
Implementing a National Cancer Clinical Trials System for the 21st Century
Second Workshop by the American Society of Clinical Oncology and Institute of Medicine
The National Clinical Trials Network (NCTN) supported by the National Cancer Institute (NCI) has played an integral role in cancer research and in establishing the standard of care for cancer patients for more than 50 years. Formerly known as the NCI Clinical Trials Cooperative Group Program, the NCTN is comprised of more than 2,100 institutions and 14,000 investigators, who enroll more than 20,000 cancer patients in clinical trials each year across the United States and internationally.
Monica Bertagnolli, professor of surgery at Harvard Medical School, chair of the Alliance for Clinical Trials in Oncology, and chair of the Institute of Medicine (IOM) workshop planning committee, noted that cancer mortality in the United States is falling. Bertagnolli said that the NCTN has contributed substantially to this reduction in cancer mortality over its 56-year legacy. However, she added that “the world has changed in many, many ways, and it has become incredibly more complex and challenging to do the kind of work that we want to do.” At the same time, the promise of cancer research has never been greater, she said.
John Mendelsohn, chair of the IOM National Cancer Policy Forum (NCPF) and director of the Khalifa Institute for Personalized Cancer Therapy at the University of Texas MD Anderson Cancer Center, opened the workshop with a brief overview of the 2010 IOM consensus report titled A National Cancer Clinical Trials System for the 21st Century: Reinvigorating
the NCI Cooperative Group Program (IOM, 2010b). Recognizing the recent transformative advances in cancer research that necessitate modernization in how cancer clinical trials are run, as well as inefficiencies and other challenges impeding the national cancer clinical trials program, the NCI asked the IOM to develop a set of recommendations (summarized in Appendix B) to improve the federally funded cancer clinical trials system. These recommendations were published in the 2010 report. In early 2011, the NCPF and the American Society of Clinical Oncology (ASCO) held a workshop in which stakeholders discussed the changes they planned to implement in response to the IOM goals and recommendations (IOM, 2011).
Two years later, on February 11-12, 2013, in Washington, DC, the NCPF and ASCO reconvened stakeholders to report on the changes they have made thus far to address the IOM recommendations.1 At this workshop, representatives from the NCI, the NCTN, comprehensive cancer centers, patient advocacy groups, the Food and Drug Administration (FDA), industry, and other stakeholders highlighted the progress that has been made in achieving the goals for a reinvigorated national cancer clinical trials system, and discussed additional strategies to further improve the system.
This report is a summary of that workshop. An overview of key accomplishments since 2010 is shown in Box 1, and a summary of suggestions from individual participants for further improvements is provided in Box 2. A summary of NCI progress to date toward implementation of the IOM recommendations was presented by James Doroshow, director of the Division of Cancer Treatment and Diagnosis at the NCI, as shown in Table 1. The workshop agenda and statement of task can be found in Appendix A. The speakers’ biographies and presentations (as PDF and audio files) have been archived at http://www.iom.edu/Activities/Disease/NCPF/2013-FEB-11.aspx.
________________
1 This workshop was organized by an independent planning committee whose role was limited to the identification of topics and speakers. This workshop summary was prepared by the rapporteurs as a factual summary of the presentations and discussions that took place at the workshop. Statements, recommendations, and opinions expressed are those of individual presenters and participants, are not necessarily endorsed or verified by the Institute of Medicine, the National Cancer Policy Forum, or the American Society of Clinical Oncology, and should not be construed as reflecting any group consensus.
BOX 1
Overview of Key Achievements Since 2010
• Consolidated and integrated cooperative groups and operations
• Substantially reduced median time to trial activation
• Improved information technology systems
• Improved intellectual property terms for collaborative research
• Improved processes and timelines for the two NCI central institutional review boards
• Increased reimbursement to sites for large phase II studies and additional funding for select phase III trials based on complexity
• New guidance from the FDA on data collection
• New initiatives and resources to support the development of precision medicine
BOX 2
Overview of Suggestions Made by Individual Participants
• Enhance and expand collaborations among stakeholders (e.g., the NCTN, the pharmaceutical and diagnostics industries, federal agencies, and patients)
• Expand use of innovative trial designs
• Develop and validate technologies for precision medicine
• Define criteria for use of genomic and other biomarker tests
• Adequately cover the costs of tumor profiling and rebiopsy if necessary
• Create a centralized clearinghouse for annotated genetic profiles of patients’ tumors
• Ensure that endpoint measurement is free of bias in trials assessing tumor response or progression
• Assess quality-of-life issues in cancer clinical trials
• Engage patients in trial design to enhance participation
• Conduct a pilot study to assess whether reimbursing oncologists for the time it takes to inform patients about clinical trials increases patient accrual
|
|
|
| Goal 1: Improve speed and efficiency of the design, launch, and conduct of clinical trials | |
|
|
|
| Recommendation | NCI Response as of February 2013 |
|
|
|
| 1: NCI should facilitate some consolidation of Cooperative Group “front office” operations by reviewing and ranking the Groups with defined metrics on a similar timetable and by linking funding to review scores |
|
| 2: Require or facilitate consolidation of Group “back office” operations and, working with extramural community, make process improvement in the operational and organizational management of clinical trials a priority |
|
| 3: The U.S. Department of Health and Human Services (HHS) should lead a transagency effort to streamline and harmonize government oversight and regulation of cancer clinical trials |
|
| 4: NCI should take steps to facilitate more collaboration among the various stakeholders in cancer clinical trials |
|
| 5: NCI should mandate submission of annotated biospecimens to high-quality, standardized central biorepositories when samples are collected from patients in the course of Group trials and should implement new funding mechanisms and policies to support the management and use of those resources for retrospective correlative science |
|
|
|
|
|
|
|
| Goal 2: Incorporate innovative science and trial design | |
|
|
|
| Recommendation | NCI Response as of February 2013 |
|
|
|
| 6: Cooperative Groups should lead the development and assessment of innovative designs for clinical trials that evaluate cancer therapeutics and biomarkers (including combinations of therapies) |
|
| 7: NCI, in cooperation with other agencies, should establish a consistent, dynamic process to oversee development of national unified standards |
|
| 8: NCI should reevaluate its role in the clinical trials system |
|
|
|
|
| Goal 3: Improve prioritization, selection, support, and completion of cancer clinical trials | |
|
|
|
| Recommendation | NCI Response as of February 2013 |
|
|
|
| 9: NCI, Cooperative Groups, and physicians should take steps to increase the speed, volume, and diversity of patient accrual and to ensure high-quality performance at all sites participating in Group trials |
|
| 10: NCI should allocate a larger portion of its research portfolio to the Clinical Trials Cooperative Group Program to ensure that the Program has sufficient resources to achieve its unique mission |
o Increased reimbursement for high-performing sites (aimed at 40% accrual) o Need for additional infrastructure support with proposed budget increased to support better reimbursement but lower total level of accrual o Increase in core resources for genomic correlative studies |
|
|
|
| Goal 4: Incentivize the participation of patients and physicians in clinical trials | |
|
|
|
| Recommendation | NCI Response as of February 2013 |
|
|
|
| 11: All stakeholders should work to ensure that clinical investigators have adequate training and mentoring, paid protected research time, necessary resources, and recognition |
|
| 12: Health care payment policies should value the care provided to patients in clinical trials and adequately compensate that care |
|
|
|
|
| SOURCE: Doroshow presentation (February 12, 2013). | |
IMPROVING SPEED AND EFFICIENCY OF TRIALS
The first four consensus recommendations in Appendix B provided strategies to achieve the goal of improving the speed and efficiency of innovative clinical trials through reorganization of the system, by enhancing collaboration, and by streamlining and standardizing data collection and analysis. A major focus since 2010 has been on consolidating and integrating the participating cooperative groups and providing more centralized administrative and IT support and data management to improve collaboration and operational efficiency.
The cooperative groups have reorganized themselves into four groups focused on adult cancers, in addition to a preexisting group focused on pediatric cancers (see Box 3). This reorganization has been an enormous undertaking and is partly due to a new Funding Opportunity Announcement from the NCI that limited funding to five groups. The merged groups submitted proposals in response to that announcement in February 2013, and awards are anticipated in 2014. Thus, the consolidation is still a work in progress.
BOX 3
Reconfigured Groups of the NCTN
• Alliance for Clinical Trials in Oncology (consolidation of Cancer and Leukemia Group B, the North Central Cancer Treatment Group, and the American College of Surgeons Oncology Group)
• Children’s Oncology Group
• ECOG-ACRIN Cancer Research Group (consolidation of the Eastern Cooperative Oncology Group and the American College of Radiology Imaging Network)
• NRG Oncology (consolidation of the National Surgical Adjuvant Breast and Bowel Project, the Radiation Therapy Oncology Group, and the Gynecologic Oncology Group)
• SWOG (formerly known as Southwest Oncology Group)
SOURCE: Comis presentation (February 11, 2013).
The Clinical Trials Strategic Planning Subcommittee, a subgroup of the NCI’s Clinical Trials and Translational Research Advisory Committee (CTAC), is charged with helping to develop a fully integrated Clinical Trials System.
However, group leaders stressed the benefits of consolidation. Bertagnolli said, “I think it’s very important to acknowledge the tremendous and extremely positive impact that the involvement of the IOM in our enterprise has had. The initial consensus statement and the first workshop have yielded truly amazing changes that have updated the groups and allowed us to really feel confident as we go forward that the work that we do will be preserved and even strengthened.”
Robert Comis, president and chair of the Coalition of Cancer Cooperative Groups, group chair of the Eastern Cooperative Oncology Group, and professor of medicine and director of the Clinical Trials Research Center at Drexel University, concurred. He reported that the consolidation of the Eastern Cooperative Oncology Group (ECOG) with the American College of Radiology Imaging Network (ACRIN) will enable an integrated data warehouse that will include case report forms and imaging data, digital pathology, a specimen repository inventory, and “omics” information and resources, adding that the pooling of resources will enable ECOG to take advantage of ACRIN’s tremendous amount of electronic imaging data. “From the inception of the ECOG-ACRIN idea, we had always envisioned this as a great opportunity, not just for us but for the whole system,” he said.
NCTN group operations were also reorganized into five major hubs:
1. Statistics and data management centers
2. Radiation therapy and imaging core services centers
3. Integrated translational science centers
4. Lead academic participating sites
5. The Canadian Collaborating Clinical Trials Network
All groups will contribute to and use the resources of the newly established integrated translational science centers, Comis noted. The goal is to develop integrated next-generation sequencing, advanced imaging, immunobiology, biorepositories with clinically annotated specimens, and reference labs. These centers will offer a platform for sustained, cutting-edge scientific effort and enhance interactions across groups and with cancer centers, he stressed.
The NCI has also expanded its Cancer Trials Support Unit to enable centralized administrative and regulatory functions for clinical trials. It now
offers 24/7 centralized Web-based patient registration; provides educational materials for patients, nurses, and physicians; and offers regulatory support, financial management, accrual reimbursement, and protocol coordination, as well as other types of support.
Previous studies indicated that a substantial contributor to the inefficiency of cancer clinical trials has been the length of time between when a trial concept is first proposed and when it is approved and activated, said Doroshow. Prior to 2008, it often took more than 2 years to activate a phase III trial and nearly that long for early-phase trials as well. However, several changes have substantially reduced the median time to trial activation, with a 30 percent improvement for early-phase trials and a 50 percent improvement for phase III trials (see Figure 1) (Abrams et al., 2013).
These time-saving changes include setting aggressive timelines for implementing clinical trials that provide not only optimal target dates, but also absolute cutoff dates, after which a trial cannot be activated. The NCI also established a new website that tracks all phases of a protocol’s life cycle, created new positions to manage protocol development, and implemented uniform templates for protocol development and reviewers’ comments.
Other major contributors to the shortened trial activation time include improved processes for the two NCI central IRBs—one for adult trials and one for pediatric trials—and updated consent templates. These changes slashed the time from protocol receipt to trial approval by a central IRB from a median of about 4 to 5 months in 2008 to only 3 weeks in 2012. As of 2013, all NCTN trials are required to use the central IRBs (with waiver exemptions possible for sites demonstrating similar local IRB review timelines).
“This will decrease a lot of needless busy work that results from having hundreds of institutions review the same protocols,” Doroshow said. It will also facilitate more clinical trials of rare cancers by enabling rapid approval of a trial as patients with these rare diseases are encountered in the clinic, he added. “Now that we’re going to have these small and molecularly defined populations, it’s rather critical that institutions have the ability to open trials when they find the right patients, because we probably will have many more trials with such small populations,” said Jeffrey Abrams, associate director of CTEP in the Division of Cancer Treatment and Diagnosis at the NCI.
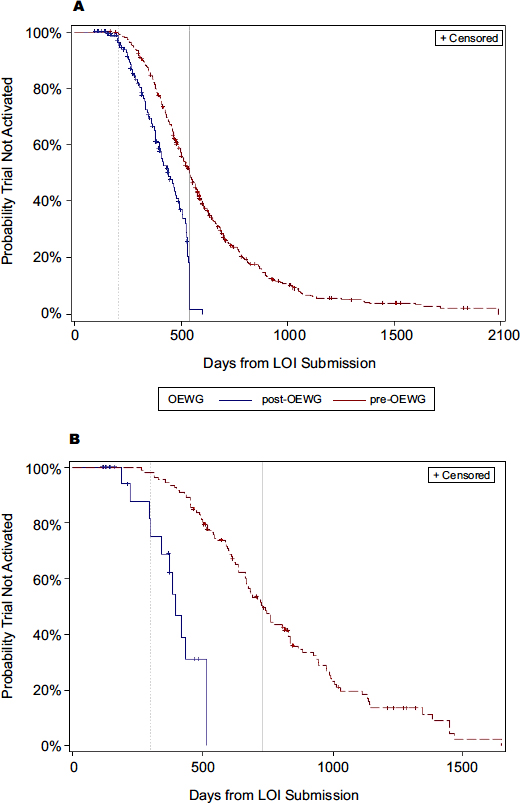
FIGURE 1 Timeline comparison of trial activation, historical versus post-implementation of the recommendations from the Operational Efficiency Working Group, April 2010 to August 2012.
NOTE: A = early-phase studies; B = phase III studies; LOI = letter of intent; OEWG = Operational Efficiency Working Group.
SOURCES: Doroshow presentation (February 11, 2013) and Abrams et al. (2013). Reprinted with permission from the Journal of the National Cancer Institute.
However, the time needed for acquisition of the drug being tested and approval from industry sponsors to begin testing it is still delaying trial activation, Doroshow noted. Despite a 30 percent improvement in early-phase trial activation times, he said, “We have to do better in interacting with pharma and getting approvals for these trials in a timely manner. We have to get these trials open in about 6 to 7 months to be appropriately timed for what our industry partners expect.”
Information Technology Improvements
The elements of the new common IT data management system the NCI implemented for the NCTN have generated multiple benefits as well (see Box 4). The new Medidata Rave Web-based remote data entry system, initiated in April 2011, enables the user to record patient information using standard forms customized for each study. “The most remarkable effort has been to implement a uniform clinical trials management system across this network with 3,000 sites,” said Doroshow. “There is no country and no pharmaceutical organization that has a uniform clinical trials data management system that unites so many sites.” (See also the “Partnering with Industry” section.)
One of the IOM recommendations under the broad goal of improving the speed and efficiency of clinical trials was to improve collaboration among stakeholders, including within NCTN groups and between the NCTN and industry, disease foundations, and patient advocacy organizations.
Renaud Capdeville, vice president of oncology global development at Novartis Pharma AG, described several advantages to industry collaboration with the NCTN. The NCTN groups and pharmaceutical industry organizations have complementary skills that can be leveraged to deliver innovative trials, he said, and the extensive network of academic and community practices within the NCTN makes it easier to conduct clinical trials on rare diseases. “Cooperative groups can reach out to patients quickly,” Capdeville noted.
