
Tools for Generating and Synthesizing Evidence
Important Points Emphasized by Individual Speakers
- Test development needs to be rigorous, using meaningful and well-designed studies, proper statistical analysis, independent external validation, and interdisciplinary expertise.
- The identification and dissemination of best practices for the entire pathway of test development ensures that everyone understands the test development process.
- Clinical utility needs to receive earlier and more intense focus, with more education about how to interpret the results.
- Appropriate control groups are important to determine whether a biomarker distinguishes a group that benefits from a treatment.
- Studies of clinical utility should be conducted in settings that are relevant to more real-world clinical decisions.
- Focusing more on value than on cost-effectiveness in assessments of molecular diagnostics will enable analyses to be descriptive in addition to prescriptive and will allow consideration of the full context of care.
- The collection of blood and tissue from every cancer patient, including those who die, could greatly advance research.
Five speakers covered diverse aspects of the development of methodologies and tools that, as the statement of task put it, are related to demonstrating the evidentiary requirements for clinical validity and clinical utility that meet the needs of all stakeholders. Discussions included guidelines for test development, the role of comparative-effectiveness research (CER) in demonstrating clinical utility, statistical techniques, cost-utility analyses, and innovation mechanisms in small companies. Common themes included the need for clearly defined standards for analyses, the importance of context in determining clinical utility, and the importance of access to welldocumented biospecimens.
Debra Leonard, professor and vice chair in the Department of Pathology and Laboratory Medicine and director of the Clinical Laboratories at Weill Cornell Medical Center, summarized the findings of a recent Institute of Medicine (IOM) report titled Evolution of Translational Omics: Lessons Learned and the Path Forward (2012b). The report was written by an IOM committee in response to the development of gene-expression array tests at Duke University that were said to predict sensitivity to chemotherapeutic agents. Papers written about the tests suggested that they represented a major advance and would better direct cancer therapy. Clinical trials were initiated in 2007, with the tests being used to select which chemotherapeutic agent patients would receive.
A paper by Baggerly and Coombes (2009), however, pointed to numerous errors and inconsistencies in the data and stated that the results could not be reproduced. Following a 2010 letter from more than 30 bioinformaticians and statisticians to the National Cancer Institute (NCI) urging the suspension of the clinical trials and an investigation of the test and computational models by the NCI, the clinical trials were stopped. The NCI then asked the IOM to review the situation and provide guidance for the field.
The IOM committee was charged with recommending an evaluation process to determine when omics-based tests are fit for use in a clinical trial. It also was asked to apply these criteria to omics-based tests used in the three cancer clinical trials conducted by the Duke investigators and to recommend ways to ensure adherence to the developed framework.
A Recommended Framework
An omics test is defined as being composed of or derived from multiple molecular measurements and interpreted by a fully specified computational model to produce a clinically actionable result (IOM, 2012b). The test can assess genomics, transcriptomics, proteomics, epigenetics, and
so on. Characteristics of an omics test include the use of complex, highdimensional datasets and interpretation by a computational model, with a high risk that the computational model will overfit the data. The term is not meant to apply to a single gene test or to noncomplex testing, though Leonard added that she believes the committee’s findings should apply to the development of any test.
The committee developed a recommended framework for the evaluation of omics tests from discovery to clinical use (see Figure 4-1). The framework begins with a discovery phase in which a candidate test is developed on a training set of data. Then the computational model is fully defined and locked down. The testing method and computational model are subsequently confirmed on a separate set of specimens or a subset of samples from the discovery set that were not used for training. If the test is intended for clinical development and eventual use, the data, computer code, and metadata should be made available to the public. The candidate test should be defined precisely, including not only the molecular measurements and computational model but also the intended clinical use for the test. This is standard for FDA, Leonard explained, but academic investigators tend not to think about how a test will be used in the clinic.
After the discovery phase, the committee concluded, test validation should be done under approval from an institutional review board (Jacobs et al., 1990) and in consultation with the FDA. The clinical testing method should be defined along with the analytical validation or confirmation of the analytical performance characteristics of a test. This can be done in a looped process, Leonard said, in which the test is modified to achieve the desired analytical performance. The defined test method should then be used on a validation sample set, with the intended use, assay, computational procedures, interpretation criteria, and target patient population all defined. The sample set can come from the discovery phase if the samples were from patients. If the discovery phase was done on cell lines or samples that were not like the patient samples, however, then validation needs to be done on a patient sample set. If the sample set is annotated with patient treatments and outcomes, it needs to be blinded to those doing the testing.
Once the test is defined and locked down, validation cannot be done iteratively. Rather, if the test needs to be changed, it must then begin the validation phase anew.
