
2
Rationale for Exploring Commonalities Across Neurodegenerative Diseases
Key Points Raised by Individual Speakers
• Ample rationale exists for integrated investigation of neurodegenerative disease.
• Neurodegenerative diseases show pathological overlaps. For example, many patients with a particular disease have more than one proteinopathy, and a single type of proteinopathy can be associated with multiple diseases.
• Neurodegenerative diseases have overlapping genetics. The same genotype can lead to disparate phenotypes, and the same phenotype may result from multiple genotypes.
• Formidable obstacles to research are present for all neurodegenerative diseases, including lack of biomarkers, long asymptomatic period before disease is manifest, lack of validated animal models, and costly clinical trials. These obstacles have contributed to a dearth of new therapies.
• Enhanced sharing of research findings and collaboration across disease-specific research communities could potentially help advance basic scientific knowledge about each disease and help facilitate therapeutics development, including therapeutics that may address more than one neurodegenerative disease.
Neurodegenerative diseases traditionally have been studied separately. The workshop was convened because of a growing recognition of potential commonalities across genetic and cellular mechanisms, which led to an interest in exploring these commonalities to (1) identify potential opportunities to better understand the basic science of neurodegenerative disease, and (2) develop new therapeutic approaches. However, the workshop began by asking the underlying question: Is there a rationale to justify studying neurodegenerative diseases together? Presentations and discussions examined common features across diseases, including pathological and genetic overlaps, common challenges, and practical considerations related to the infrastructure needed to study these diseases. These are examined in turn below.
COMMON FEATURES ACROSS DISEASES
Neurodegenerative diseases show pathological overlaps, noted some workshop presenters. For example, many patients with a particular disease have more than one proteinopathy, and a single type of proteinopathy can be associated with multiple diseases. Similarly, there are overlapping genetics across neurodegenerative diseases. The same genotype can lead to disparate phenotypes, and the same phenotype may result from multiple genotypes.
Pathological Overlaps
Neurodegenerative diseases are best known by their pathology. The pathological hallmarks of Alzheimer’s disease, for example, are senile plaques made of amyloid-beta (Aβ) protein and neurofibrillary tangles made of tau protein. Parkinson’s disease is best known by Lewy bodies made of the protein α-Synuclein. These two diseases and others are known as proteinopathies because they feature pathological protein accumulations thought to be responsible for neuron injury and death. Although each disease is associated with particular proteinopathies, Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, pointed out their broad phenotypic spectrum, showing that each disease frequently reveals mixed proteinopathies that overlap with proteinopathies from other neurodegenerative diseases (see Table 2-1). Although Aβ plaques and tau tangles are paradigmatic of Alzheimer’s disease, Lewy bodies typical of Parkinson’s disease are found in more than 50 percent of Alzheimer’s cases, and neuronal inclusions consisting of the protein TDP-43 are found in more than 40 percent. Similarly in dementia with Lewy bodies—a dementing disorder closely allied to Parkinson’s disease, yet with some features of Alzheimer’s—the paradigmatic α-Synuclein-rich Lewy bodies are accompanied by Aβ plaques in
TABLE 2-1 Pathological Hallmarks and Their Protein Components in Neurodegenerative Diseases
| Neurodegenerative Disease | Pathology | Component Proteins |
| Alzheimer’s disease | Senile plaques Neurofibrillary tangles Lewy bodies Neuronal inclusions |
Aß amyloid Tau α-Synuclein TDP-43 |
| Parkinson’s disease | Lewy bodies | α-Synuclein |
| Amyotrophic lateral sclerosis | Neuronal inclusions | TDP-43 FUS/TLS SOD1 |
| Huntington’s disease | Neuronal intranuclear inclusions | Huntingtin |
| Dementia with Lewy bodies | Lewy bodies Senile plaques Neurofibrillary tangles |
α-Synuclein Aß amyloid Tau |
| Frontotemporal diseases | Neuronal and glial inclusions | Tau TDP-43 FUS |
| Multiple system atrophy | Glial cytoplasmic inclusions | α-Synuclein |
| Prion diseases | Senile plaques | PrP protein |
NOTES: FUS/TLS = fused in sarcoma/translocated in liposarcoma; PrP = prion protein; SOD1 = superoxide dismutase 1; TDP-43 = TAR DNA-binding protein 43.
