
Key Points Raised by Individual Speakers
• Misfolded proteins appear to act as seeds or templates to cause misfolding of the same proteins from their native, soluble state into oligomers. The oligomers eventually coalesce to form insoluble aggregates that are found in all major neurodegenerative diseases.
• The protein aggregates appear to be transmissible by some type of cell-to-cell spread in vivo along anatomically connected pathways. The aggregates might become toxic to the cells in the pathway and lead to disease.
• Transmissibility can be interrupted by administration of antibodies to pathogenic proteins, suggesting immunization as a treatment strategy. Immunization requires that the target protein be found extracellularly.
The progressive accumulation of protein aggregates is the pathological hallmark of many neurodegenerative diseases (see Table 2-1 and Chapter 3). The question is what initiates the process of protein aggregation and subsequently enables it to progress and ramify through distinct pathways of the nervous system. The prevailing model for transmission within the nervous system is known as protein seeding or corruptive protein templating (Jucker and Walker, 2011; Lee et al., 2011). The “seeds” are the small amounts of
misfolded protein that are self-propagating: When they come into direct contact with native protein they convert it to the misfolded form, a process that leads to formation of oligomers; as oligomers accumulate they eventually coalesce into insoluble aggregates consisting of amyloid fibrils, which bear their characteristic beta-pleated sheet conformation (see Figure 4-1). The seed is the transmissible agent. The model is borrowed from what is known about the formation of misfolded amyloid proteins known as “prions” that are responsible for aggregating and spreading transmissible
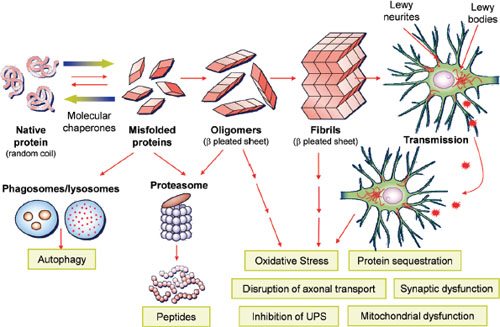
FIGURE 4-1 α-Synuclein mediated neurodegeneration. This illustrates hypothetical processes whereby normal α-Synuclein is converted into pathological α-Synuclein that fibrilizes and deposits into Lewy bodies/Lewy neurites of affected neurons in the brains of patients with Parkinson’s disease (PD)/PD with dementia/dementia with Lewy body. Genetic abnormalities and poorly understood environmental factors may accelerate this process. Normal quality control systems (chaperones, ubiquitin proteosome and phagosome/lysosome systems) that prevent/reverse protein misfolding or eliminate misfolded proteins are overwhelmed. Remarkably, recent data suggest that the progression of PD and related disorders may be linked to the cell-to-cell spread of pathological species of α-Synuclein as illustrated in the upper right of the figure. The toxic consequences of pathological α-Synuclein are illustrated in the lower right of the figure.
NOTE: UPS = ubiquitin-proteasome system.
SOURCE: Trojanowksi, 2012. Adapted from Lee and Trojanowski, 2006 (adapted figure printed with permission from Neuron).
spongiform encephalopathies. The amyloid aggregates in transmissible spongiform encephalopathies are structurally similar to those found in neurodegenerative disease, but their constituent amyloid proteins are different. In neurodegenerative disease the proteins are, most commonly, Aβ amyloid, tau, and α-Synuclein, whereas with prion diseases, the disease proteins are different pathological strains of prion proteins.
The term “transmissible,” for the purpose of this workshop summary, is not synonymous with infectious. The term refers here to cell-to-cell spreading along pathways within the brain, as opposed to spreading or infectivity between individuals. There is no evidence as yet that pathogenic proteins in neurodegenerative diseases are infectious and thus spread between individuals. In contrast, prion diseases are not only transmissible across cells of the brain, but they also can be spread within and across species by direct contact of biological fluids, according to human epidemiology studies. The epidemiology shows that common neurodegenerative disorders do not exhibit infectivity, emphasized John Trojanowski of the University of Pennsylvania. This is a fundamental difference between the two types of diseases. Ignorance of this distinction can be highly injurious to people with neurodegenerative disease who may be needlessly stigmatized if they are erroneously deemed to have an infectious disease.
This chapter summarizes workshop presentations on the prion-like mechanisms by which protein aggregates might spread through the central nervous system. It then discusses the transmissibility of specific aggregated proteins in relation to certain neurodegenerative diseases, before examining the potential success of passive immunization to counter transmission within the brain.
