
SESSION 1
WATER TREATMENT: ADVANCED PHOTOOXIDATION PROCESSES
Richard G. Zepp
U.S. Environmental Protection Agency
Athens, Georgia
Rapporteur's Report
Absorbed solar radiation provides the primary driving force for the various chemical, physical, and biological processes that oxidize or reduce substances in the environment. Among these processes are photooxidations that play an important role in cleansing the environment of the waste materials derived from human activities. These oxidations involve an array of direct and indirect photoreactions that are induced primarily by the ultraviolet component of solar radiation. Taking a cue from nature, scientists and engineers have made significant progress during the past few years in optimizing the usage of photochemical reactions to remove undesirable impurities in contaminated water. These advances were exemplified by the nine papers that were presented in the water treatment session of the workshop.
David Ollis introduced the session by pointing out that oxidation techniques for treatment of contaminated water initially involved the usage of ultraviolet light from artificial lamps or the addition of some oxidant such as ozone. Ozonation is an old technique that is still being widely used in Europe to treat drinking water. These techniques generally result in partial oxidation of organic pollutants. Recent techniques that achieve more complete oxidation have involved (1) combinations of ultraviolet radiation and oxidants (ozone, hydrogen peroxide) in homogeneous systems or (2) heterogeneous photocatalytic systems that combine near ultraviolet radiation (320 to 390 nm range) with light-activated oxidation catalysts such as titanium dioxide. Current results presented at the workshop indicate that additions of ozone and/or hydrogen peroxide to the catalyst systems further enhance oxidation efficiencies.
Systems Considered
Systems specifically discussed at the workshop included UV/ozone, UV/hydrogen peroxide, UV/ozone plus hydrogen peroxide, and heterogeneous photocatalysis involving semiconductors. The first three techniques all employed far ultraviolet radiation (< 280 nm) whereas the last involved near UV from artificial light sources or solar radiation. Both the UV/ozone and the UV/peroxide systems are currently being widely used to remove trace contaminants such as trichloroethylene (TCE) and tetrachloroethylene (PCE), but the other techniques are being rapidly developed and will likely see wide application in the near future. Other technologies discussed at the workshop that have seen usage at the laboratory or pilot scale include vacuum UV irradiation, Fenton chemistry, dye photosensitization, and photocatalysis using new semiconductors with lower energy bandgaps. Some scientists and engineers have referred to all of these treatment procedures as "advanced oxidation technologies." Hydroxyl radicals are involved as intermediates in most of these procedures.
Factors Affecting Treatment Efficiencies: Kinetic Considerations
Many of the presentations relied on the use of various kinetic studies to evaluate the treatment methodologies. The rate of oxidation of the trace contaminant was used to judge the effectiveness of the technique. The rate (conversion per unit time) of any type of photoreaction depends upon the rate of light absorption by the photoactive species or ''chromophore,'' Ia, and the quantum efficiency of the reaction, ![]() (Equation 1).
(Equation 1).
The rate of light absorption is affected by the spectral overlap between the light source and the spectrum of the chromophore. The average light absorption rate in a system is proportional to the incident light intensity and can be reduced by light attenuation within the system. In the various water treatment systems discussed in the workshop, ozone and hydrogen peroxide absorb UV radiation strongly in the far UV region (< 280 nm), but only very weakly in the middle (280 to 320 nm) and near UV (320 to 390 nm) region. Ground-level solar radiation includes ultraviolet radiation only in the middle and near ultraviolet, with the cutoff at about 295 nm. Thus, at least under "one-sun" irradiations, light absorption and contaminant oxidation rates induced by photolysis of ozone and hydrogen peroxide by solar radiation are generally considered to be too low for practical usage in treatment processes. (However, it should be noted here that solar ozone photolysis is the key natural process in initiating organic oxidations in the troposphere.) On the other hand, commonly used photocatalysts absorb strongly in the near UV, and thus rapidly absorb sunlight as well as radiation from black lights or other near UV sources. Photocatalytic systems thus appear to be better suited for solar applications.
As discussed by William Glaze, Gary Peyton, and Jack Zeff, UV/ozone and UV/hydrogen peroxide treatment systems have maximized light absorption rates by using lamps that emit far UV light, e.g., low pressure mercury arc lamps. Apparently, concentrated solar radiation has not been employed in conjunction with ozone and/or hydrogen peroxide, although it seems that it may be worthwhile to take a closer look at this option.
Commonly used photocatalysts such as titanium dioxide (anatase) absorb strongly in the near UV region. Hussain Al-Ekabi reported the successful use of near UV lamps to activate photocatalytic contaminant removal, whereas Craig Turchi, Mark Mehos, and Jim Pacheco all described efforts to use concentrated solar radiation in heterogeneous photocatalytic treatment systems (more details below).
The quantum yield for reaction is an important parameter that describes the fraction of absorbed radiation that results in reaction. Despite this fact, as emphasized by David Ollis, all too often information about quantum yields is omitted from treatment studies, making it very difficult to compare results from one system to another. Glaze pointed out that, when the contaminant is very dilute, frequently the case in the types of treatment situations considered at the workshop, the quantum yield and rate are usually directly proportional to contaminant concentration; that is, a pseudo-first order rate expression applies. As the contaminant concentration increases, however, the quantum yield and rate become independent of contaminant concentration. Similarly, in the case of photocatalysis, the rate becomes independent of concentration with increasing contaminant concentration, as clearly demonstrated by Pacheco's data for photocatalytic systems at the Sandia National Laboratories.
The quantum yield is affected by "primary" photoreactions, which directly ensue from an excited state, as well as by secondary reactions, such as free radical chain processes, recombinations, and scavenging reactions that follow the primary photoreaction. The discussions by Glaze and Peyton demonstrated that useful information about the secondary reactions in
photochemical treatments can be obtained by studies of thermal reactions such as ozone oxidations under alkaline conditions. Like the light absorption rate, quantum yields can depend on light intensity, often in some inverse fashion, as demonstrated by two Solar Energy Research Institute scientists, Arthur Nozik and Turchi, in their photocatalysis presentations. Such decreased quantum yields with increasing intensity place limits on the enhancement of treatment processes that can be achieved using concentrated photons.
In addition to light absorption rates and quantum yields, net rates of oxidation in treatment systems can be affected by mass transfer. In a system with a nonuniform light field (caused by strong light attenuation of system geometry, for example), mass transport to the photic zone can become rate limiting. Moreover, in heterogeneous systems, particularly those with immobilized catalysts, mass transport to the catalyst surface can be rate limiting. As discussed by Turchi, mass transport limitations can lead to a lack of rate dependence on light intensity in some reactor systems, obviously an important consideration in designing concentrated solar treatment systems.
Treatments Using UV, Ozone, and/or Hydrogen Peroxide
Glaze, Peyton, and Zeff presented evidence that combinations of ultraviolet light (< 300 nm) and ozone, hydrogen peroxide, or ozone/hydrogen peroxide are effective techniques for the trace contaminant oxidations in water. In such systems, organic oxidations occur by several potential mechanisms some of which actually involve dark reactions:
-
Most compounds that have unsaturated chemical bonding, e.g., aromatic hydrocarbons, ketones, and polyenes, absorb the 254 nm light that is used in the UV/ozone procedure. For such compounds, direct photolysis may be an important loss mechanism, although UV light screening by the ozone would greatly reduce the rate of this type of reaction compared to the rate in pure water. In addition to direct photolysis, two other types of processes potentially can result in organic oxidations.
-
Direct thermal reactions between ozone and certain organics may be an important oxidation pathway in the system. Ozone, however, is a highly selective oxidant. Most organic pollutants are not rapidly oxidized by direct reaction with ozone.
-
Free radicals, produced by photochemical and/or thermal processes in the UV/ozone, UV/hydrogen peroxide, or UV/ozone/hydrogen peroxide systems, make a major contribution to the organic oxidations that occur. Direct photolysis of ozone in water leads to nearly quantitative production of hydrogen peroxide. Hydrogen peroxide, hydroperoxide ions, or hydroxide ions react with ozone to produce a potent oxidant, the hydroxyl radical. As pointed out by Glaze, the combination of ozone and hydrogen peroxide at higher pH values results in oxidation of trace contaminants via hydroxyl intermediates, even in the absence of UV radiation. Direct photolysis of the hydrogen peroxide also contributes to the production of OH radicals. Hydroxyl radicals react rapidly with most organic contaminants by abstracting hydrogen atoms or by addition to aromatic systems and double bonds. Peyton noted that the efficiencies of these reactions can be enhanced in some cases by addition of substances that "promote" the yield of hydroxyl radicals via enhanced formation of transients such as superoxide and hydrogen peroxide that react with ozone to form OH radicals.
Whatever the mechanism for these reactions, practical studies at the pilot and larger scale have demonstrated their utility in cleaning up contaminated water. Glaze pointed to on-line treatment systems for treating DBCP-contaminated groundwater in the San Fernando Valley, California; removal of triazine herbicides from groundwaters in Europe; and even removal of the objectionable taste and odor of algal exudates such as geosmin by UV/hydrogen peroxide treatment in some U.S. drinking water treatment systems. Zeff likewise provided a number of "real-world" examples of the usage of UV, ozone,
and hydrogen peroxide to clean up contaminated water samples at reasonable costs. Contaminants removed include perchloroethylene, toluene, and other aromatic or aliphatic compounds.