Sandra Horning, senior vice president and global head of clinical development of hematology/oncology at Genentech, expanded on the advantages of industry-NCTN collaboration by noting that collaborative clinical trials offer a lower-cost financial model and tap into the operational capabilities
BOX 4
Common IT Data Management System (CDMS)
Electronic tool(s) or processes that support:
• Data collection: remote data capture
• Data coding: standard libraries—common toxicity criteria
• Data management: discrepancy, delinquency, communication, correction, and preparation of data for analysis
Core benefits of CDMS on NCI-supported multicenter trials:
• Reduces training costs and overall cost of data management
• Reduces risk of data delinquency and/or discrepancy
• Reduces time/effort to correct/complete data
• Reduces delays in obtaining science and safety results and improves trial management and decision making
Other benefits of CDMS on NCI-supported multicenter trials:
• Supports/complements transformation of groups into a new “network” program
• Meets FDA and other federal requirements for e-data capture, security, and transfer
• Promotes data sharing
• Sets the stage for further infrastructure improvements, such as integration with expedited serious adverse event reporting, remote auditing, and electronic filing for FDA reports
SOURCE: Doroshow presentation (February 11, 2013).
of the NCTN. These collaborations provide industry with access to patient populations within the NCTN, as well as its disease and scientific expertise, its critical mass of U.S. trial specialists, and its innovation in product use and study design. In addition, collaborations with NCTN can enhance an industry’s scientific credibility, Horning added. But what ultimately drives industry-NCTN collaborations is “a mutual respect and trust, and passion for science and improving patient outcomes,” she said.
Hans-Georg Eichler, senior medical officer at the European Medicines Agency, also stressed the advantages of collaborations among stakeholders when it comes to fostering innovations in drug regulation (see also the “Regulatory Issues” section). “Collaborations can be very effective in
stimulating innovation, not only in the technology field but also in the policy field, because if one silo says we should go this way, the other silo will immediately say ‘no, we’re not going there because it wasn’t invented here.’ Bring those two silos together in the first place and you will probably have more success than otherwise,” he said.
Several speakers, including Doroshow and Abrams of the NCI, reported that significant progress has been made in facilitating NCTN-industry collaborations. Doroshow pointed out that the NCI has harmonized all its guidelines for programs engaged in the conduct of clinical trials, so that the appropriate incentives are in place for collaboration among investigators in different programs. In addition, the NCI, in collaboration with the CEO Roundtable on Cancer, developed START clauses for company and academic collaborations to speed clinical trial negotiations (NCI and CEO Roundtable on Cancer, 2008).
The NCI has also revised its IP option on all CTEP CRADAs relating to drug development. The IP option clarifies the rights to diagnostics or other IP that might result from studying biomarkers and tissues in the trial. “If a diagnostic is discovered in one of our trials, the company that provided the drug and is our collaborator does not have the first right to that diagnostic. The investigator retains that right. On the other hand, there is no blocking of the IP so that the company would have to pay royalties every time its drug was used if a regulatory authority said the drug had to be used with that companion diagnostic,” Abrams explained.
Instead, each collaborator receives a non-exclusive, royalty-free, worldwide license for research purposes only, and a non-exclusive, royalty-free worldwide license to disclose and promote inventions as necessary or as required by a regulatory authority to be used with a drug. For alternate uses or dosing schedules for agents being tested in a clinical trial, companies are granted a non-exclusive, royalty-free, worldwide license for commercial purposes. But companies can still negotiate a co-exclusive or exclusive license for such IP.
The NCI also established IP terms for investigational multi-agent combination trials, which are becoming increasingly common in cancer research. For such studies, each collaborator receives a non-exclusive, royalty-free, worldwide license for all purposes, including commercial purposes of any combination IP. Companies can still negotiate a co-exclusive or
exclusive license for a collaborator’s IP pertaining to the agent. To help stem trial startup delays due to IP issues, the NCI set a new absolute deadline of 6 months for CRADA negotiations with industry sponsors.
Edward Benz, director of the Harvard Cancer Center at the Harvard School of Medicine and president of the Dana-Farber Cancer Institute, said that he appreciates the value of this new approach, and noted that
the usual way we’ve set up these intellectual property agreements is based on the bet that you’re going to get My Fair Lady instead of the play that never makes it to Broadway. Everybody protects jealously that potential big hit, but getting upfront research support in return for licensing terms that are friendlier to the pharmaceutical partner is actually a better deal. The value of the research support and the chance you have to make an impact with present-day support in exchange for a discount down the road in the IP arrangement, we think, is a much wiser way to approach it, and it’s been part of allowing us to have better relationships with the pharmaceutical industry.
Abrams added that collaboration with industry is more likely “now that we have done a number of things to make sure the data quality for the NCTN system is quite high and comparable to what’s achieved when industry does a study on their own. Our new IT data management system is really state-of-the-art for collecting quality data and is critical to being able to go to the FDA and support the needs of our company collaborators.”
The Web-based IT system also facilitates contracts with research agencies because “it doesn’t really matter which group is leading the trial anymore,” Abrams said. He added that “it will probably allow us to meet all the new FDA requirements that are upcoming for secure electronic data capture and transfer, and will enable our different cooperative groups to collaborate on additional scientific projects much more easily and make the data available to other people outside the groups more easily.” The IT system also facilitates the systematic and all-inclusive reporting of adverse events from Grade 1 to Grade 5, which is necessary for the drug registration trials of industry sponsors.
New aggressive timelines for getting clinical trials under way are also encouraging more industry partners to participate, Abrams pointed out. “Our industry colleagues have told us that ‘time is money.’ We can’t sit around waiting a very long time for NCI studies to get up and running.”
Genentech has had several productive clinical trial collaborations with NCTN, Horning noted, and she offered several lessons learned from those collaborations. For example, one clinical trial collaboration between the NCTN and Genentech, a study of paclitaxel with or without
bevacizumab in metastatic breast cancer, fell short of FDA data quality standards The same study raised FDA concerns about investigator bias, which was addressed retrospectively through a radiological independent review facility (IRF). Further research by the FDA and independent groups indicated that although reader discordance at the patient level was common, there was no evidence of systematic investigator bias for the progression-free survival (PFS) endpoint (Amit et al., 2011; FDA, 2012a). In addition, these studies found the potential for IRF bias through informative censoring. These results led the FDA to propose that when PFS is used as an endpoint for clinical trials on agents for solid tumors, a random audit by an IRF could avoid some of the missing data issues and mitigate informative censoring, while reducing the cost and burden of more complete IRF reviews.
According to Horning, this example serves to illustrate that industry and NCTN partners must prospectively clarify regulatory requirements to satisfy global regulatory authorities when they collaborate on clinical trials, because most drugs are registered and marketed globally. There also should be prospective agreement between a clinical trials group and an industry sponsor of a registration trial regarding data collection and curation; safety reporting and access to records; and communications, publications, and presentations; all of which should ensure that data are of high quality, reported in a timely fashion, “fit for purpose,” and compliant with regulatory requirements, Horning said. (A more extensive discussion of this trial design issue is described below in the “Regulatory Issues” section.)
Ultimately, the data collected must be adequate to reliably assess whether an investigational agent has a good risk/benefit ratio when added to or used in place of a known standard of care, Horning noted. Safety assessments need to include enough data to assess whether there are subsets of patients for whom the risk/benefit ratio is different, she added, and critical safety data must be integrated with efficacy data.
Horning observed that because of lessons learned from previous industry-NCTN collaborations, NCTN trials with registration potential have become more “industry-like” in terms of data standards, costs, and timelines, characteristics that have increased the likelihood of regulatory approval.
Changes in how NCTN studies with registration potential are conducted include
• ensuring that safety data have onset and resolution dates;
• providing more complete safety data rather than just targeted adverse event data;
• using an internally consistent database with symmetrical data collection on both arms;
• documenting why physicians or patients stop therapy;
• having procedures in place to minimize missing forms and fields; and
• reconciling expedited adverse event reports with the clinical adverse event database.
“The data management improvements address key industry considerations for quality, timeliness, and cost,” Horning said. She also appreciated the NCI’s revised IP stipulations in the CRADAs, which recognize the value to industry of annotated specimens and what they can reveal in the current era, in which predictive diagnostics have become more essential to drug development and therapeutic approval.
In addition, Horning applauded the shortened timeline the NCTN has recently instituted between concept submission and trial activation. However, she noted that there is still room for improvement in the relatively long time the NCTN takes to prioritize which trials get the final green light to go forward—a delay due to numerous discussions among investigators, groups, and NCI steering committees. In contrast, Horning said, this process is much more streamlined in Europe, where such decisions are often made at a single meeting, without as much deliberation among the various parties involved.
Horning also stressed the need for the NCTN to collaborate with global partners and satisfy global regulatory bodies. A prespecified plan for selective data collection must be agreed upon not only by the FDA but also by other relevant global health authorities, and more effort should be made to harmonize international requirements for global registration trials, Horning said. “Trials are now done globally for global registration,” she added.
Debasish Roychowdhury, senior vice president of global oncology at Sanofi, suggested that the NCTN should consider not just European collaborators but also those in other countries, such as China.
This may require ensuring that the patient populations in clinical studies represent the diverse ethnic populations that will eventually use the new drug should it be approved for the international market. However, Rachel Sherman, program specialist at the Center for Drug Evaluation and Research (CDER) Office of Medical Policy at the FDA, noted that the FDA accepts trials with no patients from the United States and has approved drugs based on such studies, but sponsors must show that those studies are applicable to the U.S. standard of medical care.
Richard Pazdur, director of CDER’s Office of Oncology and Hematology Drugs at the FDA, also stressed the international scope of drug testing and marketing. “All of the trials that come to the FDA at the present time are international trials. For the NCTN to be relevant for the next decade or so, they are going to have to address the issue of how they play into not just the national cancer trial system, but the international cancer trial system, especially as we take a look at rarer and rarer subsets of diseases. The pharmaceutical firms have already realized this and are doing trials internationally,” he said.
Capdeville described the RATIFY2 trial, an innovative, global phase III trial that Novartis is conducting in collaboration with CALGB (Cancer and Leukemia Group B; now part of the Alliance for Clinical Trials in Oncology). RATIFY is testing a multitarget kinase inhibitor called midostaurin, which preclinical studies showed is especially effective at inhibiting the FLT3 tyrosine kinase. Mutations in this kinase are associated with poor survival in acute myeloid leukemia (AML).
After midostaurin had shown clinical activity in wild-type and FLT3-mutated AML in phase I and II trials, Novartis wanted to launch a phase III trial to test the drug in AML patients with activating FLT3 mutations. According to Capdeville, they decided to collaborate with CALGB for this trial because the group had done previous studies documenting the prognostic significance for mutated FLT3 in AML and had the scientific expertise to run the trial. It was advantageous for CALGB to collaborate with Novartis, he noted, because the rareness of the FLT3 mutation in AML would require a large, global multisite study to acquire enough patients. “This was beyond what CALGB could deliver in itself, so there was this potential synergy with the global operational infrastructure of Novartis that could bring together multiple cooperative groups and centers,” Capdeville said.
RATIFY is a simple randomized phase III study. CALGB was respon-
________________
2 Randomized AML Trial In FLT3 in <60 Year olds.
sible for writing the protocol for the trial, with input from 12 other participating cooperative groups (in the United States and internationally). CALGB owns the database and is responsible for reviews by a Data and Safety Monitoring Board on a regular basis. CALGB has sponsored the trial in North America and Novartis has sponsored the trial outside North America. CALGB and the other cooperative groups share accountability for FLT3 testing. A joint clinical trial team oversees day-to-day operations. If the study is successful, Novartis will submit its findings for regulatory review.
The main challenge in the trial has been to detect the FLT3 mutation in tumor samples before patients receive chemotherapy, given the clinical urgency of the diagnosis, Capdeville noted. That testing is completed within 3 days of sampling at 1 of the 10 central labs in each of the main cooperative groups. There is a common protocol for consistency and periodic cross-validation of the test sample among laboratories.
After meetings with the FDA, trial leaders decided that a companion diagnostic would be developed at a later stage in drug development, with a bridging study aimed at showing concordance between the clinical trial assay and the companion diagnostic version of the assay. This required patients to consent to their samples being used not only for the phase III clinical trial, but also for the later bridging study, as well as central storage of all tumor samples.
The study is ongoing, but Capdeville listed several lessons that have already been learned from the collaboration:
• Keep the data flow as simple as possible.
• Foster open and transparent collaboration between industry and the cooperative groups, “which takes some time so the two understand each other well and expectations are well aligned,” Capdeville said.
• Be open to using a slightly different process than normal. “We can’t just use the Novartis SOP [standard operating procedure] or only the cooperative group SOP, so there has to be dialogue on this,” Capdeville pointed out.
• Preserve the independence of scientific and academic oversight on the study while balancing industry’s needs.
• Involve a range of disciplines, including technical as well as scientific expertise.
Partnering with Cancer Centers
Benz reported that the Dana-Farber Cancer Institute recently reorganized its oncology research program to better enable clinical translation. Prior to the reorganization, which began in 2002, most of the cancer research resources were devoted to disease programs and centers that were anatomically focused. These centers were the sites of Dana-Farber’s clinical trial activity, except for phase I studies, which stemmed from the institute’s Early Drug Development Center. Although the centers were multi-disciplinary, including surgery, radiation, oncology, nursing, pharmacology, and medical oncology, “they were becoming somewhat siloed around their particular cancers that they were interested in,” said Benz.
Recognizing that this setup was slowing the pace of clinical translation, Dana-Farber made a number of changes. One change was to recognize that the ultimate end product “wasn’t papers published in Nature or other academic metrics, but the goal would be to bring things into clinical practice,” Benz said. The restructuring aimed to implement project management principles that would translate discoveries into clinical benefit while preserving the culture of independent discovery. Dana-Farber faculty identified 12 areas ripe for translation, such as genomics, vaccines, and systems biology, and created “integrative research centers” around each, Benz reported.
Although some of these integrative research centers, such as the Center for Cancer Genome Discovery and the Center for Functional Cancer Epigenetics, focus on research pursuits grouped according to the primary method or technology being employed for discovery, Benz stressed that they are not traditional core facilities. Instead, they provide both a technology platform and an intellectual hub, and membership in each crosses departmental boundaries.
The centers are girded by business rules and accountability. The faculty leader of each center is charged with developing a business plan that explicitly includes milestones and deliverables over 5 years. The financial plan requires the center to be self-sufficient within the same time frame. “Whether it is mouse modeling, lead molecule development, or other projects, the expectation is that the work will move something closer to a clinical application,” Benz said.
To incentivize faculty to lead the centers, Dana-Farber created opportunities for responsibility and career advancement, and provided seed funding to start center activities. “We created a place in the institution where scientists with this kind of background and orientation had a home and could
make a contribution with professional upward mobility,” Benz stressed. “We realized that we had capabilities that were scattered across various labs and needed to be organized into programs,” he said. For example, a Cancer Chemical Biology program was started with three recruited faculty who had synthesized materials in their own laboratories. These newly discovered agents were about to enter or were already in clinical trials.
One of the centers, the Belfer Institute for Cancer Sciences, aims to bridge the gap between academia and industry. Researchers at the Belfer Institute have expertise in preclinical models, including genetically engineered mice, primary tumor xenografts, and short-term tissue cultures. They are also well versed in biomarkers and clinical biomarker assays and have access to clinical specimens through academic collaborations with a broad network of investigators. The Belfer Institute has been partnering with large pharmaceutical firms such as Merck and Sanofi to identify and validate new drug targets and delineate a clinical path for drugs in clinical trials. “The Belfer creates an interface where faculty or any kind of external partner can bring a target molecule and get the studies done that they need to do to decide if it should end up in an early-phase clinical trial,” Benz said.
Dana-Farber has a more traditional and longstanding partnership with Novartis that enables Dana-Farber researchers to receive 2-year research grants from Novartis on topics of interest to the company. The researchers have the freedom to publish their findings. “A lot of oncology drugs in Novartis’s pipeline have moved through that pipeline a little faster thanks to these partnerships,” Benz said.
