
TOPOGRAPHIC DIVERSITY OF FUNGAL AND BACTERIAL COMMUNITIES IN HUMAN SKIN67
Keisha Findley,68Julia Oh,68Joy Yang,68Sean Conlan,68Clayton Deming,68Jennifer A. Meyer,68Deborah Schoenfeld,69Effie Nomicos,69Morgan Park,70NIH Intramural Sequencing Center Comparative Sequencing Program,71Heidi H. Kong,69,*and Julia A. Segre68,*
Traditional culture-based methods have incompletely defined the microbial landscape of common recalcitrant human fungal skin diseases, including athlete’s foot and toenail infections. Skin protects humans from invasion by pathogenic microorganisms and provides a home for diverse commensal microbiota (Marples, 2012). Bacterial genomic sequence data have generated novel hypotheses about species and community structures underlying human disorders (Grice and Segre, 2012; Human Microbiome Project Consortium, 2012; Pflughoeft and Versalovic, 2012). However, microbial diversity is not limited to bacteria; microorganisms such as fungi also have major roles in microbial community stability, human health and disease (Peleg et al., 2010). Genomic methodologies to identify fungal species and communities have been limited compared with those that are available for bacteria (Ollive et al., 2012). Fungal evolution can be reconstructed with phylogenetic markers, including ribosomal RNA gene regions and other highly conserved genes (James et al., 2006). Here we sequenced and analysed fungal communities of 14 skin sites in 10 healthy adults. Eleven core-body and arm sites were dominated by fungi of the genus Malassezia, with only species-level classifications revealing fungal-community composition differences between sites. By contrast, three foot sites—plantar heel, toenail and toe web—showed high fungal diversity. Concurrent analysis of bacterial and fungal communities
________________
67 Reprinted by permission from Macmillan Publishers Ltd. Nature. Findley, K. F. et al. 2013. Topographic diversity of fungal and bacterial communities in human skin. Nature 498(7454):367-370. Copyright 2013.
68 Genetics and Molecular Biology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
69 Dermatology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
70 Dermatology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
71 NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, Rockville, Maryland 20852, USA.
* These authors contributed equally to this work.
demonstrated that physiologic attributes and topography of skin differentially shape these two microbial communities. These results provide a framework for future investigation of the contribution of interactions between pathogenic and commensal fungal and bacterial communities to the maintenance of human health and to disease pathogenesis.
Since Hippocrates first described oral candidiasis in 400 BC, scientists have sought to explore the roles that commensal and pathogenic fungi and microbial communities have in human health and disease (Ghannoum et al., 2010; Paulino et al., 2006). Culture-based studies have reported Malassezia, Rhodotorula, Debaromyces, Cryptococcus, and, in some sites, Candida as fungal skin commensals (Roth and James, 1988). Cutaneous fungal infections affect 29 million North Americans, but the role of dermatophytes in common toenail infections can be difficult to characterize using culture-based studies (Bickers et al., 2006). For other common skin disorders, such as seborrheic dermatitis (dandruff), fungal involvement remains incompletely understood (Gaitanis et al., 2012; Saunders et al., 2012). Difficulty in establishing growth conditions (Larone, 2002; St-Germain and Summerbell, 2011) contribute to challenges to rapidly identify and direct treatment against pathogenic fungi.
To compare culture- and DNA-sequence-based identification of human skin-associated fungi, we obtained parallel samples from four skin sites of adult healthy volunteers (Supplementary Fig. 1 and Supplementary Table 1). We characterized isolates by morphological features and molecular markers. In total, we cultured more than 130 fungal isolates: 62 Malassezia (species globosa, restricta, and sympodialis [Gioti et al., 2013; Saunders et al., 2012]), 25 Penicillium (species chrysogenum and lanosum), and 19 Aspergillus (species candidus, terreus, and versicolor) (Supplementary Table 2). Five or fewer Alternaria, Candida, Chaetomium, Chrysosporium, Cladosporium, Mucor, Rhodotorula, and Trichophyton isolates were cultured.
