
HOST DEFENSE AND IMMUNOMODULATION OF MUCOSAL CANDIDIASIS
Paul L. Fidel, Jr.,33,† and Mairi C. Noverr33,‡
Introduction
Candida albicans is a commensal organism of the oral cavity and gastrointestinal tract of most human adults as well as the genital tract of adult females. Point prevalence studies show asymptomatic colonization by C. albicans in the vagina of 15–25 percent of healthy women and in the mouth and gastrointestinal tracts of up to 60 and 90 percent of healthy adults, respectively (reviewed in Fidel, 2002). However, C. albicans can also be a pathogen of these same mucosal tissues and produce infections in both immunocompetent and immunocompromised conditions. Oropharyngeal candidiasis (OPC) encompasses infections of the hard and soft palate, tongue, buccal mucosa, and floor of the mouth, and can present as reddened patches (erythematous) or white curdlike lesions (pseudomembranous). Chewing and swallowing can be difficult and painful under these conditions. Infections can be acute or recurrent, and are common in immunocompromised patients, especially those infected with HIV. Candida-associated denture stomatitis (DS) is the most prevalent form of oral candidiasis, affecting up to 65 percent of otherwise healthy denture wearers (Budtz-Jorgensen et al., 1975; Daniluk et al., 2006). DS is characterized by inflamed oral mucosa, halitosis, and burning or bleeding of the infected mucosa (reviewed in Webb et al., 1998). Vulvovaginal candidiasis (VVC) affects a significant number of women predominantly in their reproductive years (Kent, 1991; Sobel, 1988, 1992). An estimated 75 percent of all women will experience at least one episode of acute VVC in their lifetime with another 5–10 percent developing recurrent VVC (RVVC) (Sobel, 1988, 1992). VVC involves infections of the vaginal lumen as well as the vulva. Symptoms include burning, itching, soreness, an abnormal discharge, and dyspareunia. Signs include vaginal and vulvar erythema and edema. Acute VVC has several known predisposing factors including antibiotic and oral contraceptive usage, hormone replacement therapy, pregnancy, and uncontrolled diabetes mellitus (Kent, 1991; Sobel, 1988, 1992). RVVC is multifactorial in etiology, but it is usually defined as idiopathic with no known predisposing factors in the majority of those affected (Sobel, 1988, 1992).
________________
33 Center of Excellence in Oral and Craniofacial Biology, Louisiana State University Health Sciences Center School of Dentistry, New Orleans, LA 70119.
† Email: pfidel@lsuhsc.edu
‡ Email: mnover@lsuhsc.edu
Prior to the HIV epidemic, host defense against C. albicans at these mucosal sites was largely considered one-dimensional. Although general anatomical distinctions were always understood in some capacity, the concept that “all mucosa are equal” was often implied for mucosal candidiasis whereby discussions of host defense at one mucosal site were generally applied to other mucosal sites as well. This was primarily due to the lack of large populations of individuals with mucosal candidiasis at any one geographical site that could be studied in-depth. OPC was relatively uncommon until transplantation and other forms of therapeutic or disease-associated immunosuppression (i.e., HIV/AIDS) became more evident. Yet numbers of patients to study at any one place remained quite small. VVC has always been common, but never taken very seriously as a mucosal fungal infection due to anecdotal causes and treatment regimens. Candida-associated DS that occurs in immunocompetent denture wearers was always known, but not well studied from an immunological or host-response perspective. This lack of research interest in DS was extended further as OPC became more prominent in HIV disease.
Prior to in-depth analyses of specific patient populations the majority of mucosal Candida infections were thought to be associated with some form of IgA antibody or humoral immune deficiency. However, antibody deficiency was difficult to demonstrate (Kirkpatrick et al., 1971; Mathur et al., 1977; Odds, 1988); therefore, it was the syndrome of chronic mucocutaneous candidiasis (CMC) that stimulated the next series of explanations. Because CMC was considered to be due to a deficiency in peripheral (blood) T cell–mediated immunity (Kirkpatrick, 1984; Kirkpatrick and Sohnle, 1981; Kirkpatrick et al., 1970, 1971), most other mucosal Candida infections were also considered to be caused by a similar T cell deficiency despite site specificity and individual prevalence.
The HIV epidemic created large numbers of individuals with severe immunosuppression and a significant increase in mucosal candidiasis. Accordingly, mucosal candidiasis became synonymous with OPC or esophageal candidiasis because they were being diagnosed most often. In fact, esophageal candidiasis is considered an AIDS-defining illness (Klein et al., 1984; Macher, 1988; Phelan et al., 1987). As much as VVC was considered common in HIV-infected women and a possible AIDS defining illness at one point (Burns et al., 1997; Duerr et al., 1997; Spinillo et al., 1994), it soon became clear that vaginal candidiasis, while common, was really no more common in HIV-positive women than in HIV-negative women (Clark et al., 1995; Imam et al., 1990; Leigh et al., 2001; Rhoads et al., 1987; Schuman et al., 1998b; White, 1996). This finding together with basic immunology research that was becoming of age for VVC (reviewed in Fidel and Sobel, 1996) began to suggest that factors associated with susceptibility to oral and vaginal candidiasis were different. Following some additional studies in HIV-infected persons with OPC, immunological data too began to follow the physiological anatomical distinctions and revealed that host defenses against Candida at the oral and vaginal mucosa were unique, distinct, and independent
(Leigh et al., 2001). As a result, it is now considered that all mucosa are not equal for mucosal Candida infections and that host defenses at the various mucosal sites need to be studied independently and exclusively at the local level with respect to cells and soluble immune factors.
The vast data accumulated over the past 15 years showing distinct T cell percentages or composition at different mucosal sites (e.g., vagina, gastrointestinal tract), especially in the intraepithelial layers, antigen presenting cells (e.g., dendritic cells, macrophages, Langerhans cells), or B cells (Fidel et al., 1996b; Hladik et al., 1999; Ibraghimov et al., 1995; Inghirami et al., 1990; Itohara et al., 1990; Johansson et al., 2000; Nandi and Allison, 1991) have wholly supported the concept that mucosal sites are indeed independent and unique relative to host immune reactivity. Thus, a clear divergence of immunity at mucosal sites has become evident. There is perhaps more dogma with reference to host defense against OPC (protection by CD4+ Th1-type responses), yet other nonconventional host defenses are also critically important. On the other hand, natural host defense against VVC does not appear to include T cells or any type of adaptive immunity (reviewed in Fidel Jr., 2007), but relies on innate immunity for both protection and susceptibility to infection (reviewed in Fidel and Noverr, 2012).
Host Defense Against Oropharyngeal Candidiasis
The majority of data suggest that CD4+ Th1-type cells are critical for host defense against OPC. Clinically, OPC is most common in HIV+ persons when CD4+ cell numbers drop below 200 cells/µl (Greenspan et al., 2000; Nielsen et al., 1994; Rabeneck et al., 1993; Schuman et al., 1998a). Within vitro immune analyses, peripheral blood mononuclear cells (PBMC) from most individuals, including HIV+ persons, respond to Candida antigens with Th1-type cytokines (Kunkl et al., 1998; Leigh et al., 2001). Together these results suggested that the Candida-specific T cells themselves were not becoming defective with immunosuppression, but that a threshold number of CD4+ T cells is required to protect the oral cavity against infection by this commensal organism. Below this threshold number of cells, other systemic and/or local immune mechanisms must function exclusively. Resistance and susceptibility to OPC then depends on the status of these alternative immune mechanisms.
Several aspects of local immunity have been evaluated clinically. In support of the Th1/Th2 dichotomy concept, it was reported that HIV- individuals had Th1/Th0 cytokines in their saliva, whereas HIV+ individuals had primarily Th2-type cytokines, which was more profound in those patients with OPC (Leigh et al., 1998). More recently, a distinct protective role for Th17 cells and associated cytokines has been identified for OPC (Conti et al., 2009). In fact, Th17-type responses may be more critical to normal protection than Th1-type responses. Yet like Th1-type responses, Th17-type responses, that are largely also CD4 T cell dependent, will be lost during HIV disease.
Lymphocytes have also been examined specifically in the OPC lesions. While both CD4+ and CD8+ cells have been identified (Romagnoli et al., 1997), we reported an accumulation of CD8+ T cells at a considerable distance from Candida that is located superficially at the outer epithelium (Myers et al., 2003), suggesting a role for CD8+ T cells against infection, but with a potential dysfunction in cell trafficking that promotes susceptibility to OPC. Increases in mRNA for several CD8 cell-associated cytokines (IL-2 and IL-15) and chemokines (IP-10, RANTES, and MCP-1) (Lilly et al., 2004) supported a role for reactivity by CD8 T cells. A murine AIDS model (MAIDS) also showed a rate of 30 percent recurrent OPC with a predominance of CD8+ T cells recruited into the oral tissues (Deslauriers et al., 1997). The tissue-associated CD8+ T cells primarily possess the αβ T cell receptor (TCR) (McNulty et al., 2005). The CD8 T cells present in the lesions are activated memory T cells as evidenced by cell surface activation markers (CD69, CD45R0) that are transitioning between central and effector memory status (CD27hi, CCR7Lo, CD62LLo) (Leigh et al., 2006). Hence, the CD8 T cells appear as normal activated memory cells recruited into the oral mucosa but are inhibited or challenged from migrating through the tissue otherwise.