60 percent of cases and tau tangles in 50 percent, he said. Furthermore, experimental evidence shows that some of these proteins from the same or different neurodegenerative diseases interact with one another, resulting in the acceleration of the disease process. The fact that there is such a high degree of mixed pathology and potential for interaction, in Dickson’s view, provides rationale for studying these diseases together, and suggests that combination therapies are going to be crucial. A single therapy aimed at one proteinopathy may be found ineffective because the underlying disease has multiple proteinopathies.
Just as a single neurodegenerative disease can be associated with multiple proteinopathies, a single proteinopathy can also be associated with multiple diseases. This holds for the molecular defect four-repeat tau.1 Four-repeat tauopathy is associated with at least three clinical presentations: (1) Progressive supranuclear palsy presents with an axial rigidity
1 The 6 tau isoforms differ according to the number of repeats (3 or 4) of 18 amino acids at the C terminus.
and eye movement problems, in addition to atypical Parkinsonism; (2) corticobasal degeneration presents like a frontal lobe dementia, with focal cortical syndromes, including progressive apraxia or progressive aphasia; and (3) argyrophilic grain disease is an increasingly recognized disorder of the elderly that affects the medial temporal lobe and is associated with an amnesic cognitive impairment. Given the pathological heterogeneity, Dickson urged researchers, for therapeutic purposes, to search for “upstream targets, rather than downstream pathologies.”
Genetic Overlaps
Neurodegenerative diseases also have genetic overlaps (Bertram and Tanzi, 2005). Andrew Singleton of the National Institute on Aging (NIA) first focused on monogenic forms of neurodegenerative disease and their overlaps. The same genotype can lead to disparate phenotypes. For example, presenilin-1 (PS1) mutations, which account for the greatest fraction of early-onset familial Alzheimer’s disease, are also found in the disease spastic paraparesis. Mutations in the gene leucine-rich repeat kinase 2 (LRRK2) are found in an autosomal dominant form of Parkinson’s disease, yet also in several other diseases such as progressive supranuclear palsy, amyotrophy, and multiple system atrophy. Similarly, a recently identified mutation in the gene for chromosome 9 open reading frame 72 (C9orf72) is the most common cause of familial amyotrophic lateral sclerosis (ALS). The mutation is also present in frontotemporal dementia (FTD) and clinically-diagnosed Alzheimer’s disease. There are enough people with this particular mutation, noted Singleton, that researchers need to come together to begin to look for modifiers that explain phenotypic differences. A centralized DNA and data repository supported by some type of consortium, he said, would go a long way to advance the field.
There are also overlaps in risk loci, Singleton said. The microtubule-associated protein tau (MAPT) gene is shared by Parkinson’s and progressive supranuclear palsy. The gene for α-Synuclein is shared by multisystem atrophy and Parkinson’s disease. In his experience, Singleton said that the vast majority of risk loci associated with disease are not associated with protein coding changes. They are more likely to be associated with a change in basal expression of the protein, an effect on splicing, or an effect at a particular point in time. There are numerous ways, in short, that risk loci mediate their effects.
This sample of genetic overlaps speaks to the need to look across neurodegenerative disease, as opposed to focusing solely on one disease without regard to implications for others, said Singleton. For future research, he pressed for harmonization of methods for both pathology and genetics, such as the same genetic platforms and the same genetic readout. “I think
we are used to bringing data together, certainly within disease. The next challenge lies in bringing data together across diseases and bringing together groups that wouldn’t necessarily work with each other.” The bottom line, he said, is that each approach requires standardization and collaboration across disease communities.
Besides genetics and pathology, other overlaps occur in the mechanisms of disease pathogenesis. These overlaps were the prime focus of the workshop and are covered in ensuing chapters: protein aggregation, transmissibility within the central nervous system, mitochondrial dysfunction, and RNA processing errors.
COMMON CHALLENGES
Many speakers and workshop participants pointed to several well-recognized challenges to research posed by neurodegenerative disease. These challenges contribute to the low number and flat growth of new drug approvals for neurodegeneration (see Figure 2-1). Challenges cited by various workshop participants are described briefly below.