PRION DISEASES AND THEIR TRANSMISSION
Both transmissible spongiform encephalopathies and neurodegenerative diseases are marked by insoluble protein aggregates made up of misfolded protein. Transmissible spongiform encephalopathies—also known as prion diseases—have been studied for decades because of their unique form of transmission by protein templating. The process by which prions propagate is similar to the process that underlies formation and generation of amyloid aggregates in neurodegenerative disease. Thus, prion diseases are important to study in order to understand how neurodegenerative disease might spread within the brain, according to Claudio Soto of the University of Texas Medical School at Houston.
The transmissible spongiform encephalopathies are a group of fatal neurodegenerative diseases. They include Creutzfeldt-Jakob disease in humans, bovine spongiform encephalopathy in cattle, and scrapie in sheep. Although clinical manifestations differ among them, their pathological characteristics are similar, including extensive spongiform degeneration,
widespread neuronal loss, synaptic alterations, brain inflammation, and accumulation of protein aggregates that are toxic to cells (Diaz-Espinoza and Soto, 2010).
The propagation of prion disease, both within and across species, is by misfolded prion proteins, which are the “infectious” agents. The term “prion” was coined to denote a proteinaceous particle that is infectious. Evidence that the misfolded prions are the infectious agents has been accumulating for decades. The most definitive evidence, according to Soto, comes from two studies published during the past 7 years. The first showed that injection of highly purified prion protein, which had been amplified in vitro, caused disease in normal animals (Castilla et al., 2005). The second showed a similar result by injecting purified recombinant prion protein (Wang et al., 2010). Altogether, there have been fewer than 700 cases of prion transmission to humans, with most cases caused by ingestion of beef from cows with bovine spongiform encephalopathy (Jucker and Walker, 2011).
Soto reviewed the basic features of prion proteins and their infectivity. Prions are protease-resistant, amyloid-like β-pleated sheets of diverse sizes ranging from small oligomers to large aggregates. They can be acquired by different routes of exposure—including blood transfusion, intracerebral injection, oral ingestion, or intraocular and intranasal routes—but not through skin contact or inhalation. They are highly resistant to common sterilization procedures, including extremely high temperatures, ultraviolet (UV) radiation, treatment with detergents, and proteases. Soto described the seeding as an exponential process that begins slowly. There is a long lag phase during which monomers attempt to form stable oligomers. Once this occurs, there is a swift elongation phase during which oligomers act as seeds to form a far greater number of amyloid aggregates (Soto et al., 2006). In other words, the process begins slowly, but reaches a tipping point at which time there is a sudden acceleration in forming aggregates made up of amyloid fibrils. The process is accelerated by the addition of exogenous seeds. Misfolded prion proteins, oligomers, and aggregates are, in his view, “seeding competent,” but the fine details of the seeding and transmission process are still elusive. Better understanding of the seeding process, in his view, will yield dividends for biomarker development and therapeutics for neurodegenerative disease.
Soto subsequently described his research on Aβ amyloid. His team has shown that Aβ amyloid can be transmitted to an animal by direct injection of human brain extracts from Alzheimer’s disease cases. The formation and accumulation of aggregates increases progressively with time, and the aggregates are localized to brain areas distant from the injection site (Morales et al., 2011).
Several participants emphasized that members of the public need to
understand that Alzheimer’s disease is not infectious in conventional ways, that is, through inhalation or skin contact. A recent study also found no evidence for human-to-human transmission of neurodegenerative disease– associated proteins in recipients of cadaveric human growth hormone (Irwin et al., 2013). One participant relayed his experience when a newspaper article implied that Alzheimer’s disease was infectious. It engendered many calls to Alzheimer’s organizations expressing confusion and fear. If scientific findings are not properly qualified, people may needlessly shun and stigmatize victims of neurodegenerative disease and their family caretakers—those already devastated by the disease.
TRANSMISSIBILITY OF SPECIFIC AGGREGATED PROTEINS IN SELECT NEURODEGENERATIVE DISEASES
This particular portion of the workshop focused on Aβ amyloid and tau transmission in Alzheimer’s disease, and α-Synuclein transmission in Parkinson’s disease. In describing the choice to focus on these specific topics, Trojanowski, chair of this session, noted, “We could have done more, but we as a group thought it was helpful to focus on areas that had made the most progress in cells and animal models.”