To explore fungal diversity with culture-independent methods, we prepared DNA from clinical swabs, and polymerase chain reaction (PCR)-amplified and sequenced two phylogenetic markers within the ribosomal RNA region: 18S rRNA and the intervening internal transcribed spacer 1 (ITS1) region (Bruns et al., 1992; James et al., 2006; Schoch et al., 2012). We generated a custom ITS1 database based on sequences deposited in GenBank to classify sequences to genus level with greater than 97% accuracy (Supplementary Table 3). 18S rRNA sequences were classified using the SILVA database (Quast et al., 2013). We determined the relative abundance of fungal genera of the occiput (back of head), nare (nostril), plantar heel and retroauricular crease (behind the ear). The genus Malassezia predominated in the retroauricular crease, nare and occiput; this was consistent across 18S rRNA and ITS1-characterized samples. Plantar heel was the most diverse site with representation of Malassezia, Aspergillus, Cryptococcus, Rhodotorula, Epicoccum, and others (Supplementary Fig. 2). ITS1 sequencing
enabled greater genus-level taxonomic resolution, reflecting the specificity of the genomic region and richness of the molecular database. Based on technical and analytic advantages, we selected the ITS1 region for subsequent sequencing and analyses of fungal diversity.
We generated more than 5 million ITS1 sequences from 10 healthy volunteers (from each of whom 14 skin-site samples were taken) (Supplementary Table 4). Both Ascomycetous and Basidiomycetous fungi were identified as normal skin flora. The genus Malassezia predominated at all 11 core-body and arm sites: antecubital fossa, back, external auditory canal, glabella, hypothenar palm, inguinal crease, manubrium, nare, occiput, retroauricular crease and volar forearm (Figure A16-1). We explored Malassezia species-level resolution with a taxonomic data set that we developed based on reference ITS1 sequences and our human-associated Malassezia isolates. Pairwise comparisons of these Malassezia ITS1 sequences showed sequence identity within species to be greater than 91%, and identity between species to be 70 to 88%. These Malassezia sequences served as references within the phylogenetic placer (Matsen et al., 2010) program to classify approximately 80 to 90% of Malassezia sequences per skin site to species level. Species-level identification revealed fungal specificity between body sites (Figure A16-1). M. restricta predominated in external auditory canal, retroauricular crease and glabella, and M. globosa predominated on back, occiput and inguinal crease. Sites such as nares, antecubital fossa, volar forearm and hypothenar palm were characterized by multiple species (M. restricta, M. globosa, and M. sympodialis). In total, we identified 11 of the 14 known Malassezia species among skin sites, suggesting that human skin is colonized with a wide range of Malassezia. Based on species-level resolution, we observed that fungal diversity is more dependent on body site than individual subject. ITS1 sequences also matched Candida species tropicalis, parapsilosis, and orthopsilosis, and Cryptococcus species flavus, dimennae, and diffluens, which are considered to be part of the normal human flora and to be possible pathogens in wounds or immunocompromised patients (Larone et al., 2002).