The committee recommended that the test be discussed with the FDA prior to the validation studies to learn what the FDA would want to see for approval of the test. The test development and validation should be performed in a Clinical Laboratory Improvement Amendments (CLIA)-accredited clinical laboratory if the test is intended to direct patient management. The CLIA laboratory should design, optimize, validate, and
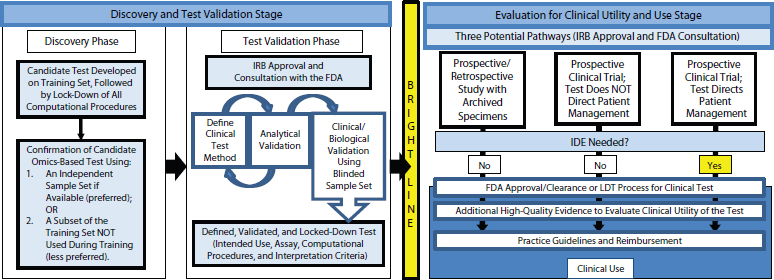
FIGURE 4-1 The recommended framework for the evaluation of omics tests extends from discovery to clinical use.
NOTE: FDA, U.S. Food and Drug Administration; IDE, investigational device exemption; IRB, institutional review board; LDT, laboratory-developed test.
SOURCE: IOM, 2012b.
implement the test under the current clinical laboratory standards, and CLIA requirements should be met by each laboratory in which the test will be performed for the clinical trial.
Pathways to Clinical Utility
During the final stage—which is separated by a bright line from discovery and validation—the test is evaluated for clinical utility and use. Clinical utility is not assessed by the FDA or in the LDT process, so the committee recommended that the process of gathering evidence to support clinical use begin before the test is introduced into clinical practice.
Three potential pathways are available for developing evidence of clinical utility, Leonard observed. Prospective or retrospective studies can be conducted using archived specimens from previously conducted clinical trials. Also, prospective clinical trials can be performed where either the test does or does not direct patient management. Whether test results direct patient management affects both the design of the prospective clinical trials and where the test is done. Regardless of the chosen method, the study or trial should receive approval from the institutional review board, and the FDA should be consulted. For investigators conducting a clinical trial in which the test will be used to manage patient care, the committee recommended that they communicate early with the FDA regarding the process and requirements of an investigational device exemption. Omics-based tests should not be changed during the clinical trial without a protocol amendment and discussion with the FDA. A substantive change to the test may require restarting the study, noted Leonard.
If supportive evidence is generated using these pathways, FDA approval or clearance can be sought or the test can be further developed as an LDT, said Leonard. Evidence can continue to be generated during this period to facilitate coverage and reimbursement discussions with payers and adoption into clinical practice guidelines.
Concluding Observations
The test development pathway is segmented, and different parts of it are done by different groups, especially in the academic environment, Leonard noted after describing the committee’s recommendations. But oftentimes the groups do not fully comprehend the impact they have on each other. The IOM report, by describing the entire pathway of test development, defines best practices so that everyone can understand the interrelatedness of the test development process.
Unfortunately, the report does not look in depth at the barriers to
the recommended pathway, Leonard noted. In addition to the cost of the clinical trial, test validation and development is expensive, and how this expense will be covered is not clear. Also, there is a lack of availability and access to annotated specimens and datasets. The National Institutes of Health (NIH) does not routinely fund the maintenance and biobanking of specimens along with associated clinical data. Finally, there is no process for establishing whether a test will be covered or the level of payment that will be received for a test.
Leonard also suggested the idea of a clearinghouse that holds data gathered from various sources to determine whether a product has clinical utility. These data could be used both in guidelines development and in deciding whether to cover or not cover the clinical use of a molecular diagnostic.
THE ROLE OF COMPARATIVE-EFFECTIVENESS RESEARCH
One way to build the evidence base for decision making in cancer genomic medicine is through CER, said Andrew Freedman, chief of the Epidemiology and Genomics Research Program’s Clinical and Translational Epidemiology Branch at the NCI. CER is intended to create evidence for decision making by finding out “what works” in health care. According to the IOM (2009, p. 13), “CER is the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition or to improve the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policymakers to make informed decisions that will improve health care at both the individual and population levels.”
One problem with CER is that it typically focuses on average treatment effects. Some treatments may have a significant effect across a broad population but may in reality only benefit particular patients and not others. Alternatively, a treatment may not reach significance and be considered ineffective when looked at across a large population, but in fact work for a certain subpopulation. The failure to recognize the heterogeneity of treatment effects can undermine the interpretation of the clinical trial results and the generalizability of those findings to patient populations. In cancer genomics, the goal is to figure out, for patients with a similar diagnosis, what tumor markers or genomic markers predict who will respond to treatment, who will not respond to treatment, and who will have adverse effects.