Benz summarized what Dana-Farber has learned from its restructuring and its industry partnerships in terms of what it takes to move things more quickly from bench to bedside. “You need a great group of investigators, and they have to be a mix of basic, clinical, and translational scientists who want to collaborate and interact in this more team- or goal-oriented scientific application,” Benz said. He stressed that these investigators have to be situated in a place where there is expertise and understanding of what the clinical problems are. “They can’t be purely clinical centers, but must also have many of the features of a strong academic partner,” he said. Another necessary ingredient is technical expertise, and “a broad interface that allows multiple points of entry, exit, and reentry for this process of partnering and translation outside of the immediate sphere,” Benz noted.
There are also many potential benefits for the NCTN in partnering with disease-specific foundations, as well as with more general foundations, Benz pointed out. Margaret Anderson, executive director of FasterCures, agreed, noting that venture medical philanthropy is a growing area. Venture philanthropy not only funds novel, high-risk research that bridges disciplines, institutions, and ideas, but also taps strong scientific expertise to guide its efforts, she said. “Venture medical philanthropy groups tear down some of the collaboration barriers due to the silos that exist in medical research,” Anderson added. Disease foundations also tend to have a great deal of oversight in whether the funds they provide are being well spent. “The hallmark of all these groups is if they are going to put a dollar down on the table for any activity, they’re going to monitor that money and look at how it is being spent and what the outcome is,” she said.
Some medical philanthropy organizations run entire trials themselves at various institutions, or support clinical centers that do so. For example, she said, the Multiple Myeloma Research Consortium has 16 member institutions and has initiated 30 trials, which launched 60 percent more quickly and enrolled patients 10 percent more quickly than industry trials. Eight of these drug studies are in their final stages. Venture medical philanthropies can also bankroll industry endeavors that prompt pharmaceutical firms to develop treatments for a specific disease they are not already inclined to explore on their own, Anderson added. She highlighted the recent approval of Kalydeco, the first drug to target the cause of cystic fibrosis—the protein product of a faulty gene (CFF, 2012; FDA, 2012e). This drug resulted from a longstanding collaboration between the Cystic Fibrosis Foundation and industry, in which the foundation provided much of the seed capital needed to launch the clinical development of the drug.
Anderson stressed that one should consider not only financial capital but also human capital when evaluating whether collaborations furthered by venture medical philanthropy will be productive and valuable. If patients trust advocacy organizations and foundations, they will be more willing to participate in the research they sponsor; these organizations “are really changing the game of clinical trial recruitment,” Anderson said. She noted that the Michael J. Fox Foundation for Parkinson’s Disease has its own “trial finder,” which had 14,000 patient volunteers and 200 clinical trials in its database within 10 months of being launched in April 2012. “It’s absolutely critical that you have clinical trial matching like this because oftentimes
patients are not going to be finding out about these trials from physicians,” Anderson said. She added that the clinical center associated with the Translational Genomics Institute has 60 percent of patients participating in clinical trials, compared with the national average of 3 percent.
“Venture medical philanthropy is fixing the leaks in the clinical trial pipeline that are diverting the stream of patients from such trials,” Anderson concluded. Improving the efficiency of trials will encourage broader participation by both physicians and patients.
In addition, many disease foundations have longstanding relationships with FDA staff, which “really paves the way for things to go more quickly. They do regulatory de-risking,” Anderson said. “They lay the groundwork for determining and answering the questions the FDA needs answered to start looking at approvals in this space.”
Anderson ended her presentation by stressing that “now’s the time to start to look at efficiencies and ways to potentially leapfrog things forward and think about how we can change this, because the bottom line is that if we’re not patients already, we’re going to be.”
An Example of Collaboration in Cardiovascular Research
David Sabatine, associate professor of medicine at Harvard Medical School and associate physician of cardiovascular medicine at Brigham and Women’s Hospital, described the TIMI Study Group, which he chairs. The TIMI Study Group,3 which was named for its first trials on thrombolysis in myocardial infarction, is an academic research organization based at Harvard’s Brigham and Women’s Hospital dedicated to advancing the knowledge and care of patients with cardiovascular disease and its risk factors. Since 1984, TIMI has conducted 65 clinical trials at more than 4,000 sites across 6 continents. More than 8,000 investigators have participated in TIMI trials, which have enrolled more than 300,000 patients to date. Most TIMI trials are sponsored by industry and enroll between 15,000 and 25,000 patients per trial.
Sabatine explained how the TIMI Study Group operates and how it collaborates with industry in conducting clinical trials. He noted that pharmaceutical companies choose to work with TIMI because it offers experienced and skilled research scientists, clinical trialists, and project managers.
________________
3 See http://www.timi.org.
“These trials are so big, they are like a jumbo jet—they’re very hard to steer so you need a lot of expertise to fly them,” Sabatine said.
Another advantage of TIMI’s infrastructure, Sabatine noted, is that “it brings all the necessary parts together under one roof. The principal investigator works very closely with the project director, who works on a daily basis to ensure all aspects of the trial are integrated. So, if we wanted to have a high rate of adjudication of events, for example, someone can take care of that by walking down the hall and talking to one of his or her colleagues.” The TIMI Study Group also offers core services that include a safety desk; trial hotline; biomarker, genetics, and electrocardiography core laboratories; a clinical events committee; and a quality-assessment team.
Monitoring of the trials is typically done by contract research organizations hired by industry sponsors, but the monitors are trained by the TIMI Study Group on the disease state and the study protocol. “We sort of take charge of them, but they aren’t on our payroll,” Sabatine said.
TIMI physicians include
• clinicians on the staff at Brigham and Women’s Hospital;
• global principal investigators for trials who come from the faculty at Harvard Medical School and dedicate between 75 and 80 percent of their time to research; and
• clinical trialists (TIMI investigators), who are highly experienced in the design of clinical trials and work daily with the senior project director on trial implementation.
The TIMI Study Group also focuses on ensuring adequate training and communication for participating sites, according to Sabatine, and has a trial hotline, staffed 24/7, that responds to all medical and operational inquiries.
TIMI project managers generally have more than 10 years of experience in running megatrials with more than 10,000 patients. “There’s a special skillset for running such large trials and our project managers have the experience that is required and that even industry may lack,” Sabatine noted. TIMI staff also apply their expertise to develop an appropriate trial design that includes the right patient population, drug dose, and endpoints. This effort is aided by the TIMI Working Group’s online databases of electronic patient records collected during its trials. Researchers can tap that database to refine inclusion and exclusion criteria for their trials. “We work very closely with the sponsor beforehand, using our databases to give
information on what might be the right enrichment factors for the trial,” Sabatine said.
As Figure 2 illustrates, each TIMI trial has a joint management team with members from both industry and TIMI, including key physicians, study chairs, and sponsor representatives, who meet biweekly.
In addition, a joint working group focused on operational issues meets weekly. Often, there is also joint management of the blood samples collected in the trial. The samples are typically split between TIMI and the sponsor, according to Sabatine.
Open and frequent communications between sponsors and TIMI gird the success of its collaborations with industry, he noted. “We have very frank conversations at the beginning of any potential marriage with our sponsors that set the boundaries and reinforce respect for and trust in the
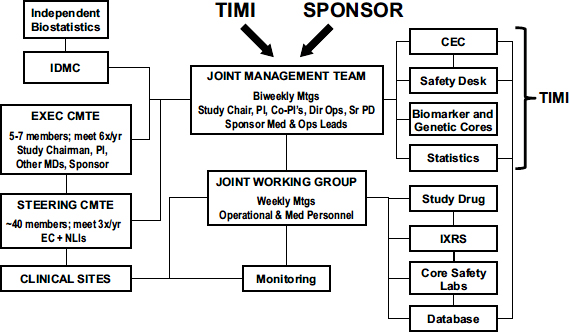
FIGURE 2 TIMI trial organization.
NOTE: CEC = Clinical Events Committee; Dir Ops = Director of Operations; EC = executive committee; EXEC CMTE = executive committee; IDMC = Independent Data Monitoring Committee; IXRS = interactive voice recognition/website system; MDs = medical doctors; Med = medical; Mtgs = meetings; NLI = national lead investigator; Ops = operations; PI = principal investigator; Sr PD = senior project director; STEERING CMTE = steering committee; TIMI = Thrombolysis In Myocardial Infarction; x/yr = times per year.
SOURCE: Sabatine presentation (February 11, 2013). Reprinted with permission from TIMI. Not for reproduction without permission from TIMI.
area of expertise each group will have,” Sabatine stressed. He added that during a trial, there are ongoing discussions with industry sponsors about all aspects of the study, including protocol design and the statistical analysis plan. “We maintain a dialogue with our sponsors throughout the half a decade that we work together,” Sabatine said.
Regarding legal agreements for the TIMI-industry collaborative trials, Sabatine noted that because of the long track record TIMI has with a number of companies, “we don’t need to reinvent the wheel but just specify the scope of work for a particular project.” He added that Harvard has strict rules that give TIMI some ownership of the data and the ability to publish results.
TIMI relies on industry support. Most TIMI trials are funded by industry, Sabatine said, “and it’s my job to keep the trials coming” to ensure financial support of TIMI’s infrastructure.
In response to a question about what motivates clinicians to participate in TIMI trials, Sabatine noted that participating study sites are not dedicated solely to TIMI trials: “There’s no pledge of allegiance to TIMI, but they do tend to work with us frequently because we spend a lot of time designing trials that are high-quality and high-profile.” The findings of many of the studies are published in prestigious scientific journals. Meaningful physician-to-physician contact also motivates doctors to participate in the research, Sabatine added. “If there’s a question from the site, they can talk to the TIMI investigator who’s spending 75 to 80 percent of his or her time on the trial,” he said.
Participating physicians are rewarded by the scientific success of the trial, according to Sabatine, and are also given financial compensation for their time. Sponsors determine how much participating physicians are paid per patient enrolled in the trial. “We advocate for the highest, most reasonable amount, but the sponsor ultimately determines the dollar value and that ends up being constant for all the sites,” Sabatine said.
The Timeline for Cancer Drug Development
To provide a perspective on the challenges involved in conducting efficient cancer clinical trials, Joseph DiMasi, director of economic analysis at the Center for the Study of Drug Development at Tufts University, presented data on clinical development and approval times for cancer drugs. He showed that antineoplastic drugs have long development timelines compared with most other therapeutic classes, and that development times are
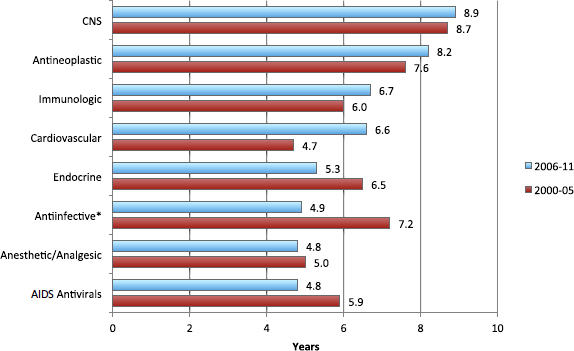
FIGURE 3 Clinical development times vary by period and across therapeutic classes, 2000-2011.
NOTES: CNS = central nervous system; *excludes AIDS antivirals.
SOURCE: DiMasi presentation (February 11, 2013).
increasing. Clinical development times for cancer drugs between 2006 and 2011 were 8.2 years on average, vs. 7.6 years between 2000 and 2005 (see Figure 3). In addition, only 13 percent of anticancer compounds that enter the clinical testing pipeline actually get approved. More detailed analysis revealed that drugs for blood cancers are nearly four times more likely to be approved than those for solid tumors, and that the risk of drug development failure varies significantly by cancer type, but not by molecule size.
DiMasi also showed that despite long development times, the number of anticancer drugs approved in 2012 was more than twice the average annual rate for the previous decade. In contrast, drug approvals for all other therapeutic classes decreased or remained essentially flat during the same time frame (Kaitin and DiMasi, 2011). The number of cancer drugs entering the clinical pipeline between 1993 and 2004 has also markedly increased, DiMasi said. “This is further evidence of increasing interest in cancer drug development, despite all the problems with this development alluded to thus far,” he concluded.
The time from first submission of a new drug or biologic application to FDA approval also varies by drug category, with shorter times for antineoplastic drugs than for most other therapeutic drug classes (in contrast
to the overall clinical development time). About 80 percent of that time comprises FDA review of the application, and the remaining 20 percent comprises sponsor responses to FDA requests. Between 2006 and 2011, approval times for cancer drugs decreased by half compared to what they had been between 2000 and 2005 (0.6 vs. 1.2 years). Although more oncology drugs are on fast-track, accelerated approval programs than compounds in other therapeutic classes, that special designation was not linked to shorter approval times by either the FDA or the European Medicines Agency (EMA), DiMasi noted. “Oncology drug development is challenging, and we need efficiency improvements to lower cost, speed development and regulatory review, and to reduce risk in this critical therapeutic class,” he concluded.
As a preface to discussing the IOM recommendation to incorporate innovative science and trial design in cancer clinical trials, one session of the workshop was devoted to exploring the latest advances in “precision” medicine (also referred to as “personalized” medicine) and the challenges in implementing these new technologies in clinical care. Levi Garraway, principal investigator and associate physician at Brigham and Women’s Hospital and assistant professor of medicine in the department of medical oncology at the Dana-Farber Cancer Institute, began this session by pointing out that for several major tumor types, including melanoma and breast, colorectal, lung, ovarian, and brain cancers, about half of those tumors harbor at least one identifiable genetic alteration that fosters tumor growth or survival and is “actionable.” He said that an actionable alteration is one that can be targeted by approved or experimental drugs, or one that suggests the inappropriateness of treatment using particular agents. “That alteration is not just for the cancer biologist to get excited about, but is actually something that may evoke, in the back of a clinician’s mind, a decision or a different choice of care,” Garraway stressed.
Many agents that target genetic pathways in cancer have already entered clinical trials. Garraway said, “We have all the ingredients needed to practice precision medicine. There is not just one, but multiple drugs in development targeting multiple components in these genetic pathways. We now have the technology to look for those genetic alterations that will enable us to match the right tumor to the right drug.”
But such matching is currently easier said than done, several speakers
pointed out. “It’s like the genomic super highway is meeting the bike path of clinical medicine,” Garraway said, due to both scientific and logistical challenges. These challenges include acquiring tumor samples, the genetic heterogeneity of those samples, developing innovative clinical trial strategies that can apply the genetic findings, and addressing issues related to quality control and reimbursement for biopsies and tests.
Among the logistical challenges is the need to obtain consent from patients to submit to biopsies and extensive genetic testing of their tumors and to appropriately counsel them about what the results mean. Walter Curran, executive director of the Winship Cancer Institute of Emory University and professor and chair of radiation oncology at Emory University School of Medicine, noted that the NCTN has trials that require patients to submit a tumor tissue sample in order to enroll. “But it’s still a work in progress for those trials where it’s not required for registration, and the more our tumor banks provide community physicians with kits that allow their staff to [submit a tumor tissue sample] more readily, the better,” he said.
Peter Adamson, chair of the Children’s Oncology Group (COG) and chief of the Division of Clinical Pharmacology and Therapeutics at the Children’s Hospital of Philadelphia, said that his cancer center obtains tissue for the majority of its studies and analyzes that tissue to determine which trial a patient might be eligible for. COG also aims to have a tissue sample submitted at the time of diagnosis for every child they treat with cancer. Charles Blanke, chair-elect of SWOG and professor of medicine at the Oregon Health and Science University Knight Cancer Institute, added, “This is a real culture change. Ten years ago, if you had mandated a biopsy, the IRB would have said, ‘Absolutely not.’ Now these patients are not only asking for biopsies when they go on trial, they also want to be biopsied when they progress. At SWOG we’ve been able to collect specimens on 86 percent of patients in our trials that just requested, rather than required, the samples.” Robert Comis noted that about 90 percent of patients enrolling in ECOG-ACRIN trials consent to having their tissue sampled and stored. “We all have huge banks of tissue and tremendous opportunities to use our annotated specimens, and those findings can be correlated with images that are integrated into the same system,” he said.