The issue of cellular migration was addressed through the study of homing receptor/adhesion molecules and chemokine receptors. Chemokine receptor expression was similar in HIV+OPC+ persons and HIV+OPC- persons (Lilly et al., 2006). Cellular migration is controlled by chemokine receptors as well as cell-associated heterodimeric homing receptors (integrins) and reciprocal tissue-associated adhesion molecules. Some homing receptor/adhesion molecule interactions (α4β7/MAdCAM; α4β1/VCAM-1) govern migration of cells out of blood and into mucosal tissues, while others govern migration through mucosal tissues (αeβ7/E-Cadherin). The CD8+ T cells present in both OPC- and OPC+ tissue had positive integrin expression with varying combinations, but no discernible difference in OPC- versus OPC+ tissue. In contrast, MAdCAM expression was significantly increased in OPC+ tissue in support of the increased presence of T cells, while E-cadherin was significantly decreased in OPC+ tissue (McNulty et al., 2005). The increase in MAdCAM is consistent with the migration of CD8+ T cells into the oral mucosal tissue. Likewise, the reduced E-Cadherin provides an explanation for the inability of the CD8+ T cells to migrate to the outer epithelium and represents a putative dysfunction in those with OPC lending to the susceptibility to OPC.
Based on the cross-sectional data with the CD8 T cells and E-cadherin (reviewed in Fidel, 2006), an important next step was to conduct a longitudinal analysis of CD8+ T cells in the tissue of those HIV+ patients who have not acquired OPC, have had sporadic cases of OPC, or who have recurrent OPC. The main objective was to confirm the cross-sectional findings and determine whether the reduction in E-cadherin in those with OPC was permanent or transient. Oral swabs were taken every 2 weeks and biopsies every 2 months to track changes in the CD8+ T cells and adhesion molecule expression before, during, and after
OPC episodes. In total, the results showed that while OPC- patients with low oral fungal burden revealed an unremarkable presence of CD8+ T cells and normal E-cadherin expression, OPC- patients who had increased oral Candida colonization (indicative of a potential preclinical OPC condition), had higher numbers of CD8+ T cells throughout the tissue with normal E-cadherin expression. In patients with an acute episode of OPC where CD8+ T cells were accumulated at the epithelial-lamina propria interface together with reduced E-cadherin expression, evaluation of E-cadherin following successful antifungal treatment revealed a return to normal expression (Quimby et al., 2011). These results suggested that under conditions of CD4+ T cell deficiency, CD8+ T cells typically migrate to the site of a preclinical infection via normal expression of E-cadherin and that reduced E-cadherin expression in those with OPC is not permanent. The reduction in E-cadherin that we have observed in patients with OPC is consistent with two independent studies showing that Candida can cleave or degrade E-cadherin through secretory aspartyl proteases (SAPs) (Frank and Hostetter, 2007; Villar et al., 2007). Hence, the reduction in E-cadherin may be a virulence mechanism for Candida that when adherent to the epithelium, promotes subsequent invasion. We postulate that increases in Candida levels lead to increased SAP-mediated degradation of E-cadherin, which creates an environment more conducive to infection and invasion and the onset of OPC.
The metabolic state of the epithelial cells is important in understanding the interaction of Candida with E-cadherin, as is the cellular localization of the epithelial cells. Filler and coworkers have shown that an adhesin (Als3) on C. albicans hyphae can bind E-cadherin on metabolically active epithelial cells, which induces endocytosis (Phan et al., 2007). Thus, it appears that Candida can either degrade or use E-cadherin. We hypothesize that these two conditions can and do occur depending on the location of the epithelial cells. If Candida gains access to basal epithelial cells that are metabolically active it will use E-cadherin to gain intracellular access through endocytosis. But if Candida remains on the apical epithelium it will likely degrade E-cadherin to occupy and superficially attach to the outer epithelium rather than be released from the epithelium inside metabolically inactive sloughing cells.
In any event, it appears that Candida has the ability to modulate E-cadherin rather than it being modulated (transient or permanent) by a host-dependent mechanism. If so, immunotherapeutic strategies directed towards restoring normal E-cadherin expression would allow CD8+ T cells to migrate to the outer epithelium for optimal effector function. Current studies are focusing on the mechanism of action by CD8+ T cells. Interestingly, treatment with IFN-γ (Bodasing et al., 2002) or the presence of protease inhibitors (PIs) in antiretroviral therapies (ART) (Cassone et al., 2002) have been successful for reducing/eliminating OPC. Additionally, IFN-γ has recently been shown to inhibit the Candida-mediated degradation of E-cadherin in vitro (Rouabhia et al., 2012). Thus, it is possible that PI and IFN-γ functioned in part by directly (IFN-γ) or indirectly (PI via inhibition of
SAPs) allowing restoration of E-cadherin and normal migration of CD8 T cells for effector function.
In contrast to cell-mediated immunity by T cells, humoral immunity does not appear to play a role in protection against or susceptibility to OPC. There is no evidence to date that a deficiency in Candida-specific antibodies is present in HIV+ persons that could account for the increased prevalence of OPC (Millon et al., 2001; Wozniak et al., 2002; Wray et al., 1990).
Innate cellular defenses also play a role in host defense. Our laboratory has studied the role of epithelial cells for a number of years. These studies show that oral epithelial cells can inhibit up to 80 percent of the Candida growth in vitro by a fungistatic mechanism via cell contact by intact, but not necessarily live epithelial cells, through an acid-labile protein receptor (Nomanbhoy et al., 2002; Steele et al., 1999, 2000, 2001; Yano et al., 2005). Vaginal epithelial cells have an identical antifungal property, although weaker (~40 percent inhibition) compared to oral epithelial cells (Nomanbhoy et al., 2002; Steele et al., 2000). Analysis of oral epithelial cells in HIV+ persons showed significantly reduced activity by cells from patients with OPC compared to that from patients without OPC, providing support as an innate protective mechanism against infection (Steele et al., 2000). Additionally, epithelial cells produce both cytokines and chemokines in response to Candida, which may contribute to the innate and/or adaptive immune response (Dongari-Bagtzoglou and Kashleva, 2003a,b; Rouabhia et al., 2002; Schaller et al., 2002; Steele and Fidel, 2002).
In subsequent studies a primary objective was to identify the antifungal effector moiety on oral epithelial cells. Accordingly, the acid treatment (that abrogates the antifungal activity) was used as a tool to accomplish this objective. The strategy involved extracting surface associated glycoproteins from the epithelial cells treated with and without periodic acid (PA). The extracted epithelial cell surface proteins were evaluated for antifungal activity. Results demonstrated that proteins extracted from PBS-treated, but not PA treated cells, were capable of inhibiting the growth of Candida similar to intact epithelial cells. These data are consistent with the fact that live epithelial cells are not required despite the need for cell contact. For identification of the effector moiety, the extracted glycoproteins were incubated with Candida, and any bound epithelial proteins were then eluted from Candida and separated by SDS-PAGE. Because this method could potentially result in contamination of samples with Candida cell surface-associated proteins, we additionally labeled the epithelial cell surface proteins with biotin prior to the acid treatment and protein extraction. This allowed for the use of Western blots to focus only on epithelial cell proteins. Results revealed two protein bands (33 and 45 kDa) eluted from Candida blastoconidia or hyphae that are present in PBS-treated, but not periodic acid (PA)-treated epithelial cells. Proteomic analysis showed that the ~45kDa protein for the PBS-treated cells was an actin molecule, whereas the ~33 kDa protein was annexin-A1. Annexin-A1 is a viable candidate for the effector molecule as it functions through signaling
cascades to inhibit cellular processes including growth (Croxtall et al., 2003; Liu et al., 2007). Further studies showed that annexin-A1, but not actin (as expected) was present on the epithelial cell surface and immunoprecipitation of annexin-A1 from the extracted proteins abrogated the antifungal activity (Lilly et al., 2010). Thus, annexin-A1 is considered to be the primary effector moiety responsible for the epithelial cell antifungal activity. Current studies are focused on the mechanism of action.
The static antifungal activity by mucosal epithelial cells can be considered an interesting symbiotic relationship. The host benefits by the lack of inflammation in the presence of the yeast, or significant invasion of tissue. Commensalism is maintained via annexin-A1 activity that we speculate signals the yeast to shut down its growth. The benefit for Candida is that it sacrifices its growth for protection against other immune responses that might otherwise kill it. This is representative of a form of immune evasion where no “danger signals” are elicited by the presence of Candida.
Based on these data, we propose that that several secondary lines of defense are important for protection against OPC when the primary defense by CD4+ cells are below the protective threshold (summarized in Figure A9-1). These include CD8 T cells and oral epithelial cells. In HIV+ persons with <200 CD4 cells/µl and OPC-, it is postulated that although the CD4+ T cells are below the protective threshold, epithelial cells have activity against Candida to hold it in check, and CD8+ T cells are able to migrate to the outer epithelium to aid in protection. On the other hand, in those susceptible to OPC, the CD8+ T cells are inhibited from migrating to the outer epithelium because of reduced E-cadherin together with reduced levels of epithelial cell anti-Candida activity resulting in bouts of OPC. When the mechanisms surrounding E-cadherin degradation/restoration, CD8+ T cell activity, and annexin-A1 antifungal activity are fully understood, each could be exploited to reverse susceptibility and/or enhance resistance or protection against OPC.
Host Defense Against Vulvovaginal Candidiasis
For VVC/RVVC, studies from a mouse model of vaginal candidiasis and many clinical studies evaluating women with RVVC over two decades have revealed a general lack of protection by local or systemic adaptive immunity (reviewed in Fidel and Noverr, 2012). It is postulated that this diversion from dogma is the result of the commensal relationship evolving to avoid frequent inflammatory responses at a reproductive site. Instead, resistance and susceptibility to RVVC and VVC is now considered to involve innate immunity acting at the vaginal mucosa.