The methods used in CER are typically the same as in traditional genomic studies (see Table 4-1), but CER has a different orientation in that it encourages the stakeholders—including patients, clinicians, and payers—to prioritize research to help deliver the answers they need. The stakeholders identify the questions that would generate the needed evidence, and a
TABLE 4-1 Comparative-Effectiveness Research Versus Traditional Studies of Genomic Tests for Cancer
| Feature of Research | Comparative-Effectiveness Research | Traditional Studies |
| Priority of study among alternatives | Determined by multiple stakeholders, using criteria such as disease burden or cost, lack of information, variability in care | Opportunity as dictated by expert assessment of emerging technology |
| Study design | Retrospective or prospective analysis | Retrospective analysis of existing tumor specimens; occasional prospective analysis of observational data |
| Comparisons | Direct comparisons of new therapy with usual care | Direct comparisons of competing therapies, often not considering usual care |
| Topics | Prevention, treatment, monitoring, and other broad topics | In most cases, prediction of narrow effects such as serious drug interactions, response to treatment, tumor recurrence |
| Perspectives | Multiple, including clinician, patient, purchaser, and policy maker | Clinician and patient |
| Study populations and settings | Representative of clinical practice | Highly selected |
| Data elements | Patient characteristics, quality of life, safety of treatment, resource use and costs, patients’ preferences | Patient characteristics, clinical end points |
| Funding | “Coverage with evidence development” programs, public–private partnerships | Private investors, research grants from federal sources such as the National Institutes of Health |
SOURCE: Ramsey et al., 2011.
synthesis of the evidence then informs clinical practice. CER emphasizes new therapies in usual care rather than in highly selected clinical trials, because the studies need to be relevant to clinical practice and not look just at clinical end points. Important considerations include quality of life, resources used, costs, and patient preferences, among other factors.
Types of Randomized Controlled Trials
In a randomized controlled trial (RCT), patients are randomized into two groups on the basis of either the treatment or the genomic test or marker. Freedman focused particularly on several varieties of RCTs as sources of evidence for both traditional genomic studies and CER. He divided RCTs into explanatory RCTs,1 adaptive clinical trials, pragmatic clinical trials, and cluster randomized trials (Meyer, 2011). For example, the demonstration that HER2-positive patients benefited from treatment with Herceptin compared with others was the classic example in cancer genomics of an explanatory RCT (Smith, 2001).
Adaptive clinical trials are “learn-as-you-go trials” where the biomarker, the treatments, or both are changed as the trial progresses and more results become available, said Freedman. One or more decision points are built into the trial design for analysis of outcomes and associated patient or disease characteristics to identify subgroups that are responding favorably.
Pragmatic clinical trials—also called practical clinical trials, effectiveness trials, or large simple trials—are designed to help decision makers choose between options for care in routine clinical practice. These trials include a broad range of health outcomes, including morbidity, quality of life, symptom severity, and costs and are similar in many ways to explanatory trials. A purely pragmatic trial is not necessarily looking for regulatory approval of efficacy; rather, it is trying to figure out what works in the real world. It may not have an ideal experimental setting and is aimed more at normal practice. Generally, such trials have broader eligibility criteria that are not highly restrictive. An example is the ongoing 4,000-patient RxPONDER trial that is looking at the use of Oncotype DX to predict chemotherapeutic benefit for patients with estrogen receptor (ER)-positive/ HER2-negative breast cancer where the cancer has been detected in one to three lymph nodes (Wong et al., 2012). Patients are being randomized to receive either chemotherapy and endocrine therapy or just endocrine therapy in order to identify the best cutoff point for use of the Oncotype DX recurrence score. It has elements of not only a traditional explanatory RCT but also a pragmatic trial. It convened a stakeholder group to identify
______________
1 Explanatory RCTs examine an intervention under ideal circumstances while pragmatic clinical trials assess interventions under real-world circumstances.
end points and the elements that would make the trial more relevant to current practice (Ramsey et al., 2013). The setting is reasonably representative of general clinical practice, as opposed to a more ideal clinical trial setting. A fairly simple intervention is being compared with usual care, and insurance firms are helping to pay for some of the tests. The outcomes are the cutoff point and disease-free survival, but quality of life, decision making, and cost-effectiveness are also being examined.
Cluster randomized controlled trials are a type of pragmatic trial in which social units or clusters rather than individuals are randomly allocated to use of a treatment. For example, such a trial can be randomized on clinical practices, where some practices deliver the intervention and some do not. Relatively few of these have occurred in oncology, especially with biomarkers.