But it can still be problematic to discern which genetic findings from tumor tissue should be conveyed to patients. David Solit, associate profes-
sor at the Human Oncology and Pathogenesis Program in the Department of Medicine at Memorial Sloan-Kettering Cancer Center, also noted that “eventually, we’re going to find inherited genetic alterations that predispose patients to cancer, and the question is how much of that information has to be returned to the patients, and do we need medical geneticists and counselors to be involved in this process if we find this inherited risk?” In addition, sometimes the testing reveals genetic flaws not currently targeted by drugs on the market or by experimental agents being tested in clinical trials to which the patient has access. Other times, appropriate trials may be available, but patients and their physicians may not be aware of them, Solit said.
Another potential challenge is to have adequate biopsy tissue available for genetic profiling, not only at the time of diagnosis, but also when a tumor progresses, in order to assess the cause of treatment resistance and how to target that resistance. “There’s a lack of tissue for some patients. About 15 percent of patients don’t have any clearly accessible tissue you can use for profiling,” said Solit. Others may not have enough tissue for the multitude of tests that may need to be done by multiple labs, added Vincent Miller, senior vice president of clinical development at Foundation Medicine, Inc. Researchers continue to identify relevant new genetic alterations, and the tests for those changes continue to be developed. “The worst situation we may be placed in as the clinician is, we only have two slides left to do four different tests. Choosing which tests to do in that circumstance is like Russian roulette,” Miller said.
Solit also noted that the genetic heterogeneity of tumor cells can pose sampling conundrums, and that metastatic lesions may harbor different mutations than the primary tumor. For example, he described a study of paired primary and metastatic tumors from melanoma patients in which mutations in the BRAF4 gene were relatively common in both the primary and metastatic samples, while mutations in the PTEN5 gene were rare in the primary tumors and common in the metastatic tumors.
There also should be adequate reimbursement for the effort involved in biopsying or rebiopsying tumors for the purpose of genetic profiling, one workshop participant stated. Hospitals are not always willing to pay for that expense, nor are providers or insurers. The participant suggested that the NCI provide the financial resources for such biopsies in clinical
________________
4 Human homolog B of v-raf (Rapidly Accelerated Fibrosarcoma viral oncogene).
5 Phosphatase and tensin homolog, a tumor suppressor gene.
studies. Marshaling adequate resources for the profiling itself is another major financial challenge.
Several speakers stressed the lack of quality control for genetic profiling. “We need credentialed assays,” Solit stressed. Miller added, “We need to develop common criteria for the testing we accept for our clinical trials.” Lisa McShane, mathematical statistician at the Biometric Research Branch in the Division of Cancer Treatment and Diagnosis at the NCI, noted that often there is variability in results from laboratories doing the same test, which could be an argument for central testing, although this could raise logistical problems. She pointed out that more than a dozen years after the first test for HER2 (human epidermal growth factor receptor 2) was used to predict the response of breast cancer patients to the drug Herceptin, there is still a debate about the best way to measure HER2. McShane added that how specimens are handled and processed can also affect the results of the tests. “How can we make sure that when your sample is taken, it is treated appropriately?” she asked.
A major scientific challenge to precision medicine for cancer is interpreting the large data sets that result from genomic sequencing and distinguishing driver mutations from passenger mutations. “The number of data points per patient is skyrocketing, and we need clinical data interpretation algorithms that are capable of addressing this,” said Garraway. Often, there are genetic variations of unknown significance for a particular tumor, but for which there is a targeted drug. In these instances, in vitro models or clinical tests could help assess if the genetic change is significant for that patient.
Genomic sequencing reveals a large of number of genetic alterations in tumors, some of which are what Garraway called “mountains,” or common alterations, but many more of which are “hills,” or mutations that occur less frequently. “The hills outnumber the mountains, and they’re critically important” in determining the treatment of individual patients, Miller said. “The number of clinically relevant alterations in a single patient is low, but the number of clinically relevant alterations across the disease state is high,” he explained. “Some genes are altered only in 1 to 5 percent of [a certain histological type of] tumor and often in a non-predictable fashion, arguing
for broad-based tests and nimble trials that can accommodate patients with these rare mutations.”
Solit noted that one patient whose tumor was assessed using whole genome sequencing had 19,000 mutations because of a defect in a DNA repair pathway. Because she responded to the combination of drugs given to her in a phase I study, Solit was able to determine that this defect was a driver mutation, even though it was not part of the standard 300-gene assay normally used to detect relevant mutations. But often, he said, the drivers are difficult to ascertain.
Garraway added, “One reason we discover the same driver mutations over and over again is because we recognize them, but there are always recurrently mutated genes that we don’t recognize.” He noted that once more tumor genomes are sequenced, clinically annotated, and compiled in a central database, more rare mutations will be identified as being driver mutations. “It is still humbling how little we know,” Garraway said.
Solit stressed that “the way you figure out if these genetic changes are drivers will not be in the lab. You need a clinical link that will tell you that particular mutation was important. This is opposed to what we’ve been doing, which is to sequence 150 or 200 genomes and then find the most common mutations and validate them in the laboratory.”
In his presentation, Solit showed that the rare clinical remissions that patients experience after being treated with investigative agents can signal driver mutations that, although rare in patients with a particular type of cancer, may also occur in several other types of cancers and therefore offer cross-disease treatment opportunities. When Solit has encountered these “exceptional responders,” genomic sequencing of the patients’ tumors has revealed rare mutations in genes that are part of the genetic pathways targeted by the drugs with which they were treated (Kaiser, 2013). These rare mutations explain why a few patients responded so favorably to a treatment when most of the patients in their study cohort, all of whom had the same histological type of tumor, did not. “It was not so surprising that these patients responded—the surprise was that they had this mutation we didn’t know about before,” Solit said.
Given the value of such discoveries, several speakers suggested that there is a need to develop and validate profiling technologies so that all cancer patients can have their tumors genetically profiled and receive cancer treatments matched to their profile. “Each patient’s treatment needs to be informed by an understanding of the molecular changes driving his or her disease—and what you don’t look for, you won’t find,” Miller said.
In his presentation, Miller described the approach developed by Foundation Medicine, a cancer diagnostics company that provides clinical laboratory services intended to help physicians tailor cancer therapy based on genomic analysis of each patient’s tumor.6 Using next-generation sequencing (NGS), Foundation Medicine sequences the coding region of more than 200 cancer-related genes, as well as 48 introns in 20 genes frequently rearranged in human cancer. “The premise of our test is to [look at the approximately 1 percent of genes] that are unambiguously implicated to be somatically altered in human cancer and study the heck out of them both in breadth, by looking broadly across tumor types, and in depth, by obtaining tremendous coverage,” Miller said.
According to Miller, Foundation Medicine’s assay is optimized for use with fine needle aspirations, core biopsies, and malignant effusions, and often identifies alterations that would never have been tested for because they are not the common mutations found in a given patient’s particular histological type of cancers. He said that Foundation Medicine translated research-grade NGS into its clinical cancer diagnostic assay by doing extensive analytic validation, which he said demonstrated the high accuracy and reproducibility required for clinical use. Miller said the assay can identify base substitutions with a sensitivity of greater than 99 percent for minor allele frequency greater than or equal to 5 percent, small insertions or deletions with a sensitivity of greater than 98 percent for minor allele frequency greater than or equal to 10 percent, and copy number alterations (amplification or homozygous deletion) with a sensitivity greater than 95 percent. He added that the specificity of the assay is greater than 99 percent (Yelensky et al., 2013).
After tumor samples have been tested, Miller explained, experts at Foundation Medicine integrate the genomic information into a report in a format understandable for both physicians and patients. “Our approach has been to link genome technology, clinical oncology, cancer biology, and information science to make this test applicable to routine clinical practice,” Miller said. Sometimes Foundation Medicine’s assay reveals aberrations that could potentially be targeted by drugs that are in clinical trials, but the trials may not be available locally, and patients are not always entered into trials, Miller noted.
Foundation Medicine also collaborates with several pharmaceutical companies to identify biomarkers in clinical trials. Some of these trials are
________________
“rescue” trials for drugs that failed previous clinical trials because of a lack of enrichment of the study population with patients most likely to respond to the drug. Others are longitudinal studies aimed at uncovering causes of treatment resistance that develop over time. Foundation Medicine also works with pharmaceutical firms to conduct prospective studies in which individuals are assigned to a line of therapy based on the presence or absence of one or more alterations in a gene or a series of genes, Miller said.
Solit’s findings from exceptional responders in clinical trials also suggest the need for an innovative clinical trial design that tests the same agent on people with a wide range of cancer types, he stressed. He proposed conducting what he called a “basket” study, in which a treatment for a specific genetic defect in a tumor is tested on multiple cancer types, each of which is put into its own basket, or arm, of the study. For example, a BRAF inhibitor may be tested on a small group of colorectal patients while simultaneously being tested on a small group of lung cancer patients or ovarian cancer patients, all of whom have a BRAF mutation and are under the same phase II clinical trial umbrella. “There are more than 400 different histological subtypes of cancer, but you can’t open up 400 BRAF inhibitor studies. However, you can open up one basket study that potentially will bring all these patients in to answer a particular hypothesis,” said Solit.
Solit noted that some researchers are proposing to set up “an umbrella of basket studies, which would allow these studies to be brought out in a modular way to participants in a network such as the NCTN or some other large network without requiring you to go through each individual IRB for each disease.” The advantage of such a setup would be the data collected from the studies, which would be valuable to patients, doctors, payers, and regulatory authorities, he added. “We want to be able to capture this data because if patients are just being treated ad hoc in the community based upon a commercial laboratory giving them a result and no data is then being disseminated, other patients with the same mutation are not going to benefit by the information gained from treating previous patients. We need a new design to do this and a group that’s willing to lead that effort,” Solit stressed. Miller agreed, adding, “Use of a broad, robust testing platform in concert with an effective ‘master’ clinical trials network should accelerate accrual to trials, minimize off-label use, and allow patients access to agents more likely to be effective for them.”
Sometimes, a basket study may discover tissue differences that affect response to a drug targeting a specific mutation—for example, a BRAF inhibitor may not work as well in a colon cancer tumor with a BRAF mutation as it does in a melanoma with the same mutation. But if a basket trial shows this variation, it is still valuable, clinically relevant information, Solit noted. In addition, any arm/basket in a trial could be amended during the study to test a different treatment if the first 15 patients with a particular type of cancer do not respond, for example.
In response to a question regarding how many patients have to be screened to discover the rare mutations needed for a basket trial, Solit replied, “It is my hope that we’re going to rely on the entire country screening patients and identifying those with the mutations targeted in the basket trial. The screening protocol should be separated from the treatment protocol. Otherwise, we’re not going to be able to find enough patients for the clinical trials we want to do.”
For patients whose tumors have multiple drivers, it will be difficult to decide how to fit them into basket studies that offer a treatment targeting only one of the drivers. George Sledge, chief of oncology in the Department of Medicine at Stanford University, pointed out that one study of 100 breast cancers found 40 drivers in 73 different combinations, and most of those drivers occurred with low frequency (Stephens et al., 2012). Solit acknowledged that this is a problem that current basket studies are not designed to address, but he added, “First we need to get data on the single agent and then if we find that all the patients with a specific doublet mutation don’t respond, we need to start looking into that combination. The only way to study that is to start sorting through it clinically, and we could capture some low-hanging fruit and help a lot of patients in the short term just by doing these basket studies.” Miller agreed, adding, “There’s not a trial for everybody, but from looking at the data, 80 percent of patients could fit into a study of treatments that target doublet or triplet genetic drivers.”
McShane noted that basket trials could be logistically efficient and innovative “because we have a big screening protocol that brings everybody into the same front door and then directs you to separate, individual, single-arm trials.” But she cautioned that it will be critical to consider what the right endpoints are for basket trials. A response endpoint might not be appropriate for a targeted therapy that results in stable disease rather than dramatic tumor regression. But using PFS as an endpoint can be problematic if the genetic aberration or other biomarker used to select treatment is also associated with a more indolent course of disease or slower progres-
sion. McShane suggested that randomization of patients within the same biomarker subgroup could address this potential source of bias because it would distinguish prognostic from predictive effects. For example, patients could be randomized to receive standard therapy with or without the new targeted agent.
McShane stressed that “innovation is great, but we have to make sure the innovative methods still answer the questions that need to be answered.” She gave examples of other innovative biomarker-based clinical trial designs (Freidlin et al., 2010), including a biomarker enrichment design used to identify breast cancer patients likely to respond to Herceptin. In this design, patients whose tumors were positive for HER2 were randomized to be treated with Herceptin or to standard therapy. The control arm was necessary because HER2 is a negative prognostic marker for breast cancer.
But this trial design meant the results could not predict how patients without the HER2 marker would do when treated with Herceptin. “We’re still debating this because the enrichment design doesn’t really let you know what’s going on with the marker-negative patients,” McShane said.
To “hedge their bets” in this type of situation, researchers can conduct a biomarker-stratified design in which all patients are tested and then randomization is stratified by test results, McShane said. Patients who test positive or negative for the biomarker are both randomized to receive the new therapy or standard therapy. However, it is difficult to accrue patients to such a trial because the test results are not used to select therapy.
McShane also noted that there are multi-arm trial designs for non-biomarker-guided therapies. These can be statistically efficient by enabling reuse of a control arm, thereby reducing the number of patients needed to answer scientific questions. Adaptive designs are another innovative type of trial that use interim monitoring to determine when to stop the trial, when to increase the number of patients put on treatment arms that appear to be more effective, or when to drop ineffective treatment arms. “These can all be handled statistically—it just has to be pre-specified in the analysis plan,” McShane said.
McShane also pointed out that joint phase II/III trials are increasingly being employed to avoid bottlenecks in trial development and patient accrual. In these trials, phase II patients are seamlessly followed in phase III, assuming that the phase II results were reasonably promising. But it’s important to choose the right endpoint for progressing into the phase III portion in such trials. A popular endpoint in this type of study is pathological complete response, but a recent study of breast cancer did not find
a strong association between a pathological complete response and overall survival (Cortazar et al., 2012). “This is just a word of caution that we can be innovative and make assumptions, but we have to be doing some reality checks to make sure that what we’re going after is the right thing,” said McShane.
Garraway described an innovative model for clinical translation of precision medicine, known as Can Seq. The Dana-Farber Cancer Institute, working with the Broad Institute, recently launched this prospective trial, which conducts whole exome sequencing on the tumors of every patient in the trial. Researchers use the exome sequences to provide a list of variants with potential clinical implications, i.e., those that are prognostic, indicate what drugs might be effective, or suggest dose. This list is reviewed by a committee comprised of experts in oncology and genetics, who then compose and send a report with the most clinically relevant information to the treating oncologist. The trial is enrolling patients with metastatic lung, colorectal, prostate, and breast cancers. Garraway described one example from the trial in which exome sequencing revealed that a lung cancer patient did not have any of the standard variants often tested for in lung cancer tumors. But he did have an atypical KRAS7 mutation, so the patient was enrolled in a clinical trial of an agent that targets the KRAS signaling pathway.
Abrams also described two innovative trials currently in the planning stage at the NCI’s Cancer Therapy Evaluation Program. The first, the Adjuvant Lung Cancer Enrichment Marker Identification in Sequencing Trial, which goes by the acronym ALChEMIST, will be testing erlotinib, which targets mutations in the EGFR (epidermal growth factor receptor) gene, and crizotinib, which targets rearrangements in the ALK (anaplastic lymphoma kinase) gene, in patients with early-stage lung cancer. Patients with adequately available tissue will have their tumors tested for the two types of mutations and will be randomized into the ECOG-ACRIN erlotinib trial or the Alliance crizotinib trial.