The general lack of a role for systemic and/or local adaptive immunity against vaginal candidiasis is considered to be due to multiple putative immunoregulatory mechanisms. These include significant constitutive concentrations of
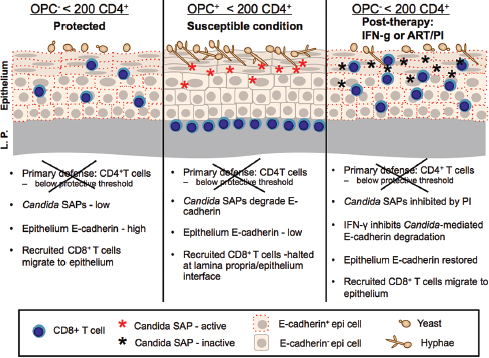
FIGURE A9-1 OPC: Protection, susceptibility, and results of treatment with ART/PI or IFN-g. Proposed immune function of the oral mucosa in HIV+ individuals with and without OPC and < 200 CD4 cells/ml. Left frame depicts the protective mechanisms associated with the HIV+ OPC– individual colonized with Candida albicans when the primary defense of CD4+ T cells is below the protective threshold (200 cells/μl). The secondary defense consists of CD8+ T cells migrating to the outer epithelium under normal expression of E-cadherin and functional oral epithelial cell anti-Candida activity that together keep Candida in check and prevent symptomatic infection. Th1 cytokines in saliva may also act in protection against infection, along with cytokines in tissue. Center frame depicts the condition of OPC. In this scenario, when CD4+ T cells are below the protective threshold, CD8+ T cells are inhibited from migrating to Candida for effector function due to a transient reduction in E-cadherin via Candida-associated SAPs, and the epithelial cells have reduced capacity to inhibit Candida, resulting in Candida overgrowth and symptomatic infection. Reduced Th1 cytokines in saliva may also contribute to the susceptibility. Right frame depicts potential treatment strategies that reduce/eliminate OPC via the restoration of E-cadherin followed by the normal migration of CD8+ T cells for effector function. This can be done directly by IFN-γ or indirectly by PI in ART that inhibits Candida-associated SAPs.
SOURCE: Yano, J., M. C. Noverr, and P. L. Fidel, Jr. 2012. Cytokines in the host response to Candida vaginitis: Identifying a role for non-classical immune mediators, S100 alarmins. Special issue: Cytokines in fungal immunity. Cytokine 58:118-128, Fig. 2, 124.
TGF-β (Taylor et al., 2000), resident γ/δ T cells (Wormley et al., 2001), reduced homing receptors on activated T cells in the draining lymph nodes to promote T cell entry (Wormley et al., 2001), lack of effector function by infiltrating polymorphonuclear neutrophils (PMNs) (Black et al., 1998; Fidel et al., 1999), presence of CD25+ Treg cells (Wormley et al., 2001), and a strong presence of plasmacytoid dentritic cells (LeBlanc et al., 2006).
In more recent years dramatic progress in our understanding of host susceptibility to VVC has been attributed to the development of a human live challenge system. In the live challenge study, resistant (no previous VVC episode) or susceptible (with infrequent VVC episodes) women were evaluated for the natural history of infection following intravaginal inoculation with live C. albicans (Fidel et al., 2004). Results revealed that protection, as evidenced by asymptomatic vaginal colonization with Candida, occurred in the absence of any inflammatory response consistent with the antifungal activity by epithelial cell annexin-A1 (Lilly et al., 2010). In contrast, symptomatic infection was accompanied by a heavy vaginal cellular infiltrate consisting of PMNs. In addition, PMN infiltration scores assessed from these women positively correlated with vaginal fungal burden although the hyphal (pathogenic) form of Candida could be present in both asymptomatic and symptomatic conditions (Fidel et al., 2004). Finally, when tested in an in vitro PMN migration assay, vaginal lavage fluid from women with symptomatic infection showed increased PMN chemotactic activity compared to lavage fluid from those with asymptomatic colonization, suggesting a chemotactic factor or factors (e.g., cytokines, chemokines) had been produced and secreted into the vaginal cavity in response to the Candida challenge (Fidel et al., 2004). Notably, evaluation of common proinflammatory cytokines and chemokines (G-CSF, TNF-α, IL-1, IL-6, IL-8, and IL-17) in lavage fluids from the study failed to identify candidates for the PMN chemotactic factors (reported in Yano et al., 2010). Moreover, Candida itself fails to directly induce PMN chemotaxis or through any organism-derived factors (Fidel, unpublished observations). Hence, organism virulence or virulence factors alone do not appear to be a major player in the PMN response. These important data from the live challenge study reshaped our knowledge of the immunopathogenesis of VVC/RVVC. Accordingly, based on the evidence showing the strong involvement of innate components of host immune responses during VVC, the paradigm has shifted into the current concept that both resistance and susceptibility to VVC are associated with innate immunity.
To explain the relationship between host susceptibility to VVC and the vaginal presence of Candida¸ the hypothesis was proposed that symptomatic infection is acquired when the number of Candida organisms achieves a threshold that varies between women depending on the sensitivity of vaginal epithelial cells to Candida and may ultimately determine the clinical outcome (reviewed in Fidel, 2007). Based on the hypothesis, we postulate the following:
1. In women with RVVC, vaginal epithelial cells are extremely sensitive to Candida and respond by secreting a danger signal (rather than typical antifungal activity via annexin-A1) that promote PMN infiltration after exposure to low numbers of Candida. These women are susceptible to recurrence by small increases in organism numbers (i.e., shortly following disruption or cessation of maintenance antifungal therapy).
2. In women with infrequent history of VVC due to any of the known predisposing factors (e.g., oral contraceptives, hormone replacement therapy, antibiotic usage), vaginal epithelial cells are less sensitive to Candida and have a higher threshold for Candida. These women remain asymptomatic following exposure to moderate numbers of Candida (with typical antifungal activity by epithelial cell annexin-A1), but if the numbers rise following the use of antibiotics or estrogen, the threshold will be breached and a similar danger response will result in a symptomatic condition.
3. In women with no history of VVC, their vaginal epithelial cells are insensitive to even large numbers of Candida and hence the threshold is rarely breached. Rather, these women likely benefit greatly from the annexin-A1-mediated antifungal activity via epithelial cells for a continued commensal state of Candida. In any event, the PMN response rarely, if ever, occurs and these women remain asymptomatic. The results of the live challenge study clearly support this hypothesis. In women with no history of vaginitis, 90 percent remained asymptomatic following inoculation. In contrast, 55 percent of women with infrequent episodes of vaginal candidiasis due to oral contraceptives, hormone replacement therapy, or antibiotic usage became symptomatic following inoculation. Moreover, if those women were tested in their susceptible state (e.g., using oral contraceptives, hormone replacement therapy, or antibiotic usage), 90 percent became symptomatic (Fidel et al., 2004). Clinical cross-sectional and the live challenge natural history study also supports the protective effects of epithelial cell annexin-A1; women with RVVC or who were susceptible to symptomatic VVC following inoculation had lower epithelial cell antifungal activity compared to women without RVVC or who simply became asymptomatically colonized following inoculation (Barousse et al., 2001, 2005). At present, it is unknown what factors are involved in establishing the level of epithelial cell sensitivity to Candida, but is presumed to be a genetic predisposition. Indeed, some polymorphisms have been identified in women with RVVC (Babula et al., 2005; Ferwerda et al., 2009). On the other hand, differences in susceptibility to infection could not be demonstrated for various haplotypic strains of mice (Black et al., 1999; Calderon et al., 2003; Clemons et al., 2004; Yano et al., 2010).
In parallel with the clinical studies examining innate immune factors against VVC, the mouse model also showed an erratic presence of vaginal PMNs during
infection (Black et al., 1998; Fidel et al., 1999; Yano et al., 2010). Although PMNs were identified to be the most predominant leukocyte population in vaginal lavage fluid irrespective of time postinoculation, their presence never resulted in correlates to reduced vaginal fungal burden and thus was presumed to be due to the pseudo-estrus condition (Fidel and Sobel, 1999; Fidel et al., 2000). In fact, the presence of the PMNs was quite variable with 60–70 percent of inoculated mice having substantial PMNs in the vagina, while the remainder had little to no vaginal PMNs.
Consequently, a series of studies were conducted to formally classify the PMN response in various conditions. Indeed, similar to clinical observations, data showed that a heavy vaginal PMN presence occurred consistently in a high percentage of inoculated animals irrespective of the haplotype or the duration of infection without affecting fungal burden (Yano et al., 2010). Of note, previous studies testing effects of estrogen concentrations, inocula and strains of C. albicans also showed similar PMN infiltration patterns (Fidel and Sobel, 1999; Fidel et al., 1996a, 2000). In light of symptomatic/asymptomatic conditions observed in inoculated women from the live challenge study, the high PMN and low PMN responses in mice were classified to be simulating the symptomatic and asymptomatic conditions of VVC, respectively (Yano et al., 2010). Although previous attempts to quantify clinical signs and symptoms of vaginitis in mice (e.g., redness, swelling, irritation, scratching) were unsuccessful and therefore could not be used as correlates to the PMN infiltration, vaginal PMNs levels appear to be rigid criteria based on the association with the clinical symptomatology of VVC. Accordingly, when tested in an in vitro PMN migration assay, results showed, similar to clinical studies, that vaginal lavage fluids from mice with high PMNs (symptomatic) had increased PMN chemotactic activity compared to those from a low PMN (asymptomatic) condition, suggesting the presence of a similar chemotactic factor or factors secreted in response to Candida (Yano et al., 2010). Taking into account previous reports showing a lack of strong vaginal cytokine/chemokine responses in humans and mice in VVC, identification of the putative PMN chemotactic factors was crucial to further uncovering immune processes associated with susceptibility to symptomatic disease. The mouse model of VVC was then exploited to further dissect the innate mechanisms associated with the immunopathology of VVC.