Observational Trials Versus Randomized Controlled Trials
Observational studies are a valuable and complementary approach to generating evidence, said Freedman. These studies are nonrandomized and include retrospective analysis of biospecimens from RCTs, retrospective and prospective cohort designs, studies based on registries, and studies with case-control designs. The strength of the evidence increases in the progression from observational studies to pragmatic RCTs to explanatory RCTs. External validity generally gets stronger in the opposite direction, however. Trade-offs, therefore, have to be made in the decisions of which type of study to use.
Freedman proposed several sets of criteria that can be used to make these decisions. RCTs may be more suitable to determine comparativeeffectiveness in genomic medicine in the following situations:
- When decisions require the highest level of certainty.
- When detecting small or modest differences in the results of treatment or testing.
- When ensuring high levels of internal validity (by controlling for selection bias, patient compliance, and other confounding factors).
- When accessible biospecimens are required for all participants.
- When detailed information on outcomes is needed.
- When genomic markers are incorporated in the design.
- When examining complex testing of multitherapy treatments.
Observational studies may be more suitable in the following situations:
- When study populations are not represented in RCTs, as with patients distinguished by age, comorbidities, or medications.
- When larger studies and diverse populations are needed, especially when looking at rare outcomes or the analysis of subgroups.
- When long-term follow-up is needed.
- When an RCT is not ethical or feasible.
- When testing or treatments are used off label.
- When comparing outcomes from multiple treatment regimens.
- When detecting larger differences in the results of treatment or testing.
- When confirming results from RCTs.
- When generating hypotheses to be tested in RCTs.
- When study results need to be generalizable.
- When study results are needed quickly.
- When treatment adherence differs.
An RCT is neither desirable nor feasible in every circumstance, said Freedman. High-quality observational study designs and evidence of underlying biological mechanisms can contribute to the evidentiary framework. For example, large prospective cohort studies with very large effects and compelling data can make a convincing argument for clinical utility even if the evidence was only generated with observational studies. The major concerns about using observational studies to inform clinical utility are that they can be poorly designed, their findings may be difficult to replicate, reliable outcome measures may be difficult to obtain, and they may be subject to bias and confounding through such factors as selection, response, adherence, attrition, or misclassification.
These limitations have not been lost on epidemiologists, and several groups are now working on guidelines to ensure high-quality studies that can be replicated. With bias and confounding, for example, new techniques such as instrumental variables or propensity score matching can adjust for the nonrandomization of subjects studied. Also, sensitivity analysis can assess the variability in results.
Considering the Context
Health policy decisions have to take into consideration the clinical context, the type of genomic application, the quality and availability of evidence to assess a marker’s benefits and risks, and the risk to patients that a wrong decision could pose, Freedman said. Clinical utility is particularly difficult to determine because it encompasses the context in which the application is being used. Strong evidence is critical, but situations in which the benefits outweigh the risks often have to be weighed on a case-by-case basis.
A comprehensive approach is needed to resolve questions about the
clinical utility of genomic applications, said Freedman. He offered the following suggestions:
- Future research should consider more outcomes measures and be conducted in settings that are relevant to more real-world clinical decisions.
- A multitude of stakeholders having a role in evidence generation could lead to better studies.
- New strategies involving transformation of the research infrastructure to “learning systems” could allow continual additions to the knowledge base.
- Any changes to the evidentiary framework should uphold rigorous best-research practice standards.
- Collaborations among cancer centers are essential, particularly to investigate rare cancers.
- Clear priorities for CER could ensure that limited resources are used to resolve the most compelling questions.
- An evidentiary framework needs to articulate the minimal evidence necessary before clinical application is warranted.
DESIGNING STUDIES TO EVALUATE BIOMARKERS FOR CLINICAL APPLICATIONS
Lisa McShane, senior mathematical statistician in the Biometric Research Branch, Division of Cancer Treatment and Diagnosis, at the NCI, provided a statistician’s perspective on the issues associated with the development of genomic-based tests. There are “widespread problems in the literature,” she said, and everyone associated with the development of these tests needs to understand how to ensure the quality and interpretability of the studies evaluating them.
Molecular diagnostics have a range of potential roles in medicine (see Figure 4-2). McShane focused on two of those roles: as a prognostic indicator and as a predictive indicator. Prognostic indicators are molecular signatures associated with clinical outcomes in the absence of therapy or with a standard therapy that all patients are likely to receive. In the latter case, an untreated population generally will not exist.
Predictive indicators are molecular signatures associated with the benefit or lack of benefit—or potential harm—from a particular therapy relative to other available therapies. In the simplest case, a patient group with a particular biomarker may benefit from a therapy while a group without that biomarker may not benefit.