Approximately 8,000 patients will need to be screened to find the 600 to 800 patients with mutations needed for the trial. These patients will be followed for 5 years. All EGFR and ALK testing will be done in a laboratory certified under the Clinical Laboratory Improvement Amendments (CLIA),8 and the rest of the tissue will be sent to investigators for the
________________
7 Human homolog of the Kirsten rat sarcoma viral oncogene.
8 See http://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html (accessd May 14, 2013).
Cancer Genome Atlas, who will conduct genomic sequencing, transcriptome determinations, and methylation analysis. Patients will consent to be recontacted about the option of doing a second biopsy if their cancer recurs to determine subsequent molecular changes in the tumor tissue.
The second trial, the Master Protocol in Advanced Lung Cancer, will have a phase II/III basket trial design. Multiple new therapies will be tested simultaneously in patients with non-small-cell lung cancer, and the study will be a registration trial for the new therapeutics. Patients will be assigned to an experimental treatment or a standard-of-care control arm on the basis of their molecular profile. If a new drug shows clinical benefit in patient populations that test positive for a specific biomarker, then the biomarker test could be evaluated for FDA clearance simultaneously with the drug evaluation, Abrams said. Initially, patients will be treated with a monotherapy, but combinations that target more than one molecular defect in the tumors could conceivably be part of the trial design, he added.
“We were hoping the NCTN would provide a platform for these kinds of studies and in fact changes have occurred that allow this new science to go forward much [more easily] than it could have in the past,” Abrams said. “Hopefully, we will have collaborations with our company partners that will enable us to have a group of drugs that could target mutations in the same trial,” he noted. Miller added that in order for such trials to be successful, companies have to be willing to engage in precompetitive collaborations.
Several participants at the workshop said that although it is encouraging to see the NCTN offering innovative trials girded by molecular screening, finding an appropriate clinical trial for a patient who has undergone molecular profiling is still a major barrier. “Genomic profiling is becoming more readily available, but a bigger obstacle to delivering optimal care to patients is actually getting the right drug,” said Richard Schilsky, chief medical officer at ASCO. “Doctors are scurrying around trying to find drugs that are suggested by these tests, whether it’s for purposes of a clinical trial or for off-label use.”
Roy Herbst, professor of medicine and pharmacology and chief of medical oncology at Yale Cancer Center and Smilow Cancer Hospital, added that in the ALChEMIST trial, only about 15 percent of patients who have their tumors screened will be eligible for treatment in the trial. “What’s going to happen with the other 85 percent?” he asked. Abrams responded that he hopes to have the NCI’s Cancer Center for Genomics do molecular profiling on the patients not eligible for treatment in the ALChEMIST trial. Bertagnolli added that the Alliance has made a com-
mitment to long-term outcome follow-up on all of the patients screened in the study, so there will be a mechanism for identifying recurrences in the entire population and an opportunity to identify secondary trials for those patients who recur.
As Doroshow reported, the NCI has other new initiatives aimed at furthering precision medicine and fostering innovative science in the clinical trials it supports, including
• developing an integrated national biospecimen bank for the NCTN as well as a shared IT infrastructure to enhance specimen inventories, clinical annotations, and access;
• developing a common process for requesting biospecimens banked from NCI-supported clinical trials; and
• revising the RFA for the National Specimen Banks for NCTN groups (U24 cooperative agreement grants) to include common operating procedures for samples collected from the NCTN and other NCI-supported trials.
In addition, the NCI initiated its Biomarker, Imaging, and Quality of Life Studies Program to ensure that critical correlative studies could be incorporated in a timely manner into phase III and large, multi-institutional phase II trials during the process of concept development. Between 2008 and 2013, 24 of 88 submitted concepts incorporating integral and integrated biomarker, imaging, quality-of-life, and cost-effectiveness analysis studies have been supported, for a total commitment of more than $30 million. The supported studies include those assessing the relevance of HER2 in esophageal cancer, translocation of 1p:19q in glioma, positron emission tomography/computed tomography imaging of prostate cancer, and the Oncotype DX test in breast cancer.
The NCI has also restructured its early experimental therapeutics program for phase I and II trials. The restructured program has more integration of its cancer biology, translational, and clinical components, which are supported by centralized technologies, including clinical data, biostatistical, and diagnostics cores. The diagnostics core can provide molecular characterization, and the biostatistical core can provide novel and fit-for-purpose trial
designs. The early phase program is “now a network that is team science– based and has substantially more resources,” noted Doroshow.
However, Comis stressed that “we need a comprehensive strategy for next-generation sequencing because right now, we’re all off on our own making deals with companies and academic institutions.” Several participants suggested creating a centralized clearinghouse for the annotated genetic profiles of patients’ tumors so they can be matched to appropriate clinical trials when available or to facilitate use of the data in retrospective studies.
In such a clearinghouse, “you track everything about the patient—their demographics, treatment data—and you do extensive genomic analysis. That way you can learn more about the prognosis and the results of standard treatment in small, rare genetic subsets of patients. Another advantage is that the patients are prescreened so they can be easily enrolled into clinical trials that are developed later,” said Blanke. Patricia Ganz, distinguished university professor at the University of California, Los Angeles, Schools of Medicine and Public Health and director of cancer prevention and control research at the Jonsson Comprehensive Cancer Center, added a plea that any such clearinghouse fully annotate specimens and include information such as concomitant medications, health behaviors, environmental exposures, and other relevant host factors that might influence the outcome for cancer patients.
IMPROVING PRIORITIZATION, SELECTION, SUPPORT, AND COMPLETION OF TRIALS
The IOM consensus report emphasized the need for sufficient funding and resources to support an effective and efficient national clinical trials system, as well as the need to prioritize trial concepts. The IOM recommended that the NCI allocate a larger portion of its research portfolio to the NCTN to ensure that it has sufficient resources to achieve its unique mission (IOM, 2010b). Although the NCI and NCTN budgets have been flat in recent years (and have declined when adjusted for inflation), Doroshow said that the NCI recognizes the need to adequately support the system and its investigators. The NCI has also been reorganizing the review, selection, and prioritization of NCTN protocols to take into account strengths and gaps in trial portfolios, as well as consideration of what trials are best suited for conduct by the NCTN as opposed to industry.
The NCI has since increased reimbursement to sites for patients on large phase II studies, and also provides additional funding for select phase III trials based on their complexity. However, this necessitates a decrease in the total number of patients enrolled in NCTN trials, because overall NCI funding for the program has not increased. The NCI also provides additional funding for critical biomarker, imaging, and quality-of-life studies, Doroshow reported, and has made changes in the funding model for new RFAs. These changes increase reimbursement for high-performing sites that have at least 40 percent accrual to their trials, and increase the core resources for genomic correlative studies.
Identifying Strengths and Gaps in Trial Portfolios
Scientific Steering Committees (SSCs) appointed by the NCI evaluate and approve trial concepts that are judged to be scientifically sound and clinically important. The newly formed NCTN Working Group was established to assist the NCI in the prioritization and selection of the NCTN clinical trials. This group, which is co-chaired by George Sledge and Robert Diasio, William J. and Charles H. Mayo Professor and director of the Mayo Clinic Cancer Center, respectively, reports back to the NCI’s CTAC.
The working group is charged with assessing the strength and balance of the active trial portfolio within each disease area and across all types of cancer and recommending improvements based on emerging scientific opportunities, portfolio strengths and gaps, and high-priority or evolving clinical needs. The working group will also review and assess the clinical trials evaluation process and results by periodically assessing the quality of completed trial outcomes, the operational performance of the SSCs, and the efficiency of clinical trials. Another task of the working group is to provide strategic advice to enhance NCTN clinical trial operations, such as collaboration and timeliness.
Twenty-eight extramural members from key stakeholder groups comprise the NCTN Working Group, including NCTN group chairs and statisticians, Community Clinical Oncology Program (CCOP) principal investigators, cancer center directors, steering committee chairs, patient advocates, translational scientists, NCI leadership, and Cancer Control Research Base principal investigators. “This is the first time that NCI or any group external to NCI has had the opportunity to look at the whole trial
portfolio. The ability to digest and assimilate the whole disease group and to also make comments across the disease entities has never really occurred in the history of the Cooperative Group Program or the Clinical Trials Program of NCI,” Diasio said. The working group will also consider summary information from other major ongoing trials outside of the NCTN for each disease area, such as industry or international trials.
The working group has developed metrics for evaluating trials that quantify factors such as feasibility, clinical importance, scientific contribution, relative cost, number of patients required, and appropriateness for the NCTN program (see Box 5).
The feasibility criterion includes not just the time and cost to implement a study but also accrual difficulty. “It was disappointing to see greater accrual in certain studies, but very poor accrual in others. This obviously is not a good use of NCI funding or time and it’s important to evaluate upfront the feasibility of studies,” Diasio said. For the clinical importance criterion, the working group came up with the concept of life-years saved to discern benefit to the patient or the population.
BOX 5
Criteria for Evaluating NCTN Trials
• Feasibility (accrual difficulty, time and cost to implement at sites)
• Clinical importance (importance of the study question relative to the state of the science in the disease; benefit per patient and for the population [e.g., life-years saved]; benefit in light of disease context)
• Scientific contribution (tests important scientific concepts or proof of principle, importance of integral or integrated correlative study questions)
• Relative cost/resources (total number of patients required, length of study [accrual and follow-up])
• Appropriateness for NCTN program (understudied/rare disease, understudied populations, trials to optimize a technique, combination trials, international academic collaborations, and contribution to public tissue and data resources)
SOURCE: Diasio and Sledge presentation (February 11, 2013).
At the time of the workshop, the working group had evaluated the NCTN trial portfolios for patients with leukemia and lymphoma, and gastrointestinal, colorectal, breast, and genitourinary cancers. One general finding of the working group was that there was considerable variability in the balance of strong and weak studies across disease groups. Although this variability could be due to differences in scientific advances or therapeutics developed for each group, there was concern that the variability might stem from a lack of standard format for the preparation and submission of trial concepts, or from differences in the approach and guidance given by the various disease-specific SSCs.
As an example of the kinds of information gained from such reviews, Diasio noted that for breast cancer, the working group found that trials were relatively strong, addressed several important questions, were multi-disciplinary, and had a good balance between systemic and local-regional studies. But the working group thought there was still room for improvement and recommended smaller, innovative, and more nimble randomized phase II trials, such as molecularly driven trials, trials that discover or validate biomarkers, trials aimed at limiting toxicity and improving quality of life, and survivorship studies. The working group concluded that the breast cancer SSC could provide more strategic guidance for concept selection and for developing standards to improve trial design, perhaps by tapping into the resources of other groups, such as task forces, working groups, or clinical trial planning meetings.
Schilsky asked if the working group planned to publicize its assessment of gaps in the clinical trials portfolio so clinical investigators are aware of them and can take appropriate steps to fill those gaps. Sheila Prindiville, director of the Coordinating Center for Clinical Trials at the NCI, responded that any recommendations or conclusions from the working group will be presented at public forums convened by the CTAC. “The information will be available to the public,” she said.
Another workshop participant pointed out that the metric for life-years saved will give undue representation to studies done on common rather than rare cancers. “A very tiny advance in a really common disease will give you a much bigger pickup of years of life saved than a fairly large advance in a rare disease, and certainly rare diseases deserve help, too. So, we wouldn’t want this to be the only metric that’s valuable in terms of looking at these trials,” the participant said.
Another point of discussion centered on the higher cost of innovative trials and how cost factors were weighed in the working group reviews of
clinical trials. A smaller trial that has more robust use of biomarkers may be more costly up front for each patient enrolled, but may be more cost-effective than a large trial with thousands of patients followed for long periods of time. “Maybe going from the emphasis being on larger phase III studies to more nimble phase II studies will allow more of those studies that focus on innovative science to be implemented,” said Diasio. Comis noted that this emphasis appeared to be the trend; his analysis of NCTN trials found that the number of complex phase II trials is increasing, while the number of phase III trials is decreasing. Diasio added that “one of the advantages of having this overview of all the trials is to bring the molecular and pathway-driven studies forward for our strong consideration at the beginning.”
Sherman stressed in her presentation that the NCI must give priority to high-quality trials. “We have a societal obligation to make sure every trial counts, but most trials use study designs incapable of meeting FDA standards for substantial evidence,” she said. She noted that a recent study the FDA conducted with Duke University evaluated 96,346 clinical studies registered at Clinicaltrials.gov and found that 96 percent had 1,000 or fewer participants and 62 percent had less than 100 participants, with the median number of participants per trial being 58 for completed trials (Califf et al., 2012). Only 34 percent of the interventional trials were double-blinded, and 30 percent were not randomized. Compared with other specialties, oncology trials were more likely to be single arm (62 percent vs. 24 percent), open label (88 percent vs. 47 percent), and non-randomized (64 percent vs. 23 percent) (Hirsch et al., 2013).
Reflecting on these findings, Sherman said, “The vast majority of trials done in this country are small and uninformative. We’re almost definitely not spending our resources as wisely as we should and we as a nation have to think about how we can change that trend. The worst thing to do is expose a person to a test agent and not have that count. Quantity is less important than quality and, less obviously, it’s drowning out the useful information by creating too much noise.”
Lessons from the National Clinical Trials System in the United Kingdom
Richard Kaplan, associate director of the UK National Cancer Research Network (NCRN) and UK Clinical Research Network and senior scientist at the Medical Research Council Clinical Trials Unit, described how the UK NCRN operates and how it differs from the U.S. NCTN. He also noted
relevant strengths and disadvantages of the UK system compared with the NCTN.
The NCRN is a single national system for cooperative phase II and III cancer clinical trials in the United Kingdom. The NCRN manages research staff in 32 regions, is tightly linked to regional cancer treatment organizations, and supports research nurses and data managers throughout the National Health Service (NHS). The UK National Cancer Research Institute (NCRI) is a partnership of government and charity funders who jointly set policies and research priorities and coordinate needed resources for cancer research. Cancer Research UK (CRUK) is the largest UK cancer charity and the largest cancer research funder in Europe. Clinical Studies Groups (CSGs) are UK-wide single-disease committees responsible for developing studies and overseeing their research portfolios. Clinical Trials Units (CTUs) are equivalent to the coordinating and data centers in the U.S. NCTN.
In the United Kingdom, virtually all of the clinical costs for patients in cancer clinical trials are covered by the NHS. Network infrastructure is funded by the National Institute of Health Research (NIHR), the UK equivalent of the U.S. National Institutes of Health (NIH). Some CTUs receive core funding via competitive peer review every 5 years, but CSGs only have a small budget to cover meeting expenses. Each trial must achieve peer-reviewed funding from the NIHR, the CRUK, or other funders. Such funding covers CTU central costs and clinical costs other than those covered by the NHS. Increasingly, industry has funded UK clinical trials, Kaplan noted. “Half of all big trials have some form of industry support, which makes a big difference in making them go fast,” he said.
CSGs are the primary venue in which new proposals for clinical trials are developed in specific disease areas. Membership in these groups rotates and there is a competitive national appointment process for new chairs and members. In addition to clinical and scientific members, CSGs include patient and funding-body representatives. The CSGs’ main objectives are to oversee existing studies, consider new research questions and develop new proposals, and provide expert advice. They also interface with industry partners for consultation about the feasibility of clinical trials and oversee specimen resources.
Anyone outside of a CSG can apply for a grant to do a trial, which is conducted with a CTU, Kaplan noted, but “even if these studies arise from somebody who’s not on one of these CSGs, they will be fed into the CSG, where the proposal will be discussed and refined and then put through as
a sort of joint proposal.” Every CSG publishes its trials portfolio on the Internet because all of the studies are potentially open to any site in the country qualified to perform them, he added. All CSGs undergo progress reviews by their peers, including representatives from North America and Europe, every 3 years. These external peers review the research portfolio, but do not do in-depth reviews of individual current trials, and mainly focus on the membership, activity, scope, future plans, and strategic direction of the CSG.