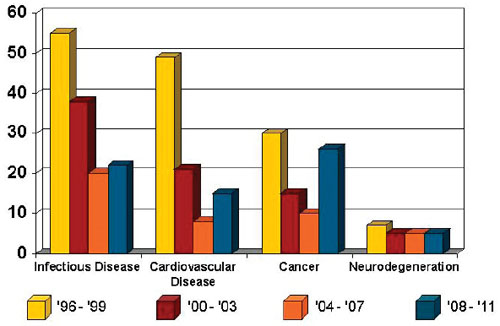
FIGURE 2-1 Drugs approved by the Food and Drug Administration from 1996 to 2011.
SOURCE: Data from Thompson Centerwatch; figure from Adrian Ivinson.
• Modeling neurodegenerative disease in animals is problematic because of species differences. Neurodegenerative disease often impairs cognition, but cognitive functioning is exceedingly difficult to model in animals.
• There is a lack of biomarkers with which to study neurodegenerative disease, especially its diagnosis. Without strong biomarkers, clinical trials are far more difficult to conduct because patients may be misdiagnosed.
• Patient heterogeneity in clinical trials often dilutes the capacity to find a medication efficacious.
• The causes of these disorders are not fully known, nor are the earliest times to intervene to modify the course of the disease.
Another daunting problem is the long preclinical period after the disease process is unleashed, but before appearance of frank symptoms, noted several participants. The disease process, as evidenced by pathological changes, can begin, in the case of Alzheimer’s disease, two decades before symptoms become manifest (Alzheimer’s Association, 2012). Once symptoms appear, they still may be too non-specific to warrant a definitive diagnosis. Yet by the time the disease is fully manifest, the global pathological damage to the brain is often so great that treating or slowing the disease course may be too late. Many argue that drug interventions are best in the asymptomatic stage while the brain is still resilient and capable of repair or compensatory changes. But administering drugs to asymptomatic individuals in clinical trials poses ethical and cost implications for the drug industry and for the Food and Drug Administration (FDA).
Drug trials, already expensive in neurological diseases, would likely be even more costly if studying asymptomatic people for whom the benefit may not accrue for years of taking drug therapy, according to several participants. Participants noted several potential barriers. For example, drug therapy could have to be administered for longer duration and the size of the trial would need to be larger. Another factor is that some patients with preclinical disease may never develop the disorder, which complicates analysis of efficacy. Furthermore, a number of costly failures loom large in deterring drug companies from making huge investments. Several large and expensive trials of antioxidants have not been successful (e.g., Galasko et al., 2012). The compounds tested had been successful in animal models, but the translation of the findings to humans was not effectively accomplished, most likely the result of species differences. Finally, pooling resources for clinical trials brings fears in drug companies of losing intellectual property, which deters investment. Amid the formidable array of challenges, as well as the economic downturn, several pharmaceutical companies have reduced their neurodegenerative disease divisions, according to press reports.
OPPORTUNITIES FOR COLLABORATION AND PARTNERSHIP
Given these challenges, Adrian Ivinson of Harvard Medical School pointed out that each of the disease communities has similar needs, such as defining more genetic risk factors for disease, elucidating genes and their contribution, defining phenotypes, understanding postgenomic modifications, identifying biomarkers, developing validated animal models, identifying and validating new drug targets, and translating results into effective clinical trials.
To meet the challenges confronting the field, Ivinson pressed for collaboration across the public and private sectors to develop clinical and pathological standards and to use them for preclinical and clinical research. He pointed out that resources—the tools, technologies, infrastructure personnel, and skill sets—are very similar, regardless of disease. In fact, he wagered that “a lab that was mostly focused on Alzheimer’s disease today … could probably switch to Parkinson’s disease tomorrow without changing much infrastructure.” He urged collaborations in the preclinical phases when intellectual property considerations are not as salient as in clinical trials. He conceded that as research moves to large-scale clinical trials, there is greater difficulty in collaboration because of intellectual property. Still, he believes collaborations are the means to achieving not just greater efficiencies, and achieving them sooner, but also achieving better research outcomes. “I think for almost every stone you turn over, you will find an opportunity to collaborate,” said Ivinson. He and other presenters acknowledged, however, that collaborative approaches to studying more than one disease have rarely been tried and may not be pertinent to all neurodegenerative diseases because of disease heterogeneity. Yet they were quick to add that the collaborative approach across diseases is worth trying where the data justify it.
