveness evaluations will become increasingly required by payers because of the need to demonstrate that a new treatment is better than the standard of care.
• What if payers were to cover a drug only if it actually works?
• Payers will progressively move toward employing coverage with evidence development.
• Can payers act to encourage patients to participate in clinical trials or even help in recruitment?
• What if payers were to co-sponsor clinical trials or provide pharmacy, lab, and outcome data for research?
• Could payers partner with industry to develop more personalized medicines faster?
Over the next 5 years, Frueh said, the answers to these questions could reshape relationships in the drug discovery and development system. For example, if payers across the board were to embrace the coverage-with-evidence-development paradigm, “that would really change the way that we’d be looking at drugs and diagnostics.”
The NCATS Pharmaceutical Collection (NPC) is a comprehensive resource of 3,800 approved and investigational medicines that was designed to facilitate the repurposing of medicines by the scientific community.1 As a recent paper states, the NPC is “a definitive, complete, and non-redundant list of all approved molecular entities as a freely available electronic resource and a physical collection of small molecules amenable to high-throughput screening” (Huang et al., 2011).
Christopher Austin of NCATS at NIH demonstrated how the NPC can be used. Drugs approved in different jurisdictions throughout the world can be accessed. Searches can look for indication, target, drug name, and so on. A search on “migraine,” for example, returned 14 drugs that are approved worldwide. Clicking on a particular drug gives the mechanism of action, known targets, the regulatory status in different countries, and other information.
To demonstrate the utility of the collection for drug repurposement, Austin cited a recent example of successfully identifying a drug that could potentially be used for the treatment of chronic lymphocytic leukemia (CLL),
![]()
1 The NPC can be accessed at http://tripod.nih.gov/npc.
which accounts for about 15,000 new diagnoses per year in the United States. In partnership with the University of Kansas and the Leukemia & Lymphoma Society, NCATS screened the NPC collection for effects against CLL patient cells as well as against cells from normal donors. Some drugs killed the CLL cells from all patients, while some killed the cells from only some of the patients. Subsets of these drugs were less effective or ineffective in killing normal donor cells. One particular drug called Auranofin was originally approved for the treatment of rheumatoid arthritis in 1984. Reverse pharmacology revealed the mechanism of action of the drug, and three clinical trial sites are now active.
The principal lesson Austin drew from this experience is that effective translation requires collaboration. The partnership benefited by the marriage of funding sources, expertise, project management, and the early incorporation of technology transfer agreements which allowed for rapid movement in establishing the trials. In fact, less than a year passed between signing the partnership agreement and the dosing of the first patients. “This is a great example of how, [through] a team effort, we were able to move forward.”
One complication in the repurposing of drugs is that about 90 percent of the drugs in the pharmaceutical collection are generic. For these drugs, paying for a registration trial to expand the indication can be a barrier. In addition, regulatory issues can impede the repurposing of on-patent or abandoned drugs. For example, one might wish to know if the new indication is related to the original mechanism of action or if it is related to an unexpected or unrelated mechanism. Or if the mode of delivery is the same. To answer such questions, it is typically the case that data are needed from the firm that originally created the drug.
Public policy changes may be necessary to encourage drug repurposing. For example, establishing exclusivity could allow the licensing of a discovery to a for-profit organization to take a drug through registration. Also, it is never too early to start thinking about reimbursement strategies, Austin said, because the goal is to get the drug to patients.
A Value Maximization Path, or ValueMaP, is under development to provide guidance in pursing drug repurposing. This guidance draws on examples of what has worked in previous projects, such as rational repurposing based on knowledge of disease pathogenesis and drug pharmacology. In addition, in selected cases computational approaches have been able to identify promising pathways or patterns (Sirota et al., 2011).
Partnerships need comprehensive and complementary expertise at every step of the process, Austin said. When the process breaks down, it often does so in the experimental medicine space, such as in the early clinical trials. Other problems have arisen when repurposing is based solely on animal models, when computational approaches are used without experi-
mental testing, and when phenotypic screens are done without a prospective plan for translating the results to humans.
Repurposing generic drugs provides a tremendous opportunity to improve human health without great additional costs, Austin concluded. But new funding paradigms may be necessary to make such drugs available to patients.










