
6
Risk Assessment
Risk assessment, as defined by Crouch and Wilson (1987), is "a way of examining risks, so that they might be better avoided, reduced, or otherwise managed." It is a process based on science whose objective is to provide criteria for policy-making. Risk assessment can be qualitative or quantitative. The development of quantitative methods for risk assessment has been most extensive for carcinogenicity, that is, estimating the potential risks of cancer associated with exposure to oncogenic chemicals and other agents. Risk Assessment in the Federal Government (NRC, 1983) and Identifying and Regulating Carcinogens (OTA, 1987) discussed the development of those methods. Compared with the work on risk assessment of neurotoxicants, the development of methods for quantitative risk assessment of carcinogens has been built on a larger body of mechanistic information and hypotheses that permits quantitative description of the cellular and molecular events involved in chemical- and radiation-induced carcinogenesis. But uncertainties still attend almost all risk assessments that involve extrapolation from acute to chronic responses, from response at high doses to response at low doses, from one route of administration to another, and from species (laboratory animals) to species (human).
In neurotoxicology, critical mechanistic information is not available, except for a few substances. Thus, risk assessment in neurotoxicology is still relatively undeveloped. Moreover, neurotoxicologic risk assessment is likely to be highly complex, compared with carcinogenicity risk assessment. Carcinogenesis at the molecular level has been proposed to involve a relatively limited set of cellular events common to the induction of all tumors; although specific carcinogens can act at different stages of carcinogenesis, the process is assumed to be fundamentally similar among tumor types and sites. In contrast, the diversity of cells and processes in the nervous system that are potentially subject to toxic action is great. Moreover, the response of those targets can depend on the developmental stage (including aging) of the organism at the time of exposure (Weiss, 1990). For example, during the cytoarchitectural organization of the human CNS after birth, neurotransmitters can function as trophic factors guiding the migration and localization of cells in brain regions; after synaptogenesis, they function primarily as information transducers (Schwartz, 1985).
An agent (such as a-methyl-p-tyrosine) that interferes with neurotransmitter synthesis during development can cause permanent alterations in regional neuronal organization; the same agent acting on the same mechanisms later in development can interrupt cell-cell communication without causing structural disarray.
No general mathematical model is likely to be appropriate for the quantitative risk assessment of neurotoxicants as a class. Defining and validating risk-assessment methods for neurotoxicants will probably require the development of a set of methods that are restricted in application to specific end points, rather than to specific toxicants. The rest of this chapter discusses some approaches to that task.
The process of risk assessment has been divided into the following stages (NRC, 1983; Cohrssen and Covello, 1989): hazard identification, dose-response analysis, exposure assessment, and risk characterization, followed by risk management. Dividing the process into those conceptual stages can assist in allocating resources and critical analyses to specific aspects of science-based policy analysis. In hazard identification, the goal is to obtain sufficient information to determine the qualitative nature of any biologic activity associated with a specific agent. Short-term tests are useful, including even those with a high rate of potentially false-positive results, such as the Ames bacterial mutation assay used in carcinogen hazard identification. When a hazard has been qualitatively identified, the next stage is to develop quantitative information on the relationship between dose and response. The relevance of the test system to predicting outcomes in humans—such as route and timing of exposure—becomes more important. Differences between conditions of the test and those anticipated to occur in human populations might be unavoidable, so implicit and explicit assumptions as to extrapolation must then be incorporated into experimental design and data analysis. Information on metabolic differences between species can also be relevant. An important outcome of the analysis is the development of sufficient information to evaluate the overall shape of the dose-response relationship (linear, sub-linear, hyperbolic, or sigmoidal), which can assist in generating inferences on likely responses outside the range of measured doses and responses. It should be noted, however, that the shape of the dose-response curve at the low end is often difficult to determine. In the third stage, exposure-assessment data are accumulated on external doses expected to be encountered by humans, including groups whose exposure or response might be expected to be greater than average (for instance, children). In some cases, information on internal dose (and toxicokinetics) can also be collected. Finally, all the preceding information—the nature of the expected outcome, the relationship between dose and outcome (response), and the expected ranges and distribution of doses from individual to individual—are incorporated into a risk characterization.