When is a prognostic test clinically useful? McShane asked. First, statistically significant does not mean clinically useful. “The literature is abso-
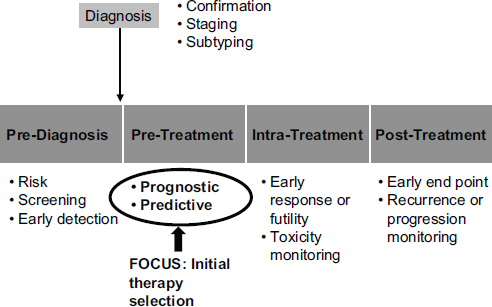
FIGURE 4-2 Potential roles for molecular diagnostics in medicine extend from prediagnosis to posttreatment.
SOURCE: Lisa McShane, workshop presentation, May 24, 2012.
lutely polluted with studies that show that a biomarker or a genomic test has some kind of prognostic information in it. But very, very few of those will ever make it into anything that is clinically useful,” she said. If a prognostic marker can split a patient population into two groups, one of which has an outcome so much better than the other that different treatment decisions are made, then that might reach the necessary level of utility. An example is the Oncotype DX test, which can identify patients who have such good outcomes after surgery that they do not need chemotherapy. A test may also split a patient population into two distinguishable groups that both have bad outcomes, however. Unless the test can guide treatment or monitoring decisions, that information may not be clinically useful. Basing clinical decisions on results from such a test may rest on very little evidence.
Test developers also can confuse prognostic and predictive markers, said McShane. A marker may be prognostic of outcomes, but a new treatment may have the same effect in people with or without the marker. This dichotomy emphasizes the importance, said McShane, of appropriate control groups to determine whether a marker is predictive and distinguishes a group that benefits from a treatment.
To determine when a predictive test is clinically useful, a treatment-by-marker interaction can be assessed, but care needs to be taken in inter-
preting the results. One should not simply rely on statistically significant p-values, McShane said. For example, a new treatment may help only people with a particular biomarker, or it may help people regardless of biomarker status but to different extents. In the latter case, it might be beneficial to give everyone the new treatment, depending on such considerations as toxicity, cost, and patient preferences.
Prospective Versus Retrospective Studies
Prospective studies to establish the clinical utility of molecular tests are much easier to design than retrospective studies, McShane observed. They can use unbiased patient cohorts and adjust for standard variables, and several design options are available to answer specific questions (Freidlin et al., 2010; IOM, 2012b). The challenges with these studies tend to be their feasibility and cost.
Retrospective studies can provide a high level of evidence if they are performed properly. For example, a prospective-retrospective study can produce high levels of evidence for the value of a test if specimens were collected carefully in a clinical trial, if a sufficient number of representative specimens were collected, if the assay was analytically validated, if an analysis plan was prespecified, and if results were validated in one or more similar but separate studies (Simon et al., 2009).
Unfortunately, McShane said, many retrospective studies are poorly conducted. There may not be sufficient specimens available from completed trials or there might not be any suitable trials to address the question of interest. The retrospective study design may be flawed. Patient characteristics may be heterogeneous, and treatments may be unknown, nonrandomized, and not standardized. Specimens may be poorly characterized and accompanied by data of uncertain quality. And the studies may be subject to misinterpretation or deficient reporting. As an example of the problems that can arise, McShane quoted from the conclusion of an American Society of Clinical Oncology update of the recommendations for the use of tumor markers in breast cancer (Harris et al., 2007, pp. 5287–5289): “The primary literature is characterized by studies that included small patient numbers, that are retrospective, and that commonly perform multiple analyses until one reveals a statistically significant result. Furthermore, many tumor marker studies fail to include descriptions of how patients were treated or analyses of the marker in different treatment subgroups.”
A major problem is that, as McShane said, “if you torture the data long enough, they will confess to anything.” For example, multiple testing of many markers, many end points, many subgroups, and so on can produce false positives. Such testing is particularly problematic when there is no prespecified analysis plan and findings are selectively reported on the
basis of statistical significance. “People will do a zillion analyses, and what they will put into the paper are the ones that came up significant,” she said.
Model overfitting is also a major problem, especially with highdimensional marker data such as those generated by omics technologies. With overly complex models that have too many parameters or predictor variables, the model will describe random error or noise instead of the true underlying relationship. Similarly, if there are many more variables than independent subjects or if the data are sparse in a high-dimensional biomarker space, a model will generally have poor predictive performance on an independent dataset.