The Alzheimer’s Disease Neuroimaging Initiative is an 8-year initiative by coordinated teams of scientists in the public and private sectors to validate biomarkers for Alzheimer’s disease (ADNI, 2010). It was showcased by several speakers as a good example of how to bring together public- and private-sector partners.
Story Landis, director of the National Institute of Neurological Disorders and Stroke (NINDS), mentioned two NINDS programs and one National Institutes of Health (NIH) Blueprint program that encourage collaboration. NINDS’s NeuroNEXT (Network for Excellence in Neuroscience Clinical Trials) is a network for conducting Phase II clinical trials for neurological conditions through partnerships with academia, private foundations, and industry (NINDS, 2013). Landis noted that this network is unusual because it has a common institutional review board and prenegotiated agreements with all the sites, with the aim of going “from pro-
tocol to patient” in 2 months. Landis also mentioned the NINDS Cooperative Program in Translational Research (U01), which is a milestone-driven funding program with the goal of taking something from proof of concept in an animal system or a culture system to an investigational new drug, investigational device exemption, 510(k), or 510(k) de novo application to the FDA within the funding period (NINDS, 2012). Finally, Landis mentioned the Blueprint Neurotherapeutics Network, which “offers neuroscience researchers a ‘virtual pharma’ to develop promising hit compounds from chemical optimization through Phase I clinical testing” (NIH Blueprint, 2013).
EXAMPLE PROMISING TECHNOLOGIES
Several speakers described promising technologies to study the complex pathogenesis of neurodegenerative disease as well as to identify new treatments. These technologies are high-throughput screening, induced pluripotent stem cells (iPSCs), and yeast as a model system. This section does not exhaustively review current promising technologies, but rather summarizes certain examples that were discussed at the workshop.
High-Throughput Screening in Primary Neurons
Autophagy and other proteostasis mechanisms are constantly in flux. Obtaining a snapshot in time through ordinary microscopy cannot be used effectively to understand such dynamic cellular processes. A new method to study dynamic changes over time within single neurons has been devised by Steven Finkbeiner and colleagues (Sharma et al., 2012). Known as robotic microscopy (RM), the high-throughput and high-content automated imaging system can acquire images rapidly and automatically with single-cell resolution and enable high-throughput applications as the cell is followed longitudinally. The longest a cell has been followed thus far is 6 months. Automated analysis programs are used to obtain anatomic and physiological features of neurons and study how these change over time in a quantitative manner. Once cells are tagged with fluorescent biosensors, RM can be used to study events such as cell-wide protein misfolding, autophagy, and trafficking. New methods are being used to study early changes in protein aggregation and to determine the effects of neurodegenerative disease-causing proteins, said Finkbeiner. Combined with statistical methods, RM can determine the extent to which a variable can predict the fate of that same neuron at a later time. This capability is being used to uncover cause-and-effect mechanisms in neurodegenerative disease, such as whether changes are pathogenic, incidental, or beneficial (Arrasate and Finkbeiner, 2005).
Induced Pluripotent Stem Cells
Creating iPSCs from humans with neurodegenerative disease presents an opportunity for studying all facets of neurodegenerative disease, according to Finkbeiner. Currently, most cellular studies are conducted in cell lines derived from animal models, rather than from humans with disease. Many lines are not even primary neurons. To study human neurons, in particular, iPSCs can be developed from skin or blood cells from people with disease and reprogrammed with transcription factors or small molecules. With specific protocols, the cells then can be differentiated into neuron subtypes of interest (e.g., motor neurons). These differentiated iPSCs can be used to study pathological functioning, identify drug targets within the affected cells, and screen drugs to predict how they might fare in human clinical trials. In an iPSC line from a patient with ALS, the mutant nerve cells had higher levels of the TDP-43 protein and shorter survival than similar lines from control lines (Bilican et al., 2012). This validated the iPSC line as an accurate model because TDP-43 is well established as the protein that forms toxic aggregates in ALS. The downsides of iPSCs are their heterogeneity at baseline and the composition of the cultures after being differentiated; their high costs; the length of time to differentiate cells into brain cells of interest; and absence of protocols for making many brain cell subtypes, Finkbeiner observed.