Photocatalytic Treatment
Titanium dioxide (anatase) is the most widely used photocatalyst in treatment systems research, due to the above-noted considerations, e.g., good spectral overlap with near UV lamps or solar radiation, high quantum efficiencies. Unless otherwise noted, all of the studies discussed in this section involved this catalyst. Photocatalytic studies are primarily at the laboratory to pilot scale phase of development. A wide variety of organic contaminants, including biologically refractory chlorinated substances such as chloroform and PCE are efficiently photomineralized in aqueous suspensions or in immobilized catalytic systems on exposure to near ultraviolet radiation. (Chloroform and PCE are stable to near UV in the absence of the titanium dioxide, and no reaction occurs in dark controls.)
The session included several discussions of the mechanism for heterogeneous photocatalytic oxidations. Arthur Nozik presented mechanistic studies in electrochemical slurry cells that provided support to the hypothesis that the photocatalyzed oxidations involve hydroxyl radicals formed via oxidation of hydroxide on the catalyst surface. As noted in the preceding section, hydroxyl radicals are potent oxidants that react rapidly with most organic contaminants. Controversy remains as to the exact mechanism(s) of these reactions, however. For example, M. Fox of the University of Texas has provided evidence that organic oxidations can proceed via direct oxidations by photochemically produced "holes" on the semiconductor surface. Glaze further noted that certain features of heterogeneous oxidation kinetics and products appear to be inconsistent with the hypothesis that "free" OH radicals are involved. The relatively minor effect of bicarbonate, a good OH radical scavenger, on oxidations reported by Pacheco, seems to argue against OH radicals as intermediates. Glaze mentioned the theory that the oxidations may involve surface-adsorbed OH radicals.
Other results presented by Al-Ekabi at this session indicated that common anions in contaminated waters had little to almost no effect on photocatalytic treatment efficiency. On the other hand, both Al-Ekabi and Pacheco found that added hydrogen peroxide enhanced the initial rate of the photocatalytic oxidations. Al-Ekabi also reported that ammonium persulfate increased the photocatalytic oxidation rate of chlorophenols. Several session participants suggested that hydrogen peroxide and persulfate enhance the oxidation efficiency in part by capturing electrons from the catalyst surface, thus reducing electron-hole pair recombination and producing hydroxyl radicals from the electron acceptor.
Efforts to move from the laboratory to pilot scale solar photocatalytic oxidations have been initiated at several laboratories. Peter Daley emphasized that treatment costs play a critically important role in the acceptance of treatment technologies. Preliminary economic assessments by Hal Link of solar photocatalytic treatment of contaminated groundwater at the Lawrence Livermore National Laboratory were somewhat discouraging, with operating costs (e.g., cleaning collector mirrors) well over $10 per 1000 gallons. However, he suggested that additional research on improving catalyst efficiency and on solar collectors can sharply reduce costs. In that regard, Mehos reported early efforts to develop efficient immobilized photocatalytic systems. These efforts have focused on developing systems with no flow rate dependence, low pressure drop, and long catalyst lifetimes. Although initial treatments in these systems had efficiencies that may compare favorably with the slurry systems, continued usage appeared to result in decreased efficiency. During subsequent discussions, Nick Serpone suggested that the reduced efficiency may be attributable to formation of titanium peroxo species on the catalyst surface.
Future Research Areas
At the end of the session several areas were identified for future research. With respect to issues most closely related to the main topic of the workshop, the use of concentrated solar photons, several research areas were identified: (1) effects of high temperatures and light intensities on treatment efficiencies, especially with the photocatalytic systems; (2) quantum efficiencies for contaminant oxidations (now almost nonexistent and badly needed for comparisons of treatments under differing conditions); (3) treatment economics for concentrated solar systems; (4) improvements in photoreactor design configuration; (5) studies of potential toxicity of oxidation intermediates under conditions that exist in concentrated solar reactors; (6) usage of concentrated solar photons for UV/ozone, UV/hydrogen peroxide, combined photocatalytic/ozone/hydrogen peroxide treatments; (7) development of improved photocatalysts that use solar photons more efficiently and are less susceptible to poisoning.
Other promising research areas identified by participants include: (1) vacuum UV irradiation (wavelengths less than 200 nm), including applications to gas phase treatment; (2) Fenton chemistry, i.e., oxidations catalyzed by the combination of iron or other metals with hydrogen peroxide; (3) dye photosensitizations, such as photosensitization by methylene blue or polymer bonded dyes.
CHEMICAL MODELS OF ADVANCED OXIDATION PROCESSES
William H. Glaze
University of North Carolina
Chapel Hill, North Carolina
Abstract
Advanced oxidation processes (AOPs) have been defined as ambient temperature processes which involve the generation of highly reactive oxyradicals, especially the hydroxyl radical. These processes show promise for the destruction of hazardous organic substances in municipal and industrial wastes and in drinking water. Four types of AOPs are considered in this paper: base-catalyzed decomposition of ozone; ozone with hydrogen peroxide; UV photolysis of ozone; and UV photolysis of hydrogen peroxide. Other AOPs include the well-known Fenton reaction (Fe[II]-catalyzed decomposition of hydrogen peroxide), semiconductor-photocatalysis, and other, less characterized processes such as cavitation, wet air oxidation, etc.
Kinetic models for AOPs are being developed based on known chemical and photochemical principles. The models take into account measured effects of radical scavengers such as bicarbonate; dose ratios of the oxidants or UV intensity; and pH. Two systems have been described in detail: 03/H2O2 and H2O2/UV. The former was discussed in detail, using two cases: oxidation of tetrachloroethylene (PCE) in a high carbonate, but otherwise pure, water matrix and oxidation of nitrobenzene at relatively high concentrations. The oxidation of PCE is well fitted to the model. The oxidation by ozone is slow and is accelerated by the addition of peroxide up to a stoichiometric ratio of the two oxidants of 1:2 (H2O2:O3). At higher ratios, the reaction rate becomes ozone mass-transfer limited. Under conditions representative of the treatment of a hard groundwater (with 4 mM bicarbonate, ozone dose rate of 4.7 × 10-6 M s-1, and peroxide dose rate of 2.5 × 10-6 M s-1) the steady state concentration of hydroxyl radical is calculated to be 2 × 10-12 M; the half-life of the first order oxidation of PCE is 2.5 min. Thus, although over 95% of the OH radicals are trapped by bicarbonate, the oxidation of a compound such as PCE is expected to be economically feasible, and a 2000 gpm plant is under construction in Los Angeles to test the process. For substrates at higher concentrations, it is shown that the reaction becomes zero order and is limited by ozone mass transfer. The effect of large amounts of oxidation byproducts is generally to use OH radicals but also to form additional peroxide, which accelerates decomposition of ozone.
The photolysis of hydrogen peroxide by 254 nm radiation has also been studied in some detail. The rate determining step is photolysis of peroxide into two OH radicals, following which the Haber-Weiss sequence occurs, accompanied by chain termination processes by species such as bicarbonate. For a typical case (Io = 0.4 W/liter; effective pathlength = 14.8 cm; PCE and bicarbonate the same as above; and [H2O2] = 1 mM), the steady state concentration of OH radicals is calculated to be 1.1 × 10-11 M. This process is suitable only when the residual peroxide is not detrimental and where there are no substrates present which absorb UV radiation in preference to peroxide, without direct photolysis. Advances in its efficiency can be achieved by the development of lower wavelength UV lamps.
List of References
1. Glaze, W.H., J.W. Kang, and D.H. Chapin. 1987. The Chemistry of Water Treatment Processes Involving Ozone, Hydrogen Peroxide and Ultraviolet Radiation Ozone: Science & Engineering 9 (4):335.
2. Garrison, R.L., C.E. Mauk and H.W. Prengle, Jr. 1975. Advanced Ozone System for Complexed Cyanides, In First International Symposium on Ozone for Water and Wastewater Treatment, R.G. Rice and M.E. Browning, eds. International Ozone Institute, Syracuse, N.Y., 1976, p. 551.
3. Prengle, H.W., Jr., C.G. Hewes, III, and C.E. Mauk. 1976. Oxidation of Refractory Materials by Ozone with Ultraviolet Radiation. In Second International Symposium on Ozone Technology, R.G. Rive, P. Pichet, and M.A. Vincent, eds. International Ozone Institute, Syracuse, N.Y., p. 224.
4. Prengle Jr., H. W., and C.E. Mauk. 1978. Ozone/UV Oxidation of Pesticides in Aqueous Solution. In Ozone/Chlorine Dioxide Oxidation Products of Organic Materials, R.G. Rice and J.A. Cotruvo, eds. Ozone Press International, Cleveland, OH. p. 302.
5. Koubeck, E. 1977. Oxidation of Refractory Organics in Aqueous Waste Streams by Hydrogen Peroxide and Ultraviolet Light. U.S. Patent No. 4,012,321.
6. Nakayama, S., K. Esaki, K. Namba, Y. Taniguchi, and N. Tabata. 1979. Improved Ozonation in Aqueous Systems. Ozone Science & Engineering 1:119.