Substantially greater diversity was observed on three foot sites (plantar heel, toenail and toe web), in both the number of genera observed and the variation between individuals (Supplementary Fig. 3 and Supplementary Table 5). The fungal profile of one of the subjects (who we will refer to as healthy volunteer 7) was notably more diverse than other participants (Figure A16-1). Healthy volunteer 7 completed a 2-month course of oral antifungal medication for a toenail infection 7 months before sampling. The remaining healthy volunteers reported no use of either oral or topical antifungal medication for at least 2 years before sampling. Healthy volunteer 7 is an outlier, but the additional genera that were identified (for example, Aspergillus and Saccharomyces) show that skin is capable of harbouring high fungal diversity. Although microbial sequencing is unable to determine causation, these data may suggest either that fungal community imbalance is associated with recurrent toenail infections or that alterations in fungal skin
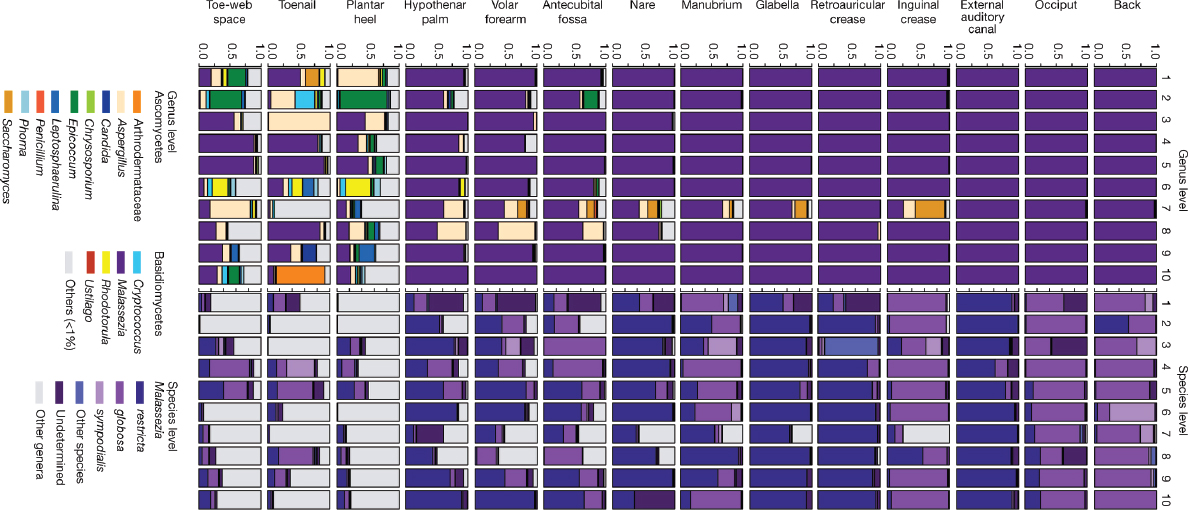
FIGURE A16-1 Relative abundance of fungal genera and Malassezia species at different human skin sites. Fungal diversity of individual body sites of healthy volunteers(1–10) was taxonomically classified at the genus level, with further resolution of Malassezia species. For all body sites, the left side of the body was used, except for the right toenail of healthy volunteer 7.
communities persist even 7 months after discontinuing antifungal medications. In comparison with culture-based analysis, ITS1 sequencing can provide a more complete view of the diversity of commensal microbiota, and also of potentially pathogenic microbiota.
To quantify and compare community similarity and taxonomic richness of skin sites, we assigned fungal sequences to taxonomic units based on genus-level phylogeny rather than percent sequence identity to obviate the high variation noted between species (Schloss et al., 2009) with the latter metric. Plantar heel was the most complex fungal site (median richness of approximately 80 genera), and other foot sites had the next highest diversity (toe web and toenail, with approximately 60 and 40 genera, respectively; Figure A16-2, Supplementary Table 6). Arm sites showed intermediate richness, ranging from 18 to 32 genera and core-body sites exhibited much lower richness, ranging from 2 to approximately 10 genera. Thus, regional location is a strong determinant of fungal richness. As observed in skin bacterial studies, left–right similarity within an individual was greater than between different individuals at the same body site (Supplementary Fig. 4 and Supplementary Table 7). To determine the temporal stability of the fungal microbiome, 6 healthy volunteers returned 1 to 3 months after initial sampling. Sites that showed Malassezia predominance at initial sampling displayed the same genus- and species-level predominance with strong community structure stability (Supplementary Figs. 5 and 6, and Supplementary Table 8). Foot sites continued to show high diversity, perhaps reflecting frequent environmental exposure.