“Funders are careful to look for whether research questions are considered important to external peer reviewers,” Kaplan said. He added that funders also “work hard to prevent too many competing large-scale trials and to decide whether the extra capacity is there for a second trial on top of one that’s already in place. The different funders coordinate behind the scenes when they receive new applications to make sure that they’re not working too much at cross purposes.”
Each regional network selects the trials it wants to support, and any trial is available as long as the site is qualified to run it. “The smaller community hospitals tend to participate in the non-interventional trials for the most part, but they do refer lots of patients elsewhere for the interventional trials,” Kaplan said. The individual trials have recently been required to meet particular time and target metrics, and some regional network funding has been explicitly linked to actual activity. “League tables” that compare performance are compiled and made public.
In 2001, when the UK clinical trials system was restructured and the NCRN was established, a major goal was to increase patient accrual to cancer clinical trials. Accrual grew from about 4 percent of all cancer patients in 2001 to 23 percent in 2011. Part of the increase is due to the availability of more non-interventional, observational, and other non-randomized studies conducted in community hospitals. Many of these sites had not previously done clinical research and undertook such studies as a way to gain experience and build up staff in order to develop the capability to do interventional trials in the future, according to Kaplan. But even in randomized trials, accrual is about 7.5 percent of patients diagnosed, he said. That percentage translates to about 19,000 patients enrolled each year, about the same number of patients enrolled in the U.S. NCTN trials, despite the United Kingdom’s smaller population of 50 million, Kaplan noted.
The NHS’s commitment to clinical research also encourages physicians to enroll their patients in clinical trials, according to Kaplan. During its annual performance reviews, the NHS assesses physicians’ participation
in the clinical trials available in their network. Peer expectations also play a role, he said. “If you are an oncologist who doesn’t participate in clinical trials that are available, it just doesn’t look good. You lose face with not only your supervisors but with your peers,” Kaplan said.
He said the most important driver of success for the whole system appeared to be “the new research staff they put in place, who were very carefully ring-fenced. The hospitals were not allowed to use research nurses for ordinary cancer care delivery.” Kaplan also noted that the size of the United Kingdom favors nationwide collaboration. In most cases, the country is just big enough to do independent large-scale trials but not big enough to do too many competing trials in most diseases.
Some of the disadvantages of the UK system noted by Kaplan include metrics that discourage clinical trials on rare diseases; the increased burden of following patients on prior trials, which interferes with the ability to start new trials; local networks tending to activate “easy” studies because some of the most important studies are the most work-intensive; and a complex system for approvals that delays trial activation.
Kaplan noted that the major cultural difference between the UK cancer research system and the U.S. system is that cancer research in the United Kingdom is viewed as being part of the health care system, rather than separate from it (as in the United States). In response to a question from a workshop participant about the ability of the UK system to support genomic and other molecular studies on cancer patients, Kaplan responded that the CRUK has proposed a major initiative to build such research capacity into the system, starting with major cancer centers that will eventually characterize every patient treated. There also is a UK Department of Health initiative to achieve the same objective. “It’s clearly seen as a priority to all of the funders,” he said.
In response to a question from Pazdur about the type of cancer trials conducted in the United Kingdom, Kaplan noted that “much of the thrust of the CSG has been new drug development, with more than half of the trials being phase III trials,” although he added that the number of phase II trials is increasing. Herbst asked how biomarker tests for patient stratification are paid for in UK cancer trials. Kaplan replied that research funders, including the arm of the government that funds the trials themselves, are currently paying for this resource. He noted that a few central labs will be handling specimens and doing molecular profiling for a large colorectal cancer study he is involved in, and although it may not be cost-effective to
do such tests yet, “we’re simultaneously trying to build the systems for it to become so.”
Studies More Appropriate for NCTN Than Industry
There was extensive discussion about which studies are more appropriate for the NCTN, vs. industry, to undertake. Doroshow noted that industry is not likely to do trials of agents for more than one histological type of cancer, so these types of trials are more appropriate for the NCTN. “So many of the advances in cancer medicine and clinical trials research have been multidisciplinary in nature and it’s not in industry’s business model to do those kinds of studies,” he said. He also quoted a former industry scientist, who said, “Even Johnson and Johnson, with all of its budget, doesn’t have access to the kind of cancer biology that the NCI supports, so if you’re going to change your system, change it in a way that allows you to optimally use that tumor biology and help bring that [knowledge] into the clinic.”
Pazdur stressed the notion that the NCTN should not do trials that can be done by industry, given that publicly funded resources support these trials. Blanke agreed, saying, “The cooperative groups have to do the research that no one else can or will do. In the old days, it was the large-scale phase III trials, but now it’s the trials on rare diseases or subsets of the common diseases. Harnessing multiple drug companies to work together is not going to come from industry voluntarily. Industry is also not likely to harness our basic scientists in drug development and conduct multidisciplinary or multimodality trials.”
But Comis said, “I don’t think we should be pigeonholed into rare tumors. I would much rather be pigeonholed to a rare biologic process, but not necessarily rare tumors.” He added, “We can be on the forefront of integrating biology into our studies and are already doing that. We have multiple studies that cross diseases, so I think we have that platform.”
But Adamson made a plea for prioritizing trials of childhood cancers, most of which are rare. “We do studies in the Children’s Oncology Group [COG] on diseases with an incidence in this country of less than 100 children a year. We need to be able to decide what is the highest priority at any given time because otherwise, we’re going to run out of patients, and by ‘we’ I mean parent patient advocates and specialists throughout the field,” he said. He added that the return on investment from NCI funding for COG and the cooperative group program is quite remarkable.
Adamson noted that in the 1950s, the 5-year survival rate for child-
hood cancers was 10 percent and now it is 80 percent. “That improvement was made entirely with NCI resources,” he said. Now, more than 90 percent of pediatric cancer patients in the United States receive treatment at COG sites and more than half are enrolled in COG trials. Still, cancer remains a leading cause of death in children, “so the work is just really beginning,” Adamson said. “I don’t think there’s any entity other than the cooperative groups that can ultimately act hand-in-hand with patients, parents, and families, as well as industry in setting the priorities,” he concluded.
Comis agreed with Adamson about the need for the NCTN to conduct studies on children with cancer. “Having the NCI prioritize pediatric studies earlier than companies did has been very helpful. Other kinds of studies that maybe industry can do but cooperative groups can do even better are novel combinations, especially when the novel agents are premarket compounds from different companies.” He added that because there are multiple mutations that often are not discerned by a typical companion diagnostic test developed by industry, “We need to have a broad-based approach to the biology of these cancers, and instead of doing a registration trial, lead the way towards what the FDA, the payer community, and the research community need to do in order to do cutting-edge research. The role of the groups is probably more important now than it ever was. The best thing for patients is what we should do.”
Nancy Roach, president of the Colorectal Cancer Coalition, pointed out that the NCTN’s ability to share data “makes it a lot easier to run multicenter trials,” as opposed to industry. She added that the NCTN should conduct biomarker-driven trials that clarify which drugs work for which molecular subtypes of tumors, now that more patients are getting their tumors sequenced. Otherwise, their doctors will give them a drug off-label, and information about the appropriateness of that drug for their tumors will be lost. Curran agreed, noting that the cooperative groups have tested a number of therapies for off-label use in randomized trials that showed a lack of benefit; as a result, these therapies are no longer offered for those indications. “This improved cost-effectiveness [of treatment] and lowered toxicity,” he said. Curran advocated for the NCTN to continue to do such off-label use studies and comparative-effectiveness research, which industry is less inclined to pursue.
INCENTIVIZING PATIENT AND PHYSICIAN PARTICIPATION
One of the IOM goals for the NCTN was to incentivize the participation of both patients and physicians in clinical trials by supporting clinical investigators and covering the cost of patient care in clinical trials. The IOM also recommended that the NCI, cooperative groups, and physicians should take steps to increase the diversity of patient accrual.
Abrams said that the number of cancer patients enrolled in NCTN trials has declined in recent years, to about 20,000 per year. Abrams reiterated that over the past 10 years, the United Kingdom was able to substantially boost the rate of patient participation in clinical trials through financial reimbursements. “We have a different system here, but if patient advocates were looking for a way to truly increase the uptake in clinical trials, that would certainly be a cause worth fighting for,” he said.
Roach suggested that there might be more patient enrollment if more clinical trials assessed quality-of-life issues important to patients, such as which cancer treatments cause the least amount of neuropathy. “Let’s not throw out the boring but high-impact studies, and do only the really cool science,” she said. She also suggested conducting a pilot project to see if reimbursing oncologists for the time it takes to inform their patients about clinical trials and covering other trial-related expenses could increase patient accrual in trials.
Recent improvements to the NCTN consent form template may facilitate patient accruals. The new template, which will be implemented in 2013, is shorter and less complex. The risks are easier to understand and are presented in a way that is meaningful to patients. In addition, different tables of risks are presented for the experimental vs. standard arms, which are grouped by regimen. Risks are described in lay terms and listed according to the body system affected.
Doroshow added that the NCI works with patient advocates in trial concept development and accrual planning, along with the cooperative groups, disease SSCs, and the Patient Advocate Steering Committee. He also stressed that the NCI tries to enhance trial participant diversity through the Minority-Based CCOPs, the Patient Navigator Research Program, and other NCI programs.
Speaking from the patient perspective, Patrick Gavin, president of Patrick Gavin R.Ph. Consulting LLC, stressed that patient involvement in
clinical trial development is key to enrolling the large number of patients needed to conduct clinical trials of innovative targeted interventions. “We as patients have the unique ability to be able to look at a proposed piece of research and tell you if a patient would be willing to participate in your experiment while we’re trying to stay alive,” he said.
For example, he said one proposed study reviewed by a patient advocate gave patients the option of being randomized to one of two treatments already available on the market. As Gavin described it, “The advocate asked the principal investigator, ‘Why would a patient or their oncologist choose to be on this trial with such totally different arms, when both treatment options exist now and he would not be bound by the randomized coin flip if he had them outside of a trial?’” This comment led to the trial being restructured, Gavin reported, adding that without input from the patient advocate, “the trial would have likely gone ahead and huge investments would have been wasted because the trial would not have accrued because local oncologists and patients would not have bought into it.”
Gavin stressed that the large numbers of patients treated in community settings are especially needed for precision medicine studies, adding,
We have to engage the broadest possible patient community in order to make clinical trials and personalized medicine possible. The nature of personalized medicine will not allow for a few thousand patients involved in answering a single question, but tens of thousands of patients answering thousands of questions. That patient community doesn’t exist if we only look to the large cancer centers. We have to be able to reach out to every hospital and other settings where patients live and convince them they need to participate in clinical trials if we are to achieve [real progress]. Patient advocates can make these essential partnerships with patients a reality.
Gavin noted that there are a number of things patient advocates can do to help boost accruals to clinical trials, including
• developing strategies that accelerate study development, activation, accrual, participation, and reporting;
• ensuring selection of trials that affect clinical outcomes for people with cancer;
• educating the public on the availability of clinical trials as a treatment option;
• developing patient-centered materials to help treating physicians discuss trials with patients; and
• publishing and disseminating clinical trial results in plain language.
“Use our patients advocate networks in addition to the National Clinical Trials Network,” Gavin concluded. “We can help you build effective trials that patients will want to join and their oncologists will want to recommend—we can help you get the message out. Involve us as early in the development process as possible,” he said.
Schilsky asked Gavin how patients can engage their oncologists to ensure that they discuss clinical trial opportunities. Gavin responded that patient advocates advise patients to talk to their oncologists about options for a clinical trial as a possible treatment option and encourage them to “do their homework” beforehand by exploring websites that list information about trials.
Increasing Physician Participation
Participants at the workshop also voiced concerns about a decline in the number of physicians willing to engage in clinical trials. Several speakers said that more mentoring and funding for young investigators are needed to maintain a critical mass of clinical investigators.
Comis noted several fellowship award programs that ECOG designed to attract new investigators, including the Young Investigator Awards. Eleven out of 15 recipients of this award during the past 15 years have entered senior positions in ECOG. All of these awards were supported by ECOG’s foundation, not federal funds, Comis said. He added that ECOG-ACRIN plans to develop a mentorship committee to formalize the fellowship awards. “Each committee chair is paying attention to bringing young investigators in, but I don’t think that’s enough, so we’re going to formalize that program,” he said.
Blanke added that SWOG also has funding opportunities for young investigators, as well as a course in which participants are flown to Seattle to learn from “the best of SWOG.” Participants are expected to develop a clinical trial protocol by the end of the course. “They not only get teaching, but a sense of excitement, and a large percentage of them move forward,” Blanke said. He added that SWOG is starting a formal mentorship program that will pair mid-level investigators with young investigators in each clinical trial. SWOG is also creating more leadership opportunities in the subcommittees for each trial. “That way, young investigators can engage in the governance and scientific direction of the group as well,” Blanke said.
Roach added that young investigators in the academic setting should be better rewarded for pursuing clinical research and engaging in collabora-
tive studies for which they may not receive much authorship recognition. Michael Caliguiri, CEO of the James Cancer Hospital and Solove Research Institute, director of the Comprehensive Cancer Center, and professor of internal medical at the College of Medicine at Ohio State University, agreed. He noted that “the ability to convince department chairs and deans at academic institutions to be more tolerant of the extramural situation and clinical research and to recognize accomplishments that aren’t traditionally measured has been a challenge. We need to come together and set some new rules in these changing times about what we regard as worthy of promotion, because that’s ultimately what keeps many of our young investigators in the game.” Doroshow pointed out that since 2009, the NCI has been providing Clinical Investigator Team Leadership Awards to promote collaborative science and to recognize outstanding clinical investigators.
Adamson added that young investigators could also be encouraged by a more efficient system that results in faster decisions, saying that
the most disheartening thing for young investigators is to invest 1 or 2 years of their career developing a clinical protocol that doesn’t get approved. We need a system where we can fail early [rather than later]. If we don’t solve that, it’s going to be hard to sit across the table from any young investigator and tell them why they should be involved. It’s one thing to invest 4, 5, or 6 months of an academic career and have an idea die. It’s very different to do it for 2-plus years, and then have to go back to the drawing board. I don’t think NCTN has addressed this.
Worta McCaskill-Stevens, Chief of the Community Clinical Oncology Program and program director for the Minority-Based Community Clinical Oncology Program at the NCI’s Division of Cancer Prevention, expanded on what has been done recently to increase patient diversity and the involvement of community practices in NCTN clinical trials. She began by describing recent changes affecting research conducted by community practices. She noted that due to financial pressures, such practices are increasingly merging and being acquired by hospitals. Consequently, investigators are “having to negotiate and see affirmation of the role of clinical trials within those systems,” she said. Stephen Grubbs, principal investigator of the Delaware Community Clinical Oncology Program and managing partner at Medical Oncology Hematology Consultants, PA, said these mergers are a major problem in the current era of tight budgets because hospital administrators often are not willing to devote resources to cancer research.
McCaskill-Stevens also noted the increasing role of molecular-based cancer care, and stressed that “the best laboratory in which care systems can be evaluated are the community settings,” where the majority of cancer patients are treated. Grubbs concurred, noting that historically, the majority of accruals for the cooperative group program have come from community-based practices. He said it is important to prepare these practices for the shift in emphasis within the NCTN to biomarker-driven phase II trials, “Without the community programs, [those trials] aren’t going to get done fast enough, so we have to make sure that program is strong as we do more phase II trials,” he said.
The CCOPs are “working to try to educate the advocacy community as well as other populations about biospecimens,” said McCaskill-Stevens. The program is evaluating infrastructure requirements at the local level for collecting and processing biospecimens, and hopes to strengthen that infrastructure accordingly by ensuring the availability of pathologists and other necessary personnel.
But Benz pointed out that reimbursement is lacking for tasks related to acquiring and testing specimens for patients in clinical trials, which go beyond routine clinical management. “Something needs to be built into the reimbursement system because the mechanisms for funding that in a community setting are almost non-existent unless they have some formal collaborative relationship with a big cancer center, which immediately shrinks down the number,” he said.