In the case of dichotomous events, such as the presence or absence of a diagnosable malignancy or death, the process results in a probability estimate that can be expressed as an increase in individual risk or, with appropriate adjustments, as an estimate of increased population risk. An approach based on a yes-no definition of outcome does not provide an accurate quantitation of events that vary in severity. In neurotoxicity, we are almost always concerned about outcomes other than death and effects that vary in severity (unless they are more or less arbitrarily defined in dichotomous terms, such as defining mental retardation solely on the basis of a critical IQ score). Because many neurobiologic functions are expressed on a continuum, it is likely that the effects of many neurotoxicants similarly fall on a continuum of severity. In modeling terms, neurotoxicants act to change the distribution of biologic properties that themselves assume a spectrum of values in a population. Gaylor
and Slikker (1990) have provided an example of a procedure that uses data on neurochemical, neurohistologic, and behavioral effects of exposure to methylenedioxymetham-phetamine (in rats or monkeys) to estimate risk as a function of dose of a potentially neurotoxic substance.
One of the most fully examined instances of this type of neurotoxicity is related to the effects of lead on cognitive function. Most measures of cognition are normally distributed around a population mean (e.g., the Stanford-Binet IQ score has a mean of 100 and a standard deviation of 15). The IQ-lowering effect of lead in children can be detected as a change in mean score (Needleman and Gatsonis, 1990). More important, however, the change in mean implies that the entire distribution of IQ scores in the exposed population is shifted. Although the change in mean IQ in lead-exposed children might seem relatively small (less than 10 IQ points in most studies and so well within the population's normal variability), an examination of the effects of such a decrease on an overall population distribution of IQ scores (Figure 6-1) reveals the potential for very important consequences of low-level lead exposure for society as a whole. The displacement of the overall curve toward lower IQ scores would reduce the proportion of children with superior IQs (i.e., 130 or more) and increase the number of children with seriously compromised intellectual potential (i.e., IQs less than 70).
Current risk-assessment methods based on carcinogenicity risk-assessment models do not capture such types of impacts, built as they are on an assumption that the end point of concern is an all-or-none (dichotomous) variable, rather than a continuous one. The problems of risk management, although not among the specific concerns of this volume, are compounded by the phenomenon of the continuum of both frequency and intensity of response related to dose. At some doses, the "most sensitive" response might be some mild form of neurotoxicity; at another, usually higher, dose, the response might be more serious, more life-threatening, or more intense.
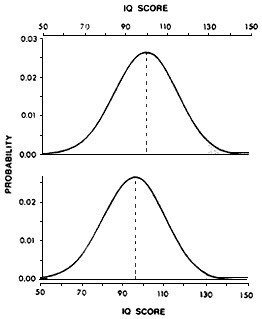
Figure 6-1 Effect of a shift in mean IQ score on the population distribution. The top figure represents a theoretical population distribution of IQ scores with a mean of 100 and a standard deviation of 15. In a population of 100 million, 2.3 million (stippled area) will score above 130. The bottom figure represents distribution of intelligence test scores with a shift of 5%, yielding a mean of 95. Here, the number of individuals scoring above 130 falls to 990 thousand, with a corresponding inflation of those scoring below 70. Source: Weiss (1990).
APPROACHES TO RISK ASSESSMENT FOR NEUROTOXICITY
Risk-assessment models ideally are based on two sources of biologically based information: statistical analyses of rich data sets and knowledge of mechanisms. Approaches involving curve-fitting require a great deal of data to develop predictions of risk with any reliability. Although, in appropriate cases, statistical analysis can be important in validating models based on hypothesized mechanisms of action, the available data are usually insufficient to resolve critical issues, such as the predicted response at exposures below those on which the data were collected. An essential part of developing risk-assessment methods is therefore the integration of biologically based models that incorporate our understanding of mechanism of action. It is important to emphasize that statistical models should conform to biologic hypotheses—that is, curve-fitting must be consistent with what is known of the biology of the affected system.