Subsequent animal studies by Yano et al. incorporated proteomic approaches in search of the putative factors responsible for the inflammatory response during the symptomatic condition (Yano et al., 2010). First, vaginal lavage fluid from mice inoculated with C. albicans was evaluated for protein expression patterns. Based on the PMN-associated symptomatology criteria, two distinct proteins at 10 kDa and 14 kDa were identified by differential expression that positively correlated with the levels of vaginal PMNs. These proteins were further analyzed by mass spectrometry for protein identification and showed a significant match for S100A8 (10 kDa) and S100A9 (14 kDa) calcium-binding proteins. Confirming
the initial protein identification, detection by Western blots and ELISA showed high abundance of S100A8 and S100A9 in vaginal lavage fluid from inoculated mice with high PMNs, but results were extremely low in fluids from those with low PMNs. Consistently, evaluation of vaginal tissues by immunohistochemistry showed elevated S100 protein expression localized at the apical surface of vaginal epithelium of mice with high PMNs, while the expression was dramatically reduced in tissues from those with low PMNs. This observation was further supported by increased S100 expression in vaginal epithelial cells at the mRNA levels, confirming epithelial cells as a primary source of S100 proteins produced under the symptomatic condition. Finally, neutralization of S100A8, but not S100A9, reduced the PMN chemotactic activity of vaginal lavage fluid in an in vitro migration assay, suggesting a role for S100A8 in mediating PMN migration during vaginal infection. Hence, at least in the mouse model, S100A8 appears to be produced by vaginal epithelial cells following interaction with Candida, and may play a key role as a PMN chemotactic factor that initiate the inflammatory response during symptomatic VVC.
S100A8 and S100A9 are small molecular weight calcium- and zinc-binding proteins of the S100 family and are also known as calgranulin A, myeloid-related protein (MRP) 8, chemotactic protein (CP) 10, and calgranulin B, MRP-14, respectively. S100A8 and S100A9 are found as monomers or heterodimeric complex called calprotectin, which has been shown to elicit antimicrobial properties to various microbial pathogens including C. albicans (Sohnle et al., 1996; Urban et al., 2009; Zimmer et al., 1995). These proteins also serve as potent chemoattractants for PMNs and participate in inflammatory processes. Due to their involvement as endogenous danger-signaling chemoattractant molecules, S100A8 and S100A9 have been termed “alarmins”, a subgroup of endogenous damage-associated molecular patterns (DAMPs) eliciting host immune responses in similar mechanisms as exogenous pathogen-associated molecular patterns (PAMPs) (Ehrchen et al., 2009). S100 alarmins are produced by phagocytes, monocytes, epithelial cells, and endothelial cells and are released at sites of inflammation (Foell et al., 2008; Gebhardt et al., 2006; Kumar et al., 2001; Ross and Herzberg, 2001; Ryckman et al., 2003). In contrast to other mucosal sites where S100 alarmins are readily detected as calprotectin (Sohnle et al., 1996; Urban et al., 2009; Zimmer et al., 1995), data from the mouse study showed that S100A8 and S100A9 were mostly present as monomers in the vaginal secretions (Yano et al., 2010). In addition, vaginal S100 alarmins exhibited no antimicrobial effect on C. albicans as evidenced by the fact that vaginal fungal burden remained unaffected by the levels of S100 alarmins detected in the vagina. However, the magnitude of PMN infiltration positively correlated with the amount of vaginal S100 alarmins (Yano et al., 2010). Recognizing the dichotomous PMN migration in inoculated animals, together with the lack of a genetic disposition of inbred mice to explain the dichotomy, other explanations were sought. Accordingly, studies showed that the alarmin trigger is based on early adherence where higher
rates of adherence are presumed to be sensed as danger by the epithelial cells and result in the triggering of the alarmin response.
Although vaginal epithelial cells appear to be a primary source of S100 alarmins during experimental VVC, PMNs also likely contribute to the S100 production once recruited into the vaginal mucosa. A proposed hypothesis is that PMNs recruited in response to the initial epithelial cell-derived S100 alarmins may initiate a second wave of S100 production, amplifying the PMN response as part of positive feedback mechanism. Figure A9-2 illustrates the complete sequence of events considered to be critical in the immunopathogenesis of VVC/RVVC. Furthermore, although the Th17 response is involved in protection against, or susceptibility to, other forms of mucosal candidiasis (Conti et al., 2009; Eyerich et al., 2008; Huang et al., 2004; Urban et al., 2009), and is a strong contributor to S100 alarmin production (Liang et al., 2006), a recent study using IL-23p19-/-, IL-17RA-/-, and IL-22-/- mice found no role for the cells or cytokines of the Th17 lineage in the S100 alarmin response during VVC (Yano et al., 2012).
Taken together, the more complete understanding of the immunopathogenesis of VVC/RVVC has important implications for diagnostics and treatment. Current diagnostics for VVC are limited. The challenge has been that Candida is commensal and present in an asymptomatic state. Positive diagnosis requires the presence of symptoms and a positive yeast culture. However, current diagnostic tests are based on the presence of organism alone. Hence, an improved diagnostic could include testing for the presence of Candida as well as symptoms using alarmins as the key biomarker for the inflammatory event. Additionally, alarmins could be key targets for immunotherapeutic strategies. One can envision blocking or neutralizing the S100 alarmins to reduce or eliminate the symptoms of VVC/RVVC. The end result would be an asymptomatic condition with Candida relegated back to a commensal state. Current studies are focused on testing the immunopathogenesis/alarmin hypothesis in clinical VVC/RVVC to provide the necessary evidence to embark on these diagnostic and immunotherapeutic strategies.
The Emerging Role of Mucosal Candida Biofilms and Host Immunity
Recently there has been a tremendous interest in the role of biofilms in infectious diseases. It is estimated that 80 percent of human infections result from pathogenic biofilms (Costerton et al., 1999). Biofilms are communities of microorganisms that are embedded in extracellular matrix (ECM), forming a complex microbial community. A unique feature of C. albicans biofilms is the morphological heterogeneity of the biofilm cells, which results in a complex 3-dimensional biofilm architecture (Chandra et al., 2001). Biofilm formation is dependent on the ability to undergo morphogenesis; mutants defective in hyphal formation in vitro are also defective in biofilm formation (Nobile et al., 2006; Ramage et al., 2002). C. albicans biofilms have a unique gene expression pattern (Yeater et al.,
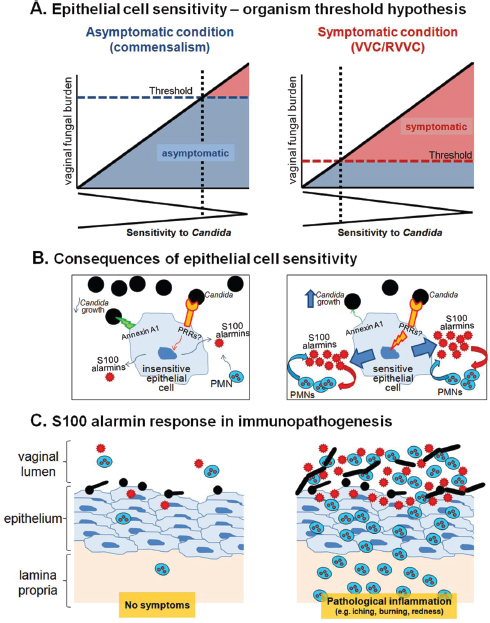
FIGURE A9-2 Schematic diagrams representing the mechanism for the effects of vaginal S100 alarmins on PMN migration during VVC. (A) Epithelial cell sensitivity–organism threshold hypothesis. In women with no history of VVC (left panel), their vaginal epithelial cells are insensitive to Candida. These women remain asymptomatic as even in the presence of high numbers for Candida (threshold number to initiate pathological response rarely breached). In women with RVVC (right panel), vaginal epithelial cells are extremely sensitive to Candida. These women are susceptible to symptomatic infection following exposure to even small numbers of Candida (low organism threshold). The thresholds represent an arbitrary organism number of the upper limit for vaginal fungal burden that
2007) and are more resistant to antifungal treatment than planktonic cells (Kojic and Darouiche, 2004; Ramage et al., 2002). There, in a clinical setting, biofilms represent a significant risk and are difficult to eradicate.