7. Berglind, L., E. Gjesing, and E. Skipperud Johansen. 1979. Removal of Organic Matter in Water by UV and Hydrogen Peroxide. In Oxidation Techniques in Drinking Water Treatment, W. Kühn and H. Sontheimer, eds. U.S. Environmental Protection Agency, EPA-570/9-79-020, pp. 510–523.
8. Brunet, R., M.M. Bourbigot, and M. Dore. 1984. Oxidation of Organic Compounds Through the Combination Ozone-Hydrogen Peroxide. Ozone Science & Engineering 6:163.
9. Duguet, J.P., E. Brodard, B. Dussert, and J. Mallevialle. 1985. Improvement in the Effectiveness of Ozonation of Drinking Water Through the Use of Hydrogen Peroxide. Ozone Science & Engineering 7:241.
10. Peyton, G.R., F.Y. Huang, J.L. Burleson, and W.H. Glaze. 1982. Destruction of Pollutants in Water with Ozone in Combination with Ultraviolet Radiation. 1. General Principles and Oxidation of Tetrachloroethylene. Environmental Science & Technology 16:448.
11. Glaze, W.H., G.R. Peyton, S. Lin, F.Y. Huang, and J.L. Burleson. 1982. Destruction of Pollutants in Water with Ozone in Combination with Ultraviolet Radiation. 2. Natural Trihalomethane Precursors. Environmental Science & Technology 16:454.
12. Peyton, G.R., and W.H. Glaze. 1988. Destruction of Pollutants in Water with Ozone in Combination with Ultraviolet Radiation. 3. Photolysis of Aqueous Ozone. Environmental Science & Technology 22(7):761.
13. Glaze, W.H., and J.W. Kang. 1988. Advanced Oxidation Processes for Treating Groundwater Contaminated with TCE and PCE: Laboratory Studies. Journal AWWA 80(5):57.
14. Aieta, E.M., K.M. Regan, J.S. Lang, L. McReynolds, J.W. Kang, and W.H. Glaze. 1988. Advanced Oxidation PRocesses for Treating Groundwater Contaminated with TCE and PCE: Pilot-Scale Evaluations. Journal AWWA 80(5):64.
15. Glaze, W.H., and J.W. Kang. 1989. Advanced Oxidation Processes. Description of a Kinetic Model for the Oxidation of Hazardous Materials in Aqueous Media with Ozone and Hydrogen Peroxide in a Semi-Batch Reactor. Industrial & Engineering Chemical Research 28:1573.
16. Glaze, W.H., and J.W. Kang. 1989. Advanced Oxidation Processes. Test of a Kinetic Model for the Oxidation of Hazardous Materials in Aqueous Media with Ozone and Hydrogen Peroxide in a Semi-Batch Reactor. Industrial & Engineering Chemical Research 28:1580.
17. Glaze, W.H., and Y. Lay. 1989. Oxidation of 1,2-Dibromo-3-chloropropane (DBCP) Using Advanced Oxidation Processes. 9th Ozone World Conference of IOA, New York City, June 3-9, 1989.
18. Somich, C.H., P.C. Kearney, M.T. Muldoon, and S. Elsasser. 1988. Enhanced Soil Degradation of Alachlor by Treatment with Ultraviolet and Ozone. Journal of Agriculture & Food Chemistry 36:1322.
19. Buxton, G.V., C.L. Greenstock, W.P. Helman, and A.B. Ross. 1988. Critical Review of Rate Constants for Oxidation of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (OH/O) in Aqueous Solutions. Journal of Physical Chemistry Reference Data 17(2):513.
20. Staehelin, J., and J. Hoigné. 1985. Decomposition of Ozone in the Presence of Organic Solutes Acting as Promoters and Inhibitors of Radical Chain Reactions. Environmental Science & Technology 19:1206.
21. Guittonneau, S., J. De Laat, M. Doré, H.P. Duguet, and C. Bonnel. 1987. Etude de la Degradation de Quelques Composes Organochlores Volatils par Photolyse du Peroxyde d'Hydrogene en Milieux Aqueux. Revue des Sciences de l'Eau 1(1–2):35.
MECHANISTIC STUDIES OF THE PHOTOCATALYTIC BEHAVIOR OF TiO2PARTICLES IN A PHOTOELECTROCHEMICAL SLURRY CELL AND THE RELEVANCE TO PHOTODETOXIFICATION REACTIONS
M.W. Peterson, J.A. Turner, and A.J. Nozik
Solar Energy Research Institute
Golden, Colorado
The general photo-oxidizing and photocatalytic properties of suspensions of semiconducting TiO2 particles are well established in the literature. Because TiO2 has the ability to completely oxidize a large number of hazardous materials to nontoxic products, it has become the benchmark semiconductor for use in a variety of schemes for the photodetoxification of contaminated water.
Because TiO2 can only absorb a small portion of the solar spectrum in the near UV (band gap 3.0 eV [rutile] to 3.2 eV [anatase]), it is desirable to shift its absorption threshold toward the red region of the spectrum, or to find alternative materials with better solar absorption characteristics that match its photocatalytic properties.
Material modification or the search for alternative materials that mimic TiO2 requires a detailed mechanistic understanding of how TiO 2 photodecomposes pollutant species. We have, therefore, undertaken a mechanistic study of photo-oxidation processes on TiO2 particles using a photoelectrochemical slurry cell.
Three types of TiO2 powders were used: 30 nm Degussa P-25 (anatase), 3 µm Fisher (anatase), and Aldrich 99.9% (rutile). Slurries were typically made at 1 × 10-4 M in a pH 1 HCl/KCl solution using 18 MO deionized water.
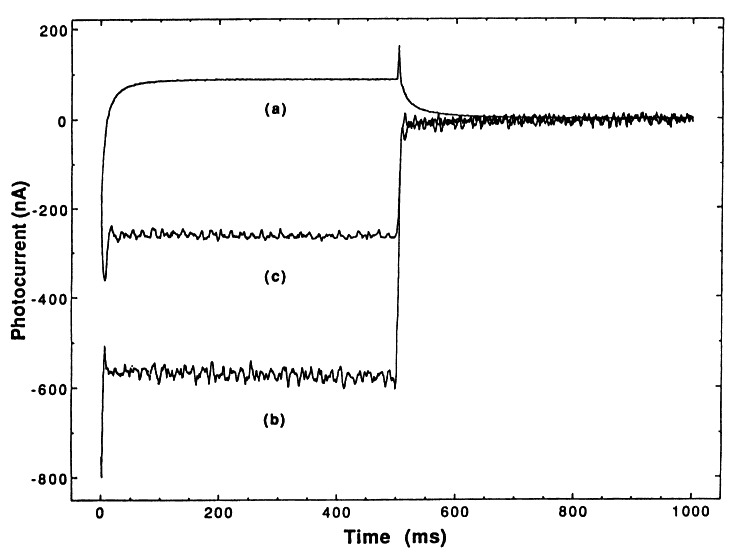
A conventional three electrode PEC cell was used that was fitted with a quartz window. Both working and counter electrodes were 0.44 cm2 Pt discs that were freshly polished before each run. The reference electrode was SCE; the cell was constantly purged with ultrapure Ar. The TiO2 was excited at 326 nm (optimum response) with a 150 W Xe arc lamp that was focused on a Jarrell-Ash 1/4 meter monochromator. The light excitation was turned on and off with a UniBliz shutter. An IBM-PC controlled the PAR 174, monitored the current, and controlled the shutter, thus allowing for collection of light-on and light-off current transients; typical flash duration was 500 ms at 1 s-1. Electrode blanks were run before and after each experiment. The potential was maintained at 0.5 V vs. SCE, which was found to be the optimum potential (i.e., low dark currents and a reasonable photocurrent).
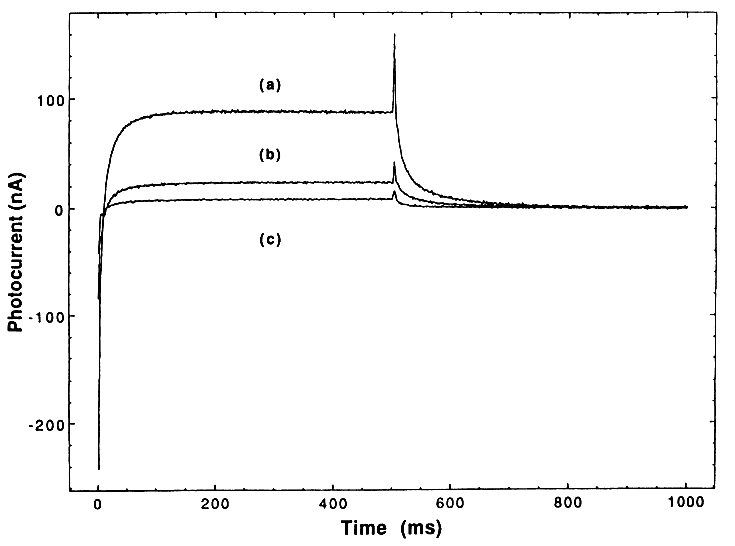
A comparison of the three transient responses for the three types of TiO2 is shown in Figure 1. It is seen that the photocurrent is initially cathodic and rapidly (within 25 ms) becomes anodic. Once steady state is achieved, the net current is anodic. However, this current is a composite of both anodic and cathodic current; this is demonstrated by the fact that an anodic current spike occurs when the light is shut off.