Aβ Amyloid Transmission in Alzheimer’s Disease
Lary Walker of Emory University described his research on the seeding process and the transmission of Aβ amyloid within the brain of Alzheimer’s animal models. Several lines of evidence reveal that the seed for Aβ amyloid aggregates and plaques is indeed Aβ amyloid. The most salient evidence comes from studying the consequences of intracerebral injections of dilute Aβ amyloid-containing brain extracts from autopsied Alzheimer’s cases into transgenic mice. The mice carry the human gene for Aβ precursor protein, from which Aβ amyloid is cleaved. One key experiment found that seeding is abolished or reduced by anti-Aβ amyloid antibodies, immunodepletion, or denaturation (Meyer-Luehmann et al., 2006). Other evidence shows that the seeded deposits are not from the injected brain extracts because there is a several months–long lag time in the formation of Aβ amyloid plaques. Finally, the seeded host must express human Aβ amyloid. Transgenic mice develop seeded Aβ plaques, whereas wild-type mice do not show any lesions after injection.
Walker’s laboratory also established that seeding stimulates Aβ deposition elsewhere in the brain. His team showed that, after hippocampal injection of Alzheimer’s disease extracts, Aβ plaques are found along axonally interconnected pathways, including the entorhinal cortex (Jucker and Walker, 2011). The opposite was the case when another team of
researchers injected the extract into the entorhinal cortex; it led to plaque formation in the hippocampus, relayed Walker. He speculated that the spreading occurred by axonal transport, but his group is only beginning to investigate the mechanisms of intracellular passage of seeds. The extract in these experiments is the supernatant of centrifuged homogenates from Alzheimer’s disease brains or from Aβ precursor protein transgenic mouse brains. Small and soluble Aβ amyloid seeds from the dilute supernatant are potent inducers of plaques, noted Walker.
Tau Transmission in Alzheimer’s Disease
Tau is a microtubule-associated protein that aggregates in its hyperphosphorylated form into neurofibrillary tangles that are a pathological signature of Alzheimer’s disease. In this disease there is a characteristic progression of neurofibrillary tangles, starting in the entorhinal cortex in prodromal stages, proceeding to the limbic areas in early to moderate stages, and finally converging on the neocortex in late stages (see Figure 4-2).
In her presentation, Karen Duff of Columbia University focused on the prodromal period when the tangles are localized to the entorhinal cortex. She and her colleagues investigated whether tau could spread to anatomically appropriate pathways in the limbic system. They developed an animal model of a transgenic mouse expressing the human pathological tau gene
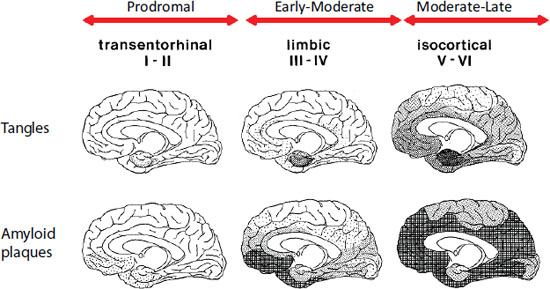
FIGURE 4-2 Plaque and tangle distribution at different stages of Alzheimer’s disease progression (Braak staging).
SOURCE: Braak and Braak, 1991.
under the control of a promoter that is specific for the entorhinal cortex. This ensured that the animal only expressed tau pathology in the entorhinal cortex. Using this model, they found that in old animals, neurofibrillary tangles appeared in three monosynaptically connected nuclei to which entorhinal neurons project: the granule cells of the dentate gyrus, the subiculum, and the CA1 region of the hippocampus (Liu et al., 2012). Tangles were not evident in nuclei immediately neighboring the entorhinal cortex. They concluded that there was propagation of tau along anatomically connected neural pathways in older animals. In younger animals, neurofibrillary tangles remained restricted to the entorhinal cortex, without any evidence of spread, which indicated an age effect (Liu et al., 2012).
Duff’s team also sought to discover the mechanism of tau spread. Both her team and another (de Calignon et al., 2012) proposed that tau is directly released into the extracellular space at the synapse as a result of degenerating axons. Once released it could be taken up by the postsynaptic neuron. Data from her in vitro work suggest that only small molecular-weight tau fibrils, not large aggregates, could be taken up by the postsynaptic neuron, she explained. Most importantly, tau’s presence in the extracellular space and not in vesicles meant to Duff that tau could be targeted there by immune therapies. Finally, using a microchamber to separate different regions of the cell, her team found that, after administration, tau is taken up by neurons and transported both retrogradely and anterogradely. In the future, she would like to employ her transgenic mouse model to study interactions between Aβ amyloid and tau in the entorhinal cortex and she would like to understand the mechanisms of cell-to-cell spread as well as the functional impact of tau pathology.
α-Synuclein Transmission in Parkinson’s and Other Neurodegenerative Diseases
α-Synuclein is a cytosolic protein thought to participate in the regulation of synaptic transmission and synaptic plasticity (Murphy et al., 2000). Amyloid fibrils of this protein constitute the major pathological signature of Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy (Lee et al., 2011), and a subset of Alzheimer’s disease cases. Virginia M.-Y. Lee of the University of Pennsylvania described two new models for studying the seeding and transmission of α-Synuclein.