Candida biofilms have been studied primarily on abiotic surfaces (Blankenship and Mitchell, 2006; Ramage et al., 2006). A large proportion of C. albicans infections involve biofilms, which can form on a variety of synthetic polymers used in medical devices (Dominic et al., 2007; Douglas, 2003; Kojic and Darouiche, 2004). Significant attention has been given to Candida biofilm formation of indwelling catheters, which can lead to life-threatening systemic infections (Crump and Collignon, 2000; Dominic et al., 2007; Kojic and Darouiche, 2004). Candida albicans is the fourth leading cause of bloodstream infections and the third most commonly isolated organism from intravascular catheters and is associated with the highest incidence of mortality (Crump and Collignon, 2000; Wisplinghoff et al., 2004). Candida biofilm development on abiotic surfaces can be divided into several growth stages, including early, intermediate, and mature (Chandra et al., 2001). During early biofilm formation, yeast cells adhere to an appropriate surface and initiate germ tube formation. The intermediate phase is characterized by continued hyphal elongation and ECM production, which consists of cell wall polysaccharides and protein (Baillie and Douglas, 2000; Nett et al., 2007). Mature biofilms consist of a yeast base, with hyphal elements encased
would initiate symptomatic infection. (B) Consequences of epithelial cell sensitivity. Under asymptomatic condition (left panel), vaginal epithelial cells are insensitive to Candida and remain unstimulated following interaction with Candida. In turn, PMN migration does not occur in the absence of S100 alarmin production. Strong cell-surface annexin A1-dependent (proposed based on oral epithelial cells) antifungal activity provides noninflammatory means to maintain Candida at the commensal state. Under symptomatic conditions (right panel), vaginal epithelial cells are extremely sensitive to Candida and exert weak antifungal activity through annexin A1. Epithelial cells become activated upon recognition of Candida via unidentified PRRs. S100 alarmins are secreted as danger signals toward which vaginal PMNs migrate through vaginal epithelium. Once in the vaginal epithelium, recruited PMNs also produce S100 alarmins as part of positive feedback mechanism to further amplify the PMN response. (C) S100 alarmin response in immunopathogenesis. PMN infiltration remains minimal in the absence of S100 alarmin production by vaginal epithelial cells, therefore, no symptom occurs (left panel). In contrast, high concentrations of S100 alarmins in vaginal epithelium trigger PMN migration to the vaginal cavity, resulting in pathological inflammation associated with the symptoms of infection. The inflammatory process enhances Candida growth and hyphal formation (right panel).
SOURCE: Adapted from: Fidel, P. L., Jr. 2011. Candida-host interactions in HIV disease: Implications for oropharyngeal candidiasis. Proceedings from the 6th World Workshop on Oral HIV and AIDS. Adv Dent Res. 23:45-49, Fig. 1, p. 47.
in ECM extending away from the surface. In addition, persister cells are present, which are a multidrug tolerant subpopulation of the biofilm (LaFleur et al., 2006). Newly formed daughter yeast cells grow out of hyphal elements and are released (dispersal), seeding new niches for biofilm formation or infection.
Candida biofilm formation on biotic surfaces has not been investigated until recently. Candida resides at mucosal surfaces as a normal inhabitant of the microbiota. Generally, mucosal disease is associated with a shift towards hyphal growth during vaginitis, oral candidiasis, and invasive GI tract infections (Bilhan et al., 2009; Cantorna and Balish, 1990; Sobel et al., 1984). This provides some evidence that biofilm formation could occur on mucosal tissues and may be associated with disease. Alternatively, biofilm growth could represent a reservoir of chronic and/or persistent colonizing organisms that serve as a source of opportunistic infection. Several animal models of infection have recently been used/established to study Candida mucosal biofilm formation including OPC (Dongari-Bagtzoglou et al., 2009), vaginitis (Harriott et al., 2010), and denture stomatitis (Johnson et al., 2010).
For VVC, in vivo and ex vivo models were recently used to examine mucosal biofilm formation by scanning electron and confocal microscopy. Wild-type C. albicans strains formed biofilms on the vaginal mucosa in vivo and ex vivo as indicated by high fungal burden and microscopic analysis demonstrating typical biofilm architecture and ECM that co-localized with the presence of fungi. In contrast, mutants in a regulator of hyphal formation (efg1/efg1) and biofilm formation (bcr1/bcr1) exhibited weak to no biofilm formation and ECM production in both models despite comparable colonization levels. This raised an interesting question: does the presence of a biofilm determine whether C. albicans behaves as a pathogen and allows the switch from commensalism and whether a biofilm influences the host response (i.e., alarmin-dependent PMN migration to the vagina)? This was recently addressed in the experimental VVC model. Unexpectedly, the presence or absence of mucosal Candida biofilm had no effect on the alarmin response. The animals, irrespective of the Candida isolate used to inoculate, had similar vaginal fungal burden, vaginal S100 alarmin concentrations, and vaginal PMNs (Lilly et al. Submitted). Hence, it does not appear that mucosal biofilm formation is critical for the immunopathogenesis of VVC/RVVC. This may be explained by the fact that symptomatic episodes of VVC/RVVC are acute and initiated more by the adherence and sensitivity to the epithelial cells than the presence of biofilm. In the general pathogenesis though, biofilm is probably more critical to treatment and clearance via pharmacologic drugs or the immune response. One might envision difficulty of drugs or immune cells/mediators (i.e., cytokines, antibodies) to penetrate the biofilm effectively to function optimally.
In the area of Candida-associated denture stomatitis, a novel contemporary rat model of DS was recently developed using a custom-fitted denture system composed of both fixed and magnetic removable plates (Lee et al., 2010). The denture system was installed against the hard palate of rats without alteration of the dental architecture. The novel design of this denture system (removable
portion) allows for longitudinal studies to evaluate the progression of the disease. Biofilm formation was analyzed on the denture and palate via scanning electron and confocal microscopy (Johnson et al., 2010). Biofilm formation on the denture occurred by week 4 postinoculation, characterized by the presence of hyphae coated with ECM. However, on the palate tissue, only blastospore colonization was observed with no clinical evidence of disease. By week 6 postinoculation, biofilm formation was observed on both the denture and the palate tissue and palatal erythema was evident. This suggests that during DS, C. albicans biofilm formation occurs initially on the denture plate, which in turn seeds the palatal tissue, resulting in mucosal biofilm formation and signs of disease. In a recent study, the biofilm deficient mutants, efg1/efg1 and bcr1/bcr1 were used to evaluate disease states in the rat model of DS. In a complete reversal of what was observed in VVC, clinical disease was only seen in animals inoculated with the wild-type strain. Hence, the presence of biofilm appears critical to pathogenesis of DS (Noverr, manuscript in preparation). This is consistent with the chronicity of the disease and the fact that experimental infections/disease occur after biofilm formation.
A model we envision to explain the pathogenesis of DS is illustrated in Figure A9-3. The denture acquires Candida first by any number of means, and a biofilm is subsequently formed. The established abiotic biofilm then proceeds to seed the mucosal tissue. Once the organism is associated with the tissue an immune response will be induced. However, due the continuous feeding and biofilm formation on the mucosa itself, the immune response can be amplified resulting in a chronic inflammatory response. Current studies are exploring the type of immune response involved and the mechanisms surrounding the response.
Conclusions
Several major points should be emphasized regarding host defense and immunomodulation against mucosal candidiasis: (1) Host defense against Candida is extremely different at different mucosal sites, including adaptive immunity by T cells playing a significant role at the oral mucosa, and innate immunity via PMNs and epithelial cells playing critical roles in the vagina mucosa. (2) The immune factors associated with protection and susceptibility are unique to each site and often involve immunomodulatory factors. Examples include alarmins in the vagina associated with susceptibility to VVC, epithelial cell-annexin-A1 in oral and vaginal tissues associated with protection, and E-cadherin and CD8 T cells in the oral cavity associated with protection. (3) The role of mucosal biofilm in pathogenesis of infection is likewise very different for each mucosa site, evidenced by current data showing no role for biofilm in susceptibility to vaginal candidiasis versus a critical role in susceptibility to denture stomatitis. Future research will undoubtedly further elucidate the mechanisms associated with protection and susceptibility to each infection, which will be critical to exploiting the immunomodulatory factors in therapeutics or targets thereof.
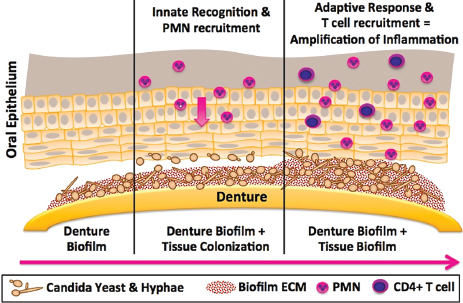
FIGURE A9-3 Proposed model of immunopathogenesis of Candida-associated denture stomatitis. The figure illustrates the central hypothesis that Candida biofilm formation on the denture (left frame) plays a significant role in denture stomatitis through the continuous inoculation of the oral mucosa from the denture (center frame) leading to tissue-associated biofilm formation and chronic erythematous inflammation due to uncontrolled innate and adaptive responses (right frame).
NOTE: PMN, polymorphonuclear neutrophil.
SOURCE: Figure courtesy of M.C. Noverr (03/2012).
References
Babula, O., G. Lazdane, J. Kroica, I. M. Linhares, W. J. Ledger, and S. S. Witkin. 2005. Frequency of interleukin-4 (il-4) -589 gene polymorphism and vaginal concentrations of il-4, nitric oxide, and mannose-binding lectin in women with recurrent vulvovaginal candidiasis. Clinical Infectious Diseases 40(9):1258-1262.
Baillie, G. S., and L. J. Douglas. 2000. Matrix polymers of candida biofilms and their possible role in biofilm resistance to antifungal agents. Journal of Antimicrobial Chemotherapy 46(3):397-403.
Barousse, M. M., C. Steele, K. Dunlap, T. Espinosa, D. Boikov, J. D. Sobel, and P. L. Fidel, Jr. 2001. Journal of Infectious Diseases 184:1489-1493.
Barousse, M. M., T. Espinosa, K. Dunlap, and P. L. Fidel, Jr. 2005. Vaginal epithelial cell anti-candida albicans activity is associated with protection against symptomatic vaginal candidiasis. Infection and Immunity 73:7765-7767.
Bilhan, H., T. Sulun, G. Erkose, H. Kurt, Z. Erturan, O. Kutay, and T. Bilgin. 2009. The role of Candida albicans hyphae and lactobacillus in denture-related stomatitis. Clinical Oral Investigations 13(4):363-368.