Mechanistically, the transients may be interpreted by the following general mechanism:
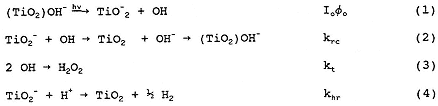
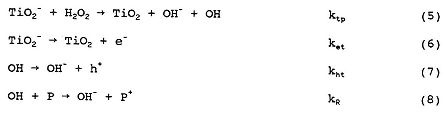
where (TiO2)OH- represents TiO2 with hydroxylated surface groups, TiO2- represents photogenerated electrons trapped on TiO2 particles, P is the reacting pollutant, and the rate constants k for reactions (1)–(8) are indicated by the respective subscripts on k that are listed next to each reaction.
Actually, the surface chemistry of TiO2 is more complicated than indicated by Equation (1) and involves the following equilibria:
where (TiO2)B represents bulk TiO2.
However, for purposes of the present paper, we just wish to indicate that upon bandgap illumination, OH radicals are formed by hole trapping on the hydroxylated surface, and trapped electrons are formed in the TiO2 particles; Equation (1) suffices for this purpose.
In the above mechanism, reaction (6) generates the anodic photocurrent and reaction (7) generates the cathodic photocurrent in the PEC slurry cell.
In order to check the proposed mechanism a series of simple experiments were performed. We believe that the cathodic photocurrent is due to hydroxide radicals in solution, and not to adsorbed OH on the TiO2. To establish this, we immobilized the TiO2 on a conducting carbon paste in a nonuniform fashion (i.e., some of the carbon paste is not coated with the TiO2). This then should lead to typical semiconductor electrode behavior for the TiO2. That is, at positive potentials in a PEC cell, if OH• radicals form only at the TiO2 surface, then they should simply act as recombination centers or result in the formation of H2O2 or O2; this would produce an anodic photoresponse. On the other hand, if OH radicals are escaping from the surface of the TiO2, they should arrive at the bare carbon surface and inject a hole resulting in a cathodic photocurrent. Our results with the immobilized TiO2 electrodes show a cathodic transient similar to the slurry cell response. Hence, we conclude that OH• radicals do escape into solution. We reject the possibility that adsorbed OH• on the TiO2 particles can contribute to the cathodic current at the carbon paste electrode because of the expected short recombination time with electrons of OH• on TiO2 compared to the diffusion time required for OH• adsorbed on TiO2 particles to reach the electrode. The lifetime of free OH• radicals in solution is controlled by their encounter with reducing agents, and is expected to be longer than their recombination lifetime as adsorbed species on TiO2.
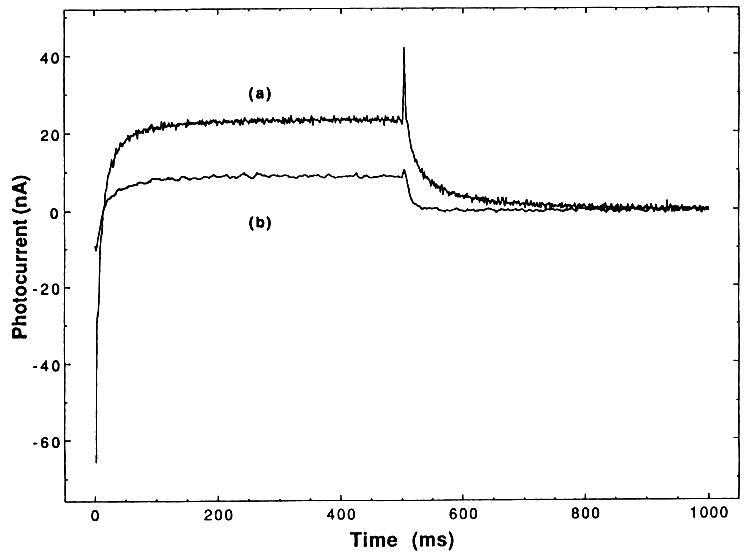
If TiO2- reacts with H2O2 as indicated in reaction (5), then the addition of small amounts of H2O2 should result in trapping all of the TiO2-, thus quenching reaction (6); only a purely cathodic photocurrent should therefore be seen. This effect occurs and is demonstrated in Figure 2 (curve b), where we have added H2O2 to make a 0.005 M slurry. Additional support for our
mechanism comes from the addition of a larger excess of H2O2. Under these conditions, we expect to also see the following reactions:

That is, we should now begin to consume OH. This effect is demonstrated in Figure 2 (curve c), where we have simply doubled the concentration of H2O2 and now see a decrease in the cathodic photocurrent.
The question of O2 vs. H2O2 in improving the kinetics of the overall photodecomposition reaction is readily addressed using the PEC slurry cell. Figure 3 demonstrates the results; it is seen that addition of O2 results in a reduction of both anodic and cathodic photocurrent (i.e., a loss of both TiO2- and OH•). This is understood by the following reaction:
together with reaction (13). Hence, while O2 serves as a good electron acceptor to complete the PEC circuit, it can also act to short the oxidation reaction of OH through reaction (13).
We also believe that a primary role for O2 in the photodetoxification process is to act as an electron sink for the photogenerated carriers. This is supported by the result that both TCE and CH2Cl2 could be decomposed to HCl in the slurry cell in the absence of 02 by using only the working electrode as the electron sink. Oxygen (O2) may also play a role as a radical scavenger in the bulk phase, and, hence, influence the kinetics by participating directly in specific chemical steps. This will depend upon the specific reactants originally present in solution.
The importance of the hydroxide radical produced by the photoexcitation of TiO2 during the photodecomposition of common organic compounds is becoming increasingly apparent. That the hydroxide radical is of importance would seem quite reasonable since its formation provides a good pathway for e--h+ charge separation by the effective scavenging of positive holes (h+). If the hydroxide radicals, rather than the positive holes themselves, are indeed the primary mode of hole transfer to the organic species, this has several interesting ramifications for photodetoxification.
One important point concerns the dependence of the rate of photodecomposition on the light intensity. If we take reactions (1), (2), (3), and (5) together with the following pollutant reactions:

and, if we consider [OH]2 kt ~ [OH] [TiO2-]krc>> [TiO2-] [H2O2 ] ktp
and assume a steady state concentration of [OH]:
then:
and
Thus, the dependence of the pseudo-first order reaction rate [OH]kp on light intensity is a function of the relative magnitude of the light intensity relative to ([TiO2-] [krc])2. At high light intensity, we would see a square root dependence, while at low intensity, we would see a linear dependence.
Such dependence of rate on light intensity has been observed in several laboratories and in one case attributed to recombination processes. Our proposed mechanism allows for a square root dependence in a more general scheme.
Conclusions
We have proposed a mechanism that describes the photoresponse of a TiO2 slurry in a PEC slurry cell. This mechanism accounts for previously observed results on photodetoxification with TiO2 involving H2O2 where both increases and decreases in the rate of pollutant decomposition can be observed. We demonstrate that for pollutant decomposition by OH the pseudo-first order disappearance of pollutant will vary as vI° for high light intensity and be linear in light intensity at low intensity. Finally we note that Equations (3), (12), and (13) represent fundamental limitations on any system that relies on OH as the primary mode of hole transfer to the pollutant.
EFFECT OF LIGHT INTENSITY ON PHOTOCATALYTIC REACTION
Craig S. Turchi
Solar Energy Research Institute
Golden, Colorado
Introduction
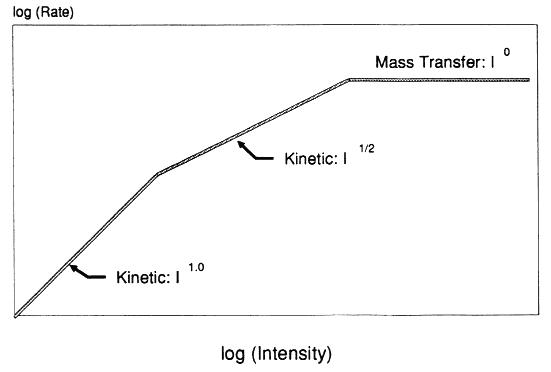
In the late 1970s photo-oxidation reactions using semiconductor photocatalysts such as titanium dioxide were identified as a novel means of completely oxidizing organic compounds to carbon dioxide. Throughout the 1980s, several research groups around the world explored the potential of this technology by photocatalytically destroying organic water pollutants in laboratory reactors. Recently, some of these research groups and U.S. Department of Energy laboratories demonstrated that sunlight can be used to drive the photocatalytic reaction. As more research effort has focused on the area of photocatalysis, it has become more important to determine the fundamental behavior of the photocatalytic reactions in order to promote economic analysis and eventual commercialization of this technology. One such parameter is the effect of ultraviolet light intensity on the observed rate of pollutant destruction.