Lee and her colleagues have developed the first neuronal model of spontaneous Parkinson’s disease. It is a hippocampal neuron primary culture to which they add non-mutated, α-Synuclein preformed synthetic fibrils (Volpicelli-Daley et al., 2011). The cultured neurons are from wild-type, non-transgenic mice. The addition of the fibrils to the neuron cell culture recruits endogenous α-Synuclein to form pathologic, insoluble Lewy bodies
and Lewy neurites. α-Synuclein becomes hyperphosphorylated and ubiquitinated, but the purpose of these modifications is unclear to her. Lewy body–like pathology forms first in axons because axons bear the highest concentrations of endogenous α-Synuclein. The pathology is propagated both retrogradely and anterogradely throughout the entire neuron. The Parkinson’s-like inclusions impair neuronal function and induce cell death. Lee said that this unique and easy-to-use primary neurons culture model will facilitate Parkinson’s research and drug discovery. She offered to share the synthetic fibrils with other researchers.
The second model she used was of M83 transgenic mice that express the human gene for pathological α-Synuclein. Her team injected preformed fibrils and lysate from symptomatic animals into the stratum and/or cortex of asymptomatic transgenic mice. The recipient mice, which normally become symptomatic with Parkinson’s-like motor impairment at 12 months of age, displayed motor impairment earlier. This finding is consistent with a previous study showing that addition of small quantities of preformed fibrils accelerates the kinetics of aggregate formation (Luk et al., 2007). At autopsy, her team found transmission of pathology throughout the entire mouse brain, including the olfactory bulb, the cerebellum, and the corpus callosum. Because pathology was evident in white-matter tracts, they believe this is the route for propagation of α-Synuclein throughout the brain. The seeded inclusions bore morphological and biochemical similarities to inclusions found in Parkinson’s disease. The animals have a shorter lifespan, dying about 100 days sooner than did untreated animals. Finally, the incubation time from injection to death is consistent regardless of the age at which the injections took place. This transgenic model represents, in Lee’s view, a new opportunity to study disease mechanisms and disease-modifying therapies. She is especially interested in determining how α-Synuclein is degraded, how endogenous seeds interact with the preformed fibrils, what enzymes are involved in phosphorylation and ubiquination, and how α-Synuclein inclusions affect cellular function prior to death.
IMMUNIZATION FOR NEURODEGENERATIVE DISEASE
This portion of the workshop dealt with research on two types of antibody immunizations, one targeted to the pathogenic protein α-Synuclein and the other to tau, to treat Parkinson’s disease and Alzheimer’s disease, respectively. Although the research is in the early stages, there have been successful treatments in animal models.
α-Synuclein Immunization for Parkinson’s Disease
The rationale for antisynuclein immunization as a treatment strategy derives from the presence of α-Synuclein in the synapse, cerebrospinal fluid (CSF), and plasma membrane, where it would be accessible to antibody attack. Dora Games of Neotope Biosciences first reiterated the evidence supporting seeding and transmission of α-Synuclein in vivo and in vitro. She also noted that decreased expression of α-Synuclein reduces behavioral deficits and pathology, whereas addition of synthetic α-Synuclein fibrils exacerbates those outcome measures (Lim et al., 2010; Luk et al., 2012).
For the study of the efficacy of immunization, Games and her colleagues selected a transgenic mouse model expressing α-Synuclein because mice develop behavioral deficits and α-Synuclein aggregates throughout the temporal cortex and hippocampus similar to what has been described in Lewy body disease. After testing several antibodies, they selected one to the C-terminus region of α-Synuclein, and conducted in vivo and in vitro studies. Upon antibody administration, they found that the antibody crossed the blood/brain barrier and localized to lysosomes. Once in the brain, the antibody reduced behavioral deficits in learning and memory, and reduced the accumulation of α-Synuclein in axons and synapses (Masliah et al., 2011). The researchers used readout antibodies to label dystrophic neurites and somatic pathologies. Using two markers, they also found that immunization preserves synaptic integrity. The combined evidence led Games and colleagues to conclude that immunization is efficacious for synucleinopathies. In response to questions, she conceded that she does not know the mechanism by which antibodies are effective. In fact, she and her collaborators are unsure as to whether the antibodies work extracellularly and/or intracellularly. Her collaborator, Eliezar Masliah, does have evidence of antibodies penetrating the neuron cell membrane. In the ensuing discussion, Games added that much needs to be learned from ongoing clinical trials of antibodies in Alzheimer’s disease. If the results, which are expected in several months, are negative, she urged investigators to evaluate why the failure occurred, whether it was the patient population, the timing of delivery, or other factors.