Model validation is essential, said McShane, yet many researchers make errors when attempting validations. In particular, as discussed by Leonard, they often use the same dataset to validate the model that they used to train it, even though such resubstitution estimates of model performance are useless (Subramanian and Simon, 2010). There are ways to do internal validation of models using data from the same sample set, but they require careful planning (e.g., Molinaro et al., 2005).
McShane also described several other common problems with evaluations of predictive tests. Randomized clinical trials adequately powered to detect treatment effects are often not sufficiently powered to establish predictive marker effects. For example, the nonsignificance of a treatment effect in a “marker negative” subgroup is often misinterpreted as no treatment effect, even though the test may not be adequately powered to exclude a treatment benefit. Also, sufficient information is sometimes not reported in studies to know whether an effect is meaningful. The p-value does not have much meaning without looking at an estimated effect size along with a measure of its uncertainty (e.g., a confidence interval).
Improving Assessments
McShane had several ideas about how to improve assessments of predictive tests. One is to place earlier and more intense focus on clinical utility, with more education about the proper interpretation. The test-development process and study design need to be rigorous, using meaningful and welldesigned studies, proper statistical analysis, independent external validation, and interdisciplinary expertise.
A biomarker study registry, as suggested by Andre et al. (2011), could aid in identifying relevant biomarker studies for overviews and metaanalyses. It also could make study protocols available, including prespecified analysis plans, and help reduce nonpublication bias and selective reporting. Several sets of guidelines exist that could encourage more complete and transparent reporting (Altman et al., 2008, 2012; McShane et al., 2005; Moore et al., 2011b).
The Worldwide Innovative Networking consortium in personalized cancer medicine has provided seed funding to establish such a biomarker study registry. The aim is to create an entity similar to the website clinicaltrials.gov. Just as journals require that a clinical trial therapy protocol be registered with clinicaltrials.gov from the start, that would happen with biomarker studies. This would provide a placeholder for studies that do not result in publication. It also would allow the distribution of prespecified analysis plans, thus addressing not only nonpublication bias but also selective reporting in published papers.
Finally, expanded access to useful specimens, including alternative sources of specimens because trial specimens are optimal but limited, would be especially useful. Specimens should be well annotated with clinicopathologic data, treatments, and clinical outcomes. Health maintenance organizations and other large health care entities could be important partners in such efforts because people move around, which makes it difficult to piece together a patient’s data for retrospective analyses.
McShane cautioned that we do not know the optimal way to collect a specimen and preserve it for every technology that might appear in the next several decades. Also, she has been involved with large biobanks of specimens that do not get used for a variety of reasons. But representative specimen collections from populations for which the tests might be used, along with data on treatment, outcome, the handling of specimens, and so on, are an important place to start. Adequate funding for collection and storage of annotated specimens from clinical trials and other carefully followed cohorts would also be very helpful, she added.
ASSESSING THE VALUE OF ONCOLOGY-BASED MOLECULAR DIAGNOSTICS
In theory, molecular diagnostics should save money or at least provide better care at lower cost. But some observers have called personalized medicine either a myth or hype, and others have declared it unaffordable, said Kathryn Phillips, professor of health economics and health services research at the University of California, San Francisco. Others have called more testing the medical equivalent of Moore’s law in computing, claiming that testing causes more visits to the doctor, which results in exponentially more visits to the doctor. “There’s a lot of debate regarding whether molecular diagnostics are really ever going to provide cost-effective care,” she concluded.
Phillips conducted an informal study using data from the Tufts registry of cost-utility analyses to learn what has already been done on the
economics of molecular diagnostics for cancer.2 More cost-utility analyses are available now than in the past, with about 14 percent focusing on cancer. The cost-effectiveness of diagnostics used to treat cancer is similar to that of other conditions. About half have a reasonable incremental costeffectiveness ratio, but only about 10 percent of the analyses demonstrate that these interventions save money, Phillips said. Another 10 percent of the studies concluded that the interventions cost more and provide less health benefits than the standard of care, and these studies generally were done before the recent increase in high-cost diagnostics and cancer drugs. “In general, new health-care interventions do not save money,” said Phillips. “They provide better health [benefits] at a reasonable cost.” Furthermore, the cost-utility analyses of 64 molecular diagnostics for cancer were even less encouraging, with 20 percent costing more and resulting in less health benefits.
Challenges to Cost-Effectiveness Analyses
Phillips listed several well-known challenges to the use of costeffectiveness analyses:
- The lack of data on effectiveness and costs.
- The need to consider the effect of the diagnostic on downstream decisions and outcomes.
- No or limited use of cost-effectiveness analyses by stakeholders.