The desired illumination intensity has a profound effect on the design of the photoreactor, particularly for solar receivers. In general, the most expensive component in a photoreactor is the light-producing or light-collecting equipment. The intensity dependence affects how diffuse or concentrated one wishes the radiation to be. If reaction rate increases with light intensity to some power greater than unity, a concentrated light beam is advantageous. Conversely, if the intensity dependence is less than I1.0, one quickly hits a point of diminishing returns by concentrating the illumination. Defining where this point occurs depends on catalyst, concentrator, and photon source costs as well as the reaction kinetics. Knowledge of the reaction rate dependence on intensity is essential for both the comparison and prediction of photocatalytic results and the design of efficient solar photoreactors. A related question of particular importance when comparing solar to UV-lamp systems concerns the relative effectiveness of different wavelength (but shorter than bandgap) photons for photocatalyst excitation. The objective of this paper is to summarize data concerning the effect of light intensity on the rate of photocatalytic degradation of organic water contaminants.
Theory
In the early 1970s Volz[1] and coworkers described the photocatalytic oxidation cycle responsible for the breakdown of TiO2-pigmented paint films. This hydroxyl radical mechanism is not widely thought to be responsible for the TiO2-photocatalyzed degradation of organic pollutants. Egerton and King[2] later examined the rate of oxidation of 2-propanol over rutile TiO2 at different illumination intensities. They proposed that oxidation of propanol was directly proportional to the concentration of positively charged holes formed by absorption of ultraviolet radiation. Egerton and King assumed the fate of photo-generated holes was either recombination or reaction with an organic molecule.
By using the steady-state approximation for hole concentration, d[h+]/dt = 0, and the assumption that electron and hole concentrations are equal, they were
able to show that reaction rate should be directly proportional to intensity at low fluxes and proportional to the square root of intensity at relatively high flux levels; that is, at high fluxes, [h+] is large, leading to
while at low fluxes organic oxidation can compete with recombination and
Because oxidation rate was assumed to be proportional to [h+], it followed that the dependence of reaction rate on light intensity should undergo transition from unity to 1/2 power as intensity increases. A detailed version of this derivation was later given by Turchi and Ollis[3]. Similar conclusions were made by Peterson et al.[4] using a mechanism based on hydroxyl radical recombination rather than electron-hole recombination.
Experimental Data
In general, available data concerning photocatalytic reaction as a function of light intensity support the theory of low-flux direct proportionality and high-flux square root dependence, yielding the behavior depicted in Figure 1. Unfortunately, because of experimental constraints, many reports cover only narrow intensity ranges, making it difficult to identify the different kinetic regimes. Egerton and King[2] displayed data covering almost six orders of magnitude in intensity. Their data showed a clear transition from I1.0 to I1/2 dependence for isopropanol oxidation over pure rutile TiO2. Data involving commercial rutile paint pigments were less definitive.
Further evidence of the transition in light intensity dependence was provided by Okamoto et al.[5] for the photocatalyzed degradation of phenol over anatase TiO2. For the particular catalyst and configuration used by Okamoto, the transition to square root behavior occurred at intensities slightly below the intensity corresponding to the ultraviolet intensity available in normal sunlight. Recent work by D'oliveira et al.[6] depicts the photocatalytic destruction rate of 3-chlorophenol undergoing a transition from linear to square root dependence on intensity over the range of 1–10 UV suns (where a UV sun is defined as 4.1 × 1019 photons (< 385 nm)/m2.s). This important result implies that concentrating solar reactors may well operate in the regime where reaction rate varies with the square root of illumination intensity.
Few other studies involving examination of light intensity have investigated a sufficient range of flux levels to witness a clear transition in flux dependence. However, the results have been consistent with the abovementioned references. For example, results at the Solar Energy Research Institute found an intensity exponent of 0.8 for trichloroethylene (TCE) degradation at intensities of 0.7–3.2 UV suns, an exponent of 0.6 for salicylate destruction at approximately 10 UV suns, and an exponent of 0.5 for formate destruction at 13–180 UV suns. These studies were carried out in different photoreactors over Degussa P25 brand titanium dioxide. Finally, work by Bahnemann et al.[7] showed TCE destruction rate to vary nonlinearly with intensity, although the absolute flux level was not given. In contrast, Peral et al.[8] reported a linear dependence on light intensity for the reaction of phenol over illuminated ZnO. Although the illumination area was not given, it seems likely from their reported reactor volume and measurement
of 2.4 × 1017 photon/s absorbed than the intensity was greater than one UV sun.
Lastly, solar experiments at Sandia National Laboratories monitored the destruction rate of TCE at different times of day (effectively different light intensities). While the data appeared to favor a direct proportionality between rate and estimated solar flux, the exact functionality was difficult to distinguish.
Conclusions
How reaction rate varies with light intensity has a major effect both on photoreactor design and on attempts to compare and model different reactor systems. Additionally, to determine the usable photon flux and to compare solar systems with UV lamps, an action spectrum is required for the photocatalyst. Consequently, economic analysis of photocatalytic reactor systems depends on knowledge of the kinetic dependence on wavelength and flux of photons. Both experimental data and theory based on photoexcitation of semiconductors suggest that the photocatalytic reaction rate will vary directly with light intensity at low fluxes and with the square root of intensity at high fluxes. Although the region of this transition is likely to vary with photocatalyst and other system properties, laboratory data imply that this transition occurs near or below the equivalent of one sun of ultraviolet flux. Therefore, although other considerations enter into a final system design (e.g., catalyst cost, mass transfer constraints, receiver/collector cost), there is an inherent kinetic advantage in using low or nonconcentrating solar reactors.
References
1. Volz, H.G., G. Kaempf, H.G. Fitzky, and A. Klaeren. 1981. ACS Sym. Series 151:163.
2. Egerton, T.A., and C.J. King. 1979. J. Oil Col. Chem. Assoc. 62:386.
3. Turchi, C.S., and D.F. Ollis. 1990. J. Catal, 122:178.
4. Peterson, M.W., J.A. Turner, and A.J. Nozik. 1990. J. Phys. Chem. (in press).
5. Okamoto, K., Y. Yamamoto, H. Tanaka, and A. Itaya. 1985. Bull. Chem. Soc. Japan, 58:2023.
6. D'Oliveira, J.-C. , G. Al-Sayyed, and P. Pichat. 1990. Environ. Sci. Technol. 24:990.
7. Bahnemann et al. 1990. Environ. Sci. Technol. (in press).
8. Peral, J., J. Casado, and J. Domenech. 1988. J Photochem. Photobiol., A; Chem. 44:209.
A COMPARISON OF ADVANCED OXIDATION PROCESSES WITH SEMICONDUCTOR-CATALYZED PHOTO-OXIDATION
Gary R. Peyton
Illinois State Water Survey
Champaign, Illinois
Introduction
Increasing awareness of the possible health effects from low levels of organic contaminants in water has prompted a resurgence of interest in the oxidation of organics in aqueous solution. Ozonation of drinking water is seeing increased usage in the United States, primarily for disinfection. Although ozone is usually thought of as a strong oxidant, it is actually quite selective in its reactions, as shown by comparison of the second-order rate constants for the reaction of ozone with acetic acid (k < 3 × 10-5 M-1 s-1) and phenol (k = 1.4 × 109 for phenoxide). In the mid-1970s, Hoigne and Bader showed that part of the oxidizing power of ozone in aqueous solution is due to the formation of hydroxyl radical as a result of ozone decomposition. Second-order reaction rate constants in the 107–1010 M-1 s-1 range indicate that hydroxyl is a powerful, indiscriminate oxidant. The Advanced Oxidation Processes (AOPs) represent an attempt to take advantage of this reactivity. The AOPs are defined as treatment processes that rely on the generation of free radicals to destroy organic contaminants.
Semiconductor-catalyzed photo-oxidation (SCPO) was introduced in the late 1970s by Bard. This class of processes has also been demonstrated to be effective on a wide range of organic contaminants, leading to an ongoing debate as to whether hydroxyl radical is responsible for organic removal in these systems.
Mechanisms and Application of AOPS and SCPO
In general, the use of AOPs involves three steps: (1) the addition of a reagent, (2) application of an initiator, which results in (3) the generation of free radicals. Oxidative free-radical treatment processes can be classified as those that require continuous initiation (CI) or those in which a propagation (P) step takes over as the primary producer of the active radical. The most common AOPs are H2O2/UV (CI), ozone/H2O2 (P/CI), and ozone/UV (P/CI). The latter two AOPs operate as propagation processes when, upon attack by OH, the species present in solution can form radicals capable of reducing dioxygen to superoxide, which then reacts with ozone to produce hydroxyl radical. Otherwise, they also act as CI processes. The SCPO apparently acts as a CI process, with the absorption of light serving as the initiation step.
The reagent in H2O2/UV treatment is hydrogen peroxide. Ultraviolet light is the initiator. The reagent in O3/H2O2, and O3/UV is ozone, while H2O2 or UV light is the initiator. Although there is no reagent per se in SCPO, oxygen or some other electron acceptor is required. The major component of the treatment cost for AOPs is usually associated with the reagent, although the initiator also represents a significant cost component. The primary reason that SCPO appears so promising is the elimination of these two cost factors. However, capital costs associated with catalyst, reactor, and collector system remain.