Black, C. A., F. M. Eyers, A. Russell, M. L. Dunkley, R. L. Clancy, and K. W. Beagley. 1998. Acute neutropenia decreases inflammation associated with murine vaginal candidiasis but has no effect on the course of infection. Infection and Immunity 66:1273-1275.
Black, C. A., F. M. Eyers, M. L. Dunkley, R. L. Clancy, and K. W. Beagley. 1999. Major histocompatibility haplotype does not impact the course of experimentally induced murine vaginal candidiasis. Laboratory Animal Science 49:668-672.
Blankenship, J. R., and A. P. Mitchell. 2006. How to build a biofilm: A fungal perspective. Current Opinion in Microbiology 9(6):588-594.
Bodasing, N., R. A. Seaton, G. S. Shankland, and A. Pithie. 2002. Gamma-interferon treatment for resistant oropharyngeal candidiasis in an HIV-positive patient. Journal of Antimicrobial Chemotherapy 50:765-766.
Budtz-Jorgensen, E., A. Stenderup, and M. Grabowski. 1975. An epidemiologic study of yeasts in elderly denture wearers. Community Dentistry and Oral Epidemiology 3(3):115-119.
Burns, D. N., R. Tuomala, B. H. Chang, R. Hershow, H. Minkoff, E. Rodriquez, C. Zorrilla, H. Hammill, and J. Regan. 1997. Vaginal colonization or infection with Candida albicans in human immunodeficiency virus-infected women during pregnancy and during postpartum period. Clinical Infectious Diseases 24:201-210.
Calderon, L., R. William, M. Martinez, K. V. Clemons, and D. A. Stevens. 2003. Genetic susceptibility to vaginal candidiasis. Medical Mycology 41:143-147.
Cantorna, M. T., and E. Balish. 1990. Mucosal and systemic candidiasis in congenitally immunodeficient mice. Infection and Immunity 58(4):1093-1100.
Cassone, A., E. Tacconelli, F. De Bernardis, M. Tumbarello, A. Torosantucci, P. Chiani, and R. Cauda. 2002. Antiretroviral therapy with protease inhibitors has an early, immune reconstitution-independent beneficial effect on candida virulence and oral candidiasis in human immunodeficiency virus-infected subjects. Journal of Infectious Diseases 185(2):188-195.
Chandra, J., D. M. Kuhn, P. K. Mukherjee, L. L. Hoyer, T. McCormick, and M. A. Ghannoum. 2001. Biofilm formation by the fungal pathogen Candida albicans: Development, architecture, and drug resistance. Journal of Bacteriology 183:5385-5394.
Clark, R. A., S. A. Blakley, J. Rice, and W. Brandon. 1995. Predictors of HIV progression in women. Journal of Acquired Immune Deficiency Syndrome and Human Retrovirology 9:43-50.
Clemons, K. V., J. L. Spearow, R. Parmar, M. Espiritu, and D. A. Stevens. 2004. Genetic susceptibility of mice to Candida albicans vaginitis correlates with host estrogen sensitivity. Infection and Immunity 72:4878-4880.
Conti, H. R., F. Shen, N. Nayyar, E. Stocum, J. N. Sun, M. J. Lindemann, A. W. Ho, J. H. Hai, J. J. Yu, J. W. Jung, S. G. Filler, P. Masso-Welch, M. Edgerton, and S. L. Gaffen. 2009. Th17 cells and il-17 receptor signaling are essential for mucosal host defense against oral candidiasis. Journal of Experimental Medicine 206(2):299-311.
Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: A common cause of persistent infections. Science 284(5418):1318-1322.
Croxtall, J. D., D. W. Gilroy, E. Solito, Q. Choudhury, B. J. Ward, J. C. Buckingham, and R. J. Flower. 2003. Attenuation of glucocorticoid functions in an anx-a1 -/- cell line. Biochemical Journal 371:927-935.
Crump, J. A., and P. J. Collignon. 2000. Intravascular catheter-associated infections. European Journal of Clinical Microbiology & Infectious Diseases 19(1):1-8.
Daniluk, T., G. Tokajuk, W. Stokowska, K. Fiedoruk, M. Sciepuk, M. L. Zaremba, D. Rozkiewicz, D. Cylwik-Rokicka, B. A. Kedra, I. Anielska, M. Gorska, and B. R. Kedra. 2006. Occurrence rate of oral Candida albicans in denture wearer patients. Advances in Medical Sciences 51(Suppl 1):77-80.
Deslauriers, N., L. Cote, S. Montplaisir, and L. De Repentigny. 1997. Oral carriage of Candida albicans in murine AIDS. Infection and Immunity 65:661-667.
Dominic, R. M., S. Shenoy, and S. Baliga. 2007. Candida biofilms in medical devices: Evolving trends. Kathmandu University Medical Journal (KUMJ) 5(3):431-436.
Dongari-Bagtzoglou, A., and H. Kashleva. 2003a. Candida albicans triggers interleukin-8 by oral epithelial cells. Microbial Pathogenesis 34:169-177.
Dongari-Bagtzoglou, A., and H. Kashleva. 2003b. Granulocyte-macrophage colony-stimulating factor responses of oral epithelial cells to Candida albicans. Oral Microbiology and Immunology 18:165-170.
Dongari-Bagtzoglou, A., H. Kashleva, P. Dwivedi, P. Diaz, and J. Vasilakos. 2009. Characterization of mucosal Candida albicans biofilms. PloS ONE 4(11):e7967.
Douglas, L. J. 2003. Candida biofilms and their role in infection. Trends in Microbiology 11(1):30-36.
Duerr, A., M. F. Sierra, J. Feldman, L. M. Clarke, I. Ehlich, and J. Dehovitz. 1997. Immune compromise and prevalence of candida vulvovaginitis in human immunodeficiency virus-infected women. Obstetrics and Gynecology 90:252-256.
Ehrchen, J. M., C. Sunderkötter, D. Foell, T. Vogl, and J. Roth. 2009. The endogenous toll-like receptor 4 agonist s100a8/s100a9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. Journal of Leukocyte Biology 86(3):557-566.
Eyerich, K., S. Foerster, S. Rombold, H. P. Seidl, H. Behrendt, H. Hofmann, J. Ring, and C. Traidl-Hoffmann. 2008. Patients with chronic mucocutaneous candidiasis exhibit reduced production of th17-associated cytokines il-17 and il-22. Journal of Investigative Dermatology 128(11):2640-2645.
Ferwerda, B., G. Ferwerda, T. S. Plantinga, J. A. Willment, A. B. van Spriel, H. Venselaar, C. C. Elbers, M. D. Johnson, A. Cambi, C. Huysamen, L. Jacobs, T. Jansen, K. Verheijen, L. Masthoff, S. A. Morre, G. Vriend, D. L. Williams, J. R. Perfect, L. A. Joosten, C. Wijmenga, J. W. van der Meer, G. J. Adema, B. J. Kullberg, G. D. Brown, and M. G. Netea. 2009. Human dectin-1 deficiency and mucocutaneous fungal infections. New England Journal of Medicine 361(18):1798-1801.
Fidel, P. L., Jr. 2002. Distinct protective host defenses against oral and vaginal candidiasis. Medical Mycology 40:359-375.
Fidel, P. L. 2006. Candida-host interactions in HIV disease: Relationships in oropharyngeal candidiasis. Advances in Dental Research 19(1):80-84.
Fidel, P. L., Jr. 2007. History and update on host defense against vaginal candidiasis. American Journal of Reproductive Immunology 57:2-12.
Fidel, P. L., Jr., and M. C. Noverr. 2012. Mucosal immunity to Candida albicans In Candida and candidiasis. Vol. 2, edited by R. A. Calderone and C. J. Clancy. Washington, DC: ASM Press. Pp. 137-154.
Fidel, P. L., Jr., and J. D. Sobel. 1996. Immunopathogenesis of recurrent vulvovaginal candidiasis. Clinical Microbiology Reviews 9:335-348.
Fidel, P. L., Jr., and J. D. Sobel. 1999. Murine models of candida vaginal infections. In Experimental models in antimicrobial chemotherapy. Vol. 2, edited by O. Zak and M. Sande. London, UK: Academic Press Ltd. Pp. 741-748.
Fidel, P. L., Jr., J. L. Cutright, L. Tait, and J. D. Sobel. 1996a. A murine model of candida glabrata vaginitis. Journal of Infectious Diseases 173:425-431.
Fidel, P. L., Jr., N. A. Wolf, and M. A. KuKuruga. 1996b. T lymphocytes in the murine vaginal mucosa are phenotypically distinct from those in the periphery. Infection and Immunity 64:3793-3799.
Fidel, P. L., Jr., W. Luo, C. Steele, J. Chabain, M. Baker, and F. L. Wormley. 1999. Analysis of vaginal cell populations during experimental vaginal candidiasis. Infection and Immunity 67:3135-3140.
Fidel, P. L., Jr., J. L. Cutright, and C. Steele. 2000. Effects of reproductive hormones on experimental vaginal candidiasis. Infection and Immunity 68:651-657.
Fidel, P. L., Jr., M. Barousse, T. Espinosa, C. Camaratti, M. Ficarra, D. H. Martin, A. J. Quayle, and K. Dunlap. 2004. An intravaginal live candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infection and Immunity 72:2939-2946.
Foell, D., H. Wittkowski, Z. Ren, J. Turton, G. Pang, J. Daebritz, J. Ehrchen, J. Heidemann, T. Borody, J. Roth, and R. Clancy. 2008. Phagocyte-specific s100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. Journal of Pathology 216:183-192.