In the discussion, one participant challenged the value of iPSCs for their immaturity and for their incapacity to model the effects of cell–cell interactions, which are highly important in neurodegenerative diseases. Another participant expressed concern about inherent variability across different iPSC lines. Finkbeiner responded that his laboratory now has approximately 50 iPSC lines and several clones from individual patients and has found that the variability is relatively small; their cell–cell interactions were recently studied in an ALS model (see Serio et al., 2013).
Yeast as a Model System
Yeast is a single-cell eukaryote with characteristics that are found in complex eukaryotic organisms. It has become a model for studying neurodegenerative disease because many of its fundamental cell pathways are relevant, such as mitochondrial gene function and autophagy, the process of self-degradation and clearance described in Chapter 3. Gregory Petsko of Weill Cornell Medical College described two crucial areas in which yeast can be useful for the study of neurodegenerative disease: mechanistic studies of pathophysiology and drug screening. These types of studies are relatively easy to model in yeast, thanks to their well-defined genome, fast growth, the fact that 80 percent of their proteins have some functional characteriza-
tion, and the ease of transfecting genes, especially human genes, into the yeast genome.
A recently published study by Petsko and colleagues highlights the utility of yeast for investigating pathophysiology of familial ALS (Ju et al., 2011). The research focused on the gene FUS/TLS, which, when mutated, is a cause of one subtype of familial ALS (Kwiatkowski et al., 2009; Vance et al., 2009). FUS/TLS is a nucleic acid binding protein that implements key functions related to RNA processing. When transfected and overexpressed in yeast, FUS/TLS mislocalizes to the cytoplasm rather than to the nucleus where it is normally found. In the cytoplasm FUS/TLS aggregates and subsequently kills the cell, thereby recapitulating the salient phenotype of ALS in motor neurons. The researchers determined that mislocalization into the cytoplasm occurs because of a defect in a region of the FUS/TLS protein that normally marks it for import into the nucleus. Petsko and colleagues then screened a library of 5,600 yeast genes to determine whether any of them could suppress FUS/TLS’s toxicity. Yeast screens are fast and inexpensive, and produce clear-cut results. Emerging from the screen were five yeast genes that suppressed FUS/TLS’s toxicity. The genes were all RNA-binding proteins. The investigators then identified one human homolog of the yeast genes and they found it, when cloned into yeast, to be effective in suppressing FUS/TLS’s toxicity. The research implicates a possible insufficiency in RNA processing or RNA quality control in mediating toxicity of FUS/TLS. “It would be hard to find this sort of thing out so easily with this kind of time scale without using a model organism as facile as yeast,” said Petsko.
RESEARCH NEEDS AND NEXT STEPS SUGGESTED BY INDIVIDUAL PARTICIPANTS
The workshop speakers identified many questions for future research and other opportunities for future action. Those related to the rationale for studying commonalities across neurodegenerative diseases are compiled here to provide a sense of the range of suggestions made (research suggestions that are specific to the topics covered in Chapters 3-6 are included at the ends of those chapters). The suggestions are identified with the speaker who made them and should not be construed as reflecting consensus from the workshop or endorsement by the Institute of Medicine.