The majority of reaction in the AOPs appears to take place in the bulk solution, while for SCPO there is accumulating evidence that diffusion and perhaps adsorption of the organic to the semiconductor surface are required. This represents a fundamental difference in the application and advantages of
the AOPs and SCPO, which carries implications connected to whether complete mineralization or simply loss of identity' of the organic compound is the treatment goal.
The SCPO processes have limitations that do not apply to the AOPs. It is important to identify these limitations. However, because of the potentially favorable economics and low energy requirements (other than solar) of SCPO processes, they remain very attractive.
Recommended Research Objectives for SCPO
-
Identify active species responsible for the destruction of organic contaminants, for the particular semiconductor catalysts that are potentially practical. This is intimately related to item 2.
-
Determine whether diffusion and adsorption to the semiconductor surface are required for destruction of the contaminant: i.e., is a radical produced which diffuses into the bulk liquid, or is it a surface state? This determines the kinetics (including competition) that will be obeyed. The nature of the reactive species is strongly related to questions such as mineralization vs. loss of identity of the contaminant; interference by scavengers such as bicarbonate, nontarget organic compounds, and natural organic material (NOM); and desirable hydrodynamics of the reactor. For example, oxidation products (in the presence of oxygen) are more polar and less hydrophobic than parent compound, and may therefore be less strongly bound to the surface, resulting in increased selectivity for parent compound.
-
Define limits of water treatment imposed by the use of solar photons, in terms of milligrams per liter of contaminant removed from a Q gallon/minute stream using a collector of area A with semiconductor of bandgap Eg with charge separation efficiency es and a solar intensity of I einsteins/m2 in different regions of the country. Also define limitations arising from reactor hydrodynamics, necessity of target compound adsorption, and completeness of mineralization required.
-
Investigate means of immobilization of the semiconductor. Early studies identified recovery of powdered semiconductor as a major cost factor; this is intimately related to reactor hydrodynamics, manufacturing processes, and, to item 5 below.
-
Investigate benefits to be gained from the application of a potential to the semiconductor, to manipulate band bending (charge separation) and physically separate oxidation and reduction steps. This may imply immobilization or growth of the semiconductor on a metal substrate.
-
Determine long-term effectiveness of SCPO with respect to: (a) loss of intrinsic catalytic activity, (b) deposition of metal oxides and organic polymers/NOM on the semiconductor surface, and (c) cleaning/regeneration of surface, if necessary.
-
Search for other semiconductors with good long-term stability and a lower bandgap energy than titanium dioxide.
ULTROX OPERATING EXPERIENCES WITH UV/OXIDATION; ECONOMICS
Jack D. Zeff
Ultrox International
Santa Ana, California
Introduction
Ultraviolet oxidation technology has had limited exposure to the various engineering disciplines despite having been commercialized almost seven years ago by Westgate Research, the predecessor company to Ultrox International. The application of this technology is steadily expanding, however, as it offers a means of solving many of the problems created by the toxic water soluble organic chemicals that are found today in groundwater, wastewaters, leachate, and drinking water supplies.
More conventional or better-known unit processes and operations such as liquid/solids separation, reverse osmosis, air stripping, biotreatment, or granular activated carbon can remove many of the toxic organics encountered today. However, these methods may eliminate the one problem only to create a waste other media. Air stripping removes VOCs from water only to discharge them into the ambient air. Reverse osmosis generates a reject stream of concentrated contaminants that must be dealt with; granular activated carbon requires either regeneration or burial, and L/S separation obviously creates sludges requiring disposal.
Therefore, it is significant that UV-oxidation, when used in tandem with some of the above-mentioned processes, or as a stand-alone treatment process, can effectively destroy or render nontoxic many of the organic chemicals found on the priority pollutant list.
Chemical oxidation without ultraviolet enhancement has been used in the treatment of waters contaminated by organic chemicals for a number of years. Potassium permanganate, chlorine, and chlorine dioxide also have been used for treating organics such as phenol and its homologs in wastewaters. Hydrogen peroxide with a catalyst such as ferrous sulfate (Fenton's reagent) has been Used for oxidizing phenol and other benzene derivatives.
There has been a need for more powerful oxidizing methods which do not produce hazardous by-products. This paper describes the experience of Ultrox International in developing and applying enhanced ultraviolet light oxidation of organic chemicals in wastewaters, drinking waters, leachates, and groundwaters. The oxidants used in these applications are ozone and hydrogen peroxide. Ultrox International was issued a process patent in 1988 covering the application of UV light, ozone, and hydrogen peroxide to a broad range of organic compounds.
Description of the UV-Oxidation Process
The design of the equipment is based on the Use of components which (1) contain very few moving parts, (2) have low pressure operation, (3) require minimum maintenance, (4) can operate intermittently in either a continuous flow or a batch mode, (5) utilize high efficiency UV lamps with a long life, and (6) use a microprocessor to control and automate the process.
The ULTROX® UV-oxidation system consists of a UV-oxidation reactor and an oxidation source — an ozone generator with an air preparation system and/or a hydrogen peroxide feed system. The reactor is fabricated from stainless steel. The UV lamps are enclosed within quartz tubes for easy replacement and are mounted vertically within the reactor. Depending upon the size of the reactor and the type of water to be treated, the reactor can have four to eight stages. Lamps are installed either in all stages or in
designated stages, depending upon the type of treatment specified. When ozone is used as the oxidant, it is introduced at the base of the stage. The ozone is dispersed through porous stainless steel diffusers. The number of diffusers needed will depend upon the type of organics being oxidized and the degree of removal required. Ozone is produced from either compressed air, dried to a-60ºF (-51ºC) dewpoint by desiccant columns, or produced from cryogenic oxygen. Up to 2% wt. ozone is generated from air, and up to 5% wt. ozone can be produced economically from oxygen.
If hydrogen peroxide is substituted for ozone, it is directly metered into the influent line to the reactor.
Within the reactor, the water flows from stage to stage in a sinusoidal path using gravity flow. When the reactor uses ozone, the residual ozone in the off-gas is decomposed back to oxygen by the use of a fixed-bed catalytic unit operating at 150ºF (66ºC). The off-gas is then vented to the atmosphere with less than 0.1 ppmw O3 (OSHA Standards).
Application of UV-Oxidation to Various Waters
Full-scale, permanent on-site UV-oxidation installation projects treat industrial groundwaters, wastewaters, or process waters. Contaminants in these waters include phenols, chlorinated solvents, hydrazine, dimethylnitrosamine, tetrohydrofuran, cyanides, and formaldehyde. Commercial systems have been designed, built, and installed to treat flows varying from 1200 gallons to 1.4 million gallons per day.
Standard equipment designs are used in all of these installations. Reactor size varies from 300 gallons to 5000 gallons. Ozone generators range from 21 to 200 pounds per day. In several cases, hydrogen peroxide is used in place of, or with, ozone.
Full-scale systems, in most cases, are automated using microprocessor control. The system usually requires periodic monitoring (once per shift or once per day). The systems are designed to operate in a batch or a continuous mode depending upon treatment requirements.
Oxidation Treatment and Operating Costs
Table 1 presents the actual costs of treating by UV-oxidation wastewater and groundwaters at various permanent industrial installations. Some of these costs are in the cents per thousand gallon range and others in the cents per gallon range. In the case of the hydrazines, a small volume of water is treated per day on a batch basis and a comparatively long reaction time is needed. Thus UV-oxidation was found to be the most cost-effective method of destroying the three types of hydrazines and the nitrosamine which is formed as a by-product by the oxidation. The UV-oxidation systems replaced a chlorination unit, which produced chlorinated organic by-products.
Table 1 Direct Operating and Maintenance Costs for UV-Oxidation at Industrial Installations
|
Type of Water |
Contaminants |
Contaminant Concentration |
Discharge to |
Volume Treated per Day |
Direct O&M Cost Range |
|
Wood treating wastewater |
Pentachlorophenol and phenol |
150 ppm |
POTW |
30,000 |
$1.25–$1.35 per 1000 gal |
|
Wood treating groundwater |
Pentachlorophenol and phenol |
5 ppm |
POTW |
86,400 |
$0.90–$1.00 per 1000 gal |
|
Fume scrubber water |
Hydrazine, monomethylhydrazine unsymmetrical dimethlhydrazine |
5 ppm |
Biotreatment plant on-site |
600–1500 |
$0.086 per gal |
|
Contaminated groundwater |
TCE, trans DCE, MeCl2 |
5 ppm |
Surface water |
300,000 |
$0.47 per 1000 gal |
|
Contaminated groundwater |
TCE, TCA, DCA, PCE, MeCl2, ViCl |
600 ppb |
POTW |
72,00 |
$0.33 PER 1000 gal |
|
Contaminated Groundwater |
THF |
1 ppm |
Ground |
216,000 |
$0.39 per 1000 gal |
|
Wastewater |
Phenol |
90 ppm |
POTW |
4,300 |
$6.48 per 1000 gal |
SOLAR PHOTOCATALYZED PROCESS ECONOMICS
Hal Link Solar
Energy Research Institute
Golden, Colorado
This presentation describes the Solar Energy Research Institute (SERI) cost estimates for solar water detoxification systems based on the best available information as of October 1990. Comparative costs are also provided for competitive conventional technologies which are presently applied in the water detoxification market. Although costs for solar photocatalytic systems are presently higher than those of competitive technologies, cost and performance improvements should lead to cost competitiveness by 1995. This presentation is based, in a large part, on a paper by Link and Turchi that has been submitted for the ASME/JSME International Solar Energy Conference, Reno, Nevada in March 1991. That paper contains a more complete description and supporting references of the issues discussed here.