Frank, C. F., and M. K. Hostetter. 2007. Cleavage of e-cadherin: A mechanism for disruption of the intestinal epithelial barrier by Candida albicans. Translational Research: The Journal of Laboratory and Clinical Medicine 149(4):211-222.
Gebhardt, C., J. Németh, P. Angel, and J. Hess. 2006. S100a8 and s100a9 in inflammation and cancer. Biochemical Pharmacology 72:1622-1631.
Greenspan, D., E. Komaroff, M. Redford, J. A. Phelan, M. Navazesh, M. E. Alves, H. Kamrath, R. Mulligan, C. E. Barr, and J. S. Greenspan. 2000. Oral mucosal lesions and HIV viral load in the women’s interagency HIV study (WIHS). Journal of Acquired Immune Deficiency Syndrome 25:44-50.
Harriott, M. M., E. A. Lilly, T. E. Rodriguez, P. L. Fidel Jr., and M. C. Noverr. 2010. Candida albicans forms biofilms on the vaginal mucosa. Microbiology. In press.
Hladik, F., G. Lentz, E. Delpit, A. McElroy, and M. J. McElrath. 1999. Coexpression of ccr5 and il-2 in human genital but not blood cells: Implications for the ontogeny of the ccr5+ th1 phenotype. Journal of Immunology 163:2306-2313.
Huang, W., L. Na, P. L. Fidel, and P. Schwarzenberger. 2004. Requirement of interleukin-17a for systemic anti-candida albicans host defense in mice. Journal of Infectious Diseases 190(3):624-631.
Ibraghimov, A. R., R. E. Sacco, M. Sandor, Z. Iakoubov, and R. G. Lynch. 1995. Resident cd4 + αβ t cells of the murine female genital tract: A phenotypically distinct t cell lineage that rapidly proliferates in response to systemic t cell activation stimuli. International Immunology 7:1763-1769.
Imam, N., C. C. J. Carpenter, K. H. Mayer, A. Fisher, M. Stein, and S. B. Danforth. 1990. Hierarchical pattern of mucosal candida infections in HIV-seropositive women. American Journal of Medicine 89:142-146.
Inghirami, G., B. Y. Zhu, L. Chess, and D. M. Knowles. 1990. Flow cytometric and immunohistochemical characterization of the gamma/delta t-lymphocyte population in normal human lymphoid tissue and peripheral blood. American Journal of Pathology 136:357-367.
Itohara, S., A. G. Farr, J. J. Lafaille, M. Bonneville, Y. Takagaki, W. Haas, and S. Tonegawa. 1990. Homing of a gamma/delta thymocyte subset with homogenous t-cell receptors to mucosal epithelia. Nature 343:754-757.
Johansson, E. L., A. Rudin, L. Wassen, and J. Holmgren. 2000. Distribution of lymphocytes and adhesion molecules in human cervix and vagina. Immunology 96:272-277.
Johnson, C. J., H. Lee, A. Yu, P. L. Fidel Jr., and M. C. Noverr. 2010. Characterization of a contemporary animal model of candida associated denture stomatitis. Oral Diseases. In submission.
Kent, H. L. 1991. Epidemiology of vaginitis. American Journal of Obstetrics and Gynecology 165:1168-1175.
Kirkpatrick, C. H. 1984. Host factors in defense against fungal infections. American Journal of Medicine 77:1-12.
Kirkpatrick, C. H., and P. G. Sohnle. 1981. Chronic mucocutaneous candidiasis. In Immunodermatology, edited by B. Safai and R. A. Good. New York: Plenum Press. Pp. 495-514.
Kirkpatrick, C. H., J. W. Chandler, and R. N. Schimke. 1970. Chronic mucocutaneous moniliasis with impaired delayed hypersensitivity. Clinical and Experimental Immunology 6:375-385.
Kirkpatrick, C. H., R. R. Rich, and J. E. Bennett. 1971. Chronic mucocutaneous candidiasis: Model building in cellular immunity. Annals of Internal Medicine 74:955-978.
Klein, R. S., C. A. Harris, C. B. Small, B. Moll, M. Lesser, and G. H. Friedland. 1984. Oral candidiasis in high-risk patients as the initial manifestation of the acquired immunodeficiency syndrome. New England Journal of Medicine 311:354-357.
Kojic, E. M., and R. O. Darouiche. 2004. Candida infections of medical devices. Clinical Microbiology Reviews 17(2):255-267.
Kumar, R. K., Z. Yang, S. Bilson, S. Thliveris, B. E. Cooke, and C. L. Geczy. 2001. Dimeric s100a8 in human neutrophils is diminished after phagocytosis. Journal of Leukocyte Biology 70:59-64.
Kunkl, A., L. Mortara, M. T. Valle, D. Fenoglio, M. Paola, A. M. Megiovanni, A. Alessandrini, G. Li Pira, G. Mazzarello, V. Del Bono, A. Canessa, D. Bassetti, and F. Manca. 1998. Recognition of antigenic clusters of Candida albicans by t lymphocytes from human immunodeficiency virus-infected persons. Journal of Infectious Diseases 178:488-496.
LaFleur, M. D., C. A. Kumamoto, and K. Lewis. 2006. Candida albicans biofilms produce antifungaltolerant persister cells. Antimicrobial Agents and Chemotherapy 50(11):3839-3846.
LeBlanc, D. M., M. M. Barousse, and P. L. Fidel, Jr. 2006. A role for dendritic cells in immunoregulation during experimental vaginal candidiasis. Infection and Immunity 74:3213-3221.
Lee, H., A. Yu, C. J. Johnson, M. C. Noverr, and P. L. Fidel Jr. 2010. Fabrication of a multi-applicable removable intraoral denture system for rodent research. JDR Discovery. In submission.
Leigh, J. E., C. Steele, F. L. Wormley, Jr., W. Luo, R. A. Clark, W. R. Gallaher, and P. L. Fidel, Jr. 1998. Th1/th2 cytokine expression in saliva of HIV-positive and HIV-negative individuals: A pilot study in HIV-positive individuals with oropharyngeal candidiasis. Journal of Acquired Immune Deficiency Syndrome and Human Retrovirology 19(3):373-380.
Leigh, J. E., M. Barousse, R. K. Swoboda, T. Myers, S. Hager, N. A. Wolf, J. L. Cutright, J. Thompson, J. D. Sobel, and P. L. Fidel, Jr. 2001. Candida-specific systemic cell-mediated immune reactivities in HIV-infected persons with and without mucosal candidiaisis. Journal of Infectious Diseases 183:277-285.
Leigh, J. E., K. M. McNulty, and P. L. Fidel, Jr. 2006. Characterization of the immune status of cd8 + t cells in oral lesions of human immunodeficiency virus-infected persons with oropharyngeal candidiasis. Clinical and Vaccine Immunology 13:678-683.
Liang, S. C., X. Y. Tan, D. P. Luxenberg, R. Karim, K. Dunussi-Joannopoulos, M. Collins, and L. A. Fouser. 2006. Interleukin (il)-22 and il-17 are coexpressed by th17 and cooperatively enhance expression of antimicrobial peptides. Journal of Experimental Medicine 203(10):2271-2279.
Lilly, E., D. J. Hart, J. E. Leigh, S. Hager, K. M. McNulty, D. E. Mercante, and P. L. Fidel, Jr. 2004. Tissue-associated cytokine expression in HIV-positive persons with oropharyngeal candidiasis. Journal of Infectious Diseases 190:605-612.
Lilly, E. A., J. E. Leigh, K. M. McNulty, S. H. Joseph, D. E. Mercante, and P. L. Fidel, Jr. 2006. Chemokine receptor expression in HIV-positive persons with oropharyngeal candidiasis. Oral Diseases 12(5):493-499.
Lilly, E. A., J. Yano, and P. L. Fidel, Jr. 2010. Annexin-a1 identified as the oral epithelial cell anticandida effector moiety. Molecular Oral Microbiology 25:293-304.
Liu, N., Y. P. Zhang, S. Han, J. Pei, L. Y. Xu, P. Lu, C. B. Shields, and X. Xu. 2007. Annexin a1 reduces inflammatory reaction and tissue damage through inhibition of phospholipase a 2 activation in adult rats following spinal cord injury. Journal of Neuropathology & Experimental Neurology 66:932-943.
Macher, A. M. 1988. The pathology of AIDS. Public Health Report 103:246-254.
Mathur, S., G. Virella, J. Koistinen, E. O. Horger, T. A. Mahvi, and H. H. Fudenberg. 1977. Humoral immunity in vaginal candidiasis. Infection and Immunity 15:287-294.
McNulty, K. M., J. Plianrungsi, J. E. Leigh, D. E. Mercante, and P. L. Fidel. 2005. Characterization of the cd8+ t-cells and microenvironment in oral lesions of HIV-infected persons with oropharyngeal candidiasis. Infection and Immunity 73:3659-3667.
Millon, L., C. Drobacheff, R. Piarroux, M. Monod, G. Reboux, R. Laurent, and D. Meillet. 2001. Longitudinal study of anti-Candida albicans mucosal immunity against aspartic proteinases in HIV-infected patients. Journal of Acquired Immune Deficiency Syndrome 26(2):137-144.
Myers, T. A., J. E. Leigh, A. Arribas, S. Hager, R. A. Clark, E. Lilly, and P. L. Fidel, Jr. 2003. Immunohistochemical evaluation of t cells in oral lesions from human immunodeficiency virus-positive persons with oropharyngeal candidiasis. Infection and Immunity 71(2):956-963.