She noted that diagnostics are complicated to analyze because of the evolving nature of the field, the complexity of the tests, the uncertainties surrounding them, and the nature of the disease. Also, because cancer can be an inherited condition, diagnostic tests may require consideration of family members. For example, a cost-effectiveness analysis of Lynch syndrome screening found that the screening is only cost-effective if family members change their behavior (Ladabaum et al., 2011). “And that’s a big if,” said Phillips. “If you are just looking at what happens to the proband, then you shouldn’t be doing Lynch syndrome screening.”
Whole-genome sequencing will be the next big dilemma, Phillips said. The complexity is far worse because of the huge amount of data available. Information may range from clinically actionable to not directly clinically actionable to unknown or no clinical significance, with various levels of risk and possible outcomes.
______________
2 The registry is available at https://research.tufts-nemc.org/cear4/Default.aspx (accessed August 10, 2012).
A Focus on Value
Phillips recommended that future analyses focus not only on costeffectiveness but also on value. Cost-effectiveness analysis is “a hard sell,” she said. Methodological concerns are common, such as defining and measuring quality-adjusted life years. Furthermore, in the United States, there is a lack of support for explicit consideration of cost.
Different frameworks for assessing value exist, said Phillips. Costeffectiveness analysis can compare one alternative to another, but without the context. Alternatively, just the benefits and risks can be compared. Still, neither of these takes into account the larger impact of the technology on the health care system. For example, a technology may have a good cost-benefit ratio but have little impact because only a few individuals are being treated. A variety of methods can be used to capture the magnitude and scope of the technology being examined, said Phillips. Also, budget impact analysis can be used to assess whether a technology is affordable, for example, within a particular health plan.
As an example of a process for assessing value, Phillips laid out potential steps for conducting a multicriteria analysis:
- Establish a decision context. What are the aims? Who are the decision makers?
- Identify options.
- Identify objectives and criteria that reflect the value associated with the consequences of each option.
- Describe the expected performance of each option against the criteria and score options.
- Assign weights for each criterion to reflect relative importance.
- Combine weights and scores for each of the options to derive overall value.
- Examine the results.
- Conduct sensitivity analyses of the results.
She noted that this type of framework could be used to make systematic decisions about new health care interventions in a way that still captures costs and benefits but does not solely focus on these variables.
Phillips pointed out that cost-effectiveness analyses based on the ideal world may not adequately reflect actual implementation. Cost-effectiveness analyses are often normative—demonstrating what should be better—when they need to be descriptive, that is, taking into account the full context of care. For example, Elkin et al. (2011) demonstrated that few costeffectiveness analyses of breast cancer diagnostics explicitly evaluated the relationships among the methods of targeting, the accuracy of the test, and
the outcomes of the intervention. The analyses tended to assume that the tests were perfect and did not consider the impact of test thresholds. As another example, Phillips (2008) found many real-world impacts on the cost-effectiveness of testing strategies. Data were missing on groups, especially the uninsured, Medicaid recipients, and minorities. Test results could be inaccurate, and some of the treatment courses did not correspond with what would be recommended by the test results. In addition, the claims and medical records for testing did not match 25 percent of the time.
Cost-effectiveness analyses are being applied to molecular diagnostics for cancer, Phillips concluded, but they raise methodological and political challenges. Focusing more on value than on cost-effectiveness will allow for headway to be made for molecular diagnostics but will also require changes in methods and public discourse. But shifting the focus to real-world analyses of value will allow decisions to be considered in the full context of care.
ADVANCING THE UTILITY OF ONCOLOGY DIAGNOSTICS
As Robert Bast did in the previous session, Noel Doheny, chief executive officer of Epigenomics, used a specific disease to make several general points about the tools available to assess the clinical utility of molecular diagnostics in oncology.
Colorectal cancer is the second largest cancer killer in the United States, causing 50,000 deaths and 140,000 new cases each year, or 15 to 20 deaths per 100,000 inhabitants (ACS, 2012). It is a disease of the developed world, with the highest prevalence in North America and Europe. Colorectal cancer is curable, though, if it is detected early enough. The 5-year survival rate for diagnosed and treated stage I or II colorectal cancer is 90 percent (ACS, 2011).
Colorectal cancer costs the U.S. health care system $17 billion per year—$7 billion in the initial year, $5 billion in the continuing-care years, and $5 billion in the last year of life (CDC, 2011). The key challenge to improving health and controlling costs, said Doheny, is changing current noncompliance with colonoscopy or stool-based screening. “If you [detect colorectal cancer] early, you never get to that last year where the costs go through the roof,” he said.