Definitions of a Representative Plant
The first step in defining costs for solar photocatalytic processes is to identify a representative plant. This plant must be configured with an awareness of the appropriate markets likely for early applications of solar catalytic systems. Through literature searches and government and industry contacts, SERI has identified three potential markets: groundwater remediation, industrial wastewater treatment, and drinking water purification. While the nation is presently supporting efforts to reduce pollution, even the Environmental Protection Agency has difficulty in estimating the potential economic impact of its new regulations. Our preliminary estimates are that compliance with new regulations could result in costs exceeding $20 million per year in each of the three markets we have identified.
Of these three markets, groundwater remediation appears to most closely match the major attributes of solar detoxification systems. The primary attributes that match near-term characteristics of solar systems are: small water flow rates, low contaminant concentrations, and relative insensitivity to intermittent operation. In addition, many of these treatment systems are expected to remain operational for 30–40 years—providing a long period to amortize the relatively high capital cost of solar systems.
Recent communications among U.S. Department of Energy laboratories have resulted in the identification of a potential site for the first solar water detoxification field test. This site is at Lawrence Livermore National Laboratory in California, where groundwater is contaminated with a variety of volatile organic compounds. The primary contaminant is approximately 400 ppb of the solvent trichloroethylene (TCE). Also present are small quantities of tetrachloroethylene, dichloroethylene, dichloroethane, and chloroform. A number of wells and treatment facilities are planned for the site. We have selected one of these treatment facilities for detailed cost comparisons.
The Livermore staff had previously estimated costs for a variety of treatment technologies for this facility as part of the feasibility study. We have built upon these existing data by estimating the potential performance and cost of a solar detoxification system designed to the same specifications. The system was designed to purify 100,000 gallons of groundwater per day.
The solar detoxification system is relatively simple. Groundwater is pumped sequentially through a filter and aerator before being introduced to the photoreactor located at the focus of a trough-type solar concentrator. The TCE is converted to water, carbon dioxide, and dilute hydrochloric acid.
Effluent is discharged to a trench from which it can reenter the underground aquifer.
Projected Costs for Solar and Competitive Technologies
Costs for the solar detoxification system are largely driven by the size of the solar concentrator array. A computer program uses kinetic data obtained from recent tests conducted at SERI and Sandia National Laboratories in solar troughs similar to those likely to be used in the representative system. The computer model projects an hour-by-hour flow rate that is a function of sun position and trough characteristics. Average daily performance is calculated based on the quantity of water treated during six representative days of the year. This average is adjusted to account for cloudy periods by using insolation data for Livermore, California. The analyst adjusts the concentrator array size to obtain the desired average daily throughput.
The cost of the concentrators was estimated using a factor of $200 per square meter of concentrator aperture area. This cost figure represents present costs for solar troughs used in thermal applications. Costs were also estimated for the photoreactors, piping, instrumentation, and other equipment. Indirect costs were estimated as a percentage of the major equipment cost using the same methodology used by the Livermore staff to estimate competitive system costs. Total capital Costs were amortized over a 20 year period using a fixed charge rate of 13.4% in accordance with the Environmental Protection Agency's analysis of conventional water treatment systems. Operations and maintenance costs were added to the amortized capital costs to obtain a total annual cost. This amount was divided by the annual quantity of water treated to obtain a levelized treatment cost in terms of dollars per thousand gallons treated.
Technologies that Livermore staff identified as providing the desired treatment capabilities at the lowest cost are granular activated carbon and lamp-driven advanced oxidation. The lamp system features a commercial unit that combines ultraviolet lamps and hydrogen peroxide to oxidize the contaminants onsite. The carbon System requires offsite regeneration of the carbon, which is included in the costs shown here.
The comparison of system costs indicate that the solar detoxification process would cost almost five times as much as the lowest cost system, the lamp system. This result is not unexpected since research and development of solar detoxification systems has so far focused on characterizing the major features of the system. Research is only now beginning to reveal how to achieve the improvements in cost and performance needed for economic parity.
Path to Cost Competitiveness
Having characterized our present knowledge of solar detoxification systems, we have identified four areas in which significant improvements should lead to competitively priced systems: lower-cost solar collectors, optimized process variables, improved catalysts, and use of a larger fraction of the solar resource. Solar collectors needed for photocatalytic processes differ from those developed for solar thermal applications. Low light concentration ratios suitable for these photochemical processes permit use of low-cost concentrator options that would be unsuitable for thermal systems. Also under consideration are nonconcentrating solar collectors. Although the design of efficient, low-cost nonconcentrating collectors may be significantly more difficult than for concentrating collectors, such 'one-sun' systems have kinetic advantages and can use diffuse light as well as direct-beam radiation.
Process variables such as pH control or addition of oxygen or other oxidants offer the potential for improvements up to a factor of two over the present, unoptimized system. Recent studies at the University of Arizona have shown that ozone can significantly improve photocatalytic reaction rates.
The largest potential for improvement is in the performance of the catalyst itself. Recent laboratory tests have shown large increases in
photocatalytic activity due to heat treatment and doping of the catalyst with appropriate materials. The SERI researchers expect to show a threefold improvement over rates presently obtained with commercially available catalysts within the next several years. Some laboratory tests have shown performance improvements as large as 60-fold.
Finally, catalysts have been identified that can potentially use lower-energy photons. Such catalysts would use a larger portion of the solar spectrum and improve system performance dramatically. For example, the rutile form of titanium dixoide is one such material that would increase the quantity of useful solar photons by a factor of 2.5.
Such improvements should result in cost competitiveness in some markets as early as 1992. Subsequent additional improvements could result in competitiveness over a broader range of applications and in areas of lower insolation by mid-to late-1990s.
Conclusions
This discussion has provided a brief overview of work which has led to the identification of likely niche markets for early application of solar photocatalytic processes. Although estimates of solar systems using present technology are higher than those of competing technologies, system improvements have been identified that should lead to cost competitiveness within a few years.
PHOTOCATALYZED REMOVAL OF MULTIPLE CONTAMINANTS FROM WATER
Hussain Al-Ekabi
Nulite
London, Ontario, Canada
The increasing level of organic pollutants in the environment has sparked public concern, so much so that immediate and innovative action is needed to address this problem.
The principal pollution treatment technologies now in use are carbon adsorption, air stripping, and bio-oxidation. These are effective in removing many organic pollutants from water streams, but they exhibit a major drawback. Both carbon adsorption and air stripping merely transfer the organic pollutants from one medium to the other. Biological treatment is relatively slow and inefficient in removing many man-made chlorinated hydrocarbons. Thus, an efficient technology is required which will not only remove but destroy the organic pollutants.
The TiO2 advanced photo-oxidation process has just recently begun to show considerable promise in removing organic pollutants from water streams. However, one of the major practical problems when using semiconductors as photocatalysts is the ''hole-electron'' recombination process. This process is extremely efficient and thus represents a major energy wasting step. For instance, the quantum yield for TiO2 photocatalytic degradation of some organic compounds has been reported to be <5%. As a result, there exists a challenging opportunity to improve the degradation rate of organic pollutants by preventing the "hole-electron" recombination process. This is an important consideration prior to commercial application of the TiO2 technology for water treatment.
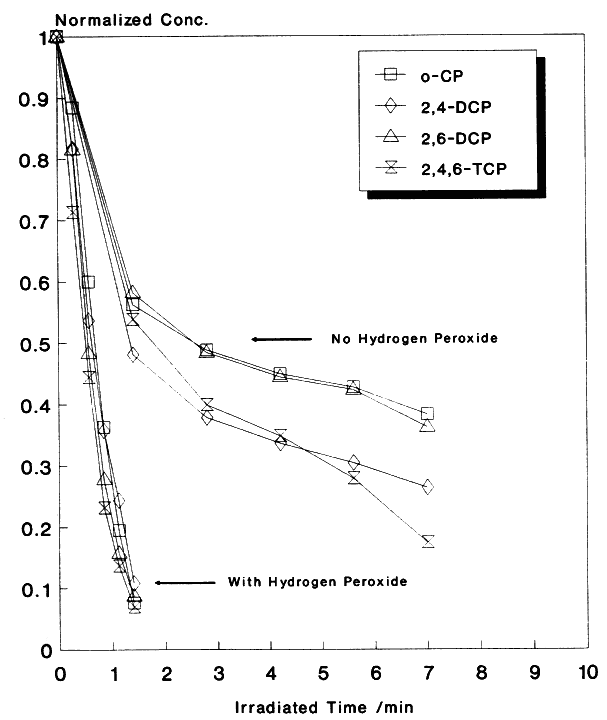
One strategy for inhibiting the "hole-electron" recombination process is to add efficient (preferably irreversible) electron acceptors to the reaction. These additives should fulfill the following criteria in order to efficiently inhibit the recombination process: (a) readily accept excess conduction band electrons or electrons from the superoxide ions (O2.- ), (b) then rapidly dissociate into harmless products, and (c) provide additional routes for the formation of OH radicals or other powerful oxidizing agents. So far we have examined four additives: oxygen, hydrogen peroxide, ozone, and persulfate and have found the results very promising. For instance, the effect of H2O2 on the degradation of a mixture of four chlorinated phenols (10 ppm each) is shown in Figure 1. Clearly, H2O2 markedly increases the degradation fate of all the components involved in this mixture. The degradation rate of a mixture of organic pollutants containing toluene, chlorobenzene, ethylbenzene, nitrobenzene, p-xylene, and o-cresol (10 ppm each) has also markedly increased when H2O2 was added to the solution prior to the irradiation. Similar results were obtained with other additives. Various experimental parameters were investigated and results will be presented.