The most commonly used approach for risk assessment of neurotoxicants, as of most noncarcinogens, is the uncertainty- or safety-factor approach (NRC, 1986; Kimmel, 1990). It is based on identifying either the lowest dose of a toxicant at which adverse effects are observed (lowest observed-adverse-effect level, LOAEL) or the highest dose at which no adverse effects are observed (no-observed-adverse-effect level, NOAEL) and then deriving a presumably safe dose by dividing by a safety or uncertainty factor. Its weakness is related to the unreliability of observation as a means of determining doses at which biologic effects actually do or do not occur. A small study might give negative results, and no study is large enough to exclude the possibility of any effect. Furthermore, the importance of subtle alterations in behavior or small neurochemical or structural changes is a matter of debate. Before undertaking tests to determine the LOAEL or NOAEL, it is important to define what is meant by "adverse effect" and to develop appropriately sensitive measures of such effects. For example, the persistence of the neurotoxic effects of small exposures to such solvents as trichloroethylene, dichloromethane, and toluene is not known, so the importance of observed decrements in performance is still under debate (Gade et al., 1988; Parkinson et al., 1990). This approach also provides no information on the slope of the dose-response curve or intensity of effect above the "safe" dose calculated (Gaylor and Slikker, 1990; Kimmel, 1990; IOM, 1991).
Thus, the determination of a LOAEL or NOAEL is strongly influenced by the sample size and design of the experiment (Gaylor, 1983; Crump, 1984). In most cases, it is difficult to determine an effect smaller than a 20% increase or decrease. As an alternative, Crump (1984) first suggested the use of a benchmark dose (BD), defined as "a statistical lower confidence limit corresponding to a small increase in effect over the background level." The increase in effect used to determine the BD would be near the lower limit of change, which can be determined with reasonable accuracy in toxicologic studies. The BD is calculated with a mathematical model; however, because BDs correspond to risks in the experimental range, they are less affected by the particular shape of the dose-response model used in the calculations. The acceptable dose is extrapolated from the BD. Using a BD, rather than the more usual LOAEL or NOAEL, avoids problems inherent in defining a negative (the NOAEL) and in the lack of precision associated with defining a LOAEL (Kimmel, 1990).
Whether a conventional approach or a BD approach is used, the dose identified is usually modified by some safety factor to yield a so-called safe dose. The incorporation of a safety factor (usually 10 raised to some power) reflects a policy decision to incorporate in a quantitative manner some of the uncertainties associated with risk assessment. In general, safety factors range
between 10 and 1,000 (NRC, 1977; Kimmel, 1990), so the risk assessment is the final estimate of dividing the BD, NOAEL, or LOAEL by 10, 100, or 1,000. Selection of the safety factor involves processes of judgment, including considerations of weight of evidence, availability of information on human response, type of toxicity observed, and probability of variations in response among susceptible groups. Some rules of thumb have been adopted to accommodate intraspecies variability, cross-species extrapolation, experimental duration, etc.; they correspond fairly well to observed variation (Dourson and Stara, 1983).