In the United States today, 100 million people are eligible for colorectal screening. Half of them get a colonoscopy; another 12 percent or so are tested by fecal tests; and the rest do not undergo any form of screening. Major impediments to screening are unpleasantness associated with stool tests; time constraints, risks, and fears associated with the colonoscopy preparation and anesthesia; and unreimbursed costs. Noncompliance is also greater among people who lack health insurance, have no other source of health care, and have not visited a doctor within the preceding year. In
addition, patients are often lulled into a false sense of security after one colonoscopy and fail to undergo subsequent testing.
Colonoscopy and fecal tests are not perfect. Colonoscopists differ in their ability to detect an adenoma, a polyp, or colorectal cancer in patients, and there are discrepancies in screening between the left and right side of the colon. Nevertheless, if everyone older than age 50 were screened regularly, as many as 60 percent of deaths from colorectal cancer could be prevented, suggested Doheny.
A Blood Test for Colorectal Cancer
The availability of a blood test could promote higher rates of screening by providing the ability to evaluate patients who would not otherwise have been screened, said Doheny. In a survey of more than 1,300 adults, 75 percent said they were more likely to get screened more frequently if a blood test were available.3
Epigenomics has been developing a blood test for colorectal screening based on a circulating marker called methylated Septin9. The test, which was going through the FDA’s premarket approval process at the time of the workshop (Vogel et al., 2006), uses real-time polymerase chain reaction to detect free-circulating tumor DNA in blood. If one of three triplicate polymerase chain reaction tests is positive, the assay is considered to be positive, and patients are sent for a colonoscopy. The intended use of the test as filed in the premarket approval application is as a qualitative assay to aid in screening for patients with average risk.
During the test’s technology development phase, Epigenomics conducted case-control studies using patients’ samples that were both positive and negative for colorectal cancer to optimize the research protocol and increase the sensitivity and specificity of the test. It then gauged the performance of the test in two prospective studies. In one of the trials, of about 8,000 people, about two-thirds had no evidence of disease, and the test found 51 cancers among this group. A “very positive dialogue” with the FDA subsequently changed the number of patients considered to have no evidence of disease, which “clarified what we needed to do in this trial,” said Doheny. At the time of the workshop, the company was getting ready to submit its clinical data later in 2012.
Epigenomics has been developing the test as an LDT, but on an interim basis. It has given licenses for the test to two laboratories, licenses that
______________
3 Results of the survey are available at http://www.prnewswire.com/news-releases/nearly-one-in-three-men-and-women-age-50-and-over-have-not-been-screened-for-colon-cancer-one-in-four-say-their-healthcare-provider-didnt-recommend-screening-124002559.html (accessed August 9, 2012).
will automatically end when the test receives regulatory approval and clearance. This procedure ensures that the laboratories switch to using the regulated product. About 1,000 tests per week are being done in the United States currently, which means that about five patients each week are being detected by a blood test as having colorectal cancer that would not have been detected if that test had not been done. “That, in our eyes, is a very positive position to be in,” said Doheny.
Remaining Challenges
Doheny discussed some of the issues that “keep him awake at night.” Imperfect standards are firmly entrenched, he said. Colonoscopies are not perfect, but they have become the gold standard. Also, incentives are misaligned for providers and payers, with current rewards disproportionately skewed toward chemotherapy.
Innovation is being driven by small companies in a difficult capital environment. Large companies tend to buy de-risked assets, and many small companies cannot afford the up-front costs of diagnostics development. Said Doheny, “It’s very difficult to build an accurate, meaningful, and valid business model.”
He also described several opportunities for improvement. Research partnerships are needed between pharmaceutical companies, diagnostic companies, and government. Although the incentives for these groups are different, they could be aligned if ways were found to combine complementary strengths, Doheny said.
Too few care approaches allow full cost clarity from first patient encounters to interventions and follow-up, Doheny observed. But several experiments, such as those being conducted by the Kaiser and Veterans Administration systems, could show a path forward.
He also suggested that the payment process could be linked through a visible mechanism to the regulatory process. In Japan, for example, when a company receives clearance on a project, a level of payment is established for each time the test is performed, which is an effective “closed loop mechanism,” according to Doheny. Similarly, rewards could be differentiated on the basis of the regulatory credential of the offering.
A registry of patients with apparent false positives is needed to demonstrate clinical utility, he said. Also, a “platinum” standard to compensate for colonoscopy variability should be identified.
The collection of blood and tissue from patients with cancer, including those who die, could greatly advance research, Doheny said. He also recommended creating an accelerated review and publication format spe-
cifically for personalized medicine assays to overcome the extended and biased review cycles in traditional publications. Finally, real-world LDT performance should be linked to FDA filings, he said.
Doheny concluded by saying that “[waiting for] perfection takes too long. Why don’t we just move ahead on some of these as appropriate?”
This page intentionally left blank.






