Enhancing Collaborations
• Develop dedicated programs and funding to identify common threads. Support work that specifically aims to evaluate therapeutic targets or therapies shown to be beneficial in one disorder in models of other neurodegenerative diseases. (Finkbeiner)
• “Establish effective links between basic scientists and clinical investigators and between academia and the pharmaceutical industry to expedite the discovery and validation of potential drug targets and the development of novel therapeutics.” (Mucke, citing the work of the Alzheimer’s Association Expert Advisory Workgroup on the National Alzheimer’s Project Act, of which he is a member [2012, p. 360])
• Create innovative programs to foster collaboration among consenting scientists. Consider allowing program officers to use supplemental support from NIH in a more flexible manner to encourage willing scientists to pursue such collaborations. (Finkbeiner)
• Harmonize pathology and genetic measures. Geneticists should work together to come up with a list of pathological and clinical standards across diseases. (Singleton)
• Catalogue ongoing efforts to produce neurodegenerative disease-related induced pluripotent stem cells. Identify who is creating them; how many exist; what their basic characteristics/homogeneity are; and how others get access. (Ivinson)
• NIA and/or NINDS should look toward funding shared/collaborative programs that support the development or enhancement of resources that could be made available to multiple investigators. These might include biomarker programs; mouse behavior (and other model systems) testing; iPS cells; shared compound libraries; and access to tissues and laser capture microdissection/nucleic acid capture and analysis. (Ivinson)
• Solicit a set of key questions that deal with the cell biology of neurodegenerative disease that can help focus the field on some of the critical basic science questions whose resolution would advance the field. (Finkbeiner)
• Share human genetics data, with the goal of data aggregation, to find truly causal gene variants that extend across populations or are population specific. (Mootha)
• Determine what accounts for selective vulnerability of neurons in neurodegenerative diseases. This question lies at the crossroads of many of the issues discussed, and involves molecular pathogenesis, genetics, cell biology, and systems biology. (Walker)
• Identify why aging is the most prevalent risk factor for neurodegenerative diseases. Specifically, determine whether a particular aspect of aging (e.g., a component of the proteostasis network) can be targeted to lower risk, or whether the diseases result from a general (and thus less tractable) decline in cellular integrity. (Walker)
Identifying Biomarkers and New Therapeutics
• Create a PPMI/ADNI2-like consortium funded by NIH and a public–private consortium of centers in academia and pharmaceutical companies to investigate mechanisms of neurodegenerative disease transmission and the potential efficacy of immune therapies as disease-modifying interventions. Unlike PPMI/ADNI, shared intellectual property (IP) could be generated and royalties derived could be distributed to all stakeholders who contribute funding and/or IP according to a predetermined formula. (Trojanowski)
• Identify new biomarkers in neurodegenerative disease. (Dunlop, Ivinson, Mehler, Mucke, Youle)
• Examine biomarkers longitudinally, which is highly valuable information for a clinical trial. (Rigo)
• Build a cohort of patients with C9orf72 mutations for a biomarker study in early or preclinical ALS/FTD. (Singleton)
• Create a catalogue of ongoing neurodegenerative disease biomarker collections and programs. This would enable researchers worldwide to locate the samples and patient records they need. (Ivinson)
• Screen novel therapies in the yeast system—extending to fission yeast—and apply to other simple well-characterized systems, such as nematodes, fruit flies, and zebrafish—thereby unifying the databases of these organisms. (Kowall)
• Complement biomarker discovery efforts with more innovative approaches to clinical trial design; for example, consider smaller trials in more homogeneous patient populations that include better biomarkers and more sophisticated neurocognitive instruments and better measures of brain function. (Mucke)
• Enforce rigorous design, analysis, and interpretation of clinical and preclinical trials. Standardization is a crucial feature wherever possible. (Games, Ivinson, Mucke)
Improving Animal Models3
• Generate experimental models that better simulate the multifactorial nature of neurodegenerative disease and use them to assess
2 PPMI refers to the Parkinson’s Progression Markers Initiative, which is a clinical study aiming to identify biomarkers of Parkinson’s disease progression. The Alzheimer’s Disease Neuroimaging Initiative (ADNI) has the same focus, but for Alzheimer’s disease.
3 For additional discussion of opportunities related to animal models, see the summary of the March 2012 Institute of Medicine workshop on improving the utility and translation of animal models for nervous system disorders. The summary is available online: http://www.nap.edu/catalog.php?record_id=13530 (IOM, 2013).
combination treatments, which may be required to defeat neurodegenerative disease. (Mucke)
• Study the effect of aging in neurodegeneration by supporting the generation of conditional transgenic mouse models in which the different pathways that contribute to progression of disease could be manipulated in a temporal manner. (Cuervo)
• Generate animal models, free from intellectual property constraints, that are fully validated and available for minimal cost to academia and perhaps at a premium cost to industry. (Youle)
This page intentionally left blank.