As noted above, statistical approaches to risk assessment should be consistent with what is known about mechanisms of action or about the biology of the affected system. The NOAEL-LOAEL approach is based on an assumption of a threshold, a dose below which an effect does not change in incidence or severity. However, the evidence of the general applicability of that assumption for all neurotoxicants is relatively weak. Even though a given neurotoxic response may require a threshold dose of a specific toxicant, other toxicants in the environment that cause the same or similar response may in effect lower the threshold dose of the specific neurotoxicant of interest. That is, a person may have a threshold of effect in a pure environment, but there may not be a threshold for a heterogeneous population in a heterogeneous environment. It is likely that some neurotoxicants have thresholds and others do not. (A lack of a threshold for a neurotoxicant makes the safety-factor approach biologically indefensible.) On the one hand, the nervous system appears to have considerable reserve capacity and plasticity, which would support the assumption of a threshold. On the other hand, it is composed of cells that are nonreplaceable, which would argue that no damage can be considered innocuous. Moreover, both susceptibility to injury and the nature of the injury can vary with developmental stage; early neurologic development includes a selective pruning of apparently excess neurons and connections (Rakic and Riley, 1983), whereas senescence includes a progressive loss of specific neurons and an apparently selective loss of function. Because the CNS develops from a limited set of cells early in development, it is appropriate to assume that exposure to neurotoxicants during critical periods of brain development can be without a threshold; data on x-irradiation-induced brain injury support this assumption (Schull et al., 1990). As shown in Figure 6-2, when in utero radiation exposure occurred during the period between 8 and 15 weeks gestation, the effects on intellectual status
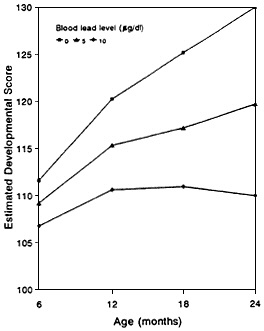
Figure 6-2 Estimated developmental scores at various ages for three blood-lead concentrations. Application of a model developed by Hattis and Shapiro (1990) to data from Bellinger et al. (1987a,b) that assumes the group is exposed to yield constant blood lead concentrations of 0, 5, and 10, µg/dL leads to estimates of developmental scores for populations of exposed children. Source: Adapted from Wyzga (1990).
were shifted to the left, possibly without a threshold. Whether the PNS and CNS differ with regard to thresholds is not known. Studies of PNS neurotoxicants, such as acrylamide, suggest a capacity for recovery through axonal regeneration, but it varies with the dose and duration of exposure; Hattis and Shapiro (1990) have investigated quantitative dose-response models as alternatives to the NOAEL-LOAEL and uncertainty-factor approach for this situation. The recent Institute of Medicine (IOM, 1991) publication on safety of seafood takes a similar quantitative modeling approach for methylmercury. In any event, the application of the NOAEL-LOAEL (or BD) approach to all neurotoxicants is unlikely to be biologically defensible.
CURVE-FITTING IN RISK ASSESSMENT FOR NEUROTOXICITY
When quantal information (the proportion of response at a given dose) is available, but little is understood about underlying biologic mechanisms, a tolerance distribution, such as the probit (log-normal) or logistic models, can be adopted to fit population data (Wyzga, 1990). Those sigmoid dose-response models assume that the distribution of individual thresholds follows the specified model, but that the population as a whole might not have a threshold.
If a substantial body of data is available, it might be possible to test goodness-of-fit of various statistical models. For most neurotoxicants, that is not now possible. Attempts have been made to fit various models to data on the PNS and CNS toxicity of lead. Data on nerve-conduction velocity in children exposed to environmental lead (through living near a smelter) as a function of blood lead concentration fit a ''hockey-stick'' type of dose-response curve (Schwartz et al., 1988). The data were sufficient to permit discrimination, in terms of goodness-of-fit, from several other models, including a logistic model and a quadratic model. In another approach to assessing lead neurotoxicity, Wyzga (1990) assumed that individual thresholds for a dichotomized measure, muscle weakness, were log-normally distributed in the population. After producing maximal-likelihood estimates of the mean and standard deviation of the threshold distribution, the distribution of blood lead concentrations was estimated. Wyzga thereby determined that the probability that a randomly chosen male from the general population would exceed the threshold and exhibit extensor muscle weakness would be 0.01. In a second analysis, Wyzga applied the model developed by Hattis and Shapiro (1990) for acrylamide exposure that compensates for past damage with the rate of repair proportional to the cumulative-damage data on children's blood lead concentrations and mental development gathered by Bellinger et al. (1987a,b). The dose-response estimation related group response to group exposure levels, and Wyzga concluded that this model, albeit only one of many alternative models that could be fitted to the data, indicates some inhibition of development at exposures that resulted in very low blood lead concentrations.