To explore the relationship between skin-associated fungi and bacteria, we sequenced the 16S rRNA gene from the same clinical samples. Consistent with previous studies (Costello et al., 2009; Grice et al., 2009), bacteria on healthy human skin were predominantly Propionibacterium, Corynebacterium, and Staphylococcus (Supplementary Fig. 7). Similar to other moist skin sites, the toenail bacteria (not surveyed previously) were predominantly Corynebacterium and Staphylococcus. Interestingly, although healthy volunteer 7 was an outlier in terms of fungal diversity and membership, the bacterial profile was normal with respect to taxonomic and ecological measures of diversity (Supplementary Fig. 7). Directed studies may help elucidate how antibacterial and antifungal therapies perturb fungal and bacterial communities.
Bacterial and fungal richness were not linearly correlated, but were instead grouped into discrete clusters of sites from the same region; arm, foot and core-body (sites from the same regions had similar bacterial and fungal richness) (Figure A16-2 and Supplementary Fig. 8). Arm sites displayed markedly greater bacterial diversity and lower fungal diversity than the foot and core-body sites. In contrast, foot sites displayed markedly greater fungal diversity with lower bacterial diversity than the arm and core-body sites. Core-body sites clustered together, and showed both lower bacterial and lower fungal diversity. These data reveal that
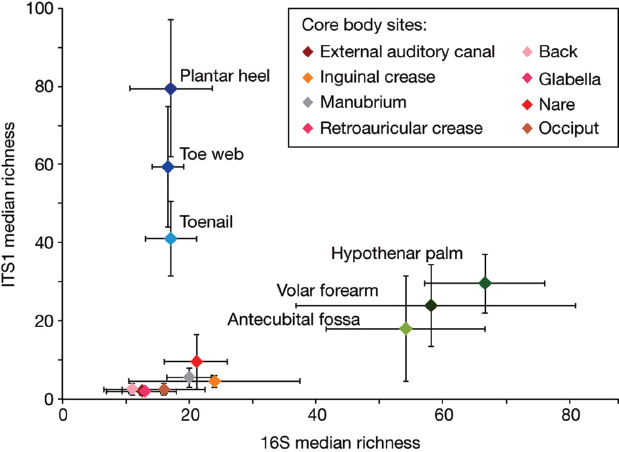
FIGURE A16-2 Median richness of fungal and bacterial genera. Median taxonomic richness (or number of observed genera) of fungal and bacterial genera at 14 body sites. Error bars represent the median absolute deviation. The values for the core body sites retroauricular crease and glabella are identical and are therefore represented by a single data point on the graph and a shared colour in the key.
the skin microbiome is complex and suggest that different characteristics shape skin bacterial and fungal communities.
Using principal coordinates analysis of community structure, we explored properties that may shape bacterial and fungal communities differentially. Consistent with previous studies (Peleg et al., 2010), bacterial communities varied in the proportion of lipophilic bacteria (Corynebacterium, Propionibacterium, and Turicella) and staphylococcal species, and grouped based on skin physiology into sebaceous, moist and dry sites (Figure A16-3 and Supplementary Fig. 9). In contrast, fungal communities were segregated more clearly by site location than physiology, with foot, arm, head and torso sites forming discrete groups. Different Malassezia species drive variation in arms, torso and head, whereas a wide range of fungal genera drive variation in feet (Figure A16-3 and Supplementary Fig. 9). Co-occurrence analysis of foot sites, based on Spearman correlation of fungal and bacterial taxonomic relative abundances (Supplementary Fig. 10), provided a preliminary evaluation of major fungal–bacterial associations. For
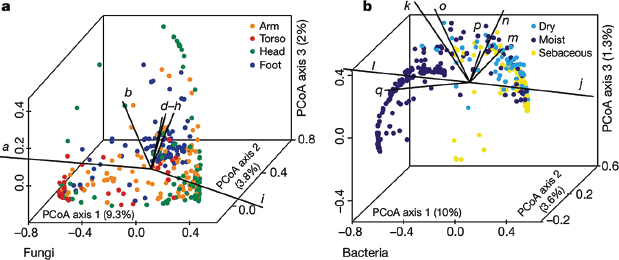
FIGURE A16-3 Forces that shape fungal and bacterial communities. a, b, Principal coordinates analysis (PCoA) of the degree of fungal- and bacterial-community similarity at 14 body sites, based on predominant genera and species. Variation in fungal communities segregated strongly according to site location, with arm, foot, head and core-body sites forming discrete groups (a). Bacterial community structure was more dependent on site physiology (b). Axes that most significantly contribute to variation and their relative lengths are shown (these are defined in Supplementary Fig. 10 legend). For fungi, M. restricta (i; ρ = 0.92) and M. globosa (a; ρ = –0.79) are primary and opposing drivers of variation based on Spearman correlation with PCoA axis 1. For bacteria, Propionibacterium (j; ρ = 0.95) contributes to sebaceous site variation, whereas Corynebacterium (l; ρ = –0.74) and Turicella (q; ρ = –0.56) are the greatest contributors at moist sites based on Spearman correlation with PCoA axis 1.