Nandi, D., and J. P. Allison. 1991. Phenotypic analysis and gamma/delta-t cell receptor repertoire of murine t cells associated with the vaginal epithelium. Journal of Immunology 147:1773-1778.
Nett, J., L. Lincoln, K. Marchillo, R. Massey, K. Holoyda, B. Hoff, M. VanHandel, and D. Andes. 2007. Putative role of beta-1,3 glucans in Candida albicans biofilm resistance. Antimicrobial Agents and Chemotherapy 51(2):510-520.
Nielsen, H., K. D. Bentsen, L. Hojtved, E. H. Willemoes, F. Scheutz, M. Schiodt, K. Stoltze, and J. J. Pindborg. 1994. Oral candidiasis and immune status of HIV-infected patients. Journal of Oral Pathology and Medicine 23:140-143.
Nobile, C. J., D. R. Andes, J. E. Nett, F. J. Smith, F. Yue, Q. T. Phan, J. E. Edwards, S. G. Filler, and A. P. Mitchell. 2006. Critical role of BCR1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathogens 2:e63.
Nomanbhoy, F., C. Steele, J. Yano, and P. L. Fidel, Jr. 2002. Vaginal and oral epithelial cell anticandida activity. Infection and Immunity 70:7081-7088.
Odds, F. C. 1988. Chronic mucocutaneous candidiosis. In Candida and candidosis. Baltimore, MD: University Park Press. Pp. 104-110.
Phan, Q. T., C. L. Myers, Y. Fu, D. C. Sheppard, M. R. Yeaman, W. H. Welch, A. S. Ibrahim, J. E. Edwards Jr., and S. G. Filler. 2007. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biology 5(3):e64.
Phelan, J. A., B. R. Salzman, G. H. Friendland, and R. S. Klein. 1987. Oral findings in patients with acquired immune deficiency syndrome. Oral Surgery 64:50-56.
Quimby, K., E. A. Lilly, M. Zacharek, K. McNulty, J. E. Leigh, J. E. Vazquez, and P. L. Fidel, Jr. 2011. Cd8 t-cells and e-cadherin in host responses against oropharyngeal candidiasis. Oral Diseases Epub ahead of print.
Rabeneck, L., M. M. Crane, J. M. Risser, C. E. Lacke, and N. P. Wray. 1993. A simple clinical staging system that predicts progression to AIDS using cd4 count, oral thrush, and night sweats. Journal of General Internal Medicine 8:5-9.
Ramage, G., K. VandeWalle, J. L. Lopez-Ribot, and B. L. Wickes. 2002. The filamentation pathway controlled by the efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiology Letters 214:95-100.
Ramage, G., J. P. Martinez, and J. L. Lopez-Ribot. 2006. Candida biofilms on implanted biomaterials: A clinically significant problem. FEMS Yeast Research 6(7):979-986.
Rhoads, J. L., D. C. Wright, R. R. Redfield, and D. S. Burke. 1987. Chronic vaginal candidiasis in women with human immunodeficiency virus infection. JAMA 257:3105-3107.
Romagnoli, P., N. Pimpinelli, M. Mori, P. A. Reichart, L. R. Eversole, and G. Ficarra. 1997. Immunocompetent cells in oral candidiasis of HIV-infected patients: An immunohistochemical and electron microscopical study. Oral Diseases 3:99-105.
Ross, K. F., and M. C. Herzberg. 2001. Calprotectin expression by gingival epithelial cells. Infection and Immunity 69(5):3248-3254.
Rouabhia, M., G. Ross, N. Page, and J. Chakir. 2002. Interleukin-18 and gamma interferon production by oral epithelial cells in response to exposure to Candida albicans or lipopolysaccharide stimulation. Infection and Immunity 70:7073-7080.
Rouabhia, M., A. Semlali, J. Audoy, and W. Chmielewski. 2012. Antagonistic effect of Candida albicans and IFN-γ on e-cadherin expression and production by human primary gingival epithelial cells. Cellular Immunology 280(1):61-67.
Ryckman, C., K. Vandal, P. Rouleau, M. Talbot, and P. A. Tessier. 2003. Proinflammatory activities of s100: Proteins s100a8, s100a9, and s100a8/a9 induce neutrophil chemotaxis and adhesion. Journal of Immunlogy 170:3233-3242.
Schaller, M., R. Mailhammer, G. Grassl, C. A. Sander, B. Hube, and H. C. Korting. 2002. Infection of human oral epithelia with candida species induces cytokine expression correlated to the degree of virulence. Journal of Investigative Dermatology 118:652-657.
Schuman, P., S. Ohmit, J. D. Sobel, K. H. Mayer, V. Greene, A. Rompalo, and R. S. Klein. 1998a. Oral lesions among women living with or at risk for HIV infection. American Journal of Medicine 104:559-563.
Schuman, P., J. D. Sobel, S. Ohmit, K. H. Mayer, A. Rompalo, A. Duerr, D. K. Smith, D. Warren, and R. S. Klein. 1998b. Mucosal candidal colonization and candidiasis in women with or at risk for human immunodeficiency virus infection. Clinical Infectious Diseases 27:1161-1167.
Sobel, J. D. 1988. Pathogenesis and epidemiology of vulvovaginal candidiasis. Annals of the New York Academy of Sciences 544:547-557.
Sobel, J. D. 1992. Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clinical Infectious Diseases 14(Suppl 1):S148-S153.
Sobel, J. D., G. Muller, and H. R. Buckley. 1984. Critical role of germ tube formation in the pathogenesis of candidal vaginitis. Infection and Immunity 44:576-580.
Sohnle, P. G., B. L. Hahn, and V. Santhanagopalan. 1996. Inhibition of Candida albicans growth by calprotectin in the absence of direct contact with the organisms. Journal of Infectious Diseases 174:1369-1372.
Spinillo, A., G. Michelone, C. Cavanna, L. Colonna, E. Capuzzo, and S. Nicola. 1994. Clinical and microbiological characteristics of symptomatic vulvovaginal candidiasis in HIV-seropositive women. Genitrouinary Medicine 70:268-272.
Steele, C., and P. L. Fidel, Jr. 2002. Cytokine and chemokine production by human oral and vaginal epithelial cells in response to Candida albicans. Infection and Immunity 70:577-583.
Steele, C., H. Ozenci, W. Luo, M. Scott, and P. L. Fidel, Jr. 1999. Growth inhibition of Candida albicans by vaginal cells from naive mice. Medical Mycology 37:251-260.
Steele, C., J. E. Leigh, R. K. Swoboda, and P. L. Fidel, Jr. 2000. Growth inhibition of candida by human oral epithelial cells. Journal of Infectious Diseases 182:1479-1485.
Steele, C., J. E. Leigh, R. K. Swoboda, H. Ozenci, and P. L. Fidel, Jr. 2001. Potential role for a carbohydrate moiety in anti-candida activity of human oral epithelial cells. Infection and Immunity 69:7091-7099.
Taylor, B. N., M. Saavedra, and P. L. Fidel, Jr. 2000. Local th1/th2 cytokine production during experimental vaginal candidiasis. Medical Mycology 38:419-431.
Urban, C. F., D. S. M. Ermert, U. Abu-Abed, C. Goosman, W. Nacken, V. Brinkmann, P. R. Jungblut, and A. Zychlinsky. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathogens 5(10):e1000639.
Villar, C. C., H. Kashleva, C. J. Nobile, A. P. Mitchell, and A. Dongari-Bagtzoglou. 2007. Mucosal tissue invasion by Candida albicans is associated with e-cadherin degradation, mediated by transcription factor rim101p and protease sap5p. Infection and Immunity 75(5):2126-2135.
Webb, B. C., C. J. Thomas, M. D. Willcox, D. W. Harty, and K. W. Knox. 1998. Candida-associated denture stomatitis. Aetiology and management: A review. Part 2. Oral diseases caused by candida species. Australian Dental Journal 43(3):160-166.
White, M. H. 1996. Is vulvovaginal candidiasis an AIDS-related illness? Clinical Infectious Diseases 22(Suppl 2):S124-S127.
Wisplinghoff, H., T. Bischoff, S. M. Tallent, H. Seifert, R. P. Wenzel, and M. B. Edmond. 2004. Nosocomial bloodstream infections in US hospitals: Analysis of 24,179 cases from a prospective nationwide surveillance study. Clinical Infectious Diseases 39(3):309-317.
Wormley, F. L., Jr., J. Chaiban, and P. L. Fidel, Jr. 2001. Cell adhesion molecule and lymphocyte activation marker expression during experimental vaginal candidiasis. Infection and Immunity 69:5072-5079.
Wormley, F. L., Jr., C. Steele, K. Wozniak, K. Fujihashi, J. R. McGhee, and P. L. Fidel, Jr. 2001. Resistance of tcr δ-chain knock-out mice to experimental candida vaginitis. Infection and Immunity 69:7162-7164.
Wozniak, K. L., J. E. Leigh, S. Hager, R. K. Swoboda, and P. L. Fidel, Jr. 2002. A comprehensive study of candida-specific antibodies in saliva of HIV-infected persons with oropharyngeal candidiasis. Journal of Infectious Diseases 185:1269-1276.
Wray, D., D. H. Felix, and C. G. Cumming. 1990. Alteration of humoral responses to candida in HIV infection. British Dental Journal 168(8):326-329.
Yano, J., E. Lilly, C. Steele, D. Fortenberry, and P. L. Fidel, Jr. 2005. Oral and vaginal epithelial cell anti-candida activity is acid-labile and does not require live epithelial cells. Oral Microbiology and Immunology 20:199-205.
