IMMOBILIZED CATALYSTS IN SOLAR CONCENTRATORS
Mark S. Mehos
Solar Energy Research Institute Golden, Colorado
Introduction
The use of solar concentrating systems as an economically viable alternative for detoxifying contaminated groundwater will require an inexpensive means by which a titanium dioxide photocatalyst can be immobilized inside a photoreactor. Early system studies indicated that due to the cost and small size of the catalyst particles, suspended catalyst systems would be unable to compete with conventional technologies such as granular activated carbon or air stripping.
In the past, researchers investigating photocatalytic methods to destroy hazardous organics relied heavily on the use of slurry reactors. In these systems, good performance is achieved by permitting the catalyst to be carried along with the contaminated water, suspended by turbulent mixing throughout the reactor. Only recently have researchers attempted to design practical photoreactors in which the catalyst is immobilized.
The key characteristics of immobilized catalyst photoreactors include catalyst performance, catalyst lifetime, mass transfer effects, and pressure drop across a typical solar photoreactor. Additionally, achieving good performance requires the need to define the optimum distribution of absorbed radiation inside a reactor to achieve peak destruction efficiency. This distribution will be a function of both the photoreactor and the concentrator designs. These characteristics will play an important role in the final design of a commercial solar detoxification system. The objective of this paper is to discuss these characteristics and to present the results of recent experiments on immobilized catalysts in solar concentrators. Conclusions will be drawn regarding the applicability of these results to the design of immobilized catalyst support systems.
Desired Characteristics
The photoreactor containing the immobilized catalyst should have the following five characteristics. First, the reactors performance, as measured by the destruction rate of the targeted organics, should approach that of slurry systems. Slurry systems are expected to have excellent destruction characteristics because of the high surface area of the submicron catalyst particles. Second, practical system designs will require that the pressure drop associated with the immobilized catalyst be as small as possible. Third, for systems in which flow rate is determined by the UV intensity, the performance of the reactor should be independent of flow rate. This characteristic is especially important if the reactor pressure drop is large. In this case, the flow rates will need to be low to avoid excessive pressure drop across the system. Fourth, in order to be economical, the cost of the catalyst should not be prohibitively high. Finally, the immobilized catalyst should maintain a high activity if it is to remain economical in the long run.
Experiments
Initial laboratory tests of catalyst support materials (glass beads and porous alumina frits) yielded encouraging results. The pressure drops associated with these supports, however, were prohibitively high and they were not considered practical for outdoor testing. A commercial catalyst-support material developed by Nulite, Inc. of New London, Ontario, was considered for initial outdoor testing since it was available in large quantities and could be configured in a way that permitted low pressure drop through the photoreactor.
The apparatus used for outdoor tests consisted of a 1.5 inch diameter borosilicate glass photoreactor containing the immobilized catalyst. The reactor was located at the line of focus of a 16 ft × 2.5 ft parabolic trough concentrator, yielding a geometric concentration ratio of 13:1. Twenty-five liters of a dilute aqueous solution of trichlorethylene (TCE) was pumped through the photoreactor, reservoir, and heat exchanger. The heat exchanger was used so that the experiments could be run under isothermal conditions.
Experiments for a number of consecutive batch runs were performed using the apparatus described above. Initial experiments resulted in performance of the immobilized catalyst approaching that of suspended catalyst experiments performed earlier. Further experiments on separate batches of the Nulite material indicated a decrease in catalyst activity following the initial high rate.
Further experiments were performed on the Nulite material to investigate the effect of flow rate on performance. Results from these experiments show a dependence of the destruction rate on the flow rate through the reactor. This effect was observed for several bolts of material tested under different configurations.
Conclusion
Experiments have demonstrated that many of the desired characteristics of an immobilized catalyst, e.g., good performance, low pressure drop, and low cost, can be achieved using a commercially available, immobilized catalyst. Experiments have indicated, however, that the activity of the immobilized catalyst decreases following a high level of initial activity. Additionally, experiments have shown a dependence of the destruction efficiency on the flow rate through the photoreactor.
The design of a reactor containing an immobilized catalyst for use in the destruction of contaminated water requires that improvements be made. Most importantly, research needs to identify causes and prevention of the observed limitations in catalyst lifetime. If the lifetime of the catalyst cannot be sustained, methods must be determined for economically regenerating the catalyst, preferably without removing the catalyst from the photoreactor. Similarly, future reactor designs must reduce or eliminate the observed dependence of destruction on flow rate. These problems are currently being investigated by in-house researchers at the Solar Energy Research Institute and Sandia and will soon be investigated through industry/university subcontracts. Resolution of these issues will increase the likelihood of the competitiveness of solar detoxification systems.
PHOTOCATALYSIS WITH LARGE-SCALE TROUGH COLLECTORS*
James E. Pacheco
Sandia National Laboratories Albuquerque, New Mexico
Abstract
Sandia National Laboratories' part in the Department of Energy's Solar Detoxification of Hazardous Waste Initiative is focused on conducting large-scale solar photocatalytic experiments. Sandia's efforts support fundamental laboratory research performed by the Solar Energy Research Institute (SERI), which will lead to the first field experiment in about one year. In Sandia's facility, water spiked with a contaminant, flows through a glass pipe reactor centered at the focus of a parabolic trough. The ultraviolet portion of the concentrated sunlight activates the titanium dioxide to form oxidizers which have the ability to decompose many organic compounds.
Sandia's facility consists of six parabolic troughs in series. These troughs were originally installed to demonstrate the potential of using solar energy to produce industrial process heat or steam for electricity generation. We modified the system by removing the original black receiver pipes and installing borosilicate glass pipes in their place to serve as a photoreactor. The troughs have aluminized acrylic film that has a reflectivity of approximately 70% in the near ultraviolet (300–400 nm). These troughs concentrate ultraviolet sunlight about 35 times. The system is equipped with two 3800 liter (1000 gallon) tanks used for reservoirs of test solution and one 3800 liter tank for holding clean water, and can be operated in either recirculating or single pass modes at flow rates up to 100 liters per minute (26 gallons per minute). The total reactor length is 218 m (720 ft) and the total aperture area of the troughs is 465 M2. Sandia's work represents the first attempt to extend the process to a practical scale. The attached figure is a photograph of the trough system.
We have conducted numerous tests with trichloroethylene (TCE), trichloroethane (TCA), salicylic acid, and textile dyes. We varied the initial concentration of TCE from 100 to 5000 parts per billion (ppb) and found that the normalized data could be described by one curve with only two parameters. We have examined the effect of intensity on the destruction of TCE by measuring the destruction rate at different times in the day. We used these measurements and combined them with models of the variation of ultraviolet with air mass along with Typical Meteorological Year (TMY) solar radiation data for a number of sites to estimate processing rates for solar detoxification systems. We also did a series of experiments to measure reaction rate of TCA alone and in the presence of TCE at concentrations of 2000 ppb. The rate of destruction of TCA was slower than the rate for TCE, implying that the design and size of a system could be governed by the slower reacting component if its concentration is significant. We found at these concentrations that the presence of one compound did not interfere with destruction of the other component. The addition of hydrogen peroxide tended to enhance the reaction kinetics although the amount required to enhance the rate significantly may not be practical. We measured the effect of added bicarbonates on the destruction of TCE and found that it slowed down the
reaction rate, implying that actual groundwater containing high concentrations (hundreds of parts per million) of bicarbonate may require pretreatment or enhancements to the process.[1]
A number of issues must be addressed before this process is field tested. Destruction rates for other common groundwater pollutants and various combinations of them should be determined. The catalyst activity lifetime needs careful examination under conditions expected to be encountered in a real-life environment. A control system will be developed which will adjust the flow rate based on available ultraviolet and will ensure that the required amount of exposure is received. A crucial issue to be resolved is the development of an immobilized catalyst, eliminating the suspension and removal of the catalyst particles. Catalyst supports are being screened at SERI and the most promising support will be tested in Sandia's full-scale trough system. The goal of the development work is to field test and commercialize the process. The SERI is currently planning a field test of the solar detoxification process at a remediation or industrial site in the early 1990s.

Figure 1 Sandia's trough facility used for solar detoxification of water experiments.
Reprinted With Permission From: Sandia National Laboratories/Solar Thermal Collector Technology


