The hockey-stick model was also evaluated for its consonance with the available information on lead's effects and on peripheral neurophysiology. As discussed by Schwartz et al. (1988), it is plausible to propose a threshold for the peripheral neurotoxicity of lead for reasons of both mechanism of action and physiology: lead inhibits synaptic release of acetylcholine through inhibition of ion-dependent neurotransmitter release, and lead reduces velocity of peripheral-nerve conduction (as measured transdermally with surface electrodes). The mechanism of action of lead thus involves events that are nonlinear: the changes in ion permeability that are associated with nerve depolarization and transmitter release involve many discrete events (e.g., channel openings, exocytotic events of electric propagation), and nerve conduction is a sum-
mation involving the function of many nerve fibers. Thus, both basic biology and mechanism of action are consistent with the results of curve-fitting in this case.
MECHANISTIC MODELS FOR RISK ASSESSMENT FOR NEUROTOXICITY
Information on mechanisms of action provides the most powerful tools for developing models for risk assessment (Silbergeld, 1990). For neurotoxicants, it is unlikely that a single mechanism or even a small number of mechanisms will be elucidated for the neurotoxicants already identified, given the variety of potential cellular and molecular targets of action. Nevertheless, in some cases, information is available on mechanisms of action of some neurotoxicants and can be used for purposes of developing quantitative approaches to risk assessment. It must be emphasized that the generalizability of such models is narrow, possibly even for similar molecules, so the approach might be useful only for assessing, case by case, the risks of the substances for which it was developed. Further developments in structure-activity analysis might permit extension of specific mechanistic models to chemicals within appropriate structural classes.
As noted in Chapter 1, many pesticides are designed to be neurotoxic to a target organism. They should be relatively nontoxic to humans in their intended uses, but will be neurotoxic to humans who are sufficiently exposed. The reason is that their mechanisms of action in target species involve biologic substrates that are also present in humans.
For example, the organochlorine pesticide dieldrin once was widely used in agriculture and in household formulations, but now is restricted in application because of its persistence and potential carcinogenicity. Dieldrin acts as a neurotoxicant by blocking the GABA channel in nerve cells (Narahashi and Frey, 1989). It is a competitive antagonist for the recognition site (receptor) associated with the channel, and it can be shown to bind to the site with a high affinity. Assuming that to be its fundamental mechanism of action, one can develop a mathematical model based on the biology of receptor-ligand interactions. Receptor-ligand binding is well described by classical mathematical models derived from Lineweaver-Burk equations of enzyme kinetics (Silbergeld, 1990). (Enzyme-substrate interactions are a class of receptor-ligand interactions, as is carcinogen-adduct binding to DNA.) As with all receptor-ligand interactions, when the concentration of the receptor is greater than that of the ligand at low concentrations of dieldrin, receptor-ligand binding is linearly related to the concentration of the ligand, dieldrin. If receptor binding is directly related to regulation of the ionophore and the function of the ionophore is critical to neuronal function, a linear model based on receptor-binding kinetics can be constructed for the neurotoxic risks associated with dieldrin. The model agrees well with electrophysiologic data. This approach provides a prediction of risk that can be validated in terms of whole-organism response to dieldrin.
It might be possible to model other neurotoxicants that act through receptor mechanisms with similar approaches to quantitative risk assessment. (Table 6-1 lists such neurotoxicants.) Obviously, these approaches describe the predicted behavior of a toxicant only at its cellular site of toxic action, e.g., relations between target-organ dose and response. For risk assessment, knowledge of pharmacokinetics—absorption, distribution, and metabolism—is relevant for estimating the overall relationship between external exposure and target-organ dose (NRC, 1987b).