example, a group of primarily Actinobacteria was anti-correlated with resident Ascomycota and Basidiomycota in contrast to a group of primarily Firmicutes and Proteobacteria that was positively correlated with these fungal taxa.
We observed that 20% (12 out of 60) of our study participants had clinical involvement (plantar-heel scaling, toe web scaling, or toenail changes), consistent with possible fungal infections (Supplementary Table 1). Of the subjects with observed clinical involvement, positive mycological cultures were obtained from toenails (two samples) and plantar heel (one sample) (Trichophyton, Penicillium, and Aspergillus). These observations are similar to the results of larger studies, which report signs of clinical involvement in up to 60% of feet of healthy individuals, and find 2 to 25% of cultures to be positive for fungi. The wide variation in prevalence of clinical involvement and positive fungal cultures is dependent on several factors, including population and climate (Cohen et al., 2005; Perea et al., 2000). As an initial inquiry into the aetiology of foot fungal disorders, we examined how observed clinical status (involved or uninvolved) at plantar heel, toe web or toenail affected fungal community structure. For uninvolved sites, interpersonal variation in community structure was highly consistent across all
foot sites. In contrast, for sites with observed clinical involvement, similarity of community structure was much higher for plantar heel but much lower for toenail (Figure A16-4). These data may suggest that there is a common fungal community shared among individuals with plantar-heel involvement, and high fungal diversity underlying toenail infections, but further studies are needed. These data sets can now be used to inform future clinical studies that examine microbial community shifts associated with fungal infections.
This systematic study clearly demonstrates that human skin surfaces are complex ecosystems, providing diverse environments for microorganisms that inhabit our bodies. Different factors determine bacterial and fungal communities, depending on the physiological properties of the skin. Malassezia species predominate on all core-body and arm sites. In contrast, foot sites display tremendous fungal diversity and markedly lower stability over time. Microbial community instability may provide an opportunity for potentially pathogenic microbes to establish disease. Plantar heels, toe webs and toenails are common sites of recurrent human fungal disease, which can be recalcitrant to treatment.
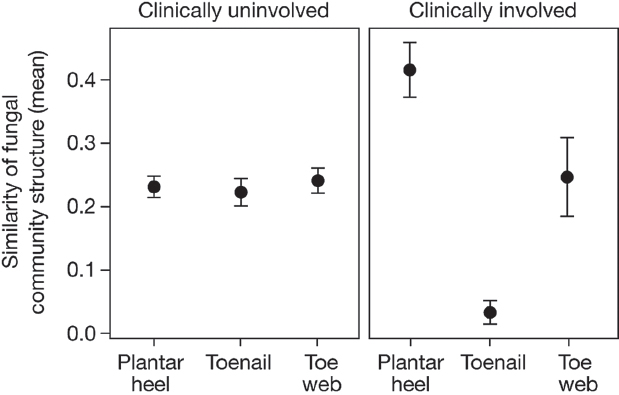
FIGURE A16-4 Clinical involvement alters shared fungal community structure. Community structure measures the type and relative abundance of each genus. A value of 1 implies identical community structure and 0 implies dissimilar structures. Among uninvolved foot sites, community structure is fairly consistent at plantar heel, toenail and toe web sites. For involved sites, plantar heel has much greater shared community structure and toenails have much lower shared community structure. Error bars represent the s.e.m.