Games raised several issues regarding the immunization study, including whether transgenic models are relevant to human disease, and she expressed concern about the lack of common standards for readouts and assays, lack of incentive for replicating studies in academic settings, and the high cost of chemistry, reagents, and in vivo support. To reduce risk in clinical trials, she argued that diagnoses should be assessed earlier, progression must be assessed faster, target engagement must be verified in early phases of development, and diagnostic and treatment biomarkers must be validated.
Tau Immunization in Neurodegenerative Disease
Tau is the main component of neurofibrillary tangles, the pathological aggregate found in Alzheimer’s disease, frontotemporal dementia, amyotrophic lateral sclerosis, and progressive supranuclear palsy. Because tau functions normally in the cytosol to stabilize microtubules and the tau tangles are intracellular, it has been widely assumed that only negligible concentrations of tau would be found in the extracellular space. That assumption has been discarded as a result of findings that reveal tau to be present in the CSF in young, healthy people, said Peter Davies of the Feinstein Institute for Medical Research. Further studies have shown tau in interstitial fluid (Yamada et al., 2011) and tau release from cultured neurons and transfected cells. The extracellular presence of tau justifies the use of antibody immunization, noted Davies.
At least four published studies show that antibodies against tau can reduce pathology and improve behavior of transgenic animals that express the mutant tau gene from humans (Asuni et al., 2007; Boimel et al., 2010; Boutajangout et al., 2010; Chai et al., 2011). Several of these studies were conducted in P301S transgenic mice. These mice are commonly used, said Davies, because they show many of the features of frontotemporal dementia and their tau pathology appears early. Using this mouse model, Davies and his colleagues revealed that antibodies directed at tau reduce pathology in the hippocampus and delay progression of disease (Chai et al., 2011). Davies reported being “very puzzled” by these findings because it is not clear to him how an extracellular reagent can exert these striking effects intracellularly. He reported spending months trying to determine if antibody penetrates intracellularly, but was unable to show it. There was discussion surrounding the question of whether antibodies worked extracellularly or intracellularly. In response to a question, Davies reported that, according to his in vitro studies, the degree of membrane depolarization does not affect the amount of anti-tau antibody levels in the culture medium.
He concluded his presentation by asking whether there is a pathogenic extracellular species of tau and what type of characteristics it has. He also raised questions about the predictability of the models to humans, especially because tauopathy, in the most common neurodegenerative diseases, is often accompanied by other proteinopathies (see Table 2-1). He urged testing in humans, and in a patient population with more pure tau pathologies, such as frontotemporal dementia or progressive supranuclear palsy.
RESEARCH NEEDS AND NEXT STEPS SUGGESTED BY INDIVIDUAL PARTICIPANTS
The speakers at the workshop identified many questions for future research and other opportunities for future action. The suggestions related to transmissibility and immunization are compiled here to provide a sense of the range of suggestions made. The suggestions are identified with the speaker who made them and should not be construed as reflecting consensus from the workshop or endorsement by the Institute of Medicine.
• Conduct epidemiology studies to directly study transmissibility of neurodegenerative disease and the rare likelihood of infectivity through surgical equipment and blood transfusions. (Trojanowski, Walker)
• Test the model of protein seeding and transmission by identifying the biophysical and cellular mechanisms by which seeds form in vivo, how they are cleared, and how they move within neurons and in the extracellular space once seeding begins to spread. (Cuervo, Walker)
• Study the proteolytic pathways by which seeded aggregates are degraded and study cellular response to seeding. (Lee)
• Determine whether transgenic animal-expressing mutant tau are relevant to humans with neurodegenerative disease. Determine whether a pathogenic extracellular species of tau exists and what type of characteristics it has. (Davies)
• Study interactions between aggregates of distinct proteins and focus on neural networks. (Duff)
• Determine if there is “cross-seeding” between misfolded proteins. (Walker)
• Before conducting clinical trials, establish target engagement, use validated diagnostic and treatment biomarkers, and use common standards for readouts and assays. (Games)
• Understand the mechanism(s) by which tau is released from neurons into extracellular fluids. (Davies)
• Develop therapies, such as immunization, that abolish transmission of misfolded amyloid proteins. (Davies, Duff, Games, Walker)
• For trials using anti-tau antibodies, test therapies in patients with pure tauopathy, such as frontotemporal dementia or progressive supranuclear palsy. (Davies)