Grubbs added that not only is a reimbursement code required for acquiring and testing biospecimens, but that code also has to be recognized by the payer. A reimbursement code is also required to reimburse physicians for the time they spend offering and explaining clinical trial options to patients, he said. “Even if the patient doesn’t actually go on a trial, clinicians still can spend a tremendous amount of time reviewing the trial with the patient and their family,” Grubbs said. Doroshow said that the NCI continues to work with the NIH and other federal agencies to help define and shape national policy on clinical trials and reimbursement, as well as to educate patients and payers regarding the benefit of clinical trials.
McCaskill-Stevens reported that at the end of 2012, the NCI integrated the CCOPs, the Minority-Based CCOPs, and the NCI Community Cancer Centers Program (NCCCP), along with the research bases for those programs, into a new program called the NCI Community Oncology Research Program (NCORP). She said this consolidated program will expand the research scope in the community setting to include not just clinical trials,
FIGURE 4 2013 site map for the NCI Community Oncology Research Program (NCORP). Circles represent NCI Community Cancer Centers Program (NCCCP) sites; squares represent Community Clinical Oncology Program (CCOP) sites, and triangles represent minority-based CCOP sites. The darker shaded states are those that have NCCCP sites.
SOURCE: McCaskill-Stevens presentation (February 12, 2013).
but also research on cancer disparities and cancer care delivery (NCI, 2012). McCaskill-Stevens noted that NCORP representatives serve on CTAC. The integral components of NCORP are the community-based oncology practices. These practices have a variety of research capacities linked to the NCTN and are provided with the support needed to participate in a collaborative research network (see Figure 4).
McCaskill-Stevens said that NCORP is a public–private partnership with a commitment to co-investment. Eligibility for NCORP community sites is based on the capacity to participate in cancer research, including
• clinical research experience (clinical trial accrual);
• cancer care delivery research infrastructure;
• available study populations; and
• senior leadership/organizational support.
Eligible community practices that participate in NCTN clinical trials have been supported with 3-year grants, but NCORP recently proposed extending those grants to 5 years in recognition of the difficulty the sites had in activating the initial grants and the challenge of preparing for another competitive renewal after only 3 years, said McCaskill-Stevens.
NCORP instituted a number of changes aimed at boosting patient accruals from community practices. In addition to transitioning to the use of the new, 24/7, Web-based oncology patient enrollment information technology of the NCTN, NCORP is developing a single portal of entry for institutions rostering with the NCTN that should improve the efficiency of the current system, which requires rostering with multiple networks. There are also ongoing discussions about how investigators can use the NCI’s Central IRB for primary review of cancer control and prevention studies run by NCORP.
NCORP has also been promoting the participation of underserved populations and incorporating disparities research questions into clinical trials and cancer care delivery research. Grubbs noted the diversity of different underserved communities, from rural populations and Native Americans to African Americans and Hispanics. “Every community has a different underserved community to deal with. There’s not one program that can encompass them all, and plenty of opportunities,” he said. But there is a lack of research on how best to enhance research participation and improve health care for underserved communities. Grubbs said he supports cancer care delivery research, but is concerned about inadequate infrastructure and resources to do such research in the community setting. Centralized databases, such as those of the Centers for Medicare & Medicaid Services and state databases for Medicaid claims, might aid in such research, he suggested.
Robin Zon, principal investigator of the Northern Indiana Cancer Research Consortium CCOP and vice president at Michiana Hematology-Oncology, PC, said that some physicians in competitive practices may not be willing to share their data unless they are de-identified and confidential. These physicians are already so time-crunched that “we don’t want to stress them by asking for more data or other information about their health care practices,” Zon said. She noted that clinical research in community practices is optional, so burdening physicians with these requests may lead them to opt out of participating in it. “We need to make things easy for them,” she concluded.
Grubbs noted that in order to take advantage of the improved IT com-
ponents of the NCTN, including its Web-based enrollment system, physicians in community practices need to have compatible electronic medical records. “But we’re finding that the hospital practices and private practices use different electronic health record systems that don’t talk to each other,” he said. Having a compatible electronic records system will require financial resources that many private practices do not have, Grubbs cautioned.
Zon also raised the issue of pay-for-performance measures, which are increasingly common among institutions and health insurers and might deter physicians from spending time on research activities. She added that the Patient Protection and Affordable Care Act does mandate coverage for clinical trial participation, but “it’s not really clear to me how to interpret that. We need to lobby to make sure such coverage becomes absolutely imperative with no grandfathering and other exceptions,” she stressed.
Under the broad goal of improving the speed and efficiency of clinical trials, the IOM consensus report included a recommendation for a trans-agency effort to streamline and harmonize government oversight and regulation of cancer clinical trials (IOM, 2010b). Another IOM recommendation specified that the NCI, in cooperation with other agencies, should establish a consistent, dynamic process to oversee the development of national, unified standards as needed for oncology research. Consequently, a number of regulatory issues were discussed at the workshop, including regulatory oversight of the diagnostic tests used in clinical trials and trials focused on small subsets of cancer patients, as well as international differences in regulations and how to harmonize them. Speakers also addressed how to avoid bias when assessing treatment response in clinical trials, and under what circumstances progression-free survival is an acceptable endpoint.
Doroshow noted that there is now a coordinated process for the development and review of trials under the FDA Special Protocol Assessment, and the NCI also established an interagency agreement with the FDA for rapid review of approved NCTN phase III treatment trials at the concept stage. He added that the NCI has been working with the FDA to coordinate early review of biomarker tests, as well as to facilitate the development of companion diagnostics and incorporation of genomic tests into clinical trials.
John Jessup, chief of the Diagnostics Evaluation Branch in the Division of Cancer Treatment and Diagnosis at the NCI, pointed out that a number of reports in recent years have highlighted the risks for patients posed by new and poorly validated diagnostics. “Investigators want to use markers, but they oftentimes have not understood the rigors of clinical assay development,” he said. Jessup added that in late 2010, the FDA Office of In Vitro Devices began to enforce its oversight authority for the safety of diagnostics used for medical decision making in clinical trials.
Biomarker tests used for medical decision making, even within a clinical trial, are required by law to be performed in a CLIA-certified laboratory. These tests include companion diagnostics, which are used in conjunction with targeted therapies, as well as molecular tests that stratify risk of disease recurrence or adverse reaction to treatment or those that indicate proper dose. Biomarker tests used solely for research purposes, including prognostic, predictive, and pharmacogenomics markers, do not need to be performed in CLIA-certified laboratories.
According to the FDA, if a biomarker test has the potential to “present serious risk to the health, safety or welfare of a research subject,” clinical investigators must get an FDA Investigational Device Exemption (IDE) in order to use the test in their studies and must collect safety and effectiveness data. However, Jessup noted, “serious risk is not well defined or quantified,” despite two FDA guidances that mention it (FDA, 2006, 2012d).
To obtain an IDE, a principal investigator is expected to document the analytical performance of the assay, including its accuracy, reproducibility, and precision, and how these characteristics translate into false positives or negatives. This information is used to demonstrate to the FDA that the risk of using the device (i.e., biomarker test) is less than the potential treatment benefit. “This means that once an assay is going to be used in a trial, even at the concept stage, it is important to be submitting information about the validity and performance of that assay as soon as possible,” Jessup stressed. He added that the NCI Cancer Diagnosis Program will assist in this process by providing templates for documentation of assay performance for immunohistochemistry, fluorescence in situ hybridization, and somatic mutation detection. But Jessup also cautioned that “a lot of the tests that seem extremely exciting may not be reproducible or accurate.”
Once assay performance data have been gathered, there is a formal pre-submission program that includes meetings with FDA staff. Jessup noted
that a recent draft guidance on presubmission for devices outlines current FDA recommendations about clinical assay development and provides information on how to contact the appropriate FDA offices (FDA, 2012a). If the results of an assay are not used in treatment decisions in a clinical trial, the test does not pose significant risk and an IDE is not required, Jessup explained. However, biomarker tests used for eligibility criteria, treatment assignment, or dose modification may require a presubmission IDE review.
In summary, Jessup suggested that investigators include in their trial protocols a section that documents the risk of false positive or false negative assay results and the potential consequences of false results in the context of the disease. Investigators should also indicate whether they think an IND (Investigational New Drug) application or IDE is required. Jessup added that the potential consequences of false results from biomarker assays should be described for patients in the informed consent documents. “The FDA wants to know exactly what patients are being told about the markers and the consequences of the assay results,” he said. He concluded that “principal investigators, assay developers and performers, and sponsors need to collaborate and partner closely.”
Garraway noted that “just because something is done in a CLIA lab doesn’t automatically mean that it’s done with high quality. It just means that it’s done the exact same way every single time.” Garraway is part of a consortium that is currently trying to define appropriate metrics for sequencing standards that could cut across various types of platforms and approaches and offer objective performance comparators (National Human Genome Research Institute, 2013).
Solit stressed that biomarker validity is more important for late-stage clinical trials. He suggested that there should be a lower bar for biomarkers used in early-stage clinical trials. “Oftentimes, patients have no other treatment options. I don’t see any risks in the trials that we’re running in advanced metastatic cancer patients, who typically have a life expectancy of less than a year,” he said. But Miller countered that “the field was set back substantially by misinterpretation of EGFR assays, so even if multiple platforms are contemplated, there should be a common playing field with minimal criteria.”
Comis noted that testing a companion diagnostic concurrently with an intervention in a registration trial can be limiting as it “locks onto a specific assay, when the whole dynamics of understanding mutations and their interactions” is continually evolving. “It can inhibit the kinds of clinical
trials we want to do, in which the biology, especially the mutations, drives the treatments tested in patients,” Comis said.
Capdeville agreed, noting that by the time a drug and companion diagnostic are approved, the field often has changed markedly, with newer genetic techniques coming to the fore. “Still, we have to start at some point when we do a clinical study, so we need informed consent that is written carefully to allow the flexibility to not only work with the assay you have at the start of the study, but to be open to some more exploratory work as better technology becomes available. It is critical that you have the ability to store samples for future testing,” he stressed.
Pazdur said that the label for a companion diagnostic merely states that it is FDA-approved. “Inherent in that is the belief that these tests will change over time and that’s why there will be bridging studies that are done that compare one in vitro diagnostic to another in vitro diagnostic. I don’t think anyone in the FDA believes that this is a static field. The grand daddy of in vitro diagnostics is estrogen and progesterone receptor testing, and that has evolved since the mid-1970s due to various bridging studies that compared different technologies,” Pazdur said.
But Mendelsohn responded that there is a lag in implementing improvements to companion diagnostic tests because the assays are paired to specific treatments. “When you check for KRAS, you have to use the approved paired assay, which we all know misses some mutations in KRAS, if you want to get paid for giving the drug. So, your philosophy hasn’t hit the practice of medicine yet, unfortunately,” he said.
Regulatory Oversight for Trials of Small Subsets of Cancer Patients
Participants discussed the appropriate regulatory oversight for trials of small subsets of cancer patients. Pazdur noted that “when there’s a very small population of patients that could benefit from a drug, it would be nearly impossible to conduct a randomized study. Instead, we would take a look at the response rate and the toxicities of the drug,” and if the benefits outweigh the risks, the drug would be approved for the indication, he said.
Pazdur added that “one has to balance out what you’re seeing in this small subgroup vs. what you’re seeing with existing therapies. If you have a drug that has an exceedingly high response rate, we would take a very kind look at that, and proceed with an accelerated approval for that indication. For rare diseases in general we have taken a very liberal policy, even when it
comes to the characterization of safety in that subgroup because it simply cannot be shown.”
Many clinical trials conducted by the NCTN assess secondary indications for approved drugs. Schilsky reported that a retrospective study conducted by ASCO in collaboration with industry used cancer clinical trial data to assess whether much of the collection and reporting of adverse events for drugs being tested for supplemental indications is necessary. A recent draft guidance from FDA (2012c) may help improve the efficiency of such trials by explicitly encouraging new and less burdensome ways of gathering and reporting adverse events and concomitant medications.
“Sometimes important safety signals can be obscured by all the additional information we’re required to report that oftentimes is not terribly informative,” Schilsky said. “We want to reduce the data collection burden on the clinical trials system so we can enhance physician participation, because one of the big obstacles we hear from doctors all the time is that there is so much paperwork involved in doing clinical trials that it’s just not worth the trouble,” Schilsky added.
The ASCO study reanalyzed multiple clinical trial toxicity databases and examined various sampling methods to determine if a more streamlined and “optimized” approach to data collection would provide sufficient safety data to support supplemental applications. This study found that capturing excess Grade 1 or 2 adverse events did not appear to add to the known safety profile, and that the probability of missing a previously unrecognized, clinically significant Grade 3 or 4 adverse event was low when the optimized data collection approach was used. In addition, review of concomitant medication databases from six trials demonstrated that no new information was gained from the summary tabulations required to be listed in the application for a supplementary new drug or biologic indication.
The resultant white paper (Abrams et al., 2009) recommended that for future supplemental trials with the appropriate qualifications, researchers need not collect
• Grade 1 or 2 adverse events (AEs) (already known);
• Grade 3 or 4 AEs in all patients;
• Stop/start dates for AEs except by cycle; or
• Concomitant medications, unless they are likely to interact with the drug being given, have an antitumor effect, or meet a specific objective of the trial (e.g., are integral to a health economics/costing study).
The analysis also indicated that a subsample of about 400 patients provided adequate probability of detecting adverse events with at least a 3 percent excess toxicity. The authors suggested that the FDA should put forth a detailed guidance document with clear directives on data collection requirements for concomitant medications and adverse events for trials of supplementary drug indications.
The FDA responded by issuing a new draft guidance in February 2012 stating that targeted safety data collection akin to what was done in the ASCO study may be appropriate when the safety profile of the drug is already well characterized from prior studies, with adverse event type and frequency being similar across multiple studies, and when the expected adverse event rates in study population are likely to be similar to what was found in previous studies. In addition, the FDA specified that targeted safety data collection may be appropriate for postmarketing studies for new indications, studies required to meet postmarketing requirements, and large outcome studies.
“We’re making great progress here. We have strong data that supports the notion that for supplemental applications there can be substantial reduction in the amounts of safety data collected without missing any important safety signals. I think that’s been acknowledged in the recent FDA draft guidance on this topic,” said Schilsky.
Sherman, of the FDA, pointed out during her presentation that the purpose of the draft FDA guidance is to help clinical trial sponsors determine the amount and types of safety data that should be collected during late-stage premarket and post-approval clinical investigations. She said this draft guidance makes clear that sponsors can use a variety of approaches to fulfill their monitoring responsibilities, and that sponsors may request different reporting formats or frequencies for adverse event reporting either by describing the method in the protocol or by requesting a waiver. Either way, Sherman stressed, alternative reporting must be agreed to by the FDA in advance of the trial launch. “Before your plan is put into place, come talk to us and make sure that everyone is happy,” she said.
Roychowdhury was more cautious about modifying adverse event
reporting. He noted that given the new mechanisms of action of many new cancer drugs, “I’m a little bit hesitant to have guidances that even for supplemental indications are going to reduce the surveillance on even Grade 1 and 2 toxicities.” He added that although collecting less safety data for clinical trials testing new indications for well-known cancer drugs may be appropriate, large companies that sponsor such trials have general SOPs for all types of therapeutic areas. “Unless the FDA and EMA come up with very clear guidances that separate oncology clinical trials from the rest, we will continue to see collection of safety data that may look to you as being superfluous and overcautious,” he said.
Schilsky acknowledged concerns about some drugs moving into clinical use with limited safety data assessment. “The sponsor and the regulatory agencies should sit down and decide what is sensible data collection given the patient population being studied, the pharmacological class of agent, the known safety profile of the drug and the objectives of the trial,” he said.