This study also investigated fungal diversity at sites of predilection for other skin disorders, including seborrheic dermatitis, tinea cruris, and subtypes of atopic dermatitis. With genomic advances, such as shotgun metagenomic sequencing, it is possible to begin to address interactions between microbes (bacterial–fungal, bacterial–bacterial, fungal–fungal) residing in these complex environments. The role of fungal commensals in educating the human immune system is gaining new appreciation in intestinal disease (Iliev et al., 2012). Further studies of healthy skin and dermatologic disorders are needed to explore these host–microbe interactions. In addition, antifungal medications, including azoles, echinocandins and amphotericin B, have potentially serious side effects such as liver or kidney damage (Perfect et al., 1992). Therefore, new treatment approaches are required to strategically target microbial dysbiosis and to combat the increasingly observed resistance against our current arsenal of antimicrobial therapies.
Methods Summary
Subject Recruitment and Sampling
This study was approved by the Institutional Review Board of the National Human Genome Research Institute (http://www.clinicaltrials.gov/ct2/show/NCT00605878) and all subjects provided written informed consent before participation. For fungal culturing studies, skin was scraped with a surgical blade and placed directly in media. For sequencing studies, swabs from skin and environmental negative controls were placed in MasterPure Yeast DNA purification kit (Epicentre) lysis buffer augmented with lysozyme. Proteinase K (Invitrogen) was added to pre-digest toenail clippings and incubated overnight with shaking at 55 uC. Steel beads (5 mm in diameter) were added to mechanically disrupt fungal cell walls using Tissuelyser (Qiagen) for 2 min at 30 Hz and then using the PureLink Genomic DNA Kit (Invitrogen). For 18S rRNA sequencing, each DNA was amplified with SR6 (59-TACCTGG TTGATTCTGC) and SR1R (59-TGTTACGACTTTTACTT) primers. For ITS1 sequencing, each DNA was amplified with adaptor plus 18SF (59-GTAA AAGTCGTAACAAGGTTTC) and 5.8S1R plus barcode primers (59-GTTCA AAGAYTCGATGATTCAC). For 16S rRNA sequencing, each DNA was amplified with adaptor plus V1_27F (59-AGAGTTTGATCCTGGCTCAG) and V3_534R plus barcode primers (59-CAGCACGCATTACCGCGGCTGCTGG).
Sequence Classification and Analyses
Sequences were pre-processed to remove primers and barcodes. Possible chimaeras were identified with UCHIME in mother (Edgar et al., 2011; Schloss et al., 2009). ITS1 sequences were classified to genus level with BLAST (basic local-alignment search tool) and the k-nearest neighbour algorithminmothur.
18S rRNA sequences were classified using the SILVA v108 database (Quast et al., 2013). 16S rRNA sequences were classified to genus level using the RDP-classifier and training set 6 (Wang et al., 2007).We curated and aligned a reference library of Malassezia ITS1 type-strain sequences retrieved from GenBank augmented with those from our human-associated fungal cultures. This curated library was used as a reference to phylogenetically place and classify ITS1 sequences to species level within pplacer (Matsen et al., 2010). Sequence placement on the reference tree was visualized in Archaeopteryx using the “guppy” command for classifications with a likelihood score of greater than or equal to 0.65. Full methods are found in the Supplementary Information.
Competing Financial Interests
The authors declare no competing financial interests.