Another mechanism of neurotoxic action could be applicable to an understanding of the consequences of cytotoxic agents that act on neural cells during development. On the basis of the principle of nonreplaceability, a
TABLE 6-1 Some Neurotoxicants That Act on Receptors
|
Receptor or Channel |
Blocker (Antagonist) |
Modulator (Agonist) |
|
Acetycholine receptor |
Lacticotoxin |
|
|
|
Erabutoxin |
|
|
|
α-Conotoxins |
|
|
|
Anatoxin-a |
|
|
|
Nereistoxin |
|
|
|
Atropine |
|
|
|
Scopolamine |
|
|
Acetycholine-activated channel |
Histrionicotoxin |
|
|
|
Amantidine |
|
|
|
N-Alkylguanidines |
|
|
Excitatory amino acid |
AP-5 (2-amino-5-phosponopentanoate) |
Oxotremorine |
|
|
AP-7 (2-amino-5-phosponopentanoate) |
|
|
|
Nephila orb web spider toxins |
|
|
|
Argiope orb web spider toxins |
|
|
|
γ-Philanthotoxin |
|
|
|
MK 801 |
|
|
|
Ketamine |
|
|
GABAA receptor or channel |
Bicuculline |
N-Methyl-D-aspartate |
|
|
Lindane |
1-Glutamate |
|
|
Dieldrin |
Kainate |
|
|
Picrotoxinin |
Quisqualate |
|
GABAB receptor or channel |
Phaclofen |
Muscimol |
|
|
|
Avermectin Bla |
|
|
|
Barbituates |
|
|
|
Benzodiazepines |
|
|
|
Ethanol |
|
Glycine receptor or channel |
Strychnine |
Baclofen |
|
Presynaptic tunnel |
β-Bungarotoxin |
α-Latrotoxin |
|
|
Botulinum toxin |
|
|
|
Tetanus toxin |
|
|
|
Taipoxin |
|
linear no-threshold model might be appropriate to estimate he effects of cytotoxic agents encountered during critical periods of development. Radiation is the classic example of such an exposure. The results of animal studies are consistent with the hypothesis of a lack of threshold for prenatal irradiation during the critical period of organogenesis of the cortex (Schull et al., 1990). The available human data are also consistent with the hypothesis (Otake and Schull, 1984; NRC, 1990). As shown in Figure 6-3, there is no clear evidence of a threshold for the effects of prenatal irradiation encountered during the critical period of fetal development, with respect to
mental retardation, in children irradiated in utero as a consequence of the atomic bombs in Hiroshima and Nagasaki.
Of greatest interest and challenge in the development of appropriate models is the possibility, discussed in Chapter 2 of this report, that early neurotoxic exposure can result in cumulative or progressive damage expressed as a late-stage degenerative disease, such as dementia or a major motor disorder. Clinical data on the parkinsonism-dementia complex on Guam and on MPTP intoxication in drug abusers in the United States indicate that such damage does occur.
Although models for cumulative damage have not been extensively developed for neurotoxicity, Hattis and Shapiro (1990) exploited the abundance of existing data on acrylamide-induced neurotoxicity in developing a model that is relevant for reversible, cumulative neurotoxic effects. Alternatively, some of the time-to-tumor models (Peto et al., 1980; Krewski et al., 1983) considered in cancer risk assessment might be relevant to such mechanisms of neurotoxicity.
Design and reporting of both animal and epidemiologic experiments could be modified in several respects to facilitate modeling of neurotoxic end points for risk assessment (Wyzga, 1990). It would be useful to conduct studies at a number of carefully measured exposure concentrations, including ones of environmental relevance. Recording of specific quantitative responses is more informative than dichotomizing or categorizing observations, and making data available in nonaggregated form would enable thorough exploration and interpretation of research data by other researchers. Summary statistics are sufficient only if the model assumed to condense the data is appropriate, and that is seldom known with certainty. Comparable sets of animal and human data should be analyzed in parallel, to establish the most effective methods of using animal data when human data are not available.
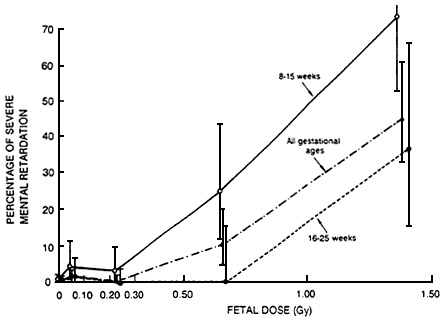
Figure 6-3 Percentage of severe mental retardation among those exposed in utero by dose and gestational age in Hiroshima and Nagasaki. Vertical lines indicate 90% confidence intervals.
Source: NRC (1990).