Progression-Free Survival Endpoints and Bias
Speakers and participants also explored how to avoid bias in assessing response to therapies in clinical trials, and whether there is a need for independent central review of imaging results when PFS is used as an endpoint. Dodd noted that the use of PFS is an area of active debate, and that in general PFS does not measure clinical benefit, nor is it a surrogate for overall survival. A trial with PFS as a primary endpoint requires strong evidence that the treatment effect is large, she said—greater evidence than would be required when overall survival is the endpoint.
Dodd pointed out that progression assessments vary by reader, with discrepancy rates in the timing and presence of progression typically greater than 30 percent. This has led to concern that there is potential for reader bias in unblinded trials due to local evaluators knowing the treatment assignment, prompting the requirement for blinded independent central review.
But Dodd’s study, published in 2008, showed that treatment effects were similar when estimated using central review or local evaluations (Dodd et al., 2008). Two more recent reviews of cancer clinical trials found more than 90 percent correlation in the hazard ratios between blinded independent central reviews and local assessments of progression of solid tumors (Amit et al., 2011; Zhang et al., 2013). “Given this, we have to ask ourselves, what is the value of central review?” Dodd said. As an alternative
to central review, she suggested using overall survival as an endpoint instead of PFS, but recognized that this is often not feasible. Dodd proposed that central review should also not be required in double-blinded studies in which the radiologists did not know the treatment assignment. Another alternative would be to audit for bias by doing blinded, independent central review on a small subset of cases.
In July 2012, an FDA advisory committee considered this issue and all committee members agreed that a prospectively defined audit approach should be considered. They advised against complete elimination of blinded independent central review, Dodd reported. She added that the EMA has some guidelines that suggest it is also open to an audit approach. “Moving forward, we need to just make sure that any method we come up with is able to identify bias,” Dodd said. “An audit using central review may be the best strategy today, but technological advances may offer alternative solutions in the future,” she stated. “We should think about ways to ensure that local reviews are blinded because the true effect of a drug on PFS may be best estimated in a double blind trial,” Dodd concluded.
But Sledge questioned the value of independent reviews. He noted that in one ECOG clinical trial, two radiologists reading the images disagreed almost half the time. “Does anyone seriously believe adding a third radiologist’s readings will be of statistical benefit?” he asked. “Why are we still even contemplating blinded independent review given that this is an experiment that’s been tried and failed?” Dodd responded,
It’s true that the discrepancy rates are shockingly high if you just look at the discordancy raw numbers, but what’s most important is the hazard ratios, which are estimates of treatment effect. What studies show is that in spite of those high discrepancy rates, the hazard ratios are in general agreement. Looking at the discrepancy rates alone is not really the answer we’re looking for. Adding another radiologist doesn’t solve the problem, but if you have two radiologists’ assessments and the treatment effects as estimated by those reads are in general agreement, that makes us feel more comfortable that there wasn’t a lot of systematic bias.
Pazdur added, “We have a regulatory obligation to make sure there isn’t bias. If the trial is truly blinded, you don’t need independent review. But most trials in oncology are unblinded because of the differential toxicity between the control and treatment arms of the trial, so one has to have some comfort that there is a true finding. That’s why we’re looking at these alternative mechanisms to ensure there is no bias.”
Pazdur gave an example of a trial of a treatment for carcinoid in which
the data safety monitoring committee recommended closure of the study for early demonstration of efficacy, based on the PFS rate, while another group reviewing the same study recommended it should be closed for futility. “That demonstrated to us that bias crept into the study,” he said, although Dodd added that carcinoids are particularly difficult cancers on which to assess progression via imaging. Dodd’s statement led Comis to suggest some studies may pose more risk of bias than others, and should have a different bias-monitoring strategy imposed on them.
Schilsky said that “if there’s any bias introduced to a study, it’s not at the level of the reading radiologist,” because it is uncommon for radiologists “to be aware of or to care about what the treatment is that’s being tested. The risk of bias comes from how the oncologist interprets that information that comes from those radiology reads.” But he added that in large clinical trials, the bias contribution of a single oncologist would be minor and wouldn’t be likely to influence the outcomes. “If all the participating physicians in the trial have a systematic bias, then that trial was doomed from the start. It’s a bigger issue than just reading the films,” he said.
Sherman stated that bias in “single trials [used to gain FDA approval] is the most concern to us because we’re making a very major decision based on a study whose findings were not replicated. Unblinded trials introduce more uncertainty, and simply hoping there isn’t bias isn’t the same thing as assessing whether or not there is.” In response to a participant who asked what industry can expect in this regard, Sherman answered, “The end of a phase II trial is the time to have that discussion about bias and monitoring with the FDA. One type of monitoring is appropriate for one study design, but in another situation it may not be. It’s never going to be a one size fits all.” Pazdur agreed, adding,
We’re dealing here with a subjective endpoint that is much different than overall survival, so we have to make certain there is no bias. You would have to guarantee that there was training on the sites of radiologists, and that there wasn’t communication between the radiologists and the treating physicians, because as we all know a doctor may go down to a radiologist to read the images. Due to discussions between the physician and the radiologist, there can be changes in interpretations and which metastatic disease sites are measured, depending on if a doctor wants the patients to continue on a particular treatment because he thinks it’s benefiting them.
Pazdur said that the FDA welcomes sponsors to suggest the least burdensome way to ensure there is no bias in their studies that use PFS as an endpoint. Francesco Pignatti, head of oncology, hematology, and diagnos-
tics in the Safety and Efficacy Sector at the EMA, said that the EMA is also open to approaches for demonstrating lack of bias. “Blinding the local evaluation is an excellent proposal. I’m also confident that the audit approach will evolve quickly as soon as we gain experience on how and when to use it. We’re trying to simplify things, so in the training and monitoring of the local evaluation, and the firewalls between radiology and clinical oncology, let’s be careful that this doesn’t result in inefficiencies,” he said.
Roychowdhury noted that some inefficiencies in clinical trials are due to excessive procedures instituted not only because of regulatory needs, but also because of paranoia on the part of sponsors about what the regulatory authorities want to see. “We cannot solve that by guidances, so how can we have more discussions between the regulatory agencies and the sponsors, especially for those on breakthrough therapies, so we can reduce the timeline of the trial?” he asked. Pignatti agreed that there can be excessive data collection for unimportant aspects due to sponsors’ lack of understanding that regulatory guidelines have flexibility and are not one size fits all.
Sherman responded that as a general rule, when in doubt, sponsors should “consult with FDA early and often to make sure [they are] collecting what’s important.” She agreed that often investigators collect an excessive amount of data or the wrong type of data because they believe such data are needed to garner an FDA approval for the treatment being tested. “Why is there this compulsion to collect every piece of data and check every box? Please come talk to us and think about how to make sure we’re making every dollar, and more importantly, every patient, count in clinical trials,” she said.
Pignatti from the EMA spoke about global regulation, including European regulation of clinical trials, how it differs from U.S. regulation, and efforts to harmonize international regulations. He noted that in Europe, the EMA is responsible for premarket evaluation of drugs as well as supervision of drugs once they reach the market. The European Union (EU) recently reviewed and revised its clinical trials legislation to take a more risk-based approach, and the EMA is trying to foster earlier and more continuous communications between regulators and sponsors. But he added that although the EMA will give advice to sponsors about trial design and conduct, that advice is optional and not binding; individual countries have not given the agency authority over design, approval, and conduct of clinical trials.
The main divergence between EU and U.S. regulation is in the area of early approval mechanisms, Pignatti said. The FDA will grant accelerated approval for a treatment for a serious or life-threatening disease based on a surrogate endpoint likely to predict clinical benefit, with confirmation of benefit in postmarketing monitoring. The new agent for an accelerated approval has to be more effective than available therapy.
The European Union, in contrast, will grant a conditional marketing authorization for a treatment that fulfills an unmet medical need for a serious, life-threatening, or orphan disease or in response to emergency threats. Even if the clinical data are not complete, authorization will be granted if researchers can show a positive benefit–risk balance, and if that benefit is confirmed with monitoring after the treatment has entered the market. “The critical difference is that in Europe, the benefit–risk must be as positive as for any other type of approval,” Pignatti explained.
In 2004, the EMA and the FDA forged a confidentiality agreement to improve dialogue between the two agencies, recognizing that both share the same fundamental public health mission. This agreement has resulted in regular and ad hoc discussions and shared activities between the two agencies “that have been very successful,” Pignatti said. “There are few differences now in our general guidances. The more difficult part is when we come to applying them, because regulators have to make decisions in the presence of uncertainty,” he concluded.
In situations with the most uncertainty (e.g., in the review of small, single-arm studies, studies done on heterogeneous populations, or early-approval applications), the two agencies can differ in their decisions, he noted. “In areas of very high uncertainty or situations where the benefit–risk balance is very close, you can continue to expect to see differences, because even having the best intentions and processes in place does not guarantee full harmonization,” Pignatti said.
For example, the FDA revoked approval of bevacizumab for breast cancer treatment because new studies did not show improvement in overall survival, but the EMA found that the benefits of bevacizumab in combination with paclitaxel outweighed its risks and approved the combination (Burstein, 2011; EMA, 2010). One study found that for 42 anticancer drugs approved by the EMA between 1995 and 2008, there were substantial differences between EMA and FDA decisions. Nearly half (47 out of 100) of the indications for these drugs had differences in approval; for 19 indications, approval was granted by only 1 agency, and 28 indications were approved by both the EMA and FDA but with different restrictions
(Trotta et al., 2011). Sixty-nine of the indications were approved first in the United States, although the time lag between FDA and EMA approvals is decreasing, Pignatti said.
According to Pignatti, another major discrepancy between FDA and EMA oversight is how the two agencies view the use of PFS as an endpoint in registration trials. Although both agencies agree that overall survival is a more clinically relevant endpoint than PFS, the EMA accepts PFS if it measures a clinical benefit, whereas the FDA tends to view it as a surrogate for overall survival. “Even within our community and our committees, this is a hotly debated issue,” Pignatti noted. This is problematic given that PFS is increasingly being used as an endpoint; one study found that between 1995 and 2000, only 21 percent of pivotal confirmatory trials used PFS as the primary endpoint, but the rate increased to 49 percent between 2006 and 2010. “The different understanding of the clinical relevance of PFS is something we definitely want to work on,” Pignatti said.
New Models for Regulation of Drug Development
Eichler, also from the EMA, described the New Drug Development Paradigms (NEWDIGS), a collaborative effort that began at the Massachusetts Institute of Technology and includes representatives from drug regulators, drug companies, payers, patient organizations, and academic institutions. NEWDIGS’s objective is to reliably and sustainably deliver new, better, and affordable drugs to the right patients faster, and to counter “Pharmageddon,” as Eichler described the current situation. “The innovation engine, particularly in the biopharmaceutical industry, isn’t humming along as it should be. Everybody is a bit disgruntled and dissatisfied, whether you’re a patient, in pharma, a provider, a payer, or a regulator,” he said.
The goals of NEWDIGS are to provide a unique, collaborative environment for innovation and learning that is creative and nonbureaucratic, to tap the entrepreneurship and collective intelligence of its participants, and to have a collaborative impact similar to what the SEMATECH collaboration had on the semiconductor industry in the 1980s.9 “NEWDIGS calls itself not just a ‘think tank’ but also a ‘do tank.’ They want to catalyze pilot
________________
9 SEMATECH is a global collaboration with the objective of accelerating the commercialization of technology innovations into manufacturing solutions (see http://www.sematech.org/corporate/index.htm [accessed May 14, 2013]).
studies in real life, not just sit and think,” Eichler explained. NEWDIGS takes a systems approach to catalyzing change by exploring the co-evolution of processes, technologies, policies, and people, he added.
NEWDIGS has developed the concept of adaptive licensing (Eichler et al., 2012) to counter some of the problems currently experienced with the regulation of new drugs. According to Eichler, these problems stem in part from the binary nature of that regulation. There is gradual learning about the effects of new drugs in a limited number of animals and people that occurs preclinically and during clinical testing. But “the next morning after that ‘magic moment’ when the new drug is approved, it’s out the door and anyone can have it and we have no idea what happens to these patients. Is that wise?” Eichler asked.
Another problem is that some patient groups are frustrated that new drugs are not offered sooner to them, while some consumer advocates maintain that more needs to be known about drugs before they enter the market. As FDA Commissioner Margaret Hamburg has noted, it has been said that the FDA has just “two speeds of approvals—too fast and too slow” (Hamburg, 2010).
To counter both problems, NEWDIGS has proposed “doing away with the magic moment and creating a number of milestones where we look at the data on the drug repeatedly over time and align the way a drug becomes available with the growing knowledge as uncertainty is progressively reduced. We can broaden the access of the drug this way,” Eichler explained. He noted that in the current regulation scheme, during post-licensing of a drug, the treatment population grows rapidly but the treatment experience does not contribute to evidence generation. With adaptive licensing, in contrast, after initial license of the drug, the number of treated patients grows more slowly due to restrictions on use, and the patient experience is captured, contributing real-world information about the safety and effectiveness of the drug (see Figure 5).
Eichler explained that adaptive licensing is a prospectively planned, adaptive approach to the regulation of drugs that has iterative phases of evidence gathering followed by regulatory evaluation and license adaptation, which can specify that the drug be withdrawn from the market or continue to be offered to patients. Adaptive licensing seeks to maximize the positive impact of new drugs on public health by balancing timely access for patients with the need to provide evolving information on benefits and harms. “Adaptive licensing is basically the tradeoff between access and knowledge,” he said.
FIGURE 5 Adaptive licensing captures more of the patient experience, contributing more real-world information about the safety and effectiveness of drugs.
NOTE: A = traditional licensing; B = adaptive licensing; RCT = randomized controlled trial.
SOURCE: Eichler presentation (February 12, 2013).
Eichler noted that similar licensing already occurs with the FDA’s accelerated approval licensing and with EMA’s Conditional Marketing Authorisation, and can be aided by pharmacovigilance tools that detect adverse reactions to drugs. He added that to achieve the full potential of adaptive licensing, licensing decisions should be aligned with coverage and prescribers’ decisions.
Adaptive licensing will require not only randomized controlled trials, but also observational studies. Although some experts are hesitant to rely on such studies, which are considered lower in the hierarchy of evidence-based research, “many regulatory decisions are already based on case studies,” Eichler pointed out. For example, the FDA may decide to withdraw a drug from the market or alter its label based on adverse event reports, which are essentially case reports, he noted.
Unlike clinical trials, which have strict conditions for patient participation, observational studies have the advantage of better detecting drug effects in the “real world,” when they are combined with other medications or influenced by concomitant conditions, Eichler added. He stressed, “We have to have the full spectrum of evidence-generation methodologies at our disposal, and you especially will need rapid learning systems in oncology where you probably have more variables than you have patients. The more information you can gather from the real world, the faster the learning experience will be.” The needs and potential benefits of a rapid learning system for cancer were described in a past NCPF workshop (IOM, 2010a).
After a day and a half of presentations, speakers and participants agreed that much has been accomplished since the publication of the IOM consensus report to improve the efficiency, innovation, oversight, and collaboration potential of the NCTN. But due to the rapidly changing nature of cancer research, challenges still remain.
“There has been an enormous amount of change in just 2 years that has been in a very positive direction,” Doroshow said. Looking forward, the NCI aims to foster an NCTN “that’s not just for treatment, screening, and diagnosis, but for control and prevention as well,” he stressed. He added, “I hope as we fund our new system, we will have a very functional platform that allows us to screen and find the patients we need for the molecular trials that are the trials of the future.” Comis added that although there are still major challenges with biomarker screening, there is a real “opportunity
to position the NCTN groups to play a critical role in the development of more targeted therapies.”
Doroshow concluded that “it’s remarkable that we can, in a financially tight time, come together to understand where the most important science is and what critical infrastructures we need to allow that science to go forward. We have done our job to modernize the system.”
This page intentionally left blank.










































