Acknowledgements
We thank J. Heitman, A. Amend, Y. Shea, M. Turner, I. Brownell, and M. Udey for helpful discussions. We thank J. Fekecs for assistance with the figures. This work was supported by the US National Institutes of Health (NIH) NHGRI and NCI Intramural Research Programs, and in part by NIH grant no. 1K99AR059222 (to H.H.K.). Sequencing was funded by grants from the NIH (1UH2AR057504-01 and 4UH3AR057504-02).
Author Contributions
K.F., H.H.K., and J.A.S. designed the outline of the study. D.S. and E.N. recruited human subjects and assisted H.H.K. in sample collection for the experiment. K.F. and J.Y. assembled and curated the fungal database. J.A.M. and C.D. prepared the clinical samples for sequencing. The members of the NIH Intramural Sequencing Center Comparative Sequencing program carried out sequencing. K.F., J.O., S.C. and M.P. analysed sequence data. K.F., H.H.K., and J.A.S. drafted the manuscript with specific contributions from J.O., J.Y., and S.C. All authors read and approved the final version of the manuscript.
Author Information
Sequence data from this study have been submitted to GenBank/EMBL/DDBJ under accession numbers KC669797–KC675175, and the Sequence Read Archive,and can be accessed through BioProject identification no.46333. Patient and sample metadata have been deposited in the controlled-access Database of Genotypes and Phenotypes (dbGaP) under study accession phs000266.v1.p1. Reprints and permissions information is available at www.nature.com/reprints. The
authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to H.H.K. (konghe@mail.nih.gov) or J.A.S. (jsegre@nhgri.nih.gov).
References
Bickers, D. R. et al. The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J. Am. Acad. Dermatol. 55, 490–500 (2006).
Bruns, T. D. et al. Evolutionary relationships within the fungi: analyses of nuclear small subunit rRNA sequences. Mol. Phylogenet. Evol. 1, 231–241 (1992).
Cohen, A. D., Wolak, A., Alkan, M., Shalev, R. & Vardy, D. A. Prevalence and risk factors for tinea pedis in Israeli soldiers. Int. J. Dermatol. 44, 1002–1005 (2005).
Costello, E. K. et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009).
Dollive, S. et al. A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol. 13, R60 (2012).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Gaitanis, G., Magiatis, P., Hantschke, M., Bassukas, I. D. & Velegraki, A. The Malasseziagenus in skin and systemic diseases. Clin. Microbiol. Rev. 25, 106–141 (2012).
Ghannoum, M. A. et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 6, e1000713 (2010).
Gioti, A. et al. Genomic insights into the atopic eczema-associated skin commensal yeastMalassezia sympodialis. MBio 4, e00572–12 (2013).
Grice, E. A. & Segre, J. A. The human microbiome: our second genome. Annu. Rev. Genomics Hum. Genet. 13, 151–170 (2012).
Grice, E. A. et al. Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 (2009).
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Iliev, I. D. et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317 (2012).
James, T. Y. et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443, 818–822 (2006).
Larone, D. H. Medically Important Fungi: A guide to identification (ASM Press, 2002).
Marples, M., ed. The Ecology of the Human Skin (Bannerstone House, 1965).
Matsen, F. A., Kodner, R. B. & Armbrust, E. V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11, 538 (2010).
Paulino, L. C., Tseng, C. H., Strober, B. E. & Blaser, M. J. Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J. Clin. Microbiol. 44,2933–2941 (2006).
Peleg, A. Y., Hogan, D. A. & Mylonakis, E. Medically important bacterial–fungal interactions. Nature Rev. Microbiol. 8, 340–349 (2010).
Perea, S. et al. Prevalence and risk factors of tinea unguium and tinea pedis in the general population in Spain. J. Clin. Microbiol. 38, 3226–3230 (2000).
Perfect, J. R., Lindsay, M. H. & Drew, R. H. Adverse drug reactions to systemic antifungals. Prevention and management. Drug Saf. 7, 323–363 (1992).