SUMMARY
The critical focus for basic and applied research in neurotoxicology remains in the primary stage of risk assessment, that is, hazard identification. Few chemicals have been adequately tested for potential neurotoxicity, and data even on many of those are still insufficient for evaluating risk. In basic research, we still need investigations to develop models for integrating molecular, biochemical, cellular, and organ-level events into rational explanations of behavior, function, and learning. Applied research is needed in both clinical and experimental disciplines, to develop appropriate methods for the detection of neurotoxicity, particularly in developing and aging organisms. Efforts in the risk assessment of neurotoxicants must focus first on improving hazard identification. Concerted efforts at improving and validating tests and increasing the vigilance and sensitivity of epidemiologic surveillance must be undertaken. With improved hazard identification, more data can be accumulated on dose-response relationships for particular neurotoxicants. The increased data base might make it possible to undertake additional exercises in statistically based modeling and curve-fitting. At the same time, further basic research in neuroscience will improve our understanding of the cellular and molecular biology of the sites of action at which toxicants affect the nervous system.
The visual and visuomotor systems hold great promise for the integration of toxicity data and fundamental neurobiology. Some chemicals affect vision, others perturb visuomotor control, and some affect both. The great increase in knowledge about the mechanics of these systems has enabled researchers to develop highly sophisticated models, including the neurobiology of the retina (e.g., Stryer, 1983), the organization of the visual cortex (e.g., Hubel and Livingstone, 1987), and the neuromotor control of visual tracking (e.g., Fromm and Evarts, 1981). At least two neurotoxicants—methylmercury (Friberg, 1977) and methanol (Politis et al., 1980)—affect visual function. However, no attempt has yet been made to test the fit of existing toxicity data on those two agents to any of the complex neurobiologic models to predict the results of small exposures or exposures at different stages of development.
Mechanistic understanding is the most powerful tool in developing models for quantitative risk assessment, for evaluating predictions of dose-response relationship outside the measurable range, and for extrapolating across species reliably. Model-building in science is important in identifying critical data gaps. Neurotoxicants have always served an important role as tools in basic neurobiology (Narahashi, 1989); and basic neurobiology is essential in neurotoxicity risk assessment. Attempting risk assessment for neurotoxicants can assist in the identification of critical data gaps to be addressed in developing and validating mechanistic models of neurotoxicity. For instance, one important general issue in neurotoxicity risk assessment is the potential irreversibility of damage. There are reasons to anticipate that neurotoxicity will be irreversible or will be reversible, depending on which components of the nervous system are affected and at what stage of development or senescence. Research is needed to identify which elements of the CNS are irreplaceable and how irreplaceability might change to affect reversibility. The research will also increase our understanding of neurologic development. Similarly, we know little about processes of aging in the brain. If some brain regions inevitably lose neurons, are they more susceptible to neurotoxic damage, particularly of the delayed type? Would such damage be appropriately estimated by a linear approach based on the assumption of additivity to background?
The greatest need in developing quantitative approaches to risk assessment of neurotoxicants is for data on neurotoxic agents. The intensive study of well-charac-
terized neurotoxicants can be of great importance, as demonstrated by recent research on lead and n-hexane. In studies of neurotoxicants, attention should be given to the concentration, duration, and timing of exposure. Especially rich data sets on specific neurotoxicants should be examined for opportunities to test goodness-of-fit (statistical) models, as has been done for lead.
Efforts should be directed to developing a broader range of biologically based models for risk assessment of neurotoxicants, with emphasis on nondichotomous events. It might be useful to examine the research on model-building that is already under way in neuroscience, particularly cognitive science. Caution should be exercised in relying on simple uncertainty-factor and threshold models for the risk assessment of all neurotoxicants under all exposure conditions in all populations.
Basic neurobiologic research (e.g., research on the neurobiology of cortical and cerebellar development and on the neuronal changes in the aging brain) should include investigation of the effects of toxicants. Research should be undertaken to further understanding of the role of pharmacokinetics in neurotoxicity so as to encourage the development of physiologically based pharmacokinetic modes such as are being developed to aid in carcinogenicity risk assessment, as well as of the differences and similarities between animals and humans with regard to neurotoxicity. Research should also be undertaken to test the utility of cellular models, such as receptor-binding models, for hazard identification and for predicting an organism's overall response.












